

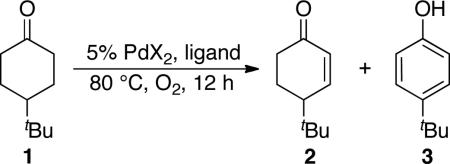
Abstract
α,β-Unsaturated carbonyl compounds are versatile intermediates in the synthesis of pharmaceuticals and biologically active compounds. Here, we report the discovery and application of Pd(DMSO)2(TFA)2 as a catalyst for direct dehydrogenation of cyclohexanones and other cyclic ketones to the corresponding enones, using O2 as the oxidant. The substrate scope includes heterocyclic ketones and several natural-product precursors.
Molecular hydrogen and oxygen are the quintessential reducing and oxidizing agents, respectively. While hydrogenation reactions are commonplace in multistep organic synthesis, aerobic oxidation reactions are seldom used. For example, numerous highly selective methods and sophisticated catalysts exist for the hydrogenation of alkenes;1 however, complementary aerobic dehydrogenation methods for alkene synthesis are unavailable2 (Scheme 1A). We recently reported a method for PdII-catalyzed aerobic dehydrogenation of cyclohexanones to phenols.3 These reactions proceed via a cyclohexenone intermediate that undergoes further dehydrogenation to the phenol under the reaction conditions (Scheme 1B). Here, we report the identification of a different Pd catalyst system that enables selective dehydrogenation of cycloketones to afford enones rather than phenols. Cyclohexenones and related α,β-unsaturated carbonyl compounds are key intermediates in the synthesis of pharmaceuticals and other biologically active compounds.4 Their preparation typically requires two or more steps5-7 and/or the use of stoichiometric reagents, such as 2-iodoxybenzoic acid (IBX)8,9 or 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ).10 Catalytic methods for aerobic dehydrogenation of ketones to enones would provide appealing, atom-economical alternatives to these stoichiometric method.
Scheme 1.

Hydrogenation/Dehydrogenation of C–C Bonds (A)and Pd-Catalyzed Dehydrogenation of Cyclohexanones (B).
The synthesis of enones via PdII-mediated dehydrosilylation of silyl enol ethers was reported by Ito and Saegusa in 1978.6a In some cases, these reactions have been achieved with catalytic PdII,6b,c but the use of ≥ 0.5 equiv of PdII is commonly required to obtain good yields of products.4b,c,11 Methods for direct PdII-catalyzed dehydrogenation of ketones have been pursued as an alternative to Saegusa reactions; however, previous examples exhibit quite limited substrate scope.12-15 Both Saegusa-type dehydrosilylation and direct dehydrogenation reactions are expected to be initiated by formation of a PdII-enolate, followed by β-hydride elimination to afford the enone product (Scheme 2).16 The resulting PdII–hydride intermediate can be oxidized by O2 to regenerate the PdII catalyst.17,18 Recent advances in PdII-catalyzed aerobic oxidation and C–H functionalization reactions19 provided useful starting points for our investigation of dehydrogenation catalysts.
Scheme 2.

Proposed Mechanism for PdII-Catalyzed Dehydrogenation of Cyclic Ketones.
Our initial catalyst screening efforts focused on the dehydrogenation of 4-tert-butylcyclohexanone 1 under relatively mild conditions: 1 atm O2, 80 °C, 12 h (Table 1).20 Use of the recently reported PdII catalyst, Pd(TFA)2/2-N,N-dimethylaminopyridine (2-Me2Npy), for conversion of cyclohexanones to phenols3 resulted in incomplete conversion and, as expected, favored formation of phenol 3 over the enone 2 (entry 1). The best previous catalyst for the conversion of cyclohexanone to cyclohexenone, reported by Tsuji and coworkers,12e forms enone 2 selectively, but only in 19% yield under these conditions (entry 2). Improved results were obtained by using catalytic Pd(OAc)2 in DMSO,21,22 affording a mixture of enone and phenol products in 63% and 14% yield, respectively (entry 3). The best results were obtained by using DMSO as a ligand (10 mol %) with Pd(TFA)2 (5 mol %; TFA = trifluoroacetate) in acetic acid (entry 7). This catalyst system led to a 91% yield of the desired enone 2. Replacing DMSO with other monodentate and bidentate ligands led to inferior results (entries 13–19; see also Table S1).23 The benefit of using DMSO as a catalytic ligand, rather than a solvent, has been observed recently in two other Pd-catalyzed aerobic oxidation reactions, including chelate-directed C–H arylation of anilides24 and oxidative amination of alkenes.25
Table 1.
Catalyst Optimization of Aerobic Oxidative Dehydrogenation of 4-tert-Butylcyclohexanone 1.a
 | |||||
|---|---|---|---|---|---|
| Entry | PdX2 | Ligand (mol%) | Solvent | 2 (%)b | 3 (%)b |
| 1 | Pd(TFA)2 | 2-Me2N-pyridine (10)/TsOH(20) | DMSO | 23 | 33 |
| 2 | Pd(TFA)2 | 5,5'-Me2bpy (5)/4Å MS | PhCl | 19 | 0 |
| 3 | Pd(OAc)2 | DMSO | 63 | 14 | |
| 4 | Pd(TFA)2 | DMSO | 34 | 56 | |
| 5 | Pd(TFA)2 | HOAc | 24 | 1 | |
| 6 | Pd(OAc)2 | DMSO (10) | HOAc | 86 | 8 |
| 7 | Pd(TFA)2 | DMSO (10) | HOAc | 91 | 8 |
| 8 | Pd(TFA)2 | DMSO (10) | Toluene | 67 | 3 |
| 9 | Pd(TFA)2 | DMSO (10) | THF | 66 | 8 |
| 10 | Pd(TFA)2 | DMSO (10) | Dioxane | 84 | 10 |
| 11 | Pd(TFA)2 | DMSO (10) | EtOAc | 30 | 6 |
| 12 | Pd(TFA)2 | DMSO (10) | PhCl | 11 | 0 |
| 13 | Pd(TFA)2 | pyridine (10) | HOAc | 55 | 2 |
| 14 | Pd(TFA)2 | 2-Me2N-pyridine (10) | HOAc | 3 | 1 |
| 15 | Pd(TFA)2 | 2-F-pyridine (10) | HOAc | 37 | 2 |
| 16 | Pd(TFA)2 | bipyridine (5) | HOAc | 0 | 0 |
| 17 | Pd(TFA)2 | 5,5'-Me2bpy (5) | HOAc | 0 | 0 |
| 18 | Pd(TFA)2 | phenanthroline (5) | HOAc | 0 | 0 |
| 19 | Pd(TFA)2 | 1,2-bis(phenylsulfinyl)ethane (5) | HOAc | 9 | 4 |
Conditions: [1] = 0.2 M (15.4 mg, 0.1 mmol), 5% PdX2 (0.005 mmol), 10% ligand (0.01 mmol), Solvent (0.5 mL), 1 atm O2, 80 °C, 12 h.
Determined by GC, external standard = tetradecane.
The high selectivity for formation of the enone with the Pd(DMSO)2(TFA)2 catalyst system is noteworthy in light of the preferential formation of phenols with a Pd(TFA)2/2-Me2Npy catalyst system.3 A comparison of time courses for reactions with the two catalyst systems (Figure 1) highlights the significant differences between the relative rates of the corresponding dehydrogenation steps (cf. Scheme 1B). Fitting of the time-course data to a simple sequential kinetic model, A → B → C,26 reveals that the first dehydrogenation step is 33-fold faster than the second step when Pd(DMSO)2(TFA)2 is used as the catalyst. In contrast, the first step is nearly 2-fold slower than the second step with the Pd(TFA)2/2-Me2Npy catalyst system.27 Further mechanistic studies are ongoing, but the observations have important implications for use of the present catalyst system in the synthesis of enones (Table 2).
Figure 1.

Comparison of kinetic profiles of Pd(DMSO)2(TFA)2- and Pd(TFA)2/2-Me2Npy-catalyzed dehydrogenation of 1. (A) Time course for catalyzed Pd(DMSO)2(TFA)2-catalyzed dehydrogenation of 1. Reaction conditions: [1] = 0.2 M (0.1 mmol), 5% Pd(DMSO)2(TFA)2 (0.005 mmol), AcOH (0.5 mL), 1 atm O2, 80 °C. (B) Time course for Pd(TFA)2/2-Me2Npy-catalyzed dehydrogenation of 1.27 Reaction conditions: [1] = 0.2 M (0.1 mmol), Pd(TFA)2 (0.005 mmol), 2-Me2Npy (0.01 mmol), TsOH (0.02 mmol), DMSO (0.5 mL), 1 atm O2, 80 °C. Internal standard = 1,4-dimethoxybenzene. Error bars represent standard deviations from 3 independent measurements.
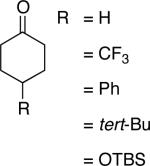
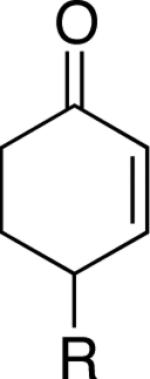
Table 2.
Pd-Catalyzed Aerobic Dehydrogenation of Diverse Cycloketones.a
| Entry | Substrate | Reaction time (h) | Temp. (°C) | Enone | Yield (%)b |
|---|---|---|---|---|---|
| 1 |
|
24 | 60 |
|
72c |
| 2 | 5 | 80 | 81 | ||
| 3 | 6 | 80 | 83 | ||
| 4 | 12 | 80 | 91 | ||
| 5 | 20 | 80 | 76 | ||
| 6 |
|
24 | 60 |
|
84c |
| 7 |
|
24 | 60 |
|
86c |
| 8 |
|
18 | 80 |
|
94 |
| 9 |
|
12 | 80 |
|
93 |
| 10 |
|
48 | 60 |
|
54d |
| 11 |
|
36 | 80 |
|
81d |
| 12 |
|
48 | 100 |
|
80 |
| 13 |
|
36 | 80 |
|
78 |
| 14 |
|
32 | 100 |
|
66 |
| 15 |
|
24 | 60 |
|
74 |
| 16 | 48 | 80 | 72e |
Reactions conditions: [substrate] = 0.2 M (0.8 mmol), [Pd(TFA)2] = 0.01 M (0.04 mmol = 5 mol %), [DMSO] = 0.02 M (0.08 mmol), solvent = HOAc (4 ml), 1 atm O2.
Isolated yield.
Ethyl acetate was used as solvent to prevent product decomposition in acetic acid, [substrate] = 0.8 M (0.8 mmol), ethyl acetate (1 mL).
[substrate] = 0.4 M (0.8 mmol), [Pd(TFA)2] = 0.02 M, [DMSO] = 0.2 M, solvent = toluene (2 ml).
In DMSO; no additional ligand.
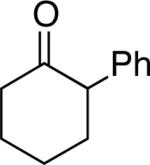
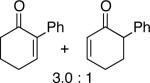
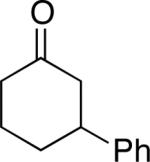
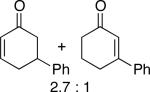
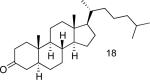
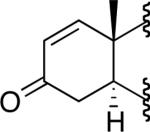
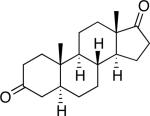
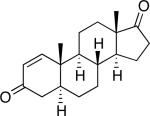
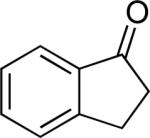
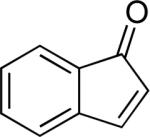
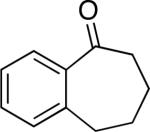
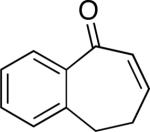
A number of 4-substituted cyclohexanone derivatives underwent dehydrogenation in good yields with the Pd(DMSO)2(TFA)2 catalyst (Table 2, entries 1–5). Substrates with electron-deficient substituents (entries 2 and 3) exhibited somewhat faster rates, and the conditions tolerated various functional groups, including trifluoromethyl and siloxy groups (entries 2 and 5). The parent cyclohexanone (entry 1) decomposed under the acidic conditions, but a good yield of enone was obtained by performing the reaction in ethyl acetate.28 Dehydrogenation of 2- and 3-substituted cyclohexanones can afford two enone regioisomers, and reactions of 2- and 3-phenylcyclohexanone proceeded with modest (~3:1) regioselectivity (entries 6 and 7). The ability to achieve highly regioselective dehydrogenation was demonstrated in the reactions of two steroid derivatives (entries 8 and 9), each of which afforded one of two possible enones in excellent yield. In both cases, the regioselectivity favored formation of the less substituted alkene. No dehydrogenation of the cyclopentanone fragment was observed in the reaction leading to 5α-androst-1-ene-3,17-dione (entry 9). The lower reactivity of cyclopentantones was also evident in the dehydrogenation of indanone, which afforded the corresponding enone in 54% yield, with toluene as the optimal solvent (entry 10). In contrast, 1-benzosuberone underwent dehydrogenation in good yield (81%, entry 11). Cycloheptanone and cyclooctanone led to a mixture of dehydrogenation products, with 2,6-cycloheptadien-1-one and 2,7-cyclooctadien-1-one formed as the major products in 26 and 25% yields, respectively, based on GC-MS and 1H NMR spectroscopic analysis.
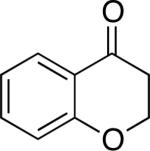
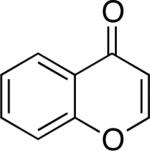
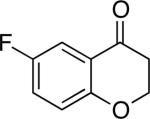
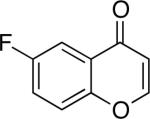
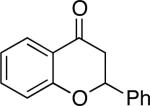
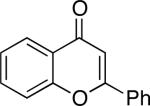
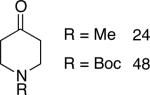
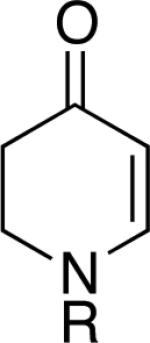
Chromones29 and flavones have important biological activity,30 and the saturated dihydrobenzopyranones are readily prepared via condensation of simple precursors.31 Aerobic dehydrogenation reactions to form chromone, 6-fluorochromone,32 and flavone33 proceeded in good yield entries 12–14 Related N-methyl- and N-Boc-piperidone derivatives underwent successful dehydrogenation to the corresponding dihydro-4-pyridone derivatives (entries 15 and 16).
Cyclic enones are common intermediates in the synthesis of natural products, and the aerobic dehydrogenation reactions described here could find broad utility in this context. For example, α,α-disubstituted cyclohexenone 4 has been used as an intermediate in the synthesis of (–)-mersicarpine. This enone was obtained in 85% yield using the Pd(DMSO)2(TFA)2 catalytic method (eq 1); the original protocol employed stoichiometric IBX and proceeded in 72% yield.34 Catalytic Saegusa-type6c and stoichiometric IBX8 oxidation methods failed in the synthesis of acyclopentene-α-dione precursor to the natural product (–)-terpestacin, and stoichiometric Pd(OAc)2 was used instead.35 Application of the aerobic Pd(TFA)2/DMSO catalyst system to this reaction afforded the enedione in 90% yield (eq 2).
 |
(1) |
 |
(2) |
In summary, we have identified a PdII catalyst system that enables direct dehydrogenation of cyclic ketones to the corresponding enones with a number of important substrates. The high selectivity for enone rather than phenol formation sharply contrasts other PdII-catalyzed dehydrogenation methods3,13 and warrants further mechanistic investigation. The ability to replace stoichiometric reagents (e.g., Br2, organoselenium reagents, and IBX) with O2 as an oxidant has important implications for large-scale applications of these methods in pharmaceutical and fine-chemical synthesis.36
Supplementary Material
Acknowledgement
We thank Prof. Clark Landis (UW-Madison) for assistance with COPASI to carry out kinetic simulations, and David Mannel (UW-Madison) for assistance with the flow-reactor. Financial support for this work was provided by the NIH (RC1-GM091161) and Eastman Chemical (summer fellowship to TD). High-pressure instrumentation was partially supported by the NSF (CHE-0946901).
Footnotes
Supporting Information Available: Reaction procedure and spectra are available free of charge via the Internet at http://pubs.acs.org.
References
- 1.a Nishimura S. Handbook of Heterogeneous Catalytic Hydrogenation for Organic Synthesis. Wiley-Interscience; 2001. [Google Scholar]; b de Vries JG, Elsevier CJ, editors. The Handbook of Homogeneous Hydrogenation. Wiley-VCH; Weinheim: 2007. [Google Scholar]
- 2.Transfer-dehydrogenation methods are currently the most effective methods for dehydrogenation of saturated carbon-carbon bonds; however, the application of these methods in organic synthesis is rare. For leading references to these methods, see the following recent review articles: Dobereiner GE, Crabtree RH. Chem. Rev. 2010;110:681–703. doi: 10.1021/cr900202j.Choi J, MacArthur AHR, Brookhart M, Goldman AS. Chem. Rev. 2011;111:1761–1779. doi: 10.1021/cr1003503.
- 3.Izawa Y, Pun D, Stahl SS. Science. 2011;333:209–213. doi: 10.1126/science.1204183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Numerous examples of cyclohexanone dehydrogenation in natural product synthesis exist. For recent examples, see: Herzon SB, Lu L, Woo CM, Gholap SL. J. Am. Chem. Soc. 2011;133:7260–7263. doi: 10.1021/ja200034b.Yokoe H, Mitsuhashi C, Matsuoka Y, Yoshimura T, Yoshida M, Shishido K. J. Am. Chem. Soc. 2011;133:8854–8857. doi: 10.1021/ja202874d.Petronijevic FR, Wipf P. J. Am. Chem. Soc. 2011;133:7704–7707. doi: 10.1021/ja2026882.
- 5.For reviews of methods for α,β-dehydrogenation of carbonyl compounds, see: Buckle DR, Pinto IL. In: Comprehensive Organic Synthesis. Trost BM, Fleming I, editors. Vol. 7. Oxford: 1991. pp. 119–149.Larock RC. Comprehensive Organic Transformations. John Wiley & Sons; New York: 1999. pp. 251–256.
- 6.For Pd-catalyzed dehydrogenation of silyl enol ethers (“Saegusa reactions”), see: Ito Y, Hirao T, Saegusa T. J. Org. Chem. 1978;43:1011–1013.Larock RC, Hightower TR, Kraus GA, Hahn P, Zheng D. Tetrahedron Lett. 1995;36:2423–2426.Yu JQ, Wu HC, Corey E. J. Org. Lett. 2005;7:1415–1417. doi: 10.1021/ol050284y.
- 7.Common methods include bromination/dehydrobromination and selenoxide and sulfoxide elimination reactions. See the following leading references: Miller B, Wong H-S. Tetrahedron. 1972;28:2369–2376.Stotter PL, Hill KA. J. Org. Chem. 1973;38:2576–2578.Trost BM, Salzmann TN, Hiroi K. J. Am. Chem. Soc. 1976;98:4887–4902.Sharpless KB, Lauer RF, Teranishi AY. J. Am. Chem. Soc. 1973;95:6137–6139.Reich H. J. Acc. Chem. Res. 1979;12:22–30.
- 8.a Nicolaou KC, Zhong YL, Baran PS. J. Am. Chem. Soc. 2000;122:7596–7597. [Google Scholar]; b Nicolaou KC, Montagnon T, Baran PS, Zhong YL. J. Am. Chem. Soc. 2002;124:2245–2258. doi: 10.1021/ja012127+. [DOI] [PubMed] [Google Scholar]; c Nicolaou KC, Montagnon T, Baran PS. Angew. Chem. Int. Ed. 2002;41:993–996. doi: 10.1002/1521-3773(20020315)41:6<993::aid-anie993>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]; d Nicolaou KC, Gray DLF, Montagnon T, Harrison ST. Angew. Chem. Int. Ed. 2002;41:996–999. doi: 10.1002/1521-3773(20020315)41:6<996::aid-anie996>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 9.For a method employing catalytic 2-iodoxybenzene sulfonate (IBS) in combination stoichiometric Oxone®, see: Uyanik M, Akakura M, Ishihara K. J. Am. Chem. Soc. 2009;131:251–262. doi: 10.1021/ja807110n.
- 10.a Walker D, Hiebert JD. Chem. Rev. 1967;67:153–195. doi: 10.1021/cr60246a002. [DOI] [PubMed] [Google Scholar]; b Buckle DR. Encyclopedia of Reagents for Organic Synthesis. John Wiley & Sons, Inc.; New York: 2010. 2,3-Dichloro-5,6-dicyano-1,4-benzoquinone. [Google Scholar]; c Bhattacharya A, DiMichele LM, Dolling UH, Douglas AW, Grabowski EJJ. J. Am. Chem. Soc. 1988;110:3318–3319. [Google Scholar]
- 11.The requirement for large amounts of PdII in these reactions may arise from inhibition of catalyst reoxidation, arising from enone coordination to PdII: Porth S, Bats JW, Trauner D, Giester G, Mulzer J. Angew. Chem. Int. Ed. 1999;38:2015–2016. doi: 10.1002/(SICI)1521-3773(19990712)38:13/14<2015::AID-ANIE2015>3.0.CO;2-#.
- 12.a Theissen RJ. J. Org. Chem. 1971;36:752–757. [Google Scholar]; b Muzart J, Pete JP. J. Mol. Catal. 1982;15:373–376. [Google Scholar]; c Wenzel TT. J. Chem. Soc., Chem. Commun. 1989:932–933. [Google Scholar]; d Park YW, Oh HH. Bull. Kor. Chem. Soc. 1997;18:1123–1124. [Google Scholar]; e Tokunaga M, Harada S, Iwasawa T, Obora Y, Tsuji Y. Tetrahedron Lett. 2007;48:6860–6862. [Google Scholar]
- 13.For a recent review of reactions of this type, see: Muzart J. Eur. J. Org. Chem. 2010:3779–3790.
- 14.PdII-catalyzed dehydrogenation of β-aryl aldehydes has been explored recently; however, the current substrate scope is rather limited: Zhu J, Liu J, Ma RQ, Xie HX, Li J, Jiang HL, Wang W. Adv. Synth. Catal. 2009;351:1229–1232.Liu J, Zhu J, Jiang HL, Wang W, Li J. Chem. Asian J. 2009;4:1712–1716. doi: 10.1002/asia.200900238.
- 15.Direct dehydrogenation of 3,3-dimethylcyclohexanone was reported recently with 1–5 mol % of an Ir-pincer catalyst and tBuethylene as the H2 acceptor. Other cyclohexanone substrates lacking gem-disubstitution undergo stoichiometric deydrogenation, forming an Ir-phenoxide product. Zhang XW, Wang DY, Emge TJ, Goldman AS. Inorg. Chim. Acta. 2011;369:253–259.
- 16.The mechanism could proceed via C- and/or O-bound Pd enolates, although β-hydride elimination is expected to occur from the C-bound enolate. For the formation of a C- bound Pd-enolate under relatively similar conditions, see: Fuchita Y, Harada Y. Inorg. Chim. Acta. 1993;208:43–47.
- 17.Recent studies suggest that aerobic oxidation of PdII-hydrides proceeds via PdII, as shown in Scheme 2. See: Konnick MM, Stahl SS. J. Am. Chem. Soc. 2008;130:5753–5762. doi: 10.1021/ja7112504.Popp BV, Stahl SS. Chem. Eur. J. 2009;15:2915–2922. doi: 10.1002/chem.200802311.
- 18.Gas update experiments show that the product:O2 stoichiometry is 2:1, consistent with Pd-mediated disproportionation of H2O2 under reaction conditions. For previous characterization of Pd-mediated H2O2 disproportionation under related conditions, see the Supporting Information for the following reference: Steinhoff BA, Fix SR, Stahl SS. J. Am. Chem. Soc. 2002;124:766–767. doi: 10.1021/ja016806w.
- 19.a Stahl SS. Angew. Chem., Int. Ed. 2004;43:3400–3420. doi: 10.1002/anie.200300630. [DOI] [PubMed] [Google Scholar]; b Gligorich KM, Sigman MS. Chem. Commun. 2009:3854–3867. doi: 10.1039/b902868d. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Chen X, Engle KM, Wang D-H, Yu J-Q. Angew. Chem. Int. Ed. 2009;48:5094–5115. doi: 10.1002/anie.200806273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.See Supporting Information for further details.
- 21.Pd(OAc)2/DMSO catalyst systems for aerobic oxidation reactions were pioneered by the groups of Hiemstra and Larock. For initial reports, see: Larock RC, Hightower TR. J. Org. Chem. 1993;58:5298–5300.van Benthem RATM, Hiemstra H, Michels JJ, Speckamp WN. J. Chem. Soc., Chem. Commun. 1994:357–359.
- 22.For mechanistic characterization of this catalyst system, see: Steinhoff BA, Stahl SS. J. Am. Chem. Soc. 2006;128:4348–4355. doi: 10.1021/ja057914b.
- 23.The other ligands, such as pyridine (entry 13), 2-F-pyridine (entry 15) and the bis-sulfoxide ligand (entry 19) were selected on the basis of their utility in other recent Pd-catalyzed oxidation reactions. See, for example: Nishimura T, Onoue T, Ohe K, Uemura S. J. Org. Chem. 1999;64:6750–6755. doi: 10.1021/jo9906734.Izawa Y, Stahl SS. Adv. Synth. Catal. 2010;352:3223–3229. doi: 10.1002/adsc.201000771.Chen MS, Prabagaran N, Labenz NA, White MC. J. Am. Chem. Soc. 2005;127:6970–6971. doi: 10.1021/ja0500198.
- 24.Brasche G, García-Fortanet J, Buchwald SL. Org. Lett. 2008;10:2207–2210. doi: 10.1021/ol800619c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McDonald RI, Stahl SS. Angew. Chem. Int. Ed. 2010;49:5529–5532. doi: 10.1002/anie.200906342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kinetic fitting was carried with COPASI software: Hoops S, Sahle S, Gauges R, Lee C, Pahle J, Simus N, Singhal M, Xu L, Mendes P, Kummer U. Bioinformatics. 2006;22:3067–3074. doi: 10.1093/bioinformatics/btl485.
- 27.The kinetics of phenol formation (Figure 1B) show that this reaction is more complicated than a simple sequential A → B → C process. Specifically, a kinetic “burst” is evident during the first catalytic turnover that leads to the early rapid conversion of cyclohexanone to cyclohexenone. The fit in Figure 1B reflects a fit of the data after this burst phase. Mechanistic studies to elucidate the origin of these observations are ongoing.
- 28.Catalyst decomposition appears to be more rapid when ethyl acetate is used as the solvent rather than acetic acid. Vigorous agitation of the reaction mixture to ensure good gas-liquid mixing, or the use of higher O2 pressure, improves the outcome. When using elevated O2 pressures, we employed a dilute mixture of O2 in N2 (9%) as the gas source to reduce flammability hazards. See Supporting Information for details.
- 29.Si D, Wang Y, Zhou YH. Drug. Metab. Dispos. 2003;37:629–634. doi: 10.1124/dmd.108.023416. [DOI] [PubMed] [Google Scholar]
- 30.Cermak R, Wolffram S. Curr. Drug. Metab. 2006;7:729–44. doi: 10.2174/138920006778520570. [DOI] [PubMed] [Google Scholar]
- 31.a Loudon JD, Razdan RKJ. Chem. Soc. 1954:4299–4303. [Google Scholar]; b Choudary BM, Ranganath KVS, Yadav J, Lakshmi Kantam M. Tetrahedron Lett. 2005;46:1369–1371. [Google Scholar]
- 32.Kurono M, Yamaguchi T, Usui T, Fukushima M, Mizuno K, Matsubara A. 1986.
- 33.Kondo T, Oyama K-I, Yoshida K. Angew. Chem. Int. Ed. 2001;40:894–897. doi: 10.1002/1521-3773(20010302)40:5<894::AID-ANIE894>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 34.Nakajima R, Ogino T, Yokoshima S, Fukuyama TJ. Am. Chem. Soc. 2010;132:1236–1237. doi: 10.1021/ja9103233. [DOI] [PubMed] [Google Scholar]
- 35.Trost BM, Dong G, Vance JA. Chem. Eur. J. 2010;16:6265–6277. doi: 10.1002/chem.200903356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dehydrogenation of substrate 1 has been carried out on a 10-gram scale using a prototype flow reactor donated to UW-Madison by Eli Lilly. See Supporting Information and the following reference for details: Ye X, Johnson MD, Diao T, Yates MH, Stahl SS. Green Chem. 2010;12:1180–1186. doi: 10.1039/c0gc00106f.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.