

Abstract
Aims
For the rapid detection of P. aeruginosa from chlorinated water and aerosols, gyrB gene-based real-time PCR assay was developed and investigated.
Methods and Results
Two novel primer sets (pa722F/746MGB/899R and pa722F/746MGB/788R) were designed using the most updated 611 Pseudomonas and 748 other bacterial gyrB genes for achieving high specificity. Their specificity showed 100% accuracy when tested with various strains including clinical isolates from cystic fibrosis patients. The assay was tested with P. aeruginosa-containing chlorinated water and aerosols to simulate the waterborne and airborne transmission routes (detection limit 3.3 × 102 CFU·PCR−1 − 2.3 × 103 CFU·PCR−1). No chlorine interference in real-time PCR was observed at drinking water level (~ 1 mg·L−1), but high level of chorine (12 mg·L−1) interfered the assay, thus neutralization was needed. P. aeruginosa in aerosol was successfully detected after capturing with gelatin filters with minimum 2 min of sampling time when the initial concentration of 104 CFU·mL−1 bacteria existed in the nebulizer.
Conclusions
A highly specific and rapid assay (2–3 hrs) was developed by targeting gyrB gene for the detection of P. aeruginosa in chlorinated water and aerosols, combined with optimized sample collection methods and sample processing, so the direct DNA extraction from either water or aerosol was possible while achieving the desired sensitivity of the method.
Significance and Impact
The new assay can provide timely and accurate risk assessment to prevent P. aeruginosa exposure from water and aerosol, resulting in reduced disease burden, especially among immune-compromised and susceptible individuals. This approach can be easily utilized as a platform technology for the detection of other types of microorganisms, especially for those that are transmitted via water and aerosol routes, such as Legionella pneumophila.
Keywords: gyrB, Pseudomonas aeruginosa, real-time PCR, water, aerosol
1. Introduction
Pseudomonas aeruginosa is a Gram-negative rod bacterium, which has a remarkable ability to adapt and thrive in a variety of environments: water (Pellett et al. 1983; Kimata et al. 2004); soil (Cavalca et al. 2000); occupational places, such as metal working fluids (Karadzic et al. 2006); clinical settings (Wolfgang et al. 2003); hospital and municipal wastewater (Schwartz et al. 2006); and industrial effluents (Karadzic et al. 2006). P. aeruginosa has been found in non-treated bottled mineral water (Hunter 1993; Naze et al. 2010), tap water (Trautmann et al. 2001), and water distribution systems (Emde et al. 1992). P. aeruginosa is a primary agent leading to otitis externa (Reid and Porter 1981), folliculitis (Chandrasekar et al. 1984), and is a common cause of severe nosocomial infection and respiratory distress (Cobben et al. 1996). Its ability to colonize in various environments and its contact with weakened populations causes several cases of community-acquired infections each year (Lavenir et al. 2007). P. aeruginosa infection is the primary and most important cause of morbidity and mortality in people with the genetic disease cystic fibrosis (CF) (West et al. 2002). These patients frequently develop chronic pneumonia after exposure to P. aeruginosa (Granstrom et al. 1984; Armstrong et al. 1995; Quittner et al. 2009).
There have been a number of outbreaks of P. aeruginosa in which water consumption and aerosol inhalation were implicated as the sources of infection. Patients infected with P. aeruginosa showed the same genotypes that were detected in tap water (Trautmann et al. 2001; Bert et al. 1998). This type of problem was resolved after the plumbing fixtures were changed (Ferroni et al. 1998). In the case of aerosol exposure, nosocomial P. aeruginosa colonization and infections were reported in mechanically ventilated patients (Doring et al. 1991; Berthelot et al. 2001; Valles et al. 2004).
The simple and rapid detection of P. aeruginosa is needed in order to more effectively protect public health. This is especially true in hospital and clinical settings where there is high risk for contamination and growth, exposure and spread, and infection of susceptible individuals. Testing that is more practical and rapid will lead to more prevalent testing and earlier detection so that intervention strategies can be implemented to reduce human exposure and infection. Any circumstances and environments that involve immune-compromised and susceptible individuals, such as CF patients, can also get benefit from it (Rutala et al. 2008). Currently, polymerase chain reaction (PCR) is most widely used because of its reliability and sensitivity although it does not provide information related to the viability of the detected P. aeruginosa (Khan and Yadav 2004; Anuj et al. 2009; Deschaght et al. 2009; Feizabadi et al. 2010; Subrayan et al. 2010). For the genetic identification and characterization of Pseudomonas species via PCR-based methods, various targets have been reported, such as 16S rRNA (Relman et al. 1992), toxA (Khan and Cerniglia 1994), oprI, oprL (De Vos et al. 1997), algD (da Silva Filho et al. 2004), and gyrB (Qin et al. 2003). The 16S ribosomal RNA (rRNA) gene is most commonly used, but it is not feasible to develop highly specific primer and probe sets using this gene due to the high similarities of the 16S rRNA gene sequences (Moore et al. 1996; Yamamoto et al. 2000). Genetic exchanges with other non-P. aeruginosa species can impact upon the specificities of assays targeting the oprI and oprL genes (Lavenir et al. 2007). PCR assays targeting the toxA and algD genes had false-negative results due to the sequence variation in highly polymorphic P. aeruginosa (Qin et al. 2003; Lavenir et al. 2007). Due to a higher molecular evolution rate and less horizontal gene transfer (Kasai et al. 1998; Anzai et al. 2000), real-time PCR assay targeting the gyrB gene has been designed and evaluated as a highly sensitive and specific assay when tested with P. aeruginosa strains and species of pseudomonads closely related to P. aeruginosa (Lavenir et al. 2007). However, we sought to improve the gyrB gene-based assay through this study because the previous P. aeruginosa gyrB-specific primers were designed based on multi-sequence alignment of 88 gyrB sequence entries within the genus Pseudomonas (Lavenir et al. 2007). At present, gyrB sequences including whole genome sequences have risen to more than 600 entries under the genus Pseudomonas in the GenBank database.
The first objective of this study was to design specific primers and probe for the detection P. aeruginosa by TaqMan real-time PCR assay targeting the gyrB gene. The second was to optimize a method of DNA extraction directly from water and aerosol samples without any enrichment of the sample. The efficient concentration and high-throughput sample preparation from water and aerosol combined with real-time PCR would provide a versatile tool for investigating P. aeruginosa occurring from nosocomial, waterborne, and airborne transmission. This methodology potentially provides timely, quantitative results that would have broad applications including source identification, exposure and risk assessment, and epidemiological investigations all supporting disease prevention.
2. Materials and methods
2.1. Bacterial strains
Seven clinical (FRD1, 6077, PAO1, X13272, CF27, 08-066, and PA14) and three environmental (MSH10, MSH3, and E2) P. aeruginosa strains were obtained from Dr. Daniel Wozniak at The Ohio State University and they were used for enumeration, recovery, and specificity tests (Table 1). P. aeruginosa 10145T was obtained from the American Type Culture Collection (ATCC, Rockville, MD, USA). P. fluorescens ATCC 49838, P. putida ATCC 49128, and P. stutzeri ATCC 17588 T were obtained from Microbiologics® Inc (St. Cloud, MN, USA). All P. aeruginosa strains were cultured on Lysogeny Broth (LB) agar or Tryptic Soy Agar (TSA) (BD Diagnostics, Sparks, MD, USA) when they were recovered from the stock cultures. Pseudomonas isolation agar (PIA) plates (BD Diagnostics, Sparks, MD, USA) were used and incubated at 25°C for 24 (water) or up to 48 hours (aerosol) when tested with samples. The 7 non-Pseudomonas bacterial type strains were purchased from the American Type Culture Collection (ATCC). Salmonella enterica subsp. enterica ATCC 19585T, Bacillus subtilis subsp. subtilis ATCC 6051T, Bacillus cereus ATCC 14579T, and Bacillus licheniformis ATCC 14580T were cultured on TSA plates. Bacteroides fragilis ATCC 25285T, Bacteroides ovatus ATCC 8483T, and Prevotella melaninogenica ATCC 25845T were prepared as described in a previous study (Lee and Lee, 2010). Stock cultures were maintained in Tryptic Soy Broth (TSB) (BD Diagnostics, Sparks, MD, USA) media supplemented with 20 % glycerol (Fisher Scientific, Pittsburgh, PA, USA) and were frozen at −80 °C until needed.
Table 1.
Sequences of amplified gyrB gene by PCR.
| Strain | Most similar reference strain by gyrB sequencing | Accession # | Similarity (%) | E value | Source |
|---|---|---|---|---|---|
| ATCC 10145T | P. aeruginosa ATCC 10145 T | AB039386 | 100 | 0.0 | Type strain* |
| MSH10 | P. aeruginosa ATCC 10145 T | AB039386 | 99 | 5e-105 | Environmental isolate, water |
| MSH3 | P. aeruginosa ATCC 10145 T | AB039386 | 93 | 1e-96 | Environmental isolate, water |
| FRD1 | P. aeruginosa ATCC 27853 | EF064840 | 97 | 2e-51 | Mucoid clinical isolate, cystic fibrosis |
| 6077 | P. aeruginosa SC-1 | FJ652724 | 91 | 2e-45 | Non-mucoid, clinical isolate, cornea |
| PAO1 | P. aeruginosa PAO1 | AE004091 | 100 | 0.0 | Non-mucoid, clinical isolate |
| X13272 | P. aeruginosa ATCC 25001 | AB039386 | 99 | 5e-51 | Non-mucoid, clinical isolate, blood |
| PA14 | P. aeruginosa ATCC 10145 T | AB039386 | 96 | 1e-111 | Non-mucoid |
| E2 | P. aeruginosa ATCC 25011 | FJ652721 | 94 | 5e-48 | Environmental isolate, tomato plant |
| CF27 | P. aeruginosa ATCC 25011 | FJ652721 | 94 | 5e-48 | Non-mucoid, clinical isolate, cystic fibrosis |
| 08-066 | P. aeruginosa ATCC 25011 | FJ652721 | 99 | 5e-63 | Non-mucoid, clinical isolate, cystic fibrosis |
ATCC, American Type Culture Collection, Manassas, Va.; other strains were received from Dr. Wozniak’s lab the Ohio State University.
2.2. Primer and probe design
A total of 611 gyrB sequences of Pseudomonas genus were retrieved from National Center for Biotechnology Information’s GenBank database (http://www.ncbi.nlm.nih.gov) and gathered into the Bioedit sequence alignment editor v.7.0 software (Hall 1999) and then aligned using Clustal X software (Larkin et al. 2007). From homologous regions, two sets of forward/reverse primers and a minor groove binder (MGB) probe were determined by Primer Express software (Applied Biosystems, Foster City, CA, USA). The specificity of the designed gyrB primers and probes was checked by comparing with 748 other non-Pseudomonas bacterial gyrB genes. In the MGB probe, FAM (6-carboxy-fluorescein) was conjugated at the 5′ end as a reporter dye.
2.3. Specificity tests
The specificity of designed primer pairs for P. aeruginosa-specific assay was evaluated using end-point PCR amplification-sequencing analyses. We tested PCR amplification by gyrB-based primers with genomic DNA templates from 11 P. aeruginosa strains, 3 non-P. aeruginosa strains, and 7 non-Pseudomonas bacterial type strains. DNA templates were extracted using a DNeasy Blood &Tissue Kit (Qiagen, Valencia, CA, USA) by following the manufacturer’s instructions. DNA concentration was measured with NanoDrop (NanoDrop Technologies, Wilmington, DE, USA) and adjusted to 10 ng per 1 PCR tube for template DNA. PCR was conducted using a MultiGene Thermal Cycler (Labnet International, Inc., Edison, NJ, USA) by incubation for 5 min at 94°C, 30 cycles of denaturation at 94°C for 1 min, annealing at 60°C for 30 s, extension at 72°C for 1 min, and final extension at 72°C for 10 min. After PCR amplification, aliquots (5 μL) of the PCR products were analyzed by electrophoresis on a 1.0% agarose gel (Invitrogen, Carlsbad, CA, USA) stained with 0.1 μg·mL−1 ethidium bromide (Fisher Bio Reagents, Fisher Scientific, Pittsburgh, PA, USA) in TBE buffer (89 mmol·L−1 Tris–borate, 2 mmol·L−1 EDTA; National Diagnostic, Atlanta, GA, USA) at 100 V for 40 min. The bands were documented with a Bio-Rad Quantity One Gel Doc system (Bio-Rad, Hercules, CA, USA) using a 1 kb DNA ladder (Invitrogen, Carlsbad, CA, USA) as a molecular weight marker. For the confirmation of the identity, all the PCR amplified products were sequenced using ABI Prism 3730 DNA analyzer (Applied Biosystems, Foster City, CA, USA) at the Plant-Microbe Genomics Facility of The Ohio State University (http://pmgf.biosci.ohio-state.edu/).
2.4. Standard curves
Standard curves for P. aeruginosa were generated using real-time PCR assay with the ABI 48-well StepOne™ real-time System (Applied Biosystems, Foster City, CA, USA). At first, 10-fold serial dilutions of P. aeruginosa cells were prepared after P. aeruginosa ATCC 10145T was grown on PIA plates. One colony of P. aeruginosa was suspended in 1 mL of sterile phosphate buffered saline (PBS; 0.14 mol·L−1 NaCl, 2.7 mmol·L−1 KCl, 10.1 mmol·L−1 Na2HPO4, 1.8 mmol·L−1 KH2PO4 [pH 7.6], Fisher Scientific, Pittsburgh, PA, USA) and aliquots of 100 μL were serially diluted (1:10 dilution) with 900 μL PBS. Aliquots of 10 μL from the serial dilution set were spread on PIA plates and incubated for 24 hours at 25°C to count colony forming units (CFU). DNA was extracted from the 100 μL serial dilution set with a QIAamp DNA Stool Mini Kit (Qiagen, Valencia, CA, USA) in accordance with the manufacturer’s instructions. From the final 200 μL of DNA extraction, 5 μL of DNA was used as a DNA template. Two gyrB-based primers were compared with a commercially available TaqMan® Pseudomonas aeruginosa Detection Kit (Applied Biosystems, Foster City, CA) using the 48-well StepOne™ real-time System. The PCR reaction consisted of a total volume of 25 μL mixture containing 5 μL of template DNA, 12.5 μL of TaqMan® universal PCR master mix (Applied Biosystems, Foster City, CA, USA), 500 nM of each primer, and 250 nM of each probe labeled with 6-carboxy fluorescein (FAM). Thermal cycling consisted of an initial cycle at 50°C for 2 min (activation of the UNG) and 95°C for 10 min (activation of the AmpliTaq Gold DNA polymerase), followed by 40 cycles of denaturation at 95°C for 30 s, and annealing and extension at 60°C for 1 min 30 s. A mixture of all PCR reagents without any template DNA was used as a negative control for each PCR reaction. Standard curves were derived by plotting threshold cycle (CT) values against the log-transformed cell counts (CFU·PCR−1) in triplicate.
2.5. Detection of P. aeruginosa in water
In order to examine possible interferences (e.g. water matrix, chlorine concentration, non-bacterial cells) on the performance of real-time PCR assay, P. aeruginosa MSH10, MSH3 and PAO1 were prepared and spiked into 250 mL of various water samples, such as PBS, swimming pool water, and tap water. The spiked solutions were filtered through a nylonmembrane filter (pore size 20.0 μm, Magna, GE Water &Process Technologies, Trevose, PA, USA) to remove any large particles while allowing passage of bacteria. After the prefiltration, the samples were filtered through a 0.45-μm-pore-size, 47-mm-diameter cellulose membrane filter (Millipore, Bedford, MA, USA) to concentrate bacteria present in the water samples. The filtered membranes were put into a sterile 15 mL disposable tube with 2 mL of sterile PBS solution and then vortexed. Mild sonication (40 ~ 45 kHz) was carried out to detach cells from the filter membrane efficiently for 1 min at 25°C (FS 20, Fisher Scientific, Pittsburgh, PA, USA). After the sonication, the resuspended cells were centrifuged at 10,000 g for 15 min at 4 °C and then the supernatant was gently removed. To remove non-bacterial cells that might be present in water samples, such as eukaryotic cells, 250 μl of somatic cell releasing agent (SRA, New Horizons Diagnostics Corp., Columbia, MD, USA) was added to the pellet. The mixture was centrifuged again in the same way and the supernatant was removed to wash out non-bacterial cells (Lee and Deininger 2010). The harvested cells were resuspended in 1.4 mL of ASL buffer (Qiagen, Valencia, CA, USA) and DNA was extracted using a QIAamp DNA Stool Mini Kit. The final eluted DNA solution was evaporated using a Savant DNA 120 SpeedVac (Savant Instruments, Farmingdale, NY, USA) and then it was reconstituted. The concentration of the DNA was determined with a NanoDrop system (NanoDrop Technologies, Wilmington, DE, USA) and the final DNA concentrate was used for real-time PCR detection.
Since chlorine is usually added in a water distribution system to suppress microbial regrowth, the effect of residual chlorine in water on the real-time PCR assay was investigated using tap water and swimming pool water samples, representing low- and high-chlorine concentrations, respectively. For this, P. aeruginosa PAO1 (tap water) and P. aeruginosa MSH 10 and MSH3 (swimming pool) were spiked into 250 mL water samples. In parallel, both types of water samples were dechlorinated with Na2S2O3 (Sigma-Aldrich, St. Louis, MO, USA) that neutralizes any residual halogens and thus prevents continuation of bactericidal action (American Public Health Association, 1992). Total chlorine was detected as 12.0 and 0.7 mg·L−1 from swimming pool and tap water samples, respectively, using Hach DR 2800 spectrophotometer (Hach, Loveland, CO, USA) as described in the manufacturer’s instructions. For neutralizing chlorine in swimming pool and tap water samples, 167 μL and 49 μL of 10 % Na2S2O3 solution was added, respectively.
2.6. Detection of P. aeruginosa in aerosol
P. aeruginosa PAO1 strain was aerosolized using a Collison nebulizer (BGI Inc., Waltham, MA, USA) operated at 3 L·min−1. The aerosol was directed into a HEPA-filtered single-pass chamber constructed of Lucite 122 × 25.4 × 25.4 cm giving a volume of 78.7 L. The chamber was placed within a 1300 Series A2 biological safety cabinet (Thermo Scientific, Asheville, NC, USA). The nebulizer was loaded with ~104 CFU·mL−1 of P. aeruginosa PAO1 in 0.9% sterile saline solution. The bacteria concentration of the nebulizer suspension was determined pre- and post- aerosol generation using the plate count method. Aerosol samples were collected using two sampling approaches. The first employed an impactor (Andersen sampler, Atlanta, GA, USA) loaded with 100 × 15 mm PIA plates and operated at a flow rate of 28.3 L·min−1. Samples were collected over varying intervals: 2, 5, 10, and 15 minutes. Upon completion of sampling, the PIA plates were removed from the impactor and were incubated for 24 hours at 25°C. After the incubation, the number of colonies were counted using a Quebec Darkfield colony counter (Reichert Products, New York, NY, USA). After taking into account the volume of air sampled, the P. aeruginosa concentration in the aerosol was calculated as CFU per cubic meter (CFU·m−3). All experiments were done in triplicate. This approach provided an estimate of the culturable P. aeruginosa.
The second air sampling method employed gelatin filters (Sartorius Stedim Biotech, Gottingen, Germany), which were loaded into polypropylene filter cassettes (47 mm diameter, Omega Specialty Instrument Co., Chelmsford, MA, USA). Sampling was conducted at a flow rate of 3L·min−1. For both methods, sampling was repeated three times across four time intervals, i.e. 2, 5, 10, and 15 minutes… Gelatin filters were retrieved from the cassettes and then mixed with 1.4 mL of ASL buffer solution, heated at 70°C for 5 minutes and vortexed. To this solution, 15 μL of proteinase K solution (Qiagen, Valencia, CA) and 1 mL of buffer AL were added. After vortexing, the solution was heated for 10 minutes at 70°C. One mL of ethanol was added into this solution and it was vortexed. The solution was transferred into a spin column that comes with a collection tube (Qiagen, Valencia, CA) and per one aerosol sample, it generated 4 aliquots with final volume of 800 μL. The columns and the collection tubes were centrifuged at 6,000 g for 2 min. The remaining steps were followed according to the manufacturer’s instructions included in the QIAamp DNA Stool Mini Kit. All plate counts and real-time PCR measurements were done in duplicate.
Concurrent with aerosol collection, the chamber air was monitored using a direct-reading laser time of flight particle spectrophotometer (Aerodynamic Particle Sizer, TSI Incorporated, Shoreview, MN, USA). This device counts and sizes particles in the size range of 0.523 ~ 19.81 μm and was set to record 20 second integrated readings.. Humidity and temperature were monitored throughout all aerosol generation runs.
2.7. Statistics
PCR-negative was given a value of 1 CFU/PCR for statistical purposes. Statistical significance was defined as p <0.05. The potential effects of water matrix and chlorine concentration on the detection of P. aeruginosa with the real-time PCR were evaluated using analysis of covariance (ANCOVA). Statistical analyses were performed using the R statistical language (R Development Core Team, 2009).
3. Results
3.1. Specificity of designed primer
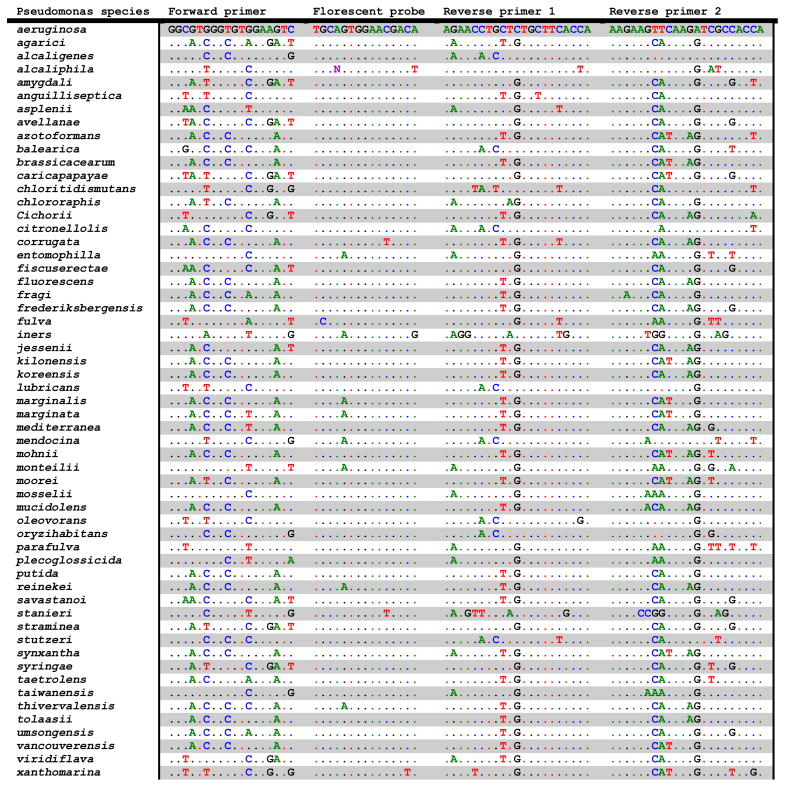
Two primer/probe pairs were designed for the P. aeruginosa specific TaqMan real-time PCR system (Table 2). One primer pair, pa722F/746MGB/899R, consisting of a forward (722F: 5′-GGCGTGGGTGTGGAAGTC-3′), a reverse (899R: 5′-TGGTGGCGATCTTGAACTTCTT-3′), and a probe (746MGB: 5′-6FAM-TGCAGTGGAACGACA-MGBNFQ-3′) allows amplifying the 190 bp gyrB gene segment of P. aeruginosa species. The other primer pair, pa722F/746MGB/788R has a different reverse primer (788R: 5′-TGGTGAAGCAGAGCAGGTTCT-3′) and the expected size of amplified product is 67 bp. The major factor for the specificity to P. aeruginosa was the forward primer (pa722F), which has the only unique sequence to P. aeruginosa among 57 Pseudomonas species. One mismatch was found from P. entomophila (AY907567) and P. mosselii (FJ418640) and two mismatches were found from P. monteilii (FJ418641), P. parafulva (FJ418638) and P. taiwanensis (FJ418634). Two gyrB-based reverse primers and a probe, pa788R, 899R and 746MGB, were regarded as less specific to P. aeruginosa than pa722F.
Table 2.
Comparison of sequence homology among the 57 species of Pseudomonas using gyrB gene and sequence information of P. aeruginosa-specific primers/probe. Dots indicate conserved sequences between P. aeruginosa and other Pseudomonas species.
|
The specificity of each gyrB-based primer pair (pa722F/899R and pa722F/788R) was evaluated with the 11 P. aeruginosa strains, 3 non-P. aeruginosa strains and 7 non-Pseudomonas strains. As expected from the sequence data, each primer pair amplified fragments of 190 and 67 bp, respectively, out of 11 Pseudomonas strains and 0 from 10 other bacteria (3 non-P. aeruginosa and 7 non-Pseudomonas bacteria) (data not shown). Real-time PCR results showed that all 11 strains of P. aeruginosa generated positive fluorescent signals between 15 to 20 cycles, while the 3 non-P. aeruginosa and 7 non-Pseudomonas strains produced no signals until 40 cycles. From the obtained PCR products from the 11 P. aeruginosa, sequencing analysis was carried out to check P. aeruginosa identification using pa722F/899R and they were all identified as gyrB genes of P. aeruginosa.
3.2. Real-time PCR and standard curves
A series of experiments were conducted to compare the results obtained using our gyrB-based pa722F/746MGB/899R, pa722F/746MGB/788R, and other P. aeruginosa detection kit (TaqMan® Pseudomonas aeruginosa Detection Kit, Applied Biosystems, Foster City, CA, USA) (Figure 1). P. aeruginosa genomic DNAs were extracted from serial dilutions of P. aeruginosa ATCC 10145T of which the concentration was determined as colony forming units (CFU) to determine the detection limit and the quantitative range of the designed real-time PCR assay using each primer set. The standard curves demonstrated that the two gyrB assays and one kit assay were linear over the range 2.3 × 102 – 5.0 × 106 CFU·PCR−1, corresponding to a threshold cycle (CT) ranging from 17.0 to 36.2 cycles. The minimum quantification limits of the assays for pa722F/746MGB/788R, pa722F/746MGB/899R, and reference kit were 3.3 × 102, 2.3 × 103, and 2.3 × 102 CFU·PCR−1, respectively. As shown in Figure 1, the gyrB-based pa722F/746MGB/788R assay also enabled detection of the genomic DNA at an average of 4.54 cycles sooner than the assay with pa722F/746MGB/899R. Due to the observed increased sensitivity and shorter detection times, the pa722F/746MGB/788R primers and probe set were selected for use in subsequent water and aerosol qPCR studies. Assuming a constant number of gyrB gene copies per cell, the calculated calibration curve could be used to convert the assessed CT value of water and aerosol samples into cell counts in our study.
Figure 1.

Standard curves generated using short- (
 ), long- (
), long- (
 ) gyrB-based primers and a commercial kit (
) gyrB-based primers and a commercial kit (
 ).
).
3.3. Detection of P. aeruginosa in water
In the case of the directly-extracted DNA from filters through which P. aeruginosa-spiked samples were filtered, the possible interferences of the natural water matrix, filter material, and the presence of other bacteria were not detected in the real-time PCR detection of P. aeruginosa. The fluorescence signals with the normal shape of amplification plot were observed and the CT values from filtered membranes were determined and compared with the standard curves. The results demonstrated that the presence of P. aeruginosa in the membrane produced similar CT values when detected from pure culture by the gyrB-based assay (data not shown).
Analysis of covariance (ANCOVA) was performed with the fitted curves from pure culture samples and the spiked samples (membrane filtration) in order to determine whether the slope and intercept between these two curves were similar or not. The comparison of the slopes of both equations using ANCOVA indicated no significant difference (P = 0.53) between the two systems in the efficiency of the designed primer regardless of the presence of filter and water matrices. It also indicated that the entire procedure of real-time PCR detection including DNA extraction and purification effectively removed possible interference that may have been present in the water and membrane matrices. A variety of negative controls including sterile MilliQ water (Milliport Corp., Bedford, MA, USA), membrane filters, and 7 non-Pseudomonas produced no false cross-reaction in TaqMan real-time PCR detection (data not shown).
After neutralization, satisfactory dechlorination was carried out since the resulting total chlorine concentrations were observed at 0.0 mg·L−1 in all the samples. No significant difference was found between the tap water (0.7 mg·L−1) and neutralized tap water (0.0 mg·L−1) in terms of the performance of real-time PCR (Table 3). Both CT values and appearances of the two amplification plots were similar between tap water and dechlorinated tap water. Results indicate that a low chlorine concentration (0.7 mg·L−1) did not cause interference in real-time PCR with the same DNA templates. In the case of highly chlorinated water, such as swimming pool water, results indicated that a high chlorine concentration (12.0 mg/·L−1) significantly interfered with the PCR. However, treatments with and without somatic cell releasing agent (SRA) did not show significant differences in PCR (CT) (Table 3).
Table 3.
Interference of high and low chlorine on real-time PCR detection of P. aeruginosa. For comparison, another set of samples were neutralized before real-time PCR. SRA (somatic cell releasing agent) was used to remove possible non-bacterial cells present in pool samples and to adjust pH. CT values are shown and converted CFU/PCR values are provided in parenthesis. Tests were done in duplicate.
| Swimming pool water (12 mg·L−1 of total chlorine) | Neutralized swimming pool water (0 mg·L−1 of total chlorine) | |||
|---|---|---|---|---|
| P. aeruginosa strain used for spiking | No SRA treatment | SRA treatment | No SRA treatment | SRA treatment |
| MSH10 | 31.67 ± 0.26 (1.6 ± 0.2 × 103) | 32.72 ± 0.00 (0.9 ± 0.0 × 103) | 18.24 ± 0.01 (1.8 ± 0.0 × 106) | 17.71 ± 0.08 (2.4 ± 0.1 × 106) |
| MSH3 | 32.47 ± 0.23 (1.1 ± 0.1 × 103) | 30.24 ± 0.01 (3.4 ± 0.0 × 103) | 22.69 ± 0.10 (1.8 ± 0.1 × 105) | 22.82 ± 0.03 (1.7 ± 0.0 × 105) |
| Tap water (0.7 mg·L−1 of total chlorine) | Neutralized tap water (0 mg·L−1 of total chlorine) | |||
| PAO1 | 25.34 ± 0.12 (4.4 ± 0.3 × 104) | 26.60 ± 0.06 (2.3 ± 0.1 × 104) | 25.99 ± 0.08 (3.2 ± 0.1 × 104) | 26.17 ± 0.07 (2.9 ± 0.1 × 104) |
3.4. Detection of P. aeruginosa in aerosol
For simulating P. aeruginosa transmission through bioaerosol, series of experiments of aerosol generation, collection and detection were performed. We developed an efficient procedure of bioaerosol capture, DNA extraction and concentration, and real-time PCR detection with good sensitivity that can rapidly detect the aerosolized target bacteria. An impinger works by bringing in air through an inlet and then the air was transmitted through a liquid medium where the air particles are trapped in the liquid. During our experiments, the liquid impinger media was divided into two sub samples to test bacterial concentration using a culture method and real-time PCR. During the preliminary experiments optimizing the entire procedure of bioaerosol detection, we found that the running time up to 30 minutes generated the best recovery of the aerolized P. aeruginosa, both measured with plate count and real-time PCR under our experimental conditions. Extending the aerosol capturing time longer than 30 minutes did not necessarily increase the bacterial recovery. After 15 minute collection, the level of recovered P. aeruginosa remained almost constant. A total of 4 air samples were collected each from the gelatin filters (real-time PCR) and from the impactors (plate count). Real-time PCR results were 5.8 × 104, 7.5× 104, 1.1 × 105, and 1.4 × 105 CFU·filter−1 at the air collection time of 2, 5, 10, 15 min, respectively, (Figure 2). The plate count results were 5.3 × 104, 2.0 × 105, 1.4 × 105, and 9.3 × 104 CFU·mL−1, respectively. The P. aeruginosa number before and after the experiments were 1.8 × 105 and 1.2 × 105 CFU·mL−1, respectively. At the same time, results from the laser particle spectrophotometer indicated a mean particle number concentration of ~1.0 × 102 to 5.0 × 102 particles·cm−3 (= 1.0 × 108 – 5.0 × 108 particles·m−3) with the mode in the particle size distribution occurring in the range of 1 – 3 μm particle size, which was regarded as the size range of bacteria.
Figure 2.

The levels of P. aeruginosa captured using gelatin filters and quantified with real-time PCR (pa722F/746MGB/788R). CT values were converted to CFU using the standard curve.
4. Discussion
This is a study to develop a novel P. aeruginosa specific real-time PCR assay to detect P. aeruginosa from water and aerosols using a combination of sampling optimization and TaqMan real-time PCR. The gyrB gene of P. aeruginosa was targeted for designing new primers because the gyrB gene, encoding a type II topoisomerase, has been known as a better candidate for the detection and identification of bacterial species (Fukushima et al. 2002; Lan et al. 2008). The sequences of gyrB gene of P. aeruginosa with other species of Pseudomonas revealed higher divergence in their sequences than 16S rRNA genes in our in silico analysis.
Two sets of primer and probe based on gyrB gene were developed to amplify 190 and 67 bp and they were designated as pa722F/746MGB/899R and pa722F/746MGB/788R, respectively. As shown in Table 1, E-value indicates exceptional specificity of the gyrB gene in detecting P. aeruginosa. E-value is a parameter that describes the statistical significance threshold for reporting the number of likelihood matches when being searched against the sequences available in Genbank database. The lower E-value means the more significant matches exist when compared with the sequences in the Genbank database (http://www.ncbi.nlm.nih.gov/blast/html/blastcgihelp.html). In general, E-value lower than 10−8 indicates highly acceptable sequence matches. Thus, the value (0 – 2× 10−45) in this study supports exceptional specificity of the gyrB gene-based assay in P. aeruginosa detection.
End-point PCR results showed 100% specificity for the detection of P. aeruginosa and the sequencing analysis confirmed that all amplified PCR products were P. aeruginosa. The results of real-time PCR showed that this assay could accurately discriminate and quantify P. aeruginosa from other bacteria including 3 non-P. aeruginosa (P. fluorescens, P. putida and P. stutzeri). Five species, P. entomophila, P. mosselii, P. monteilii, P. parafulva, and P. taiwanensis, showed only 1 or 2 bp mismatches when they were compared with the sequences of pa722F primer and showed 2 mismatches when compared with the reverse primer (pa788R) (Table 2). For discriminating these closely related 5 species from P. aeruginosa, a stringent control of annealing temperature can be an easy solution because the combination of pa722F and pa788R primer provides at least 3 mismatches and it can lower melting temperature (Tm) of the primer-template. In addition, the alternative reverse primer (pa899R) has at least 4 mismatches with these 5 species, so it can also provide high specificity to differentiate P. aeruginosa from other closely related species.
The detection limit of quantification using the pa722F/746MGB/788R primer and probe set was 3.3 × 102 CFU·PCR−1. This detection sensitivity was comparable to other study (ca. 102 CFU) (Motoshima et al. 2007). False positive and cross amplification was not observed. In order to compare the efficiency of newly developed system with the commercial kit, ANCOVA was performed with a null hypothesis: the slope of standard curve from pa722F/746MGB/788R assay is the same as the one from the commercial kit. The null hypothesis was not rejected, which means the slopes from the two systems are not significantly different (P > 0.05). It has been known that, only the ‘slope’ of regression lines of standard curves weigh in when determining the real-time PCR efficiency (E = 10[−1/slope] where E is efficiency) (Rasmussen 2001). Thus, it can be concluded that the newly designed assay is regarded as sensitive as the commercial kit and its use is warranted.
Possible factors that can influence on real-time PCR detection of P. aeruginosa were investigated: membrane filters, water matrix, presence of other bacteria and eukaryotic cells. It was found that all these factors did not interfere with the real-time PCR, except high chlorine level. High level of chlorine, such as swimming pools, appeared to interfere with the PCR. Thus, neutralization of the chlorine was needed before DNA extraction. However, chlorine concentration in water at low level, such as tap water, did not show interference in real-time PCR, so an additional neutralization step with sodium thiosulfate can be discarded. SRA was used to lyse and remove non-bacterial eukaryotic cells that might be present in swimming pool samples (Lee and Deninger 2010). There was no significant difference in CT values between the swimming pool water samples treated with SRA and without SRA, thus it implies that there were not many eukaryotic cells in the samples and the use of SRA may be more useful for handling hot tub or whirlpool samples that have more human cells.
To our knowledge, this is the first study to measure aerosolized P. aeruginosa using gelatin filter for bacterial capture and detecting with gyrB-based real-time PCR. Collection on the gelatin filters were especially conducive to direct DNA extraction. Upon aerosolization from a nebulizer loaded with 104 CFU·mL−, we were able to detect P. aeruginosa (5.8 × 104 CFU/filter) after a short 2 min sampling interval. soon as 2 min, when After 15 min of sampling recoveries about doubled to 1.4 × 105 CFU·filter−1. Plate count results show that the culturable counts were lower by 33% than the numbers estimated from the real-time PCR results. This discrepancy might be attributed to the use of impactors during collection of bacteria from the air. It might result in desiccation, injury and stress to the bacteria (Buttner et al. 1997).
In conclusion, the gyrB gene real-time PCR assay showed high sensitivity and specificity and can be used for quantitative detection of P. aeruginosa in water and air. The turnaround time for water and aerosol testing was less than 2 and 3 hours, respectively. The quantification limit was approximately 3.3 × 102 CFU·PCR−1. The estimated cost was about $ 10 per sample. This approach could be used to study the source of expulsion of P. aeruginosa in water and aerosol and has potential in monitoring of P. aeruginosa in clinical cases. It can also be applied for clinical environments (e.g. surgical intensive care unit, respiratory system) and indoor environments (e.g. fountains, mist) where high infection risks exist.
For further validation testing with larger number of environmental water and aerosol samples is recommended. A general limitation of PCR-based assays in using a single target gene is that it can produce potential false-positive or negative (Qin et al. 2003). For this, multiplex real-time PCR targeting more than one genetic marker, together with gyrB marker, would be suggested for further improvement minimize potential false-positive or negative results..
Acknowledgments
The project described was supported by Award Number UL1RR025755 from the National Center for Research Resources, funded by the Office of the Director, National Institutes of Health (OD) and supported by the NIH Roadmap for Medical Research. Dr. Daniel Wozniak was supported by Public Health Service grants AI061396 and HL058334. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Research Resources or the National Institutes of Health. We would like to thank Dr. Eric Lutz for the help in running the bioaerosol chamber and Jonathan Lutz for his feedback on the manuscript.
Footnotes
Note: Nucleotide sequence data reported are available in the DDBJ/EMBL/GenBank databases under the accession numbers HQ425710 to HQ425720
References
- American Public Health Association. Standard methods for the examination of water and wastewater. Washington, D.C: American Public Health Association, Inc; 1992. [Google Scholar]
- Anuj SN, Whiley DM, Kidd TJ, Bell SC, Wainwright CE, Nissen MD, Sloots TP. Identification of Pseudomonas aeruginosa by a duplex real-time polymerase chain reaction assay targeting the ecfX and the gyrB genes. Diagn Microbiol Infect Dis. 2009;63:127–131. doi: 10.1016/j.diagmicrobio.2008.09.018. [DOI] [PubMed] [Google Scholar]
- Anzai Y, Kim H, Park JY, Wakabayashi H, Oyaizu H. Phylogenetic affiliation of the pseudomonads based on 16S rRNA sequence. Int J Syst Evol Microbiol. 2000;50(Pt 4):1563–1589. doi: 10.1099/00207713-50-4-1563. [DOI] [PubMed] [Google Scholar]
- Armstrong DS, Grimwood K, Carzino R, Carlin JB, Olinsky A, Phelan PD. Lower respiratory infection and inflammation in infants with newly diagnosed cystic fibrosis. BMJ. 1995;310:1571–1572. doi: 10.1136/bmj.310.6994.1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bert F, Maubec E, Bruneau B, Berry P, Lambert-Zechovsky N. Multi-resistant Pseudomonas aeruginosa outbreak associated with contaminated tap water in a neurosurgery intensive care unit. J Hosp Infect. 1998;39:53–62. doi: 10.1016/s0195-6701(98)90243-2. [DOI] [PubMed] [Google Scholar]
- Berthelot P, Grattard F, Mahul P, Pain P, Jospe R, Venet C, et al. Prospective study of nosocomial colonization and infection due to Pseudomonas aeruginosa in mechanically ventilated patients. Intensive Care Med. 2001;27:503–512. doi: 10.1007/s001340100870. [DOI] [PubMed] [Google Scholar]
- Buttner MP, Willeke K, Grinshpun SA. Sampling and analysis of airborne microorganism. In: Hurst CJ, Knudsen GR, editors. Manual of Environmental Microbiology. Washington, DC: American Society for Microbiology; 1997. pp. 629–640. [Google Scholar]
- Cavalca L, Di Gennaro P, Colombo M, Andreoni V, Bernasconi S, Ronco I, Bestetti G. Distribution of catabolic pathways in some hydrocarbon-degrading bacteria from a subsurface polluted soil. Res Microbiol. 2000;151:877–887. doi: 10.1016/s0923-2508(00)01155-4. [DOI] [PubMed] [Google Scholar]
- Chandrasekar PH, Rolston KV, Kannangara DW, LeFrock JL, Binnick SA. Hot tub-associated dermatitis due to Pseudomonas aeruginosa. Case report and review of the literature. Arch Dermatol. 1984;120:1337–1340. [PubMed] [Google Scholar]
- Cobben NA, Drent M, Jonkers M, Wouters EF, Vaneechoutte M, Stobberingh EE. Outbreak of severe Pseudomonas aeruginosa respiratory infections due to contaminated nebulizers. J Hosp Infect. 1996;33:63–70. doi: 10.1016/s0195-6701(96)90030-4. [DOI] [PubMed] [Google Scholar]
- da Silva Filho LV, Tateno AF, Velloso Lde F, Levi JE, Fernandes S, Bento CN, et al. Identification of Pseudomonas aeruginosa, Burkholderia cepacia complex, and Stenotrophomonas maltophilia in respiratory samples from cystic fibrosis patients using multiplex PCR. Pediatr Pulmonol. 2004;37:537–547. doi: 10.1002/ppul.20016. [DOI] [PubMed] [Google Scholar]
- De Vos D, Lim A, Jr, Pirnay JP, Struelens M, Vandenvelde C, Duinslaeger L, et al. Direct detection and identification of Pseudomonas aeruginosa in clinical samples such as skin biopsy specimens and expectorations by multiplex PCR based on two outer membrane lipoprotein genes, oprI and oprL. J Clin Microbiol. 1997;35:1295–1299. doi: 10.1128/jcm.35.6.1295-1299.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deschaght P, De Baere T, Van Simaey L, Van Daele S, De Baets F, De Vos D, et al. Comparison of the sensitivity of culture, PCR and quantitative real-time PCR for the detection of Pseudomonas aeruginosa in sputum of cystic fibrosis patients. BMC Microbiol. 2009;9:244. doi: 10.1186/1471-2180-9-244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doring G, Ulrich M, Muller W, Bitzer J, Schmidt-Koenig L, Munst L, et al. Generation of Pseudomonas aeruginosa aerosols during handwashing from contaminated sink drains, transmission to hands of hospital personnel, and its prevention by use of a new heating device. Zentralbl Hyg Umweltmed. 1991;191:494–505. [PubMed] [Google Scholar]
- Emde KME, Smith DW, Facey R. Initial investigation of microbially influenced corrosion (MIC) in a low-temperature water distribution-system. Water Res. 1992;26:169–175. [Google Scholar]
- Feizabadi MM, Majnooni A, Nomanpour B, Fatolahzadeh B, Raji N, Delfani S, et al. Direct detection of Pseudomonas aeruginosa from patients with healthcare associated pneumonia by real time PCR. Infect Genet Evol. 2010;10(8):1247–1251. doi: 10.1016/j.meegid.2010.08.008. [DOI] [PubMed] [Google Scholar]
- Ferroni A, Nguyen L, Pron B, Quesne G, Brusset MC, Berche P. Outbreak of nosocomial urinary tract infections due to Pseudomonas aeruginosa in a paediatric surgical unit associated with tap-water contamination. J Hosp Infect. 1998;39:301–307. doi: 10.1016/s0195-6701(98)90295-x. [DOI] [PubMed] [Google Scholar]
- Fukushima M, Kakinuma K, Kawaguchi R. Phylogenetic analysis of Salmonella, Shigella, and Escherichia coli strains on the basis of the gyrB gene sequence. J Clin Microbiol. 2002;40:2779–2785. doi: 10.1128/JCM.40.8.2779-2785.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granstrom M, Ericsson A, Strandvik B, Wretlind B, Pavlovskis OR, Berka R, Vasil ML. Relation between antibody response to Pseudomonas aeruginosa exoproteins and colonization/infection in patients with cystic fibrosis. Acta Paediatr Scand. 1984;73:772–777. doi: 10.1111/j.1651-2227.1984.tb17774.x. [DOI] [PubMed] [Google Scholar]
- Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser. 1999;41:98–98. [Google Scholar]
- Hunter PR. The microbiology of bottled natural mineral waters. J Appl Bacteriol. 1993;74:345–352. doi: 10.1111/j.1365-2672.1993.tb05137.x. [DOI] [PubMed] [Google Scholar]
- Karadzic I, Masui A, Zivkovic LI, Fujiwara N. Purification and characterization of an alkaline lipase from Pseudomonas aeruginosa isolated from putrid mineral cutting oil as component of metalworking fluid. J Biosci Bioeng. 2006;102:82–89. doi: 10.1263/jbb.102.82. [DOI] [PubMed] [Google Scholar]
- Kasai H, Watanabe K, Gasteiger E, Bairoch A, Isono K, Yamamoto S, Harayama S. Construction of the gyrB Database for the Identification and Classification of Bacteria. Genome Inform Ser Workshop Genome Inform. 1998;9:13–21. [PubMed] [Google Scholar]
- Khan AA, Cerniglia CE. Detection of Pseudomonas aeruginosa from clinical and environmental samples by amplification of the exotoxin A gene using PCR. Appl Environ Microbiol. 1994;60:3739–3745. doi: 10.1128/aem.60.10.3739-3745.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan IU, Yadav JS. Real-time PCR assays for genus-specific detection and quantification of culturable and non-culturable mycobacteria and pseudomonads in metalworking fluids. Mol Cell Probes. 2004;18:67–73. doi: 10.1016/j.mcp.2003.09.004. [DOI] [PubMed] [Google Scholar]
- Kimata N, Nishino T, Suzuki S, Kogure K. Pseudomonas aeruginosa isolated from marine environments in Tokyo Bay. Microb Ecol. 2004;47:41–47. doi: 10.1007/s00248-003-1032-9. [DOI] [PubMed] [Google Scholar]
- Lan J, Zhang XH, Wang Y, Chen J, Han Y. Isolation of an unusual strain of Edwardsiella tarda from turbot and establish a PCR detection technique with the gyrB gene. J Appl Microbiol. 2008;105:644–651. doi: 10.1111/j.1365-2672.2008.03779.x. [DOI] [PubMed] [Google Scholar]
- Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- Lavenir R, Jocktane D, Laurent F, Nazaret S, Cournoyer B. Improved reliability of Pseudomonas aeruginosa PCR detection by the use of the species-specific ecfX gene target. J Microbiol Methods. 2007;70:20–29. doi: 10.1016/j.mimet.2007.03.008. [DOI] [PubMed] [Google Scholar]
- Lee CS, Lee J. Evaluation of new gyrB-based real-time PCR system for the detection of B. fragilis as an indicator of human-specific fecal contamination. J Microbiol Methods. 2010;82:311–318. doi: 10.1016/j.mimet.2010.07.012. [DOI] [PubMed] [Google Scholar]
- Lee J, Deininger RA. Real-time determination of the efficacy of residual disinfection to limit wastewater contamination in a water distribution system using filtration-based luminescence. Water Environ Res. 2010;82:475–478. doi: 10.2175/106143009x12487095237035. [DOI] [PubMed] [Google Scholar]
- Moore ERB, Mau M, Arnscheidt A, Bottger EC, Hutson RA, Collins MD, et al. The determination and comparison of the 16S rRNA gene sequences of species of the genus Pseudomonas (sensu stricto) and estimation of the natural intrageneric relationships. Syst Appl Microbiol. 1996;19:478–492. [Google Scholar]
- Motoshima M, Yanagihara K, Fukushima K, Matsuda J, Sugahara K, Hirakata Y, et al. Rapid and accurate detection of Pseudomonas aeruginosa by real-time polymerase chain reaction with melting curve analysis targeting gyrB gene. Diagn Microbiol Infect Dis. 2007;58:53–58. doi: 10.1016/j.diagmicrobio.2006.11.007. [DOI] [PubMed] [Google Scholar]
- Naze F, Jouen E, Randriamahazo RT, Simac C, Laurent P, Bleriot A, et al. Pseudomonas aeruginosa outbreak linked to mineral water bottles in a neonatal intensive care unit: fast typing by use of high-resolution melting analysis of a variable-number tandem-repeat locus. J Clin Microbiol. 2010;48:3146–3152. doi: 10.1128/JCM.00402-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellett S, Bigley DV, Grimes DJ. Distribution of Pseudomonas aeruginosa in a riverine ecosystem. Appl Environ Microbiol. 1983;45:328–332. doi: 10.1128/aem.45.1.328-332.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin X, Emerson J, Stapp J, Stapp L, Abe P, Burns JL. Use of real-time PCR with multiple targets to identify Pseudomonas aeruginosa and other nonfermenting gram-negative bacilli from patients with cystic fibrosis. J Clin Microbiol. 2003;41:4312–4317. doi: 10.1128/JCM.41.9.4312-4317.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quittner AL, Modi AC, Wainwright C, Otto K, Kirihara J, Montgomery AB. Determination of the minimal clinically important difference scores for the Cystic Fibrosis Questionnaire-Revised respiratory symptom scale in two populations of patients with cystic fibrosis and chronic Pseudomonas aeruginosa airway infection. Chest. 2009;135:1610–1618. doi: 10.1378/chest.08-1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team. R: a language and environment for statistical computing. R Foundation for Statistical Computing; Vienna, Austria: 2009. [Google Scholar]
- Rasmussen R. Quantification on the LightCycler. In: Meuer S, Wittwer C, Nakagawara K, editors. Rapid Cycle Real-time PCR, Methods and Applications. Heidelberg: Springer Press; 2001. pp. 21–34. [Google Scholar]
- Reid TM, Porter IA. An outbreak of otitis externa in competitive swimmers due to Pseudomonas aeruginosa. J Hyg (Lond) 1981;86:357–362. doi: 10.1017/s0022172400069114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Relman DA, Schmidt TM, MacDermott RP, Falkow S. Identification of the uncultured bacillus of Whipple’s disease. N Engl J Med. 1992;327:293–301. doi: 10.1056/NEJM199207303270501. [DOI] [PubMed] [Google Scholar]
- Rutala WA, Weber DJ. the Healthcare Infection Control Practices Advisory Committee (HICPAC) [last accessed on 6 June 2011];Guideline for disinfection and sterilization in healthcare facilities, 2008. 2008 Available at: http://www.cdc.gov/hicpac/pdf/guidelines/Disinfection_Nov_2008.pdf.
- Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- Schwartz T, Volkmann H, Kirchen S, Kohnen W, Schon-Holz K, Jansen B, Obst U. Real-time PCR detection of Pseudomonas aeruginosa in clinical and municipal wastewater and genotyping of the ciprofloxacin-resistant isolates. FEMS Microbiol Ecol. 2006;57:158–167. doi: 10.1111/j.1574-6941.2006.00100.x. [DOI] [PubMed] [Google Scholar]
- Subrayan V, Peyman M, Lek Yap S, Mohamed Ali NA, Devi S. Assessment of polymerase chain reaction in the detection of pseudomonas aeruginosa in contact lens-induced severe infectious keratitis. Eye Contact Lens. 2010;36:201–203. doi: 10.1097/ICL.0b013e3181e3efa3. [DOI] [PubMed] [Google Scholar]
- Tamura K, Dudley J, Nei M, Kumar S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24:1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- Trautmann M, Michalsky T, Wiedeck H, Radosavljevic V, Ruhnke M. Tap water colonization with Pseudomonas aeruginosa in a surgical intensive care unit (ICU) and relation to Pseudomonas infections of ICU patients. Infect Control Hosp Epidemiol. 2001;22:49–52. doi: 10.1086/501828. [DOI] [PubMed] [Google Scholar]
- Valles J, Mariscal D, Cortes P, Coll P, Villagra A, Diaz E, et al. Patterns of colonization by Pseudomonas aeruginosa in intubated patients: a 3-year prospective study of 1,607 isolates using pulsed-field gel electrophoresis with implications for prevention of ventilator-associated pneumonia. Intensive Care Med. 2004;30:1768–1775. doi: 10.1007/s00134-004-2382-6. [DOI] [PubMed] [Google Scholar]
- West SE, Zeng L, Lee BL, Kosorok MR, Laxova A, Rock MJ, et al. Respiratory infections with Pseudomonas aeruginosa in children with cystic fibrosis: early detection by serology and assessment of risk factors. JAMA. 2002;287:2958–2967. doi: 10.1001/jama.287.22.2958. [DOI] [PubMed] [Google Scholar]
- Wilson VL, Tatford BC, Yin X, Rajki SC, Walsh MM, LaRock P. Species-specific detection of hydrocarbon-utilizing bacteria. J Microbiol Methods. 1999;39:59–78. doi: 10.1016/s0167-7012(99)00098-6. [DOI] [PubMed] [Google Scholar]
- Wolfgang MC, Kulasekara BR, Liang X, Boyd D, Wu K, Yang Q, et al. Conservation of genome content and virulence determinants among clinical and environmental isolates of Pseudomonas aeruginosa. Proc Natl Acad Sci U S A. 2003;100:8484–8489. doi: 10.1073/pnas.0832438100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto S, Kasai H, Arnold DL, Jackson RW, Vivian A, Harayama S. Phylogeny of the genus Pseudomonas: intrageneric structure reconstructed from the nucleotide sequences of gyrB and rpoD genes. Microbiology. 2000;146 (Pt 10):2385–2394. doi: 10.1099/00221287-146-10-2385. [DOI] [PubMed] [Google Scholar]