

Summary
Fluorescence loss in photobleaching experiments and analysis of mitochondrial function using superoxide and redox potential biosensors revealed that mitochondria within individual yeast cells are physically and functionally distinct. Mitochondria that are retained in mother cells during yeast cell division have significantly lower redox potential and higher superoxide levels compared to mitochondria in buds. Retention of mitochondria with lower redox potential in mother cells occurs to the same extent in young and older cells, and can account for the age-associated decline in total cellular mitochondrial redox potential in yeast as they age from 0–5 generations. Deletion of Mmr1p, a member of the DSL1 family of tethering proteins that localizes to mitochondria at the bud tip and is required for normal mitochondrial inheritance, produces defects in mitochondrial quality control and heterogeneity in replicative lifespan (RLS). Long-lived mmr1Δ cells exhibit prolonged RLS, reduced mean generation times, increased mitochondrial redox potential and lower mitochondrial superoxide levels compared to wild-type cells. Short-lived mmr1Δ cells exhibit the opposite phenotypes. Moreover, short-lived cells give rise exclusively to short-lived cells, while the majority of daughters of long-lived cells are long lived. These findings support the model that the mitochondrial inheritance machinery promotes retention of lower-functioning mitochondria in mother cells and that this process contributes to both mother-daughter age asymmetry and age-associated declines in cellular fitness.
Keywords: aging, organelle inheritance, roGFP, reactive oxygen species, mitochondrial redox state, senescence factors
Introduction
An intuitive concept in human experience is that babies are born young, largely independent of the age of their parents. The finding that a similar mother-daughter age asymmetry also occurs in the budding yeast Saccharomyces cerevisiae gave rise to the model that age determinants are asymmetrically distributed during yeast cell division, which allows for continued aging of mother cells and rejuvenation of daughter cells (Mortimer & Johnston 1959; Egilmez & Jazwinski 1989; Kennedy et al. 1994; Sinclair & Guarente 1997). In support of this, oxidatively-damaged proteins, mitochondria with low membrane potential (Δψ) and extrachromosomal rDNA circles were identified as senescence factors that are retained in mother cells (Sinclair & Guarente 1997; Lai et al. 2002; Aguilaniu et al. 2003). Conversely, ROS has been linked to mother-daughter age asymmetry, and the activity of cytosolic catalase, an antioxidant, is increased in daughter cells after cytokinesis and separation from their mother cells (Nestelbacher et al. 2000; Aguilaniu et al. 2003; Heeren et al. 2004; Erjavec & Nyström 2007; Erjavec et al. 2008; Eisenberg et al. 2009). Sir2p, the founding member of the Sirtuin family of age-regulating proteins, is required for asymmetric distribution of aging determinants and mother-daughter age asymmetry (Kaeberlein et al. 1999; Aguilaniu et al. 2003; Erjavec et al. 2007).
Segregation of mitochondria on the basis of Δψ and of an oxidatively damaged mitochondrial protein has been linked to mother-daughter age asymmetry (Lai et al. 2002; Klinger et al. 2010). Moreover, there are links between mitochondrial ROS and aging in yeast and other cell types (Miquel & Economos 1979; Sun & Tower 1999; Schriner et al. 2005; Klinger et al. 2010; Lam et al. 2011). Deletion of the mitochondrial MnSOD SOD2 or CCCP treatment increase ROS and decrease yeast chronological lifespan (Longo et al. 1996; Stöckl et al. 2007), while reduction of mitochondrial ROS production by overexpression of SOD2, deletion of the nuclear protein MRG19p or manipulations that increase respiration result in increased chronological lifespan, RLS or both (Jiang et al. 2000; Fabrizio et al. 2003; Harris et al. 2003; Barros et al. 2004; Bonawitz et al. 2007; Lavoie & Whiteway 2008; Mittal et al. 2009).
While chronological lifespan extension by increased respiration is well documented, analysis of the role of respiration for RLS extension by calorie restriction yielded conflicting results (Lin et al. 2002; Lin et al. 2004; Kaeberlein et al. 2005; Lavoie & Whiteway 2008). The role of mitochondrial metabolic activity in RLS in yeast is also a matter of debate. Indeed, deletion of mitochondrial DNA, which encodes respiratory chain components, has variable effects on lifespan in different yeast strains (Kirchman et al. 1999; Heeren et al. 2004; Kaeberlein et al. 2005). Similarly, deletion of mitochondrial metabolic genes that have been implicated in lifespan control in C. elegans has no effect on aging in yeast (Smith et al. 2008).
Here, we studied the role of mitochondrial inheritance in lifespan control and mother-daughter age asymmetry in budding yeast. We find that mitochondria within individual yeast cells are variable in superoxide levels and redox potential. In addition, we obtained evidence that mitochondria with higher superoxide levels and lower redox potential are preferentially retained in mother cells and that this process may contribute to the age-associated decline in mother cell fitness. Finally, we find that a mutation that affects mitochondrial quality control during inheritance compromises lifespan control and mother-daughter age asymmetry.
Results
Mitochondria in individual yeast cells are physically and functionally distinct
To determine whether mitochondria in budding yeast are heterogeneous in function, we assessed mitochondrial redox potential using a redox-sensing GFP-variant (roGFP1) (Hanson et al. 2004) and mitochondrial superoxide using dihydroethidium (DHE) (e.g. (Lam et al. 2011)). In roGFP1, a native cysteine is mutated and novel cysteines are introduced near the chromophore (C48S, S147C, Q204C). Disulfide formation between these cysteines in oxidizing environments promotes protonation of the GFP chromophore, which increases excitation at 400 nm and decreases excitation at 490 nm. The ratio of fluorescence upon excitation at 400 and 490 nm indicates the extent of roGFP1 oxidation and is independent of roGFP1 protein levels. Targeting of roGFP1 to mitochondria in HeLa cells revealed that the mitochondrial matrix in these cells is highly reducing, with a midpoint potential of −360 mV (Dooley et al. 2004).
We generated a plasmid-borne fusion protein, mito-roGFP1, that consists of roGFP1 fused to the signal sequence of a mitochondrial matrix protein (ATP9) and is expressed under control of a strong constitutive promoter. Mito-roGFP1 is targeted quantitatively to mitochondria and has no obvious effect on mitochondrial morphology or distribution (Fig. 1A). Equally important, mito-roGFP1 undergoes rapid, reversible ratiometric changes in fluorescence in response to oxidizing and reducing agents (Fig. S1).
Fig. 1. Individual yeast cells display heterogeneity in mitochondrial redox state and ROS levels.
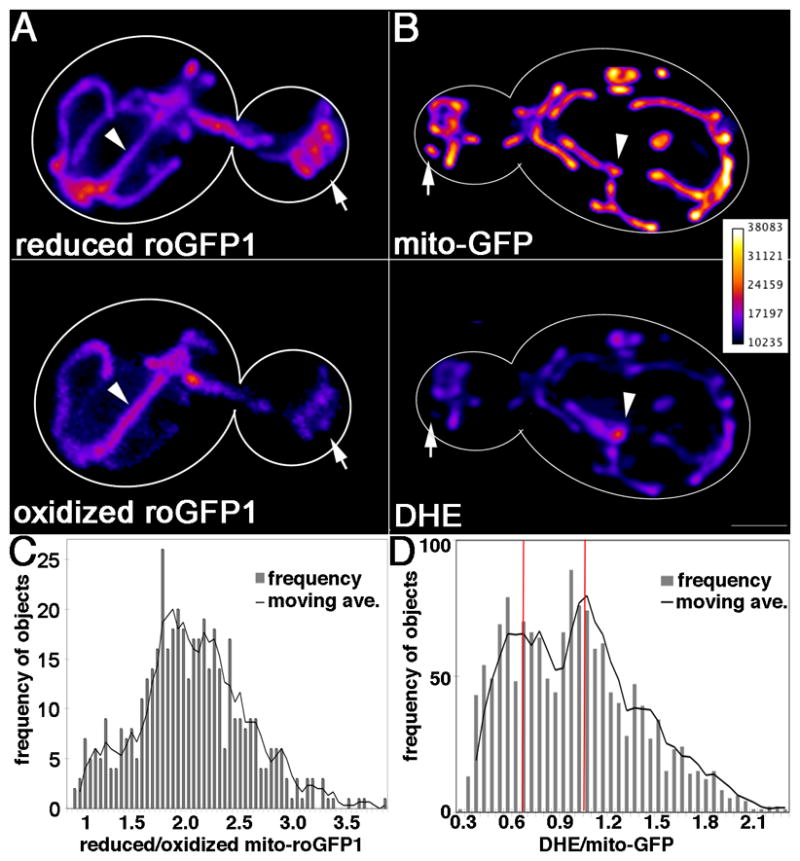
A–B) Maximum projections of mid-log phase wild-type cells that either express mito-roGFP1 (A) or express mito-GFP and are strained with DHE (B). Images shown are representative of >200 cells examined. Reduced mito-roGFP1: fluorescence at λex=490 nm. Oxidized mito-roGFP1: fluorescence at λex=400 nm. Lighter colors reflect higher fluorescence intensity (scale at right). Cell outlines are shown in white. Arrows: mitochondria with higher redox potential (i.e. high levels of reduced roGFP signal and reduced levels oxidized roGFP signal) (A), or reduced superoxide (low DHE staining) (B). Arrowheads: mitochondria with low redox potential (A) or high superoxide (B). Bar = 1 μm. C–D) Histograms of R/O mito-roGFP1 (C) and DHE/mito-GFP (D) observed in mitochondrial objects separated by at least 1 voxel in deconvolved, thresholded fluorescence images of cells expressing mito-roGFP1 or mito-GFP and stained with DHE. Mitochondrial redox potential and superoxide levels are heterogeneous. Superoxide levels fall into two general classes; the red lines indicate maximum frequencies of each superoxide level class based on three-point moving average. Solid black line: three point moving average. n = [blank] cells for C. n= 20 cells for D.
To visualize mitochondrial ROS in living cells, wild-type yeast cells that express mitochondria-targeted GFP (mito-GFP) were stained with DHE. Mitochondrial superoxide as a function of mitochondrial mass was determined by comparing the fluorescent signal from DHE to that of mito-GFP. In wild type yeast, DHE localizes to structures labeled with mito-GFP (Fig. 1B), and undergoes changes in fluorescence in response to treatment with oxidizing agents and ROS scavengers (SFig. 1).
Both biosensors reveal variability in mitochondrial function within individual yeast cells. Using mito-roGFP1, we detect some mitochondria that are highly reduced and other mitochondria that are more oxidized (Fig. 1A–B). Similarly, mitochondria within the same cell exhibit strong or weak staining with DHE (Fig. 1C–D), and therefore have high and low superoxide levels, respectively. We observed a good fit with a Gaussian distribution of mitochondrial redox potential (Anderson-Darling goodness-of-fit statistic for a normal distribution fit: 0.959, p value = 0.015). In contrast, analysis of the ratio of DHE to mito-GFP revealed an apparent bimodal distribution of mitochondrial superoxide levels: mitochondria with higher superoxide levels (DHE/mito-GFP ratio around 1.1) and mitochondria with lower superoxide levels (DHE/mito-GFP ratio around 0.7) (Fig. 1B and 1D).
If mitochondria within individual cells are functionally distinct, they are expected to be physically distinct. We tested this using fluorescence loss in photobleaching (FLIP) experiments on yeast in which GFP is targeted to the mitochondrial matrix. In FLIP, a small area is photobleached repeatedly. As diffusion brings fresh fluorophores into the targeted area and photobleached fluorophores out of the targeted area, fluorescence is lost from all fluorophores that localize to the same membrane-bound compartment.
We repeatedly photobleached a 0.5 μm2 spot on mitochondria in the mother cell and visualized GFP-labeled mitochondria by laser scanning confocal microscopy (Fig. 2). The fluorescence of mito-GFP in the targeted area was lost within < 2 sec. Subsequently, fluorescence was also lost from some mitochondria in the mother cell. However, fluorescence persisted in other mitochondria in the mother cell and in all of the mitochondria within the bud. These findings indicate that mitochondria in the mother cell can be discrete, physically distinct entities. They also show that mitochondria in the bud are physically distinct from mitochondria in the mother cell. Thus, mitochondria within individual yeast cells can be physically and functionally distinct.
Fig. 2. Mitochondria in the mother cell are physically distinct.
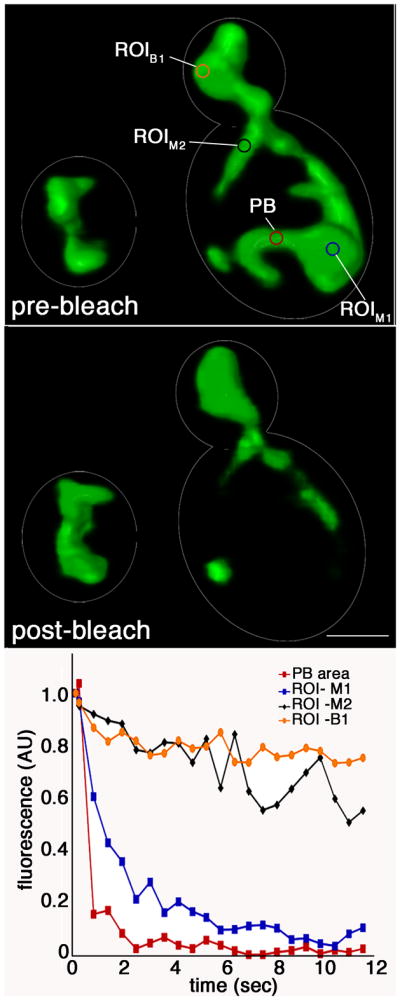
A 0.5 μm2 area was photobleached repeatedly in mitochondria in the mother cell of wild-type yeast expressing matrix-targeted mito-GFP. A cycle consisting of 125 ms photobleaching and 250 ms imaging was repeated for a total of 12 sec. A) Shaded volume projections of cells before (left) and after (right) photobleaching. PB: photobleached zone. ROI B1, ROI M1, ROI M2: regions of interest in the bud and the mother cell, respectively. B) Mean fluorescence intensity of mito-GFP as a function of time in the photobleached zone (PB area; red), a region of a mother cell mitochondrion that exhibits a loss of fluorescence (ROI-M2; blue), a region of a mother cell mitochondrion that does not exhibit loss of fluorescence (ROI-M1; black) and a region of mitochondria that accumulate in the bud tip and do not exhibit loss of fluorescence (ROI-B1; orange). Images and data are representative from analysis of >25 cells examined.
Mitochondria with lower redox potential are preferentially retained by mother cells
If mitochondria are a mother-daughter age asymmetry determinant, then mitochondria that are functionally distinct should be asymmetrically distributed during yeast cell division. Analysis of DHE/mito-GFP ratios shows a small but statistically significant increase in the level of mitochondrial ROS in mother cells compared to buds (Fig. 3A). Thus, mitochondria with lower ROS are preferentially inherited by daughter cells and mitochondria with higher ROS are retained in mother cells.
Fig. 3. Mitochondria with higher superoxide levels and lower redox potential are retained in mother cells in young and old yeast cells.

A) Quantitation of DHE/mito-GFP of mitochondria in mother cells and buds of mid-log phase yeast cells (n= 57) were obtained as for Fig. 1. Asterisks denote significant differences. Mitochondrial superoxide levels were higher in mother cells compared to buds (p = 0.015). B) ConA-594-labeled cells were propagated in glucose-based media, and stained with Calcofluor white. Upper panels show a young cell at t = 1 hr of growth that has uniform ConA-594 labeling and one Calcofluor-labeled bud scar. Lower panels show an older cell at t = 17 hr of growth that has non-uniform ConA-594 labeling and several Calcofluor-labeled bud scars (arrows). Images shown are maximum projections of deconvolved z-series. C) Quantitation of R/O mito-roGFP1 in buds and mother cells at 0, 2 and 5 generations of replication (n = 35, 31 and 17, respectively) was carried out as for Fig. 1. Asterisks denote significant differences; *: p= 0.00025, **: p=0.003, ***: p=0.04. Old cells have lower mitochondrial redox potential compared to young cells. D) The decrease in mitochondrial redox state from 0–5 generations modeled by the equation (R/O)n=0.94n(R/O)n=0. Grey: decline in mitochondrial redox potential predicted by the model. Black: observed mitochondrial redox potential. Inset: decline of mitochondrial redox potential predicted by the model from 0 to 48 generations, the maximum RLS of the wild-type yeast strain. The red line marks theoretical mitochondrial redox state at 22 generations, the average RLS of the wild-type strain used.
To determine whether mitochondria are also segregated on the basis of their redox potential, we measured R/O mito-roGFP1 as a function of replicative age. Replicative lifespan (RLS) is the cumulative number of mitotic divisions that a cell can undergo and is used as a model for aging in cell division-competent cells that undergo reproductive senescence. We developed a method to detect old cells in mid-log phase cultures by fluorescence microscopy, based on a widely used biotinylation method (e.g. (Lam et al. 2011)). Mid-log phase cultures were pulse-labeled with Alexa 594-Concanavalin A (ConA-594), which stains the cell wall, and propagated in media without ConA-594. As a result, daughter cells produced after the ConA-594 pulse are unstained, and older, ConA-594-stained cells are readily distinguished from young cells. Finally, to determine the age of ConA-594 labeled cells as a function of time of growth, we used Calcofluor white to visualize bud scars (Fig. 3B; SFig. 6).
We do not detect import of mito-roGFP into cells > 5 generations, presumably because the mito-roGFP has high turnover rates in mitochondria and mitochondrial protein import declines with age. Therefore, we measured mitochondrial redox potential in cells as they age from 0–5 generations, about 23% of the mean RLS for our wild-type yeast cells. We find that mitochondrial redox potential declines with age (Fig. 3C, n=73). This finding is consistent with a recent report that age-associated increases in ROS levels are also detectable in young yeast cells that are 5–7 generations in replicative age (Lam et al., 2011).
We also detect a subtle but statistically significant decrease in the redox potential of mitochondria in mother cells compared to buds as cells age from 0–5 generations of replicative age. Though the difference in mitochondrial redox potential in mother cells and buds is small, we obtained evidence that retention of mitochondria with lower redox potential in mother cells can contribute to mother-daughter age asymmetry (Fig. 3D). We developed a mathematical model for age-associated declines in mitochondrial redox potential based exclusively on the retention of less fit mitochondria in mother cells as they age from 0–5 generations: (R/O)n=0.94n(R/O)n=0, where R/O is R/O mito-roGFP1 and n is the number of cell divisions a cell has undergone (Fig. 3D). The age-associated decline in total cellular mitochondrial redox potential predicted by this model fits well with the observed decline in mitochondrial redox potential in mother cells from 0–5 generations of replicative age. Moreover, extrapolation of the model from young to old cells predicts a decline in mitochondrial redox potential that is compatible with the RLS of the yeast strain used for these studies: the redox potential of wild-type cells at mean (22 generations) and maximum (48 generations) RLS is 27.3% and 5.5% of that observed in newborn cells, respectively (Fig. 3D, inset).
Together, our findings indicate that the machinery for mitochondrial inheritance has the capacity to retain mitochondria with lower redox potential in mother cells. They also indicate that retention of these lower functioning mitochondria in mother cells can be responsible for the decline in mitochondrial redox potential that occurs as yeast cells age.
Deletion of MMR1 alters replicative lifespan and mother-daughter age asymmetry
If mitochondrial quality control during inheritance affects cell fitness, then mutations that compromise mitochondrial inheritance should affect lifespan. Many proteins are required for normal mitochondrial inheritance in yeast. However, the vast majority of these proteins have been implicated in other processes including actin dynamics and function, endocytosis, as well as mitochondrial morphology, DNA maintenance, mRNA trafficking or protein assembly (Peraza-Reyes et al. 2010). One exception is Mmr1p. Mmr1p is a member of the conserved family of DSL1 tethering proteins. It localizes to mitochondria in the bud tip and is required for mitochondrial inheritance to buds (Itoh et al. 2004). Indeed, mutation of Mmr1p together with Gem1p, a miro-like protein, results in severe defects in mitochondrial inheritance and triggers a checkpoint that blocks cytokinesis (Garcia-Rodriguez et al. 2009).
Deletion of SIR2 results in a decrease in mean RLS from 22 to 14 generations and maximum RLS from 48 to 30 generations (n=76; p = 8.7 × 10−9 for mean RLS). Deletion of MMR1 has a profound effect on RLS (Fig. 4A). mmr1Δ mutant clones give rise to two subpopulations of cells (Fig. 4B); separating the subpopulations into two data sets suggests that they are statistically different (p = 2.1 × 10−13). One subpopulation exhibits premature loss of replicative capacity compared to wild-type cells (p = 1.0 × 10−31 for mean RLS). The majority of these cells fail to produce offspring, and the few that do are replication-competent for a maximum of 5 rounds of cell division (n=51). The other subpopulation exhibits a RLS that is 36% greater than that observed in wild-type cells (p = 0.004 for mean RLS), with a mean and maximum RLS of 30 and 52 generations, respectively (n=51) (Fig. 4A). The observed alteration in RLS in mmr1Δ mutants is not a consequence of genetic background, the method used for generating synchronized cells before RLS measurement or loss of metabolic activity (Fig. S2, Table S1, Fig. S5). Thus, a mutation in MMR1 that inhibits accumulation of mitochondria in the bud results in defects in lifespan determination and the production of cells with either shortened or prolonged RLS.
Fig. 4. Deletion of MMR1 affects mother-daughter age asymmetry.
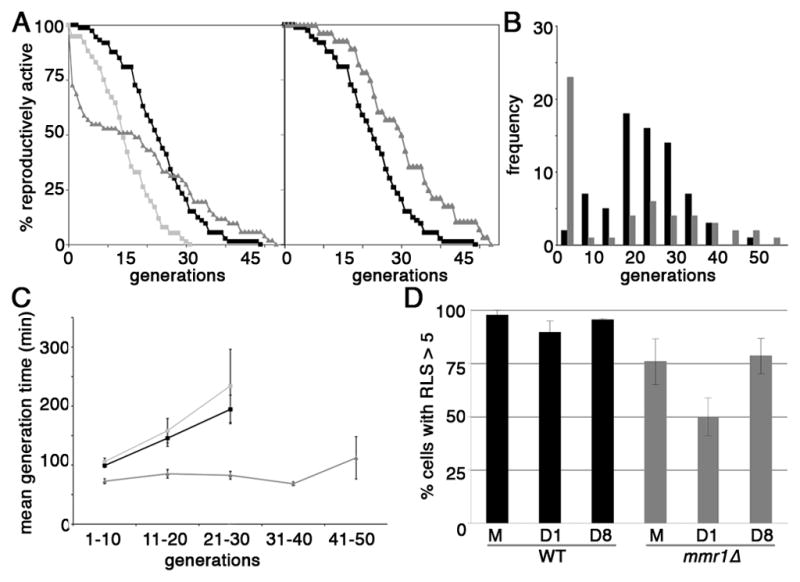
(A) Left panel: RLS of BY4741 (ν; n = 73), mmr1Δ (σ; n = 51) and sir2Δ (ν; n = 76) cells were measured as described in Materials and Methods, using pheromone treatment to distinguish virgin mother cells from their mothers. The data shown is pooled from 3 independent experiments. Right panel: RLS of wild-type cells (ν) and the long-lived subpopulation of mmr1Δ (σ) cells. Short-lived mmr1Δ cells, which have a RLS < 5 generations, were removed on the basis of their lifespan distribution histogram (Fig. 4C). The remaining cells were corroborated to be long-lived by their clustering in a plot of RLS versus mean generation time from 0–10 generations (Fig. S3). B) Histogram of the RLS distribution of wild-type (black) and mmr1Δ (grey) cells. C) Mean generation time for wild-type (BY4741) (ν; n = 91), sir2Δ (ν; n = 55) and long-lived mmr1Δ cells (σ; n = 43) as a function of replicative age (generation). The data shown is pooled from 2 independent experiments. Generation time is the time from the last cell division to mother-bud separation, the latter of which was assessed by physically separating mothers from buds using a micromanipulator on a dissecting microscope. Generation time was averaged over 10 generations. Error bars: standard error of the mean. D) Percent of cells with RLS > 5 generations in mother cells (M) and their first (D1) and eight (D8) buds in wild-type and mmr1Δ cells. The data are pooled from 2 independent experiments, where n = 40 for each cell type studied in each experiment.
Long- and short-lived mmr1Δ mutants also exhibit a phenotype associated with aging: altered generation time. Early studies revealed that aging mother cells take increasingly longer times to progress through the cell cycle compared to young cells (Mortimer & Johnston 1959; Egilmez & Jazwinski 1989). Under our growth conditions, the mean generation time of wild-type cells is 90–110 min during generations 1–10, and increases to 200 min during generations 21–30 (Fig. 4C). sir2Δ cells also exhibit an age-associated increase in generation time. In contrast, the generation time of long-lived mmr1Δ cells is less than that of young wild-type cells. Moreover, it does not increase from generations 1–40, and exhibits a modest increase to 110 min only after 41–50 replications. This phenotype is also not a consequence of genetic background or synchronization method (Fig. S2). Finally, short-lived mmr1Δ cells exhibit a mean generation time that is 10-fold greater than that of wild-type cells during the first 1–10 generations (Fig. S4). Thus, long-lived mmr1Δ cells exhibit a delay in the increase in mean generation time as a function of age, a phenotype associated with young cells, and short-lived mmr1Δ cells exhibit an increased mean generation time, a phenotype associated with old cells.
Next, we studied the effect of deletion of MMR1 on mother-daughter age asymmetry. A widely used method to assess this process is to compare the mean RLS of daughter cells and their mother cells (Lai et al. 2002; Shcheprova et al. 2008). However, since mmr1Δ daughter cells have either long or short RLS, the mean RLS does not provide information regarding short-or long-lived mmr1Δ cells. Therefore, we assessed mother-daughter age asymmetry in mmr1Δ cells as the frequency of producing short-lived daughter cells from the first (D1) and eighth (D8) daughter cells from virgin mother cells (M) (Fig. 4D). Only 2–11% of wild-type D1 and D8 cells have RLS <5 generations (n=75, 71 and 60, respectively). In contrast, there are significantly more short-lived cells in mmr1Δ mother cells (24%, n = 77) and their D1 (50%, n = 66) and D8 (22%, n = 58) daughters compared to wild-type cells. Moreover, the incidence of short-lived cells is higher in D1 daughters compared to their mothers and D8 daughters. This difference likely reflects the fact that short-lived mmr1Δ cells do not produce D8 daughters and only produce short-lived daughters (see below). Thus, deletion of MMR1 results in defects in mother-daughter age asymmetry. In contrast to wild-type daughter cells, who are born with their full replicative potential, the replicative potential of D1 daughter mmr1Δ cells is lower than that of their mothers.
mmr1Δ cells exhibit changes in mitochondrial quality control
To determine whether the changes in lifespan and mother-daughter age asymmetry in mmr1Δ cells are linked to defects in mitochondrial quality control during inheritance, we studied mitochondrial redox potential and ROS in short- and long-lived mmr1Δ cells. As a first step, we found that unbudded cells in mmr1Δ mid-log phase cultures exhibit characteristics of short-lived mmr1Δ cells (reproductive incompetence and increased mean generation times) and budded mmr1Δ cells have mean generation times consistent with long-lived mmr1Δ cells (Fig. 5A). Consistent with this, mmr1Δ cell cultures exhibit higher levels of unbudded cells (36%) compared to wild-type cell cultures (18%) (Table S1). Cellular metabolic activity, as assessed using FUN-1, is similar in unbudded and budded mmr1Δ and wild-type cells (Table S1). Thus, we identified methods to identify cell fractions that are enriched in short- and long-lived mmr1Δ cells in mid-log phase yeast cultures.
Fig. 5. Mitochondrial fitness correlates with lifespan.

A) Left panel: Percentage of unbudded wild type (black; n = 39) and mmr1Δ (grey; n = 40) cells that generate 5 or more offspring. Right panel: percentage of unbudded (U) and budded (B) wild-type (black; n = 157) and mmr1Δ (grey; n = 158) cells with a generation time ≤ 120 min. B) Maximum projections of ratiometric images of R/O mito-roGFP1 in mid-log phase wild-type and mmr1Δ cells. Colors reflect the intensity of ratio of reduced-to-oxidized mito-roGFP1 (scale at lower left). Cell outlines are shown in white. Upper and lower panels: budded and unbudded cells, respectively. Images shown are representative from analysis of >400 cells. C) Quantitation of R/O mito-roGFP1 of mitochondria in unbudded (U) and budded (B) wild type (black; n = 243) and mmr1Δ (grey; n = 173) cells, measured as described in Fig. 2. Asterisks denote significant changes. Mitochondrial redox potential is higher in budded mmr1Δ cells compared to unbudded mmr1Δ cells and in budded wild type cells compared to unbudded mmr1Δ cells (**p = 0.0003, *p= 0.0113). D) Maximum projections of mito-GFP and DHE of budded (upper panels) and unbudded (lower panels) wild-type and mmr1Δ cells. Colors reflect the intensity of fluorescence (scale at lower right). Cell outlines are shown in white. Images shown are representative from analysis of >100 cells. E) Quantitation of DHE/mito-GFP of mitochondria in unbudded (U) and budded (B) wild-type (black, n=79) and mmr1Δ (grey, n=80) cells, measured as described in Fig. 2. Asterisks denote significant changes. Mitochondrial superoxide production is lower in budded compared to unbudded mmr1Δ cells, and lower in budded mmr1Δ cells compared to wild-type cells (**p = 0.007, *p= 8 × 10−13).
The total cellular mitochondrial redox potential of unbudded and budded wild-type cells are similar (Fig. 5B–C). In contrast, the mitochondrial redox potential of budded, largely long-lived mmr1Δ cells is greater than that of unbudded, largely short-lived mmr1Δ cells. Moreover, the mitochondrial redox potential of wild-type cells is greater than that of unbudded, largely short-lived mid-log phase mmr1Δ cells. We obtained similar results upon analysis of mitochondrial superoxide levels in WT and mmr1Δ cells (Fig. 5D–E). Superoxide levels of budded, largely long-lived mmr1Δ cells are greater than those of wild-type cells and of unbudded, largely short-lived mmr1Δ cells. Thus, mitochondrial fitness in mmr1Δ cells, as assessed by redox potential and superoxide levels, correlates with cellular fitness and lifespan.
Further characterization of mmr1Δ cells revealed a correlation between mitochondrial superoxide levels and cell fitness in buds of long-lived mmr1Δ cells. First, we found that short-lived mmr1Δ cells always give rise to short-lived cells (Fig. 6A). Conversely, although long-lived mmr1Δ cells can give rise to short- and long-lived daughter cells, the majority of the daughter cells produced from long-lived mmr1Δ cells are long-lived (Fig. 6A). These findings are consistent with a non-genomic heritable cellular constituent(s) within mmr1Δ cells that affects the RLS of their daughter cells.
Fig. 6. Daughter cell fitness and mitochondrial inheritance in mmr1 Δ cells.
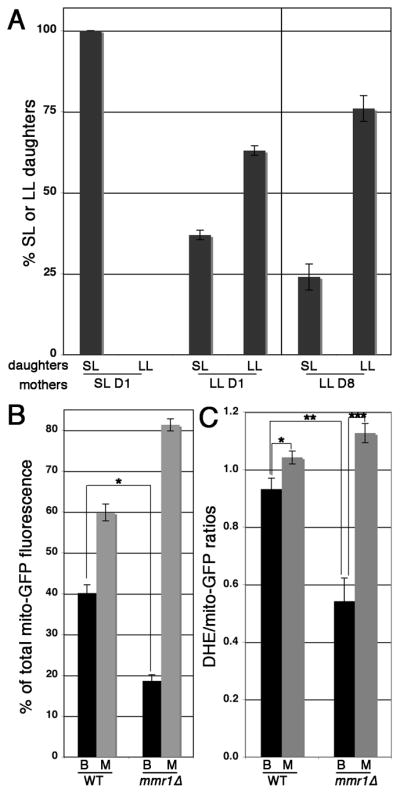
A) The 1st and 8th daughter cells produced from the same virgin mother cell were isolated, and the RLS of these cells and of the daughters produced from these cells was determined as for Fig. 4. Short-lived cells (SS) were identified as cells with RLSs < 5 generations, while long-lived cells (LL) were identified as cells with RLSs > 5 generations. The graph shows the percent SL or LL daughter cells produced from short- or long-lived D1 or D8 mother cells. Short-lived mmr1Δ cells give rise to only short-lived daughter cells while long-lived mmr1Δ cells give rise to short- or long-lived daughter cells. The data are pooled from 2 independent experiments, where n = 40 for each cell type studied in each experiment. B) Mitochondria were visualized using mitochondria-targeted GFP in wild-type cells (BY4741) and mmr1Δ cells, and the amount of GFP- label in mitochondria in mother cells and buds was measured in cells in which the bud is >60% of the size of the mother cell. The relative mitochondrial volume was determined by calculating integrated voxel intensity in thresholded, deconvolved wide-field z-series of mitochondria-targeted GFP. Deletion of MMR1 results in a decrease in the amount of mitochondria that are inherited by daughter cells (p = 1.3 × 10−10). The data shown is representative data from 3 experiments (n = 79). C) Quantitation of DHE/mito-GFP of mitochondria in the buds (B) and mother cells (M) of wild-type (WT) and long-lived mmr1Δ cells, measured as described in Fig. 1. While mitochondrial superoxide levels are lower in bud compared to mother cells in both cell types (*p = 0.02; **p = 4 × 10−7), mitochondrial superoxide levels in the buds of long-lived mmr1Δ cells is significantly lower than that observed in wild-type cells (***p = 0.001).
In addition, we find that deletion of MMR1 results in defects in both the quantity and quality control of mitochondrial inheritance. In contrast to wild-type cells, where daughter cells inherit 40% of the total cellular mitochondria from mothers, daughter cells of largely long-lived mmr1Δ cells inherit only 18% of the total cellular mitochondria (Fig. 6B). There is no significant difference in redox potential among mother cells and buds in largely long-lived mmr1Δ cells (data not shown). However, mitochondria that are inherited by daughters of largely long-lived mmr1Δ cells have significantly lower superoxide levels compared to their mother cells and compared to both mother cells and bud of wild type cells (Fig. 6C). These findings indicate that one important non-genomic but heritable age determinant in mmr1Δ cells is mitochondria with low superoxide levels.
Discussion
Preferential retention of mitochondria with higher superoxide levels and lower redox potential in mother cells as they age
Heterogeneity in mitochondrial Δψ has been detected in yeast, cultured animal cells, neurons and pancreatic beta cells (Collins et al. 2002; Lai et al. 2002; Wikstrom et al. 2007). We obtained the first evidence for heterogeneity in mitochondrial superoxide levels and redox potential within individual yeast cells. Using DHE as a biosensor, we find that some mitochondria within yeast contain low levels of superoxide, while others contain high levels of this ROS. Using mito-roGFP1, we find that the mitochondrial matrix of budding yeast is a reducing environment. Similar results were obtained using mito-roGFP1 in HeLa (Dooley et al. 2004). In addition, we detect heterogeneity in mitochondrial redox potential within individual cells. Quantitative analysis of DHE to mito-GFP ratios suggests that mitochondrial superoxide levels are not normally distributed. Rather, we observed two general classes of mitochondrial superoxide levels.
Fluorescence recovery after photobleaching studies (FRAP) revealed that mitochondria in HeLa cells are discontinuous, allowing them to have distinct functional properties (Collins et al. 2002). Using FLIP, we detect multiple, physically distinct mitochondria in budding yeast. Our studies also reveal that mitochondria in the bud tip can be physically distinct from mitochondria in the mother cell. This data provides a structural basis for the observed heterogeneity in mitochondrial function. It is also direct evidence that mitochondria in budding yeast are not a continuous reticulum. Recent studies indicate that mitochondria with extremely low Δψ exhibit defects in mitochondrial fusion, which would allow this population of low-functioning mitochondria to remain physically distinct from higher functioning mitochondria (Twig et al. 2008). In light of the observed heterogeneity in mitochondrial function, it is possible that there are mechanisms to ensure that low-functioning mitochondria are separated from high-functioning mitochondria in yeast.
In addition, we find that mitochondria with lower redox potential and higher ROS are preferentially retained in mother cells, and mitochondria with higher redox potential and lower ROS are preferentially inherited by daughter cells. This segregation of higher- from lower-functioning mitochondria during yeast cell division occurs at a constant rate as cells undergo replicative aging from 0–5 generations. These findings are consistent with previous observations that mitochondria with low Δψ and oxidatively damaged aconitase are preferentially retained in mother cells (Laun et al. 2001; Lai et al. 2002; Klinger et al. 2010). Collectively, this data support the model that the machinery for mitochondrial inheritance exercises mitochondrial quality control which results in segregation of higher from lower functioning mitochondria among mother and daughter yeast cells.
Equally important, we developed a method to identify cells of known RLS in mid-log phase cultures using fluorescent ConA to label the yeast cell wall, and find that mitochondrial redox potential declines with replicative age and that this decline is evident even in young cells from 0–5 generations of replicative age. This decline in mitochondrial function in relatively young cells is consistent with the age-dependent increase in superoxide levels in young yeast cells described recently (Lam et al. 2011). Moreover, we obtained evidence that retention of mitochondria with lower redox potential in mother cells can contribute to age-associated declines in mitochondrial redox potential. Specifically, we developed a mathematical model for age-associated decline in mitochondrial redox potential based on the observed decline in this process from 0–5 generations. Extrapolation of the model to old cells revealed a decline of mitochondrial redox potential from maximum to minimum levels during a timeframe that is similar to the mean and maximum RLS of the yeast strain used for these studies. Interestingly, this decline depends on the initial redox state of mitochondria in a virgin mother cell. Together, these results are consistent with a model that retention of lower functioning mitochondria in the mother cell results in a mother cell-specific decline in mitochondrial function with age.
Although mitochondria that are in buds and therefore destined for inheritance by daughter cells are higher-functioning compared to mitochondria in mother cells, mitochondria in buds also exhibit a decline in redox potential with age. This raises an interesting question. How are daughter cells born young if they inherit low-functioning mitochondria from their aging mother cells? Recent studies, which indicate that repair mechanisms are preferentially activated in daughter cells after they separate from mother cells, provide a possible explanation. Cytosolic catalase of yeast, Ctt1p, has been implicated in detoxification of mitochondrial ROS and is required for normal RLS (Nestelbacher et al. 2000; Van Zandycke et al. 2002). Erjavec and Nyström (2007) find that the Ctt1p activity increases in daughter cells after they separate from mother cells. The superior ROS management observed in newly formed daughter cells requires Sir2-dependent asymmetric protein segregation during yeast cell division. Thus, mitochondria with high ROS and lower redox potential that are inherited by daughters from aging mother cells may be repaired and rejuvenated by Ctt1p that is activated after daughter cells separate from their mother cells.
Role for the mitochondrial inheritance machinery in mitochondrial quality control and mother-daughter age asymmetry
Previous studies indicate that deletion of prohibitins, mitochondrial inner membrane proteases that play a role in protein processing and mitochondrial quality control, results in a decrease in yeast RLS and age-dependent mitochondrial segregation defects (Piper et al. 2002; Kirchman et al. 2003). To further explore the role of mitochondrial quality control in lifespan, we studied the effect of deletion of MMR1 on RLS and mitochondrial function. Mmr1p localizes to mitochondria in the bud tip and is required for normal mitochondrial inheritance (Itoh et al. 2004) and for anchorage of newly inherited mitochondria in the bud tip (our unpublished observations).
We find that deletion of MMR1 affects mitochondrial quality control and daughter cell fitness and mother-daughter age asymmetry. In contrast to wild-type cells, where daughter cells are born young, with their full replicative potential, mmr1Δ cells are either short-lived or long-lived. The long-lived mmr1Δ cells exhibit phenotypes associated with longevity and fitness (delay in the increase in mean generation time increase with age, reduced mitochondrial superoxide levels and increased mitochondrial redox potential), while short-lived mmr1Δ cells exhibit the opposite phenotypes. The slow growth of short-lived mmr1Δ is associated with oxidative damage from elevated superoxide levels and low redox potential, and not likely due to reduced mRNA translation observed in the slow-growing long-lived cells described by Delaney et al. (2011). Furthermore, we observe a link between mitochondrial redox potential and superoxide levels and cellular fitness: mmr1Δ cells with low redox potential and high superoxide levels exhibit short RLS while mmr1Δ cells with higher redox potential and lower superoxide levels exhibit long RLS. Finally, we find that all mmr1Δ daughter cells are not born young: the replicative potential of a subset of daughter cells that develop from mother cells is lower than that of their mothers.
Quantitative analysis of the heterogeneity of mitochondrial redox potential and superoxide levels within individual yeast cells provides an explanation for the extraordinary fitness and lack thereof of mitochondria in long- and short-lived mmr1Δ cells. In wild-type cells, where 40–50% of total cellular mitochondria are inherited by daughter cells, daughter cells inherit a combination of fit and less fit mitochondria, which are on average more fit than mitochondria in mother cells. In contrast, mitochondrial inheritance and quality control are compromised in mmr1Δ cells. Since daughter cells inherit only 18% of total cellular mitochondria, they are more likely to inherit a disproportionate amount of a single functional class of mitochondria (fit or less fit), which in turn affects cellular fitness and lifespan.
In the case of short-lived mmr1Δ cells, whose mitochondria have higher superoxide levels compared to mitochondria in wild type cells, all daughter cells inherit less fit mitochondria. As a result, they are less fit and have shortened RLS. In the case of long-lived mmr1Δ cells, which are endowed with mitochondria with lower superoxide levels compared to those found in wild-type cells, the probability of inheriting fitter mitochondria is greater. As a result, the majority of the daughter cells derived from long-lived mmr1Δ cells are long-lived.
Overall, our studies indicate that the machinery for mitochondrial inheritance exercises mitochondrial quality control and promotes inheritance of fitter mitochondria by daughter cells and retention of less fit mitochondria in mother cells. They support the model that mitochondria are aging determinants and that retention of mitochondria with higher ROS and lower redox potential in mother cells contributes not just to mother-daughter age asymmetry but also to age-associated declines in mitochondrial redox potential in yeast. Thus, we obtained evidence for a novel mechanism for a decline in mitochondrial function with age. Ongoing studies are designed to determine mechanisms underlying segregation of higher from lower functioning mitochondria both within individual yeast cells and during yeast cell division.
Experimental procedures
Yeast strains and growth conditions
All S. cerevisiae strains used in this study are derivatives of the wild type BY4741 strain (MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0). The mmr1Δ strain, 4139 (MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 mmr1Δ::KANMX6), and the sir2Δ strain, 3738 (MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 sir21Δ::KANMX6) have the MMR1 or SIR2 genes replaced by KANMX6 cassettes, respectively. All three strains are from Open Biosystems (Huntsville, AL). The strain RMY003 (MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 mmr1Δ::leu2) has the MMR1 gene replaced by the LEU2 gene in a BY4741 background. For labeling of mitochondria, BY4741 and 4139 strains were transformed with a centromeric plasmid containing the mitochondrial matrix targeting signal sequence of citrate synthase 1 (CIT1) fused to GFP (Garcia-Rodriguez et al. 2009). The resulting strains are TSY200: BY4741[pCIT1GFP:URA3] and TSY201: 4139[pCIT1GFP:URA3]. BY4741 cells expressing a Cit1p-GFP fusion protein from its genomic locus, 95700-YNR001C, (MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 his5+ CIT1-GFP) are from the Yeast GFP Clone Collection (Huh et al. Nature, 2003; Invitrogen, Eugene OR). The RMY015 strain (MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 his5+ CIT1-GFP mmr1Δ::leu2) has the MMR1 gene replaced by the LEU2 gene in 95700-YNR001C. Yeast cells were cultivated and manipulated as described previously (Garcia-Rodriguez et al. 2009).
To create the plasmid pmito-roGFP1, the mitochondrial-targeting signal sequence (MTSS) of ATP9 was obtained by restriction enzyme digestion of plasmid ID# B1063 or pTDT104 GAL1+preATP-9-RFP (Garcia-Rodriguez et al. 2009) with SpeI and XhoI (New England Biolabs, Ipswich, MA). These constructs were gel purified and ligated with T4 ligase (NEB, Ipswich, MA) into the Spe1 and Xho1 sites in yeast shuttle vectors Plasmid ID# 1177 or p416 GPD (Addgene, Cambridge, MA). The ligation product was used to transform DH5α-competent bacterial cells (Invitrogen, Carlsbad, CA). Plasmid clones were recovered and processed with a MiniPrep kit (Qiagen, Valencia, CA). The roGFP1 insert was amplified using PCR and forward primers 5′-AGATACGGATCCATGAGTAAAGGAGAAGAACTTTTCACTGGAG-3′ and reverse primers R 5′-AGATACCTCGAGCCATGGTACCAGCTGCAGATCTC-3′ (IDT, Coralville, IA) from pRSETB (Dooley et al. 2004). The roGFP1 PCR product and the new plasmid, which contained p416 GPD/ATP9 MTSS, were then digested with restriction enzymes XhoI and BamHI (NEB, Ipswich, MA). These products were gel purified and ligated using T4 ligase and used to transform DH5α competent bacterial cells. Plasmid clones were recovered and processed with a MiniPrep kit. To verify proper plasmid construction, the pmito-roGFP1 plasmid was sequence verified with the sequencing primer 5′-CAGCACGTGTCTTGTAGTTCCCG-3′.
Fluorescence microscopy
For visualization of mitochondria, cells were grown to mid-log phase in synthetic complete (SC) medium with or without uracil at 30°C. Live cells were mounted on glass slides, and used for no more than 20 mins for visualization. For labeling with the superoxide indicator, dihydroethidium (DHE) (Invitrogen – Molecular Probes, Eugene, Oregon), cells were incubated with 40 μM DHE (dissolved in DMSO) in fresh growth medium for 30 min at 30°C, then washed 1x with growth medium and visualized without fixation. For determination of relative mitochondrial redox state we transformed yeast with a plasmid containing the mitochondrial targeting sequence of ATP9 fused to roGFP1 (mito-roGFP1). Cells were grown to mid-log phase in SC-Ura, concentrated and visualized without fixation.
All imaging was performed as described previously (Garcia-Rodriguez et al. 2009) on one of the following microscope systems: an Axiovert 200M microscope with 100x/1.4 Plan-Apochromat objective (Zeiss, Thornwood, NY) and Orca ER cooled charge-coupled device (CCD) camera (Hamamatsu, Bridgewater, NJ); an Axioskop 2 microscope with 100x/1.4 Plan-Apochromat objective (Zeiss, Thornwood, NY) and an Orca 1 cooled CCD camera (Hamamatsu) or an Axiocam CCD camera (Zeiss, Thornwood, NY) with FITC and/or Rhodamine filter sets; an inverted AxioObserver.Z1 microscope with a 100x/1.3 oil EC Plan-Neofluar objective (Zeiss, Thornwood, NY) and Orca ER cooled charge-coupled device (CCD) camera (Hamamatsu, Bridgewater, NJ), along with an LED Colibri system (Zeiss, Thornwood, NY) including LED wavelengths at 365 and 470 nm. Hardware was controlled by Openlab, Volocity software (Perkin-Elmer, Waltham, MA) and Axiovision software (Zeiss, Thornwood, NY), respectively.
Fluorescence loss in photobleaching (FLIP)
FLIP experiments were performed on a Nikon A1R-MP laser scanning confocal microscope. Wild-type yeast expressing mitochondrial matrix-targeted GFP were mounted on agarose pads as described above. Bleaching was performed with the 488-nm laser line on a spot 0.5 μm in diameter. A cycle consisting of 125 ms photobleaching and 250 ms imaging was repeated for a total of 12 sec.
Quantitation of the fluorescence of GFP- or DHE-labeled mitochondria
Cells were incubated with 40 μM DHE in growth medium for 30 min at 30°C, washed with growth medium, and visualized without fixation. To quantify relative fluorescence of GFP and DHE in mitochondria, wide-field z-series were collected through a beamsplitter (DualView, Photometrics, Inc., Tucson, AZ) that simultaneously projects images of green and red fluorescence on a CCD camera. The filters showed negligible crosstalk under the conditions used for staining and imaging. Transmitted-light images were collected to indicate the location and size of the bud and mother cell. Channel images were aligned in ImageJ using transmitted light and cell background as guides and the Cairn Image Splitter plugin. The obtained images were digitally deconvolved by iterative restoration (100% confidence limit, 40 iteration limit). Relative mitochondrial volume was estimated by calculating integrated voxel intensity in thresholded, deconvolved wide-field z-series of mitochondria-targeted GFP. Images were deconvolved using a constrained iterative restoration algorithm (Volocity, Perkin-Elmer, Waltham, MA). Voxels were identified and quantified using Volocity Quantitation software. Bud size and zones of mother and bud were identified using corresponding transmitted-light images.
The distribution of mitochondrial superoxide relative to mitochondrial mass was determined by calculating integrated voxel intensity (F) of DHE in deconvolved z-series. The distribution of DHE or GFP in specific regions within the mother or bud was normalized as Fregion of interest/Ftotal. The F values for DHE were compared to the corresponding F values for GFP in the same region by calculating the DHE/GFP ratio. DHE/GFP ratio was calculated in cells in which the bud length was over 0.50 times the mother cell length.
To test the dynamic range of DHE, we exposed yeast expressing Cit1p-GFP from its chromosomal locus to 10mM H2O2 (Fisher Scientific, Pittsburgh, PA) or 50μM Tempol (Sigma-Aldrich, St. Louis, MO) for 30 minutes in SC-Ura. Cultures were washed twice with 1 mL of fresh media and visualized along with a control sample.
Quantitation of the fluorescence of reduced-to-oxidized mito-roGFP1
Yeast cells were transformed with plasmid pmito-roGFP1 to measure mitochondrial redox state. The relative redox state of mito-roGFP1 was determined by calculating the ratio of the integrated voxel intensity in background-subtracted, thresholded, deconvolved wide field z-series of mito-roGFP1 at excitation 470nm and 365nm, reduced to oxidized, respectively. Images were deconvolved using a constrained iterative restoration algorithm (Volocity, Perkin-Elmer, Waltham, MA). Voxels were identified and quantified using Volocity Quantitation software. Bud size and zones of mother and bud were identified using corresponding transmitted-light images.
To determine the ratio of reduced to oxidized mito-roGFP1, z-series were collected, deconvolved, background-subtracted and thresholded using Volocity software (Perkin Elmer, city, state). To calculate the reduced to oxidized ratio of mito-roGFP1, we divided the reduced channel (λex=470 nm, λem=525 nm) by the intensity of the oxidized channel (λex =365 nm, λem =525 nm). To test the dynamic range of mito-roGFP1, we incubated yeast expressing mito-roGFP1 in 10 mM H2O2 (Fisher Scientific, Pittsburgh, PA) or 10 mM DTT (Fisher Scientific, Pittsburgh, PA) for 30 minutes in SC-Ura. Cultures were washed twice with 1 mL of fresh media and visualized along with a control sample using an inverted epifluorescence microscope equipped with a Colibri LED system (Zeiss, Thornwood, NY) and Orca ER CCD camera (Hamamatsu, Bridgewater, NJ).
Replicative Lifespan Determination
RLS measurements were performed as described previously (Erjavec et al. 2008), with or without alpha-factor synchronization. Briefly, frozen yeast strain stocks (stored at −80°C) were grown in rich, glucose based solid medium (YPD) at 30°C. Single colonies of each yeast strain were suspended in liquid YPD and grown to mid-log phase at 30°C with shaking and aeration. If synchronized, cultures were treated with alpha-factor (100 μg/ml, Genemed Synthesis Inc) for 3 hrs, washed three times with water, and resuspended in water. For unsynchronized and synchronized cultures, an aliquot of the cell suspension was applied to YPD plates, small budded-cells or shmoos, respectively, were isolated and arranged in a matrix using a micromanipulator mounted on a dissecting microscope (Zeiss, Thornwood, NY). These small budded cells or shmoos were henceforth referred to as the mother cell. After their first round of replication, mother cells were removed and discarded, and their daughter cells renamed virgin mother cells. After each replication, the time and number of daughter cells produced by each virgin daughter cell was recorded until all replication ceased.
ConA-594 labeling for determination of old cells in culture
Yeast cultures were grown to mid-log phase and incubated with 100μM concanavalin A, Alexa Fluor-594 conjugate (Sigma-Aldrich, St Louis, MO) for 30 min at 30°C, washed with growth medium, and propagated in fresh medium. ConA fluorescence was used to identify old cells. Cell age was determined by Calcofluor staining of bud scars. Because calcofluor staining interferes with mito-roGFP1 imaging, redox potential was measured in separate aliquots of cells that were not stained with Calcofluor.
Statistical Methods
All p values were determined using a two-tailed Student’s t-test assuming unequal variance. Probability plots for distribution fitting, with associated Anderson-Darling goodness-of-fit statistics and p-values, were carried out using Minitab 14 statistical analysis software.
Other methods
Yeast cells were transformed using the lithium acetate method (Garcia-Rodriguez et al. 2009). Budding index was calculated by dividing the number of budding cells by the total number of cells in a mid-log phase culture grown in YPD.
Viability was determined using the FUN 1 cell stain from the LIVE/DEAD Yeast Viability Kit (Invitrogen – Molecular Probes, Eugene, OR) according to the manufacturer’s instructions. Briefly, mid-log phase cells grown on YPD were diluted to a concentration of approximately 1 × 107 cells/ml, FUN 1 was added to a final concentration of 10 μM and samples were incubated at 30°C for 40 minutes with shaking. The conversion of FUN 1 by viable cells was quantified by fluorescence microscopy. Cells with prominent fluorescent intravacuolar structures were scored as viable. Cells that lacked these structures and had diffuse green or yellow cytosolic fluorescence were scored as non-viable.
To determine the percent of cells that undergo cytokinesis in under 120 min, cells from wild-type and mmr1Δ mid-log phase cultures were isolated and arranged in a matrix using a micromanipulator mounted on a dissecting microscope (Zeiss, Thornwood, NY). Cytokinesis was determined by assessing separation of daughter and mother cell with the micromanipulator at 120 min.
Supplementary Material
Acknowledgments
We thank the members of the Pon laboratory and N. Erjavec for technical assistance and valuable discussions, and L. Medina for expert statistical analysis. This work was supported by grants from the National Institutes of Health (NIH) (GM45735 and GM45735S1) and the Ellison Medical Foundation (AG-SS-2465-10) to LP and from the NIH (1 F31 AG034835) to JRMF. The confocal microscope used for these studies was obtained using an NIH/NCRR shared instrumentation grant (1S10RR025686) to LP, and supported in part through a NIH/NCI grant (5 P30 CA13696). GM45735S1, 1S10RR025686 and 1 F31 AG034835 were issued from the NIH under the American Recovery and Reinvestment Act of 2009.
Footnotes
Supplemental data includes one table and four figures.
Author contributions
José R. McFaline-Figueroa and Jason D. Vevea contributed equally to the development of this work, including concept development, method development, data acquisition, data analysis and interpretation, and manuscript preparation. Theresa C. Swayne acquired and analyzed data, developed methods for fluorescence quantitation and contributed to editing of this manuscript. Chun Zhou constructed plasmid pmito-roGFPP1 for visualization of mitochondrial redox state. Christopher Liu and Galen D. Leung contributed to data acquisition. Istvan R. Boldogh contributed to concept development, method development and to editing of this manuscript. Liza A. Pon contributed to concept development, data analysis and interpretation and manuscript preparation.
References
- Aguilaniu H, Gustafsson L, Rigoulet M, Nyström T. Asymmetric inheritance of oxidatively damaged proteins during cytokinesis. Science. 2003;299:1751–1753. doi: 10.1126/science.1080418. [DOI] [PubMed] [Google Scholar]
- Barros MH, Bandy B, Tahara EB, Kowaltowski AJ. J Biol Chem. 2004. Higher respiratory activity decreases mitochondrial reactive oxygen release and increases life span in Saccharomyces cerevisiae; pp. 49883–49888. [DOI] [PubMed] [Google Scholar]
- Bonawitz ND, Chatenay-Lapointe M, Pan Y, Shadel GS. Reduced TOR signaling extends chronological life span via increased respiration and upregulation of mitochondrial gene expression. Cell metabolism. 2007;5:265–277. doi: 10.1016/j.cmet.2007.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins TJ, Berridge MJ, Lipp P, Bootman MD. Mitochondria are morphologically and functionally heterogeneous within cells. EMBO J. 2002;21:1616–1627. doi: 10.1093/emboj/21.7.1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dooley CT, Dore TM, Hanson GT, Jackson WC, Remington SJ, Tsien RY. Imaging dynamic redox changes in mammalian cells with green fluorescent protein indicators. J Biol Chem. 2004;279:22284–22293. doi: 10.1074/jbc.M312847200. [DOI] [PubMed] [Google Scholar]
- Egilmez NK, Jazwinski SM. Evidence for the involvement of a cytoplasmic factor in the aging of the yeast Saccharomyces cerevisiae. J Bacteriol. 1989;171:37–42. doi: 10.1128/jb.171.1.37-42.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg T, Knauer H, Schauer A, Buttner S, Ruckenstuhl C, Carmona-Gutierrez D, Ring J, Schroeder S, Magnes C, Antonacci L, Fussi H, Deszcz L, Hartl R, Schraml E, Criollo A, Megalou E, Weiskopf D, Laun P, Heeren G, Breitenbach M, Grubeck-Loebenstein B, Herker E, Fahrenkrog B, Frohlich KU, Sinner F, Tavernarakis N, Minois N, Kroemer G, Madeo F. Induction of autophagy by spermidine promotes longevity. Nat Cell Biol. 2009;11:1305–1314. doi: 10.1038/ncb1975. [DOI] [PubMed] [Google Scholar]
- Erjavec N, Cvijovic M, Klipp E, Nyström T. Selective benefits of damage partitioning in unicellular systems and its effects on aging. Proc Natl Acad Sci USA. 2008;105:18764–18769. doi: 10.1073/pnas.0804550105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erjavec N, Larsson L, Grantham J, Nyström T. Accelerated aging and failure to segregate damaged proteins in Sir2 mutants can be suppressed by overproducing the protein aggregation-remodeling factor Hsp104p. Genes Dev. 2007;21:2410–2421. doi: 10.1101/gad.439307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erjavec N, Nyström T. Sir2p-dependent protein segregation gives rise to a superior reactive oxygen species management in the progeny of Saccharomyces cerevisiae. Proc Natl Acad Sci USA. 2007;104:10877–10881. doi: 10.1073/pnas.0701634104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabrizio P, Liou L-L, Moy VN, Diaspro A, Valentine JS, Gralla EB, Longo VD. Genetics. 2003. SOD2 functions downstream of Sch9 to extend longevity in yeast; pp. 35–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Rodriguez LJ, Crider DG, Gay AC, Salanueva IJ, Boldogh IR, Pon LA. Mitochondrial inheritance is required for MEN-regulated cytokinesis in budding yeast. Curr Biol. 2009;19:1730–1735. doi: 10.1016/j.cub.2009.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson GT, Aggeler R, Oglesbee D, Cannon M, Capaldi RA, Tsien RY, Remington SJ. Investigating mitochondrial redox potential with redox-sensitive green fluorescent protein indicators. J Biol Chem. 2004;279:13044–13053. doi: 10.1074/jbc.M312846200. [DOI] [PubMed] [Google Scholar]
- Harris N, Costa V, MacLean M, Mollapour M, Moradas-Ferreira P, Piper PW. Free Radic Biol Med. 2003. Mnsod overexpression extends the yeast chronological (G(0)) life span but acts independently of Sir2p histone deacetylase to shorten the replicative life span of dividing cells; pp. 1599–1606. [DOI] [PubMed] [Google Scholar]
- Heeren G, Jarolim S, Laun P, Rinnerthaler M, Stolze K, Perrone GG, Kohlwein SD, Nohl H, Dawes IW, Breitenbach M. The role of respiration, reactive oxygen species and oxidative stress in mother cell-specific ageing of yeast strains defective in the RAS signalling pathway. FEMS Yeast Res. 2004;5:157–167. doi: 10.1016/j.femsyr.2004.05.008. [DOI] [PubMed] [Google Scholar]
- Itoh T, Toh-E A, Matsui Y. Mmr1p is a mitochondrial factor for Myo2p-dependent inheritance of mitochondria in the budding yeast. EMBO J. 2004;23:2520–2530. doi: 10.1038/sj.emboj.7600271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang JC, Jaruga E, Repnevskaya MV, Jazwinski SM. FASEB J. 2000. An intervention resembling caloric restriction prolongs life span and retards aging in yeast; pp. 2135–2137. [DOI] [PubMed] [Google Scholar]
- Kaeberlein M, Hu D, Kerr EO, Tsuchiya M, Westman EA, Dang N, Fields S, Kennedy BK. Increased life span due to calorie restriction in respiratory-deficient yeast. PLoS Genet. 2005;1:e69. doi: 10.1371/journal.pgen.0010069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeberlein M, McVey M, Guarente L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 1999;13:2570–2580. doi: 10.1101/gad.13.19.2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy BK, Austriaco NR, Guarente L. Daughter cells of Saccharomyces cerevisiae from old mothers display a reduced life span. J Cell Biol. 1994;127:1985–1993. doi: 10.1083/jcb.127.6.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchman PA, Kim S, Lai CY, Jazwinski SM. Interorganelle signaling is a determinant of longevity in Saccharomyces cerevisiae. Genetics. 1999;152:179–190. doi: 10.1093/genetics/152.1.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchman PA, Miceli MV, West RL, Jiang JC, Kim S, Jazwinski SM. Acta Biochim Pol. 2003. Prohibitins and Ras2 protein cooperate in the maintenance of mitochondrial function during yeast aging; pp. 1039–1056. [PubMed] [Google Scholar]
- Klinger H, Rinnerthaler M, Lam YT, Laun P, Heeren G, Klocker A, Simon-Nobbe B, Dickinson JR, Dawes IW, Breitenbach M. Experimental gerontology. 2010. Quantitation of (a)symmetric inheritance of functional and of oxidatively damaged mitochondrial aconitase in the cell division of old yeast mother cells; pp. 533–542. [DOI] [PubMed] [Google Scholar]
- Lai CY, Jaruga E, Borghouts C, Jazwinski SM. A mutation in the ATP2 gene abrogates the age asymmetry between mother and daughter cells of the yeast Saccharomyces cerevisiae. Genetics. 2002;162:73–87. doi: 10.1093/genetics/162.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam YT, Aung-Htut MT, Lim YL, Yang H, Dawes IW. Free Radic Biol Med. 2011. Changes in reactive oxygen species begin early during replicative aging of Saccharomyces cerevisiae cells; pp. 963–970. [DOI] [PubMed] [Google Scholar]
- Laun P, Pichova A, Madeo F, Fuchs J, Ellinger A, Kohlwein S, Dawes I, Fröhlich KU, Breitenbach M. Mol Microbiol. 2001. Aged mother cells of Saccharomyces cerevisiae show markers of oxidative stress and apoptosis; pp. 1166–1173. [PubMed] [Google Scholar]
- Lavoie H, Whiteway M. Increased respiration in the sch9Delta mutant is required for increasing chronological life span but not replicative life span. Eukaryotic cell. 2008;7:1127–1135. doi: 10.1128/EC.00330-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S-J, Ford E, Haigis M, Liszt G, Guarente L. Calorie restriction extends yeast life span by lowering the level of NADH. Genes & Development. 2004;18:12–16. doi: 10.1101/gad.1164804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S-J, Kaeberlein M, Andalis AA, Sturtz LA, Defossez P-A, Culotta VC, Fink GR, Guarente L. Calorie restriction extends Saccharomyces cerevisiae lifespan by increasing respiration. Nature. 2002;418:344–348. doi: 10.1038/nature00829. [DOI] [PubMed] [Google Scholar]
- Longo VD, Gralla EB, Valentine JS. J Biol Chem. 1996. Superoxide dismutase activity is essential for stationary phase survival in Saccharomyces cerevisiae. Mitochondrial production of toxic oxygen species in vivo; pp. 12275–12280. [DOI] [PubMed] [Google Scholar]
- Miquel J, Economos AC. Favorable effects of the antioxidants sodium and magnesium thiazolidine carboxylate on the vitality and life span of Drosophila and mice. Exp Gerontol. 1979;14:279–285. doi: 10.1016/0531-5565(79)90039-1. [DOI] [PubMed] [Google Scholar]
- Mittal N, Babu MM, Roy N. Aging Cell. 2009. The efficiency of mitochondrial electron transport chain is increased in the long-lived mrg19 Saccharomyces cerevisiae; pp. 643–653. [DOI] [PubMed] [Google Scholar]
- Mortimer RK, Johnston JR. Life span of individual yeast cells. Nature. 1959;183:1751–1752. doi: 10.1038/1831751a0. [DOI] [PubMed] [Google Scholar]
- Nestelbacher R, Laun P, Vondrakova D, Pichova A, Schuller C, Breitenbach M. The influence of oxygen toxicity on yeast mother cell-specific aging. Exp Gerontol. 2000;35:63–70. doi: 10.1016/s0531-5565(99)00087-x. [DOI] [PubMed] [Google Scholar]
- Peraza-Reyes L, Crider DG, Pon LA. Mitochondrial manoeuvres: latest insights and hypotheses on mitochondrial partitioning during mitosis in Saccharomyces cerevisiae. Bioessays. 2010;32:1040–1049. doi: 10.1002/bies.201000083. [DOI] [PubMed] [Google Scholar]
- Piper PW, Jones GW, Bringloe D, Harris N, MacLean M, Mollapour M. Aging Cell. 2002. The shortened replicative life span of prohibitin mutants of yeast appears to be due to defective mitochondrial segregation in old mother cells; pp. 149–157. [DOI] [PubMed] [Google Scholar]
- Schriner SE, Linford NJ, Martin GM, Treuting P, Ogburn CE, Emond M, Coskun PE, Ladiges W, Wolf N, Van Remmen H, Wallace DC, Rabinovitch PS. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science. 2005;308:1909–1911. doi: 10.1126/science.1106653. [DOI] [PubMed] [Google Scholar]
- Shcheprova Z, Baldi S, Frei SB, Gonnet G, Barral Y. A mechanism for asymmetric segregation of age during yeast budding. Nature. 2008;454:728–734. doi: 10.1038/nature07212. [DOI] [PubMed] [Google Scholar]
- Sinclair DA, Guarente L. Extrachromosomal rDNA circles--a cause of aging in yeast. Cell. 1997;91:1033–1042. doi: 10.1016/s0092-8674(00)80493-6. [DOI] [PubMed] [Google Scholar]
- Smith ED, Tsuchiya M, Fox LA, Dang N, Hu D, Kerr EO, Johnston ED, Tchao BN, Pak DN, Welton KL, Promislow DE, Thomas JH, Kaeberlein M, Kennedy BK. Quantitative evidence for conserved longevity pathways between divergent eukaryotic species. Genome Res. 2008;18:564–570. doi: 10.1101/gr.074724.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stöckl P, Zankl C, Hütter E, Unterluggauer H, Laun P, Heeren G, Bogengruber E, Herndler-Brandstetter D, Breitenbach M, Jansen-Dürr P. Free Radic Biol Med. 2007. Partial uncoupling of oxidative phosphorylation induces premature senescence in human fibroblasts and yeast mother cells; pp. 947–958. [DOI] [PubMed] [Google Scholar]
- Sun J, Tower J. FLP recombinase-mediated induction of Cu/Zn-superoxide dismutase transgene expression can extend the life span of adult Drosophila melanogaster flies. Mol Cell Biol. 1999;19:216–228. doi: 10.1128/mcb.19.1.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, Alroy J, Wu M, Py BF, Yuan J, Deeney JT, Corkey BE, Shirihai OS. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27:433–446. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Zandycke SM, Sohier PJ, Smart KA. The impact of catalase expression on the replicative lifespan of Saccharomyces cerevisiae. Mech Ageing Dev. 2002;123:365–373. doi: 10.1016/s0047-6374(01)00382-7. [DOI] [PubMed] [Google Scholar]
- Wikstrom JD, Katzman SM, Mohamed H, Twig G, Graf SA, Heart E, Molina AJ, Corkey BE, de Vargas LM, Danial NN, Collins S, Shirihai OS. beta-Cell mitochondria exhibit membrane potential heterogeneity that can be altered by stimulatory or toxic fuel levels. Diabetes. 2007;56:2569–2578. doi: 10.2337/db06-0757. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.