

Abstract
Dimeric pyrrole-imidazole alkaloids represent a rich and topologically unique class of marine natural products. This full account will follow the progression of efforts that culminated in the enantioselective total syntheses of the most structurally ornate members of this family: the axinellamines, the massadines, and palau’amine. A bio-inspired approach capitalizing on the pseudo-symmetry of the members of this class is recounted, delivering a deschloro derivative of the natural product core. Next, the enantioselective synthesis of the chlorocyclopentane core featuring a scalable, catalytic, enantioselective Diels–Alder reaction of a 1-siloxydiene is outlined in detail. Finally, the successful divergent conversion of this core to each of the aforementioned natural products, and the ensuing methodological developments are described.
Introduction
Within the field of natural product synthesis, alkaloids continue to be studied with great zeal and represent veritable gold mines for organic chemistry. Indeed, these molecules continually lead to the discovery of new reactivity, provide fertile testing grounds for new methodology, and exhibit profound medicinal potential. This full account traces the progression of studies that successfully led to enantioselective total syntheses of the most complex members of the marine-derived pyrrole-imidazole alkaloid family.
The isolation of the marine natural product sceptrin (4, Scheme 1) from Agelas sceptrum sponge in 1981 by Faulkner and Clardy marked the first report of a dimer of the pyrrole-imidazole alkaloid family.1 This symmetric alkaloid contains a cyclobutane core and is enantioenriched, indicating a probable enzyme-mediated, head-to-head cyclization of the natural product hymenidin (1) that is also present in the isolation extract. This report was followed by the discovery of the bicyclic, nonsymmetrical dimer ageliferin (5) in 1986 by Rinehart.2 In 1993, this family reached a new pinnacle of architectural complexity with the isolation of palau’amine by Scheuer and coworkers, which was assigned the structure 12,17-epi-13 (Scheme 1, palau’amine numbering used for all molecules herein).3 This natural product contains a fully substituted cyclopentane core woven into a dense hexacyclic architecture containing eight contiguous stereocenters and nine nitrogen atoms, and is notably lacking one of the acylpyrrole units. Over the next few years, the styloguanidines and konbu’acidins, which possessed similar hexacyclic architectures to palau’amine (12,17-epi-13) were reported, some containing the missing second acylpyrrole unit.4 The publication of the axinellamines (9–10) in 1999 by Quinn and coworkers marked the first report of tetracyclic pyrrole-imidazole alkaloids that contained a similar cyclopentane cores as palau’amine (12,17-epi-13), with the exception of the relative stereochemistry at C12 and C17.5 A few years later, massadine (11) was isolated by Fusetani et al, representing a second tetracyclic pyrrole-imidazole alkaloid with a differing mode of cyclization from that found in the axinellamines, and with a hydroxyl group instead of a chloro substituent at C17 (palau’amine numbering).6 The stylissadines, published in 2006, represent the first tetrameric members of the pyrrole imidazole alkaloids, and are apparent dimers of massadine (11).7 Insight into their genesis was revealed with the isolation of massadine chloride (12) in 2007 in collaboration with the Köck group.8 It was found that this natural product converts to massadine (11) in aqueous media, presumably via the intermediacy of “massadine aziridine”.9 It is important to note that none of the absolute configurations of these natural products are known, but that predictions have been made by Fusetani (for massadine (11))6 and Lindel, Kinnel, and Köck (for palau’amine (13)).10
Scheme 1. Biosynthesis of pyrrole-imidazole alkaloids.

The striking discrepancy that existed between the tetracyclic pyrrole-imidazole alkaloids and the originally reported structure of the hexacyclic palau’amine (12,7-epi-13, Scheme 1) was intriguing. The axinellamines (9–10) and the massadines (11–12) possessed trans-pyrrole amide side chains, matching the all-trans nature of sceptrin (4) and ageliferin (5). In contrast, the hexacyclic compounds palau’amine (12,17-epi-13), styloguanidine and konbu’acidin appeared to stand alone in the family of dimers with two inverted stereocenters at C12 and C17. However, numerous approaches towards the synthesis of the originally assigned structure of palau’amine (12,17-epi-13) continued to appear in the literature, indicating that the initial stereochemical assignment was generally accepted.
We began to question the original stereochemical assignment of palau’amine (12,17-epi-13) in 2004 during investigations into the conversion of (−)-sceptrin (4) to (−)-ageliferin (5) via a vinylcyclobutane rearrangement.11 It was reasoned that a similar conversion could happen in Nature, as sceptrin (4) is almost always the most abundant pyrrole-imidazole alkaloid found in isolation extracts which contain the more complex dimers.12 Thus, it was speculated that the genesis of the tetracyclic and hexacyclic congeners (e.g. 9–13) would not require the invocation of a separate enzymatic pathway, but simply an oxidative ring expansion of sceptrin (4) via intermediate 7, or an isomerization/oxidative ring contraction of ageliferin (5) via intermediates 6 and 7, as shown in Scheme 1. The 2-aminoimidazole in 7 could undergo further oxidation to produce “pre-axinellamine” (8), which is primed to undergo intramolecular cyclization to access each of the more intricate structures 9–13. This hypothesis was further supported by the successful conversion of ageliferin (5) into the carbogenic core of the axinellamines (9–10).13
In order for hexacyclic pyrrole-imidazole alkaloids such as palau’amine (12,17-epi-13) to fit into this hypothesis, the stereochemistry at C12 would have to be inverted to match that of sceptrin (4) and ageliferin (5). Additionally, it would not be a logical leap of faith to assume that the stereochemistry of the chlorine atom (C17) should also match that of the rest of the family (e.g. 9–12). Finally, in 2007, reports from the Köck,14 Quinn,7b,15 and Matsunaga16 groups simultaneously appeared in the literature that officially reassigned palau’amine (to structure 13) to match the axinellamines (9–10) and massadines (11–12), thus bringing uniformity to the entire family of alkaloids. These groups used 2-D NMR spectroscopy (and in one case a quantitative computational approach)14 to support the revised structure, as a crystal of these alkaloids suitable for X-ray analysis has never been acquired. An elegant synthesis of an analogue of the hexacyclic core of cis-palau’amine (12,17-epi-13) by the Overman laboratory demonstrated that the spectral data were not in agreement with those reported in the isolation papers, giving further support to the trans structure.17
While several members of the pyrrole-imidazole alkaloid family possess notable biological activity,2–5,6,7b,16,18 the primary inspiration for chemists to pursue their synthesis lies in their intriguing structures. Notably, the axinellamines (9–10), massadines (11–12), and palau’amine (13) have densely functionalized cores with eight contiguous stereocenters, high nitrogen content (ca. 1:2 N:C), and sensitive functional groups such as hemiaminals and labile halogens. Several groups have reported efforts towards the synthesis of the axinellamines (9–10)9,13,19 and the massadines (11–12),9,19c,19d,20 although the majority of synthetic approaches to these molecules have focused on palau’amine (13),9,17,19b,19c,19f,21 and there are numerous reviews on these approaches.22 Additionally, several Ph.D. theses have emerged with a wealth of unpublished chemistry.23 Several of these approaches occurred during the fourteen-year interim between the original report and the structural reassignment,9,17,19b,19c,21a–j while others have recently targeted the correct structure.19f,21k–u The first successful synthesis of one of the complex pyrrole-imidazole alkaloids emerged from our laboratory in 2008 with the completion of axinellamines A (9) and B (10),24 and was followed later that year with the synthesis of massadine (11) and massadine chloride (12).25 In 2010, palau’amine (13) finally succumbed to synthesis, confirming the structural reassignment.26 This article will recount the details of the efforts that culminated in the final racemic routes to each of these molecules. Additionally, a catalytic enantioselective route that delivered the natural enantiomer of each of the alkaloids 9–13 is reported, confirming their absolute stereochemistry to match that of sceptrin (4) and ageliferin (5).
Results and Discussion
Bio-Inspired Approach to the Cyclopentane Core
Due to the dimeric nature of the complex pyrrole-imidazole alkaloids 9–13, it would appear on first inspection that they could arise from a simple, symmetrical precursor. Initial investigations into the synthesis of the cyclopentane core of 9–13 attempted to capitalize on their pseudo-symmetry by targeting a seco-sceptrin derivative (16, Scheme 2). It was envisioned that such a precursor could be oxidatively cyclized to generate a deschloro version of the core such as 14. Although such a cyclization represents one pathway among a plethora of possibilities, there is precedent from the Horne group that 2-aminoimidazoles could be induced to react in such a manner.27 Additionally, although the formation of such a hindered quaternary center could be outcompeted by several less strained cyclization modes, the Harran group has showed promising results in similar cyclizations.21g,21j,21o The risky nature of such a step was outweighed by the rapidity with which a structure such as seco-sceptrin azide could be accessed: imidazole formation from diketone 17, which could in turn be derived from ozonolysis of Diels–Alder product 18.
Scheme 2. Bio-inspired retrosynthetic scheme based on an oxidative cyclization from the symmetric seco-sceptrin-N3 (16).

In the forward sense, a thermal [4+2] cycloaddition between dimethyl fumarate and 2,3-dimethylbutadiene followed by functional group manipulations yielded diazide 18 in 78% yield over four steps (Scheme 3). Ozonolysis and bromination provided the linear diketone 17 in 45% overall yield, which was treated with Boc-guanidine and deprotected with trifluoroacetic acid to deliver seco-sceptrin-N3 (16). With this key precursor in hand, several oxidative conditions were applied in an attempt to obtain a cyclized product. These conditions included several electrophilic halogen sources (NCS, NBS, tBuOCl), hypervalent iodine sources (PhIO, PhI(OAc)2, Koser’s reagent), and other oxidants (m-CPBA, DMDO, TFDO) in a variety of solvents and at varying pH. Unfortunately, no evidence of the desired cyclization to 14 or of the existence of the oxidized intermediate 15 was ever encountered. It appeared that the main products were oxidized 2-aminoimidazoles that subsequently captured solvent or residual water (subsequent cyclization of these intermediates could not be induced). Thermal and acidic conditions in an attempt to cyclize through a simple isomerization also failed.
Scheme 3. Attempt to secure the core of the pyrrole-imidazole alkaloids with an oxidative cascade from a linear precursor (16).

a Reagents and conditions: (a) Dimethyl fumarate (1.0 equiv), 1,3-dimethylbutadiene (1.0 equiv), neat, 135 °C, 1.5 h; (b) LiAlH4 (2.0 equiv), THF, 0 °C, 1 h; (c) methanesulfonyl chloride (4.0 equiv), pyridine, 0 → 23 °C, 1 h; (d) sodium azide (6.0 equiv), DMF, 100 °C, 2 h, 78% for four steps; (e) O3, MeOH, −78 °C, 15 min, then DMS (3.0 equiv), 23 °C, 20 h; (f) diisopropylethylamine (6.0 equiv), TMSOTf (4.0 equiv), 0 → 23 °C, then NBS (2.0 equiv), MeCN, THF, 0 °C, 15 min, 45% for two steps; (g) Boc-guanidine (10 equiv), DMF, 23 °C, 65 h, 14%; (h) TFA:DCM (1:2), 23 °C, 6 h, quant. THF = tetrahydrofuran, DMF = N,N-dimethylformamide, DMS = dimethylsulfide, TMSOTf = trimethylsilyl trifluoromethanesulfonate, NBS = N-bromosuccinimide, TFA = trifluoroacetic acid, DCM = dichloromethane.
Fortunately, aspects of the route to seco-sceptrin-N3 (16) did deliver some promising results. During the isolation of diketone 17 by column chromatography on silica gel, a significant amount of a byproduct was being formed, and was later identified as aldol product 20 (Scheme 4). It was intriguing to realize that prior to 2-aminoimidazole formation, the desired carbon–carbon bond was being formed by weak Brønsted-acid induced cyclization. After dehydration to form cyclopentene 21, exposure to Boc-guanidine followed by deprotection provided the cyclized cyclopentane 22, which bore a striking resemblence to the core found in the natural products 9–13. The cascade resulting in the formation of this product most likely involves two SN2 reactions, a conjugate addition, and a condensation to form the final 2-aminoimidazole. Unfortunately, 2-D NMR data revealed that the stereochemistry at the spiro junction at C16 was epimeric to that found in the natural products. Additionally, no functional handles were available to selectively introduce the chlorine atom needed at C17. However, these results inspired us to adapt the approach with a pre-functionalized C17, perhaps permitting elaboration to a fully functionalized cyclopentane core.
Scheme 4. Access to a deschloro core structure 22.

a Reagents and conditions: (a) SiO2, 23 °C, 12 h, 90%; (b) 2,6-lutidine (4.0 equiv), SOCl2 (2.0 equiv), DCM, 0 °C, 1 h, 30%; (c) Boc-guanidine (7.0 equiv), THF, 55 °C, 12 h; (d) TFA:DCM (1:1), 23 °C, 1 h, 22% for two steps.
Abiotic Approach to the Cyclopentane Core
The two main roadblocks encountered during the bio-inspired approach that culminated in Scheme 4 were 1) the lack of a chlorine substituent at C17 on the cyclopentane and 2) undesired stereochemical outcome at C16. Additionally, C20 would have to be oxidized to a hemiaminal, and although ample precedent for amine to imine conversion existed in the literature,28 as well as for amides to N-acylimines,29 no such chemistry had ever been applied to 4,4-disubstituted imidazolines. With these considerations in mind, a revised retrosynthesis was forged (Scheme 5).
Scheme 5. Revised retrosynthetic analysis of the axinellamines (9–10) and palau’amines (e.g. 13).a.

a R = H or acyl-2-(4,5-dibromopyrrole).
The first disconnection to dideoxy-pre-axinellamine (23, described in our biosynthetic hypothesis)22b consists of two separate (but equally important) steps: oxidation of the 2-aminoimidazole with intramolecular nucleophilic capture of the resulting iminium(s), and oxidation of C20. The former closely follows the biosynthetic hypothesis that was reviewed in 2007.22b It was believed that this oxidation occurs in Nature to make all of these natural products, and that there would be a thermodynamic bias for them to undergo similar transformations in the laboratory. It is worth noting that several other biosynthetic hypotheses have been published by other groups,3a,9,21a,21t,21u,22c,30 and that our hypothesis builds on pioneering work by Rinehart2d and Horne.27,31 The oxidation of C20 is a strategic disconnection: by saving the oxidation of C20 for the ultimate or penultimate step of the synthesis, the stability and reactivity problems associated with a hemiaminal could be avoided during the majority of the skeleton-building steps. As alluded to earlier, this disconnection was notably lacking in precedent, and thus its inclusion into the retrosynthesis was not without a significant amount of risk.
Dideoxy-pre-axinellamine (23) could arise from acylation of bis-azide 24, which could in turn be generated from the spirocyclic amino ketone 25. Taking lessons from the bio-inspired route (Scheme 2), 25 was envisioned to arise from a guanidine addition to enone 26 followed by amination. Unlike the deschloro route (vide supra), the proximal substitution at C17 has the potential to direct the aza-Michael addition, possibly avoiding the exclusive formation of the wrong spirocenter. Enone 26 could be traced back to diketone 27 via intramolecular aldol and chlorination. Finally, 27 could arise from the [4+2] cycloaddition product 28.
The synthesis of 28 via a [4+2] cycloaddition provided a unique opportunity to render the entire route asymmetric, as numerous asymmetric cycloadditions of siloxydienes have been reported in the literature (albeit rarely with a 1-siloxy substituent).32 A brief summary of the efforts to achieve an asymmetric [4+2] cycloaddition (leading to cyclohexenes 32) with TIPS diene 31 can be found in Table 1. Utilizing Corey’s oxazaborolidine catalyst 33 with trifluoromethanesulfonimide33 or aluminum tribromide34 and various dienophiles gave little to no enantiomeric excess. Evans’ ligand 34 with copper(II) triflate35 did induce some asymmetry with dienophile 35, but was not efficient enough (ca. 10% yield) to justify further optimization. The Shibasaki catalyst FujiCAPO (36)36 provided the product in excellent yield (using a TMS version of the diene), but predominantly gave the exo- stereoisomer (epimeric at C17). Promising results came when the Ishihara catalyst 3737 was employed with dienophile 35: copper(II) triflate gave excellent enantiomeric excess (96%) in nitromethane, with acceptable yield (61%). When the Lewis acid was exchanged for copper(II)trifluoromethanesulfonimide, the yield was virtually quantitative while enantioselectivities remained excellent. This procedure proved to be scalable with a minimal depreciation in yield and enantioselectivity. Additionally, the product was crystalline and X-ray crystallographic analysis secured the absolute configuration as that corresponding to the stereochemistry of sceptrin (4) and ageliferin (5). It should be noted that intermediates that were only synthesized in a racemic manner will be annotated with a (±), while those synthesized using enantioenriched material will be annotated with the sign of their optical rotation henceforth in this article.
Table 1.
Catalytic asymmetric [4+2] cycloadditions attempted on TIPS diene 31.
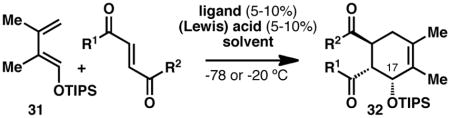 | |||||
|---|---|---|---|---|---|
| ligand | R1/R2 | metal/acid | solvent | yield | ee |
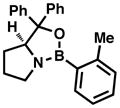 33 |
OEt | Tf2NH | DCM | 30% | <5% |
| OMe | AlBr3 | DCM | <5% | - | |
| OCH2CF3 | AlBr3 | DCM | 89% | <5% | |
|
| |||||
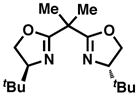 34 |
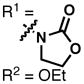 35 |
Cu(OTf)2 | DCM | 10% | −40% |
|
| |||||
 36 |
OMe | Ba(OiPr)2 + CsF | THF | 33% 66% exo |
9%a 95% |
|
| |||||
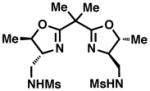 37 |
 35 |
Cu(OTf)2 | DCM | 60% | 84% |
| Cu(OTf)2 | MeNO2 | 61% | 96% | ||
| Cu(NTf2)2 | MeNO2 | 97% | 95%b | ||
| Cu(NTf2)2 | MeNO2 | 84% | 89%c | ||
| [19.2 g scale] | |||||
Reactions performed in the Shibasaki laboratory, using the TMS-version of 31. GC yields reported for this experiment.
Procedure for sub-gram scale: dienophile 35 (1.0 equiv), diene 31 (3.0 equiv), ligand 37 (0.10 equiv), Cu(NTf2)2 (0.11 equiv), MeNO2, −20 °C, 96 h, 97% isolated yield, 95% ee
Procedure for multi-gram scale: dienophile 35 (1.0 equiv), diene 31 (2.5 equiv), ligand 37 (0.050 equiv), Cu(NTf2)2 (0.055 equiv), MeNO2, −20 → 4 °C, 15 h, 84% isolated yield, 89% ee. Product could be recrystallized (hexane:2-propanol) to >99% ee.
The reduction of cyclohexene 38 (Scheme 6) to intercept the racemic route proved surprisingly difficult. Several harsh reductive conditions failed to deliver the desired diol, and instead reduced the oxazalidinone to deliver products such as 39. To circumvent this problem, 38 was treated with dodecanethiol to deliver thioester 40, which could readily be reduced with lithium aluminum hydride to intercept the racemic route (86% yield, 60 g scale). Mesylation, displacement with azide, and TIPS deprotection provided cyclohexenol 41 in 81% yield over three steps. This allylic alcohol could then be protected as the p-methoxybenzyl ether and ozonized to deliver diketone 42 (86%, two steps), which could be brominated and cyclized on warm silica gel to deliver the fully substituted cyclopentane 43. Unfortunately, attempts to avoid the protecting group interchange by carrying the triisopropylsilyl group forward were very low-yielding, presumably due to the increased steric hindrance of the TIPS group for the bromination and aldol cyclization steps.
Scheme 6. Elaboration to a fully substituted cyclopentane core.a.

a Reagents and conditions: (a) Dodecanethiol (4.4 equiv), n-butyllithium (4.0 equiv), THF, −78 °C → 0 °C, 18 h, 74%; (b) LiAlH4 (3.0 equiv), Et2O, 0 °C, 1 h, 86%; (c) methanesulfonyl chloride (4.0 equiv), pyridine, 0 °C → 23 °C, 1 h; (d) sodium azide (6.0 equiv), DMF, 100 °C, 11 h; (e) TBAF (1.1 equiv), THF, 23 °C, 1 h, 81% for three steps; (f) NaH (2.0 equiv), PMBCl (1.1 equiv), TBAI (0.1 equiv), DMF, 0 °C → 23 °C, 2 h; (g) O3, MeOH, −78 °C, 1 h then DMS (3.0 equiv), 23 °C, 16 h, 86% for two steps; (h) diisopropylethylamine (6.0 equiv), TMSOTf (4.0 equiv), 0 °C → 23 °C, 1.5 h, then NBS (2.0 equiv), THF, MeCN, 0 °C, 15 min, then SiO2, 50 °C, 16 h, 66%. TBAF = tetra-n-butylammonium fluoride, PMBCl = p-methoxybenzyl chloride, TBAI = tetra-n-butylammonium iodide.
Although seemingly straightforward, the dehydration and chlorination of 43 to the desired trihaloenone 26 proved surprisingly laborious (Scheme 7). Treatment of 43 with Brønsted acids and Lewis acids failed to deliver the dehydrated product 44, and basic conditions only led to epoxide formation to yield oxirane 45. Burgess reagent efficiently formed sulfamidate 46, but could not be further elaborated to the desired carbamoyl enone 47. In an attempt to moderate the reactivity of the alpha position, bromoketone 43 was treated with lithium chloride to provide chloroketone 48, which was equally resistant to dehydration conditions (albeit generating less decomposition products than 43). Deprotection of the p-methoxybenzyl group with trifluoroacetic acid provided diol 50, which proved resistant to dehydration, but could be reacted with thionyl chloride to yield cyclic sulfite 52 (which was resistant to elimination). Upon exposure to sulfuryl chloride, however, diol 50 underwent a cascade reaction to accomplish not only the dehydration but also an in situ chlorination with inversion, directly providing the desired chlorine at C17 with the correct stereochemistry. This reaction has been optimized to 60% yield, and can provide the desired enone 26 on multigram scale..
Scheme 7. Roadblocks en route to the trihalogenated enone 26.a.

a Reagents and conditions: (a) LiCl (3.3 equiv), DMF, 23 °C, 1.5 h; (b) TFA:DCM (1:9), anisole (2.0 equiv), 0 °C, 1 h, 72% for two steps; (c) NaOH (1.1 equiv), THF, H2O, 0 °C, 45 min, 75%; (d) Burgess reagent (5.0 equiv), benzene, 50 °C, 6 h, 95%; (e) 2,6-lutidine (3.0 equiv), SOCl2 (2.0 equiv), DCM, 0 °C, 2 h, 78%; (f) SO2Cl2 (2.0 equiv), 2,6-lutidine (3.0 equiv), DCM, 0 °C, 30 min, 60%.
Trihalo enone 26 bears close resemblance to its deschloro derivative 21 (Scheme 4), so several conditions were attempted to effect the direct conversion of 26 into the fully-substituted core 54 (Scheme 8). Unfortunately, due to the highly sensitive nature of enone 26 (which contains five highly electrophilic sites), no appreciable amounts of product were ever detected. To temper the reactivity of enone 26, a Luche reduction was carried out (thus mitigating the reactivity of three of the electrophilic sites), followed by displacement of the allylic bromide with bis-Boc-guanidine, to deliver allylic guanidine 55 in 55% yield over two steps (4.2 g scale). Upon exposure to several oxidative conditions, 55 swiftly converted to the spirocycle 56 via aza-Michael addition to the transient enone. Over 50 conditions were tried at room temperature and below, but in every case only the wrong stereoisomer at C16 was produced. In a final series of experiments, high temperature oxidations that had the potential to override the kinetic bias towards the wrong spiro isomer were explored. Indeed, exposure of 55 to o-iodoxybenzoic acid in refluxing benzene was able to provide the desired diastereomer 57 in a 1.3:1 ratio to 56 at C16 (70%, 1.8 g scale). Attempts to resubject 56 to the same reaction conditions to effect a retro-aza-Michael/aza-Michael equilibration were unsuccessful, indicating that the aza-Michael addition was irreversible under the reaction conditions.
Scheme 8. Elaboration of enone 22 to the fully substituted core 20.a.

a Reagents and conditions: (a) NaBH4 (1.0 equiv), CeCl3 • 7 H2O (0.5 equiv), MeOH, 0 °C, 15 min; (b) N,N’-bis-Boc guanidine (1.2 equiv), DBU (1.2 equiv), DMF, −20 °C to 0 °C, 4 h, 55% for two steps; (c) IBX (2.0 equiv), benzene, 83 °C, 16 h, 70%, 1.3:1 57:56; (d) N-Boc guanidine (6.0 equiv), DMF or MeCN, 23 °C, 24–48 h, 5–20%; (e) NaN(CHO)2 (1.5 equiv), TBAI (0.1 equiv), THF, 23 °C, 3 h; (f) TFA:H2O (1:1), 50 °C, 24 h, 69% for two steps, 36% 25, 33% 16-epi-25; (g) H2NCN (40 equiv), pH 5, brine, 70 °C, 4 h, 71%. DBU = 1,8-diazabicycloundec-7-ene, IBX = o-iodoxybenzoic acid.
With every desired functional group bearing the correct stereochemistry in the cyclopentane core of 57, all that was required to reach the key precursor 24 was the formation of the 2-aminoimidazole from the chloroketone. However, even this simple transformation was met with roadblocks, as several guanidine sources in a variety of solvents delivered only the base-induced cyclization product 58 (this was not a problem on the wrong C16 isomer 56, resulting in several misleading model studies). An interim solution was devised in our 2008 synthesis by utilizing mono-Boc guanidine in THF without additional base.24a An improved route that is more amenable to scale-up is reported herein. Originally, the mixture of C16 isomers (56/57) was tediously separated by preparative TLC. It was later found that exposure of 56/56 to sodium diformylamide followed by deprotection with trifluoroacetic acid allowed for facile isolation of aminoketone 25 along with its C16 isomer (69% total, 36% 25, 820 mg scale, wrong diastereomer reacts more efficiently). Exposure of 25 to cyanamide in brine produced 24 in 71% yield. The judicious choice of brine (instead of water) was critical in this transformation to avoid displacement of the C17 chlorine with water (vide infra). With access to hundreds of milligrams of the fully-substituted cyclopentane core, the first investigations into biomimetic closures to deliver the axinellamines (9–10), massadines (11–12), and palau’amine (13) could finally be explored.
Explorations on Biomimetic Closures and Formation of a Universal Retrosynthetic Plan
Upon close inspection of the natural products 9–13, it becomes apparent that there are several potentially unstable functional groups and substructures. Palau’amine (13) has a strained trans-5,5 system that is only held together by two aminal moieties. The axinellamines (9–10) contain an aminal to anchor the core, while the massadines (11–12) are held together by N,O-ketals. Despite all of these seemingly labile substructures, all of the natural products 9–13 were reported to be stable to isolation conditions and extended storage. For this reason, it was believed that any open-chain versions of these natural products would be thermodynamically inclined to form the corresponding closed structures found in Nature, as reasoned in the biosynthetic hypotheses (and outlined in Scheme 5). Therefore, the synthesis of dideoxy-pre-axinellamine (23, Scheme 5) was accomplished by the reduction of the azides in 24 with hydrogen over platinum oxide followed by acylation with dibromopyrrole 59 to provide 23 in 26% overall yield along with 17% of the mono-acylated product. Dideoxy-pre-axinellamine (23) is also available via a fully-protected route, as noted in the original communication.24a
It was expected that the oxidation of the 2-aminoimidazole in 23 would cause a spontaneous closure to one or all of the axinellamine (60) or palau’amine (61) congeners via intramolecular capture with the spirocenter nitrogen or the pyrrole-amide sidechain (massadine-type closure was not possible without C20 oxidation). Indeed, several oxidants such as NBS, NCS, MnO2, DMDO, and others were able to selectively oxidize the 2-aminoimidazole found in 23, although oxidation of the dibromopyrroles was a very common side reaction. In several cases, if a nucleophilic solvent was used, oxidized derivatives such as 62 could even be detected by spectroscopy. However, it was perplexing to observe that even though intermediates such as 62 were found, the only product that could ever be isolated from the oxidative attempts was the unsaturated cyclopentene 63. This product represented a veritable dead end: two stereocenters had been destroyed, and another reactive olefin was added to the system. Under no circumstances were any products with desired modes of cyclization found in even trace amounts, with hundreds of conditions attempted.
The failures of such a seemingly promising biomimetic finale did not come without useful reconnaissance. The use of strong oxidants resulted in overoxidation of the pyrrole moieties in addition to the desired 2-aminoimidazole oxidation. Additionally, no oxidation of C20 was ever observed, even when extensive oxidation of the rest of the molecule was taking place. Finally, closure to the complete natural product cores would not be an easy feat to accomplish, and would require careful synthetic planning. Taking these lessons into account, a detailed non-biomimetic retrosynthetic analysis was forged. The general rules for the disconnections were as follows: C20 oxidation and pyrrole installation should be saved until the latest possible step, intramolecular closure should take place in a manner that is thermodynamically biased, and the molecules’ inherent reactivity should be harnessed (avoiding the protection of functional groups). These guidelines led to the complete retrosynthesis shown in Scheme 10.
Scheme 10. Non-biomimetic retrosynthesis of the pyrrole-imidazole alkaloids to a common precursor (25).

The axinellamines (9–10) presented the most straightforward targets, as they did not necessitate the installation of the C20 hydroxy or either of the pyrrole moieties prior to core cyclization. Therefore, the installation of the pyrroles would be saved to the last step, leading to axinellamine azide (68). The next disconnection would prove crucial to the completion of each of the natural products outlined in this scheme: deletion of the C20 hydroxy group (amounting to a formal C–H oxidation in the forward direction) to give the axinellamine core structure 69. It was anticipated that this step would be far from trivial as no such oxidations were present in the literature (vide infra). Compound 69 could be drawn back to 2-aminoimidazole 24, requiring oxidation and closure with only one proximal nucleophile present to form the core structure.
Analysis of the massadines (11–12) bears resemblance to the axinellamine retrosynthesis, with one key exception: oxidation of C20 in the massadine retrosynthesis had to precede closure to the core, since the C20 hydroxyl group formed an integral part of its architecture. Thus, after removal of the pyrroles to yield massadine chloride azide 70, scission of the bridging C20 hydroxyl group would lead back to diol 71. It was anticipated that the closure from 71 to 70 would be selective at the hydroxy group (thus outcompeting axinellamine closure) under acidic conditions. Diol 71 could arise from oxidation of the 2-aminoimidazole in 72, which could in turn be reached via imidazole synthesis from aminoketone 73. Finally, in a circumstance even more challenging than the axinellamine route, the C20 hydroxy group would need to be installed on the common precursor 25, which contains a primary ammonium group.
Palau’amine (13) presented the most challenging structure by far: the installation of the pyrrole unit could not be left until the final stages of the synthesis. Additionally, biomimetic efforts towards the core (Scheme 9) had shown absolutely no promise for an oxidative closure of a pyrrole-amide sidechain onto an oxidized 2-aminoimidazole. Finally, of the two azide moieties present in the common precursor 25, one of them would need to be selectively acylated, despite their virtually identical chemical environments. These considerations led to two retrosynthetic routes to palau’amine (5) that capitalized on a strategy that would simultaneously allow the selective acylation of one of the azides and generate a structure that was thermodynamically biased to close to the natural product core upon selective acylation of one azide.
Scheme 9. Failed biomimetic approaches to the pyrrole-imidazole alkaloids.a.

a Reagents and conditions: (a) PtO2 (0.1 equiv), H2 (1 atm), TFA:H2O (1:9), 23 °C, 2 h; (b) (trichloroacetyl)pyrrole 59 (3.0 equiv), diisopropylethylamine (3.0 equiv), DMF, 40 °C, 3 h, 26% + 17% mono-acylated product; (c) NBS (1.0 equiv), MeOH, −78 °C, 26 h; (d) neat TFA, 30 °C, 24 h, 34%.
In Route 1 (Scheme 10), oxidation of C20 was reserved until the final step in order to minimize side reactions stemming from the reactive hemiaminal moiety throughout the synthesis, even though the success of such an oxidation was questionable in the presence of the pyrrole moiety found in 64. Deoxypalau’amine azide 64 could be drawn back to its constitutional isomer 65, which contained a nine-membered macrocyclic lactam that appeared by molecular models to be poised to undergo transannular cyclization upon isomerization of the 2-aminoimidazole. This strategic disconnection was designed specifically to overcome the barrier that would likely be encountered in forming the trans-5,5 core by capitalizing on a potential relief of transannular strain present in a nine-membered macrocycle to the conformationally stable (albeit still strained) bicyclo[3.3.0]octane of palau’amine (13). Thus, it was postulated that a dynamic equilibrium among conformational macrocyclic isomers of 65 would eventually lead to the thermodynamic sink of deoxy-palau’amine-azide (64) by a net, irreversible ring-chain tautomerization. A conceptually similar approach to topology control had been demonstrated in our synthesis of the kapakahines.38 It was envisioned that 65 could arise from a Staudinger ligation from 66 to form the amide bond, a strategy that was pioneered by Raines and Bertozzi,39 and has been shown to be especially effective at the formation of medium-sized rings.40 The final retrosynthetic barrier to overcome would be the attachment of the pyrrole, which could arise by a formal SNAr reaction on the bromo-2-aminoimidazole 67.
The potential complications arising from the oxidation of C20 in the last step of Route 1 led to the generation of an alternate plan. Route 2 contains the same general strategy as the ligation route with two key exceptions: the C20 oxidation would be performed at the same stage as the massadine route (24 → 73), and the elaborate Staudinger ligation would be replaced with a more traditional macrolactamization in the penultimate step. Certainly this route would present problems with chemoselectivity due to the ever-present hemiaminal, but if it could not be installed at a later stage as in Route 1, then its presence would be unavoidable.
Total Synthesis of (−)-Axinellamine A and (−)-Axinellamine B24b
To attempt a synthesis of the axinellamines (9–10), two main hurdles would have to be overcome: first, closure to the core would have to proceed where it failed on dideoxy-pre-axinellamine (23); second, a suitable oxidative reaction to install the C20 hemiaminal would need to be invented. The former could only be solved by trial-and-error, but the latter could be studied on a model system. It is worth noting that there are two inherent challenges to this type of oxidation: chemoselectivity on a complex system (with several Lewis basic atoms and multiple oxidizable sites) and redox control (the hemiaminal product has a higher potential to be oxidized than the starting material). To selectively study this redox challenge, 1,3-diazaspiro[4.4]nonan-2-imine (78) was prepared and exposed to several oxidative conditions (Scheme 11).23x
Scheme 11. Model system for the oxidation of spiro-annulated imidazolines.a.

a Reactions monitored by HPLC-MS and 1H-NMR comparison to known standards of 79 and 80.
Halogenating reagents (NCS, NBS, NaOCl, NaOBr, tBuOCl, I2) usually reacted at nitrogen, and some of the resulting N-halo species were stable enough to be observed by NMR. Attempts to eliminate these N-halo species to the desired iminium with base proved unsuccessful, causing decomposition of the substrate. Ruthenium protocols that have been successful in the oxidation of amines (RuCl3/H2O2, Ru–C/tBuOOH, RuCl2(PPh3)3/tBuOOH,) generally provided complex mixtures, with both 79 and 80 present in varying ratios. Hypervalent iodine reagents (I2O5, PhIOH(OTs), IBX, PhIO, PhI(OAc)2, HIO3) proved generally unreactive, with the exception of iodosobenzene and iodobenzene diacetate, which gave trace product in MeOH. Stronger oxidants such as potassium ferrate gave rapid overoxidation to 80. Potassium persulfate gave 79 as the major product, with some 80 and some leftover starting material. Silver(II) oxide in the presence of nitric acid gave product, but with poor reproducibility. The most promising results, however, were achieved by employing a ligand-decorated silver(II) complex: silver(II) picolinate. This complex was studied by Bacon et al. in the 1960’s,41 and was found to oxidize amines to imines (cleaved in situ to aldehydes) without extensive overoxidation.41c It was non-basic and the oxidations were performed in water (pyrrole–imidazole alkaloids are generally stable in aqueous acidic media), making it ideal for the application to the axinellamines (9–10).
However, before the conditions could be applied to the axinellamine system, a synthesis of the desired core structure 69 would need to be carried out, and is illustrated in Scheme 12. The synthesis commenced with the oxidation of 24 with dimethyldioxirane to give a transient diol, which was treated with trifluoroacetic acid to effect closure to the tetracyclic core 69 as a mixture of diastereomers (both of which would be used to generate the stereoisomeric pair of the natural products 9 and 10). It was gratifying that no elimination byproducts (analogous to the 62 → 63 conversion in Scheme 9) were observed during this cyclization. The core structure 69 was now poised to undergo oxidation at C20, but it remained to be seen if the insight gained from the model study in Scheme 11 would be compatible with such a complex system. Indeed, 69 possessed several sites that could be oxidized, as well as a hemiaminal and an alkyl halide that could complicate selective oxidation at C20. In the event, 69 was exposed to silver(II) picolinate (81) in 10% aqueous trifluoroacetic acid to deliver 68α and 68β in 43% yield over three steps. It is worth noting that this final oxidation proceeds at room temperature in an open flask in under 30 minutes, and the disappearance of the brick-red silver(II) picolinate can be observed as the reaction turns to a clear, homogeneous solution. Additionally, when the separated diastereomers of tetracycle 69 is exposed to the same conditions, yields of up to 77% can be obtained for this single step.
Scheme 12. Enantioselective total synthesis of (−)-axinellamine A (9) and (−)-axinellamine B (10).a.

a Reagents and conditions: (a) DMDO (0.35 mL of 0.09 M solution), H2O, 0 °C, 15 min; (b) TFA:DCM (2:1), 23 °C, 17 h; (c) silver(II) picolinate (3.5 equiv), TFA:H2O (1:9), 23 °C, 30 min, 43% (3:4 68α:68β); (d) 1,3-propanedithiol (15–20 equiv), Et3N (11–14 equiv), MeOH, 23 °C, 2 h; (e) (trichloroacetyl)pyrrole 59 (3.0 equiv), diisopropylethylamine (3.0 equiv), DMF, 45 °C, 6–11 h; 9 = 45%, 10 = 24%. DMDO = dimethyldioxirane.
The completion of the enantioselective synthesis of the axinellamines (9–10) required the reduction of the azides followed by acylation with a dibromoacylpyrrole surrogate. The former step proved surprisingly difficult, presumably due to complications with the highly polar, sensitive substrate. Phosphine reduction of the azides resulted in general decomposition, and hydrogenation over various metals provided inconsistent results (although platinum oxide allowed for low yields of diamine to be obtained). The best results were obtained by exposure to propanedithiol and triethylamine, providing the diamine in sufficient purity to carry forward. Selective acylation with (trichloroacetyl)pyrrole 59 provided enantioenriched axinellamine A (9) and axinellamine B (10) in 45% and 24% yield over two steps, respectively. The optical rotation and circular dichroism spectra matched both the reported data5a and natural samples provided by the Quinn laboratory, indicating that the natural enantiomer of these alkaloids had been synthesized. It should be noted that the absolute stereochemistry of these enantiomers matches that found in sceptrin (4) and ageliferin (5), lending further credence to the biosynthetic hypothesis outlined in Scheme 1.
Total Synthesis of (−)-Massadine and (−)-Massadine Chloride, and Their Epi- Analogues25
Although the retrosynthesis of the massadines (11–12) outlined in Scheme 10 bears resemblance to that of the axinellamines, it represents the product of several initial explorations that were met with failure. The main goal of these explorations was to uncover the ideal stage to install the C20 oxidation state on the massadine scaffold, and a brief summary of these efforts is shown in Scheme 13. Initial explorations involved C20 oxidation prior to 2-aminoimidazole formation, but it was quickly discovered that the C20 hemiaminal was incompatible with basic conditions necessary to install the 2-aminoimidazole. Therefore, two alternative routes were investigated: Route 1 was an attempt to install the C20 hydroxyl group and form the tetrahydropyran core prior to 2-aminoimidazole formation. This approach began with the silver(II) picolinate-mediated oxidation of α-acetoxyketone 83 to provide 84 in 54% yield. Treatment of this intermediate with aqueous trifluoroacetic acid provided the desired ether formation in 65% yield, providing three out of the four rings in the massadine core. Unfortunately, all attempts to elaborate tetrahydropyran 85 to the necessary 2-aminoimidazole were met with failure, reminiscent of tricycle 58 (Scheme 8), which could not be elaborated to the axinellamines (9–10). Therefore, oxidation of C20 was attempted with the 2-aminoimidazole already in place, as shown in Route 2. Disappointingly, over oxidation of the 2-aminoimidazole occurred prior to oxidation of C20, proving that the electron-rich heterocycle was not compatible with the silver(II) picolinate conditions.
Scheme 13. Attempts to secure both the 2-aminoimidazole and the C20 hemiaminal en route to the massadines (11–12).a.

a Reagents and conditions: (a) silver(II) picolinate (2.5 equiv), TFA:H2O (1:9), 23 °C, 30 min, 58%. b TFA:H2O (2:1), 50 °C, 20 h, 65%.
A solution was found with a two-step 2-aminoimidazole synthesis, allowing for all basic conditions to preceed C20 oxidation by beginning with aminoketone 25 (Scheme 14). The originally reported synthesis of the massadines (11–12)25 utilized a protected version of 25 under the assumption that oxidation of the primary amine would outcompete oxidation of C20 (as shown in Bacon’s original report).41c However, it was found that the optimized conditions (which employed excess trifluoroacetic acid) were sufficient to completely temper the reactivity of the primary amine, allowing for the oxidation of C20 to occur in 69% yield, delivering 73 without any protecting groups. Formation of the 2-aminoimidazole was also optimized since the original report, utilizing brine as a solvent which greatly reduced the amount of Cl-OH exchange at C17, providing 72 in 65% yield. Oxidation to the transient diol 65 with DMDO followed by acid-mediated cyclization provided massadine chloride azide 70 in 71% yield as a 1:3.7 ratio of diastereomers. Interestingly, attack from the spiroguanidine nitrogens (to form the axinellamine core) was not observed, presumably because the acidic conditions hindered their reactivity. Unfortunately, the major diastereomer was epimeric to the natural products 11–12, although it is a possibility that the alkaloids corresponding to this diastereomer exist in Nature and have yet to be isolated (note the similarity to axinellamines A and B, 9–10).
Scheme 14. Total synthesis of (−)-massadine (11) and (−)-massadine chloride (12).a.

a Reagents and conditions: (a) silver(II) picolinate (2.5 equiv), TFA:H2O (1:9), 23 °C, 30 min, 69%; (b) H2NCN (40 equiv), pH 5, brine, 70 °C, 4 h, 65%; (c) DMDO (1.3 equiv), TFA:H2O (1:9), 0 °C, 1 h; (d) neat TFA, 23 °C, 3 h, 71% (1:3.7 d.r. at C6/C10); (e) PtO2 (0.3 equiv), H2 (1 atm), TFA:H2O (1:19), 1 h, 23 °C; (f) (trichloroacetyl)pyrrole 59 (16 equiv), diisopropylethylamine (16 equiv), DMF, 23 °C, 14 h, 60% from 70α (2:1 9:8), 55% from 70β (4:1 88:89); (g) H2O, 60 °C, 4 h, quant.
The completion of the synthesis required reduction of the azides and acylation in a manner similar to the axinellamines (9–10). However, propanedithiol could not be used due to the more sensitive nature of the massadine scaffold to nucleophiles and base, and therefore hydrogenation over platinum oxide was employed. Acylation of 70β provided 6,10-epi-massadine chloride (88) and and 6,10-epi-massadine (89) in a 4:1 ratio in 55% yield (88 could be converted to 89 with mild heating in aqueous media, occurring much slower than the natural analogs 11 and 12).25 Acylation of 70α with (trichloroacetyl)pyrrole 59 provided massadine chloride (12) and massadine (11) in 60% yield as a 2:1 ratio (the conversion of 11 to 12 occurs readily in aqueous media with mild heating).8,25 The optical rotation and circular dichroism spectra of these natural products matched those of literature reports6,8 and natural samples provided by the Köck group, thus confirming their absolute configurations. This further supported the biosynthetic hypothesis outlined in Scheme 1, as the absolute stereochemistry of the massadines (11–12) matched that of the axinellamines (9–10), sceptrin (4) and ageliferin (5). It is interesting to note that the epi-series (88 and 89) have not been discovered in natural extracts (unlike to the axinellamines, where the epi-series was isolated from the same sponge).5a It is possible that 88 and 89 are natural products that have not yet been identified from natural sources.
Total Synthesis of (−)-Palau’amine
Compared to the axinellamines (9–10) and the massadines (11–12), the planned retrosyntheses of palau’amine (13) were ridden with questionable steps. The first of these steps was shared between both routes in Scheme 10: the coupling of a pyrrole moiety to the 2-aminoimidazole. During the synthesis of the axinellamines (9–10), the 2-aminoimidazole 24 was treated with electrophilic bromine sources in non-nucleophilic solvents in an attempt to induce intramolecular capture by the spirocyclic guanidine to yield 70 (Scheme 15A). However, it was found that bromination of the 2-aminoimidazole was followed by an isomerization instead of nucleophilic capture, providing products such as 67 (Scheme 10). The potential of such products to be utilized as coupling partners for transition metal mediated aminations was evaluated on model C16 epimer 90, and several conditions were tried to effect palladium42 or copper43 catalyzed amination of bromo-2-aminoimidazoles. Although these couplings were met with universal failure, a common byproduct did not escape notice: imidazolinone 91 was present in several crude reaction mixtures, presumably arising from capture of the highly active imidoyl bromide isomer of 90 with residual water at the high temperatures commonly employed during Buchwald–Hartwig and Ullmann aminations. The removal of all base, ligand, and catalyst from the reaction conditions quickly confirmed this result, delivering 91 directly with mild heating.
Scheme 15. Initial investigations into the generation of an N-coupled pyrrole.a.

a 90 (1.0 equiv), TFA:H2O, 60 °C, 24 h, 82%. b 90 (1.0 equiv), Neat pyrrole, 60 °C, 2 h, 71%. c 90 (1.0 equiv), AcOH (3.0 equiv), pyrrolidine (3.0 equiv), 40 °C, 3 h, 48%. d 90 (1.0 equiv), AcOH (3.0 equiv), benzylamine (3.0 equiv), 40 °C, 3 h, 72%.
The hydrolysis of an aryl bromide would not prove useful in the synthesis of the desired N-arylpyrrole, so the reactivity of 90 was further explored using different nucleophiles (Scheme 15B). An attempt to install a pyrrole directly unsurprisingly led to reactivity at the pyrrole C2, providing 92, which unfortunately possesses a connectivity that holds no worth in the synthesis of the natural products. A glimmer of hope was found when alkylamines such as pyrrolidine and benzylamine were employed, providing the N-coupled products 93 and 94. The answer was now clear: an alkylamine pyrrole surrogate could be coupled to the 2-aminoimidazole followed by conversion to the pyrrole after attachment. Three such surrogates are shown in Scheme 14C: the dihydropyrrole ester 95, the dihydrooxazine ester 96, and the linear primary amine 97.
The optimized synthesis of the correct C16 isomer of the bromo-2-aminoimidazole was accomplished by treatment of 24 with elemental bromine in a trifluoroacetic anhydride/trifluoroacetic acid solvent mix in 83% yield (Scheme 16). Trifluoroacetic anhydride (TFAA) was essential, mediating an in-situ trifluoracetylation of the 2-aminoimidazole to cause the desired bromination to occur. Without TFAA, a major byproduct was dihydroxylation of the 2-aminoimidazole (similar to what is seen upon treatment with electrophilic halogen sources in the presence of water). The coupling of the resulting bromide 67 with pyrrole surrogates 95 and 97 proceeded smoothly, whereas dihydrooxazine 96 failed to deliver any product. Linear amine 97 was favored for the final sequence, since mild treatment with acid could convert it directly to the desired pyrrole acid 99 in 45% overall yield from 67 (at least five transformations occur in one pot to deliver the desired product). This pyrrole acid was coupled with thiol 99, delivering the thioester 66 (48% yield), which was poised to undergo ligation upon deprotection of the phosphine with a nucleophilic amine. Several failed attempts at the ligation (with various conditions for phosphine deprotection) ultimately culminated in the treatment of 66 with DABCO in dry THF40 to deliver the 20-deoxymacropalau’amine azide 65 in 20% yield. To our delight, the macrocyclization was selective for the adjacent azide (albeit low-yielding), and the product was stable enough for 2-D NMR studies, revealing the intriguing folded structure shown with ROESY correlations. A correlation between C11 and N14 was very promising: the transannular cyclization would require proximity to exist between the 2-aminoimidazole and the amide nitrogen, and that proximity already appeared to exist in solution.
Scheme 16. Synthesis of deoxypalau’amine azide 66.a.

a Reagents and conditions: (a) TFAA:TFA (1:1), 1 h, then Br2 (2.0 equiv), 1 h, then TFA:H2O (1:1), 1 h, 38 °C, 58% (b) AcOH (3.0 equiv), 97 (3.0 equiv), THF, 38 °C, 6 h; (c) TFA:DCM (1:30 to 1:1), 23 °C, 12 h, 48%; (d) EDC (1.5 equiv), DMAP (2.0 equiv), thiol 99 (1.2 equiv), MeCN, 23 °C, 2 h, 48%; (e) DABCO (7.5 equiv), THF, 60 °C, 6 h, 20%; (f) neat TFA, 70 °C, 23 h, 38% (ca. 75% conversion). TFAA = trifluoroacetic anhydride, EDC = 3-(ethyliminomethyleneamino)-N,N-dimethylpropan-1-amine, DMAP = 4-(N,N-dimethylamino)pyridine, DABCO = 1,4-diazabicyclo[2.2.2]octane.
Unfortunately, Scheme 16 does not accurately convey the difficulty that was encountered in the several attempted conversions of macrocycle 65 to deoxypalau’amine azide 64.44 Indeed, over 50 reaction conditions were tried on very limited amounts of material, ranging from standard heat and acid to exotic, transition metal-catalyzed N-arylation attempts. It was only after considerable screening that the successful conditions of warm, neat trifluoroacetic acid were discovered to successfully acquire 64 in 38% yield. With the core of palau’amine (13) in hand, conditions for the oxidation of C20 were applied to 64 to approach palau’amine azide (100). Unfortunately, oxidation of the pyrrole completely outcompeted oxidation at C20 with any oxidant employed, from silver(II) picolinate to potassium persulfate, and several of the others mentioned on the model study in Scheme 11. Consistently, the desired increase in mass was detected by HPLC-MS, but crude NMR spectra showed that oxidation had not occurred at the spirocyclic C20 position, but instead on the pyrrole moiety. This roadblock could not be overcome, so our sights were turned away from 64 (which bore such close resemblance to palau’amine that the proton NMRs were almost identical except for one key hemiaminal peak) and towards Route 2 in Scheme 10.
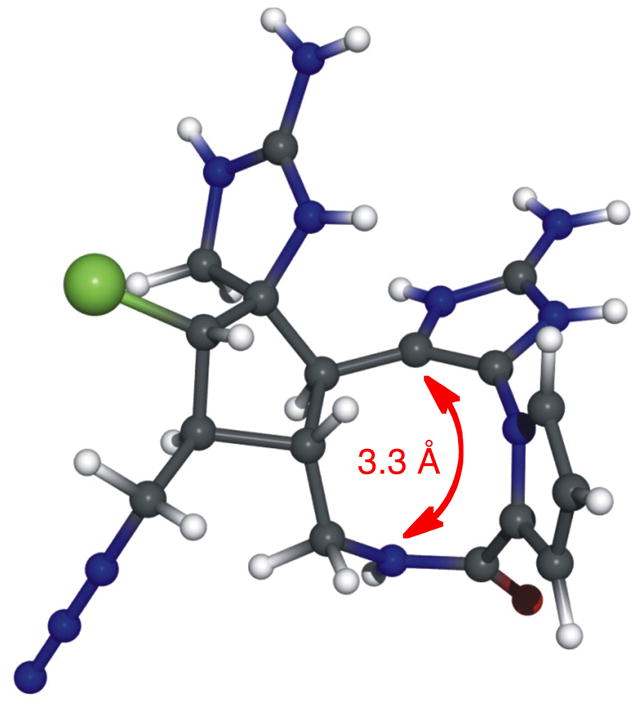
The interesting, folded structure of 20-deoxymacropalau’amine-azide (65) merited more detailed inspection, possibly providing insight into the oxidized derivative (macropalau’amine, 74, Scheme 10) that would have to be synthesized for the revised route. The fc-rDG/DDD method14,45 was applied to 65 with floating chirality to the five stereogenic centers. The basis for the conformational and configurational analysis is a ROESY spectrum from which cross-peaks are translated into interproton distances. For compound 65, we were able to obtain 19 interproton distances from a ROESY spectrum with a mixing time of 100 ms (complete details in the Supporting Information). This resulted in 91 out of 99 structures that have the same relative configuration, which are divided into two families (family I and II). Due to small configurational differences, family I was further divided into family Ia (52 structures) and family Ib (24 structures), and family Ia served as reference for the conformational analysis of 65, and an averaged structure is depicted in Figure 1. Two key interproton distances were of interest: H5/H12 (3.6 Å) and H11/H14 (2.8 Å). The proximity of these key protons was indicative of the interesting, folded nature of 65. The most telling inter-atom distance, however, was the H14/C10 distance (3.3 Å, red arrow), which indicated that even in solution at room temperature, the two atoms between which the desired transannular reaction was to occur were already in proximity with each other. The expectation was that the conformation of macrocycle 65 would exist in the analogous system with the required C20 oxidation already in place (macropalau’amine (74), Scheme 10).
Figure 1. Structural conformation of 20-deoxymacropalau’amine-N3 (65) based on the average of 52 interproton distances from a 100 ms ROESY spectrum.
The “backup” route to palau’amine (13) involved early installation of the C20 hemiaminal, diverging from the massadine synthesis at 2-aminoimidazole 71 (Scheme 17). Bromination of 66 with elemental bromine in the presence of trifluoroacetic anhydride delivered 71 in 54% yield, and attachment of the pyrrole surrogate 97 followed by pyrrole generation proceeded in 44% yield. All attempts to employ the Staudinger ligation steps used in Scheme 16 on the C20-oxidized derivative 70 failed to provide any product. This appeared to be due to the sensitive nature of the hemiaminal moiety during attempts to couple the highly nucleophilic ligation precursor 99 to the pyrrole acid found in 70. However, an even more direct route was followed in a final sequence which was carried out without purification of intermediates. First, hydrogenation of the azide moieties in 76 over palladium acetate provided diamine 75 in sufficient purity to be carried forward. The choice of palladium acetate was critical: palladium on carbon adsorbed all of the material, while platinum oxide reduced the pyrrole.
Scheme 17. Enantioselective total synthesis of (−)-palau’amine (13).a.

a Reagents and Conditions: (a) TFAA:TFA (1:1), 1 h, then Br2 (2.0 equiv), 1 h, then TFA:H2O (1:1), 38 °C, 1 h, 54%; (b) AcOH (3.0 equiv), 97 (3.0 equiv), THF, 38 °C, 6 h, then TFA:DCM (1:30 to 1:1), 23 °C, 12 h, 44%; (c) Pd(OAc)2 (1.6 equiv), H2 (1 atm), TFA:H2O (1:9), 23 °C; (d) EDC (3.0 equiv), DMF, 23 °C, 3 h; (e) neat TFA, 70 °C, 24 h, 17% for three steps.
Crude diamine 75 was subjected to several coupling reagents, 46 including (but not limited to) HATU, HBTU, TCTU, DPPA, T3P, EDC, DIC, DCC, BOP, BOP-Cl, PyAOP, PyBOP, PyBrOP, and cyanuric chloride in several combinations of polar solvents (75 was only soluble in DMF, DMSO, and water in solvent screens). Interestingly, base seemed to inhibit thes reaction (and eventually cause decomposition). However, EDC provided the desired mass of macropalau’amine 74 (420) by HPLC-MS (with or without HOBt additive), and exposure to hot trifluoroacetic acid (analogous to Scheme 16) provided palau’amine (13) in 17% overall yield for the final three-step sequence. The optical rotation and circular dichroism spectra correlated well with the published results,15 indicating that the correct enantiomer of the natural product had been synthesized, and proving its absolute configuration. The absolute stereochemistry matched that of the massadines (11–12), axinellamines (9–10), sceptrin (4) and ageliferin (5), thus tying together the entire family of pyrrole-imidazole alkaloids in uniform handedness.
Conclusion
This article has traced the evolution of the first laboratory syntheses of (−)-palau’amine (13), (−)-axinellamine A (9), (−)-axinellamine B (10), (−)-massadine (11), and (−)-massadine chloride (12). These pyrrole-imidazole alkaloids have inspired and confounded chemists for almost two decades, and have provided a driving force for creativity and innovation in our laboratory and several others. The natural enantiomer of each of these natural products was synthesized with one unifying route, which was enabled by the power of a catalytic, enantioselective Diels–Alder reaction. The absolute configurations of 9–13 were found to share common stereochemistry with the structurally simpler family members (−)-sceptrin (4) and (−)-ageliferin (5), supporting a common biogenesis such as the one proposed in 2004. The syntheses of these marine natural products have led to the development of a powerful oxidation protocol which utilizes silver(II) picolinate to selectively oxidize spiroimidazolines in complex molecular architectures without overoxidation. This chemistry also led to further exploration of silver chemistry, inspiring the development practical methods for the direct C–H arylation of heterocycles47 and quinines.48 Additionally, nuances of the reactivity of bromo-2-aminoimidazoles have been reported, including their hydrolysis and innate coupling to heterocycles and amines. Finally, a macrocyclization combined with a strategic transannular cyclization enabled the synthesis of palau’amine (13), and proved the structural revisions published in 2007. It is worth noting that the success of each of these syntheses relied heavily on the innate reactivity present in each of the advanced intermediates, which would never have been unveiled had they been shielded with excessive protecting groups.49 Access to each of these enantioenriched natural products has enabled several collaborations to investigate their relatively unexplored biological activity, the results of which will be reported in due course.
Supplementary Material
Acknowledgments
We are grateful to Mr. A. Sobel and Prof. C. Lewis for technical assistance and helpful discussions. We thank Prof. Shibasaki for assistance with asymmetric Diels–Alder reactions using his catalytic system, and Prof. Ishihara for gracious donations of his ligand and helpful discussion. We thank Prof. A. Rheingold for X-ray crystallographic measurements. We are grateful to the NSF (predoctoral fellowships to I.B.S. and R.A.R.), the Japan Society for the Promotion of Science (JSPS, postdoctoral fellowship to J.Y. and A.N.), the Natural Sciences and Engineering Research Council of Canada (NSERC, postdoctoral fellowship to I.S.Y.), the U.S. Department of Defense and the Hertz Foundation (predoctoral fellowships to D.P.O), and the German Academic Exchange Service (DAAD) for a postdoctoral fellowship to T.G. Financial support for this work was provided by the NIH/NIGMS (GM-073949), Bristol–Myers Squibb (unrestrictred research grant).
Footnotes
Supporting Information. Experimental details, spectra, and X-ray crystallography. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Walker RP, Faulkner DJ, Van ED, Clardy J. J Am Chem Soc. 1981;103:6772–6773. [Google Scholar]
- 2.(a) Keifer PA, Koker MES, Schwartz RE, Hughes RG, Jr, Rittschof D, Rinehart KL. 26th Interscience Conference on Antimicrobial Agents and Chemotherapy; 1986. [Google Scholar]; (b) Rinehart KL. Pure Appl Chem. 1989;61:525–528. [Google Scholar]; (c) Kobayashi J, Tsuda M, Murayama T, Nakamura H, Ohizumi Y, Ishibashi M, Iwamura M, Ohta T, Nozoe S. Tetrahedron. 1990;46:5579–5586. [Google Scholar]; (d) Keifer PA, Schwartz RE, Koker MES, Hughes RG, Jr, Rittschof D, Rinehart KL. J Org Chem. 1991;56:2965–2975. [Google Scholar]; (e) Williams DH, Faulkner DJ. Tetrahedron. 1996;52:5381–5390. [Google Scholar]
- 3.(a) Kinnel RB, Gehrken HP, Scheuer PJ. J Am Chem Soc. 1993;115:3376–3377. [Google Scholar]; (b) Kinnel RB, Gehrken HP, Swali R, Skoropowski G, Scheuer PJ. J Org Chem. 1998;63:3281–3286. [Google Scholar]
- 4.(a) Kato T, Shizuri Y, Izumida H, Yokoyama A, Endo M. Tetrahedron Lett. 1995;36:2133–2136. [Google Scholar]; (b) Kobayashi Ji, Suzuki M, Tsuda M. Tetrahedron. 1997;53:15681–15684. [Google Scholar]
- 5.Urban S, Leone PdA, Carroll AR, Fechner GA, Smith J, Hooper JNA, Quinn RJ. J Org Chem. 1999;64:731–735. doi: 10.1021/jo981034g.These “axinellamines” should not be confused with the structurally unrelated natural products that share their name: Bascombe KC, Peter SR, Tinto WF, Bissada SM, McLean S, Reynolds WF. Heterocycles. 1998;48:1461–1464.
- 6.Nishimura S, Matsunaga S, Shibazaki M, Suzuki K, Furihata K, Van SRWM, Fusetani N. Org Lett. 2003;5:2255–2257. doi: 10.1021/ol034564u. [DOI] [PubMed] [Google Scholar]
- 7.(a) Grube A, Köck M. Org Lett. 2006;8:4675–4678. doi: 10.1021/ol061317s. [DOI] [PubMed] [Google Scholar]; (b) Buchanan MS, Carroll AR, Addepalli R, Avery VM, Hooper JNA, Quinn RJ. J Org Chem. 2007;72:2309–2317. doi: 10.1021/jo062007q. [DOI] [PubMed] [Google Scholar]
- 8.Grube A, Immel S, Baran PS, Köck M. Angew Chem, Int Ed. 2007;46:6721–6724. doi: 10.1002/anie.200701935. [DOI] [PubMed] [Google Scholar]
- 9.The existence of “massadine aziridine” was first proposed by Professor Romo in 2005 and by our group in early 2006. We were remiss not to mention this in the previous reference. Please see the following, along with reference 11d: Wang S, Dilley AS, Poullennec KG, Romo D. Tetrahedron. 2006;62:7155–7161.
- 10.Lindel T, Jacquot DEN, Zoellinger M, Kinnel RB, McHugh S, Köck M. Tetrahedron Lett. 2010;51:6353–6355. [Google Scholar]
- 11.(a) Baran PS, O’Malley DP, Zografos AL. Angew Chem, Int Ed. 2004;43:2674–2677. doi: 10.1002/anie.200453937. [DOI] [PubMed] [Google Scholar]; (b) Baran PS, Zografos AL, O’Malley DP. J Am Chem Soc. 2004;126:3726–3727. doi: 10.1021/ja049648s. [DOI] [PubMed] [Google Scholar]; (c) Baran PS, Li K, O’Malley DP, Mitsos C. Angew Chem, Int Ed. 2006;45:249–252. doi: 10.1002/anie.200503374. [DOI] [PubMed] [Google Scholar]; (d) Northrop BH, O’Malley DP, Zografos AL, Baran PS, Houk KN. Angew Chem, Int Ed. 2006;45:4126–4130. doi: 10.1002/anie.200600514. [DOI] [PubMed] [Google Scholar]
- 12.(a) Personal communication with Professor Matthias Köck. ; (b) Assmann M, Kock M. Z Naturforsch, C. 2002;57:157–160. doi: 10.1515/znc-2002-1-226. [DOI] [PubMed] [Google Scholar]
- 13.O’Malley DP, Li K, Maue M, Zografos AL, Baran PS. J Am Chem Soc. 2007;129:4762–4775. doi: 10.1021/ja069035a. [DOI] [PubMed] [Google Scholar]
- 14.Grube A, Kock M. Angew Chem, Int Ed. 2007;46:2320–2324. doi: 10.1002/anie.200604076. [DOI] [PubMed] [Google Scholar]
- 15.Buchanan MS, Carroll AR, Quinn RJ. Tetrahedron Lett. 2007;48:4573–4574. [Google Scholar]
- 16.Kobayashi H, Kitamura K, Nagai K, Nakao Y, Fusetani N, Van SRWM, Matsunaga S. Tetrahedron Lett. 2007;48:2127–2129. [Google Scholar]
- 17.Lanman BA, Overman LE, Paulini R, White NS. J Am Chem Soc. 2007;129:12896–12900. doi: 10.1021/ja074939x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.(a) Rosa R, Silva W, Escalona dMG, Rodriguez AD, Morales JJ, Ortiz M. Experientia. 1992;48:885–887. doi: 10.1007/BF02118426. [DOI] [PubMed] [Google Scholar]; (b) Bernan VS, Roll DM, Ireland CM, Greenstein M, Maiese WM, Steinberg DA. J Antimicrob Chemother. 1993;32:539–550. doi: 10.1093/jac/32.4.539. [DOI] [PubMed] [Google Scholar]; (c) Cafieri F, Carnuccio R, Fattorusso E, Taglialatela-Scafati O, Vallefuoco T. Bioorg Med Chem Lett. 1997;7:2283–2288. [Google Scholar]; (d) Peng J, Shen X, El SKA, Dunbar DC, Perry TL, Wilkins SP, Hamann MT, Bobzin S, Huesing J, Camp R, Prinsen M, Krupa D, Wideman MA. J Agric Food Chem. 2003;51:2246–2252. doi: 10.1021/jf0207880. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Bickmeyer U, Drechsler C, Kock M, Assmann M. Toxicon. 2004;44:45–51. doi: 10.1016/j.toxicon.2004.04.001. [DOI] [PubMed] [Google Scholar]; (f) Ortlepp S, Sjoegren M, Dahlstroem M, Weber H, Ebel R, Edrada R, Thoms C, Schupp P, Bohlin L, Proksch P. Mar Biotechnol. 2007;9:776–785. doi: 10.1007/s10126-007-9029-x. [DOI] [PubMed] [Google Scholar]; (g) Rodriguez AD, Lear MJ, La CJJ. J Am Chem Soc. 2008;130:7256–7258. doi: 10.1021/ja7114019. [DOI] [PubMed] [Google Scholar]; (h) Cipres A, O’Malley DP, Li K, Finlay D, Baran PS, Vuori K. ACS Chem Biol. 2010;5:195–202. doi: 10.1021/cb900240k. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Scala F, Fattorusso E, Menna M, Taglialatela-Scafati O, Tierney M, Kaiser M, Tasdemir D. Mar Drugs. 2010;8:2162–2174. doi: 10.3390/md8072162. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Eder C, Proksch P, Wray V, van SRWM, Ferdinandus E, Pattisina LA, Sudarsono J Nat Prod. 1999;62:1295–1297. doi: 10.1021/np990071f. [DOI] [PubMed] [Google Scholar]
- 19.(a) Starr JT, Koch G, Carreira EM. J Am Chem Soc. 2000;122:8793–8794. [Google Scholar]; (b) Dransfield PJ, Wang S, Dilley A, Romo D. Org Lett. 2005;7:1679–1682. doi: 10.1021/ol0473602. [DOI] [PubMed] [Google Scholar]; (c) Tan X, Chen C. Angew Chem, Int Ed. 2006;45:4345–4348. doi: 10.1002/anie.200601208. [DOI] [PubMed] [Google Scholar]; (d) Sivappa R, Hernandez NM, He Y, Lovely CJ. Org Lett. 2007;9:3861–3864. doi: 10.1021/ol0711568. [DOI] [PubMed] [Google Scholar]; (e) Tang L, Romo D. Heterocycles. 2007;74:999–1008. [Google Scholar]; (f) Zancanella MA, Romo D. Org Lett. 2008;10:3685–3688. doi: 10.1021/ol801289b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.(a) Breder A, Chinigo GM, Waltman AW, Carreira EM. Angew Chem, Int Ed. 2008;47:8514–8517. doi: 10.1002/anie.200803284. [DOI] [PubMed] [Google Scholar]; (b) Chinigo GM, Breder A, Carreira EM. Org Lett. 2011;13:78–81. doi: 10.1021/ol102577q. [DOI] [PubMed] [Google Scholar]
- 21.(a) Overman LE, Rogers BN, Tellew JE, Trenkle WC. J Am Chem Soc. 1997;119:7159–7160. [Google Scholar]; (b) Dilley AS, Romo D. Org Lett. 2001;3:1535–1538. doi: 10.1021/ol015864j. [DOI] [PubMed] [Google Scholar]; (c) Belanger G, Hong FT, Overman LE, Rogers BN, Tellew JE, Trenkle WC. J Org Chem. 2002;67:7880–7883. doi: 10.1021/jo026282y. [DOI] [PubMed] [Google Scholar]; (d) Poullennec KG, Kelly AT, Romo D. Org Lett. 2002;4:2645–2648. doi: 10.1021/ol026140q. [DOI] [PubMed] [Google Scholar]; (e) Poullennec KG, Romo D. J Am Chem Soc. 2003;125:6344–6345. doi: 10.1021/ja034575i. [DOI] [PubMed] [Google Scholar]; (f) Katz JD, Overman LE. Tetrahedron. 2004;60:9559–9568. [Google Scholar]; (g) Garrido-Hernandez H, Nakadai M, Vimolratana M, Li Q, Doundoulakis T, Harran PG. Angew Chem, Int Ed. 2005;44:765–769. doi: 10.1002/anie.200462069. [DOI] [PubMed] [Google Scholar]; (h) Dransfield PJ, Dilley AS, Wang S, Romo D. Tetrahedron. 2006;62:5223–5247. [Google Scholar]; (i) Lanman BA, Overman LE. Heterocycles. 2006;70:557–570. doi: 10.3987/com-06-s(w)16. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Nakadai M, Harran PG. Tetrahedron Lett. 2006;47:3933–3935. [Google Scholar]; (k) Bultman MS, Ma J, Gin DY. Angew Chem, Int Ed. 2008;47:6821–6824. doi: 10.1002/anie.200801969. [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Cernak TA, Gleason JL. J Org Chem. 2008;73:102–110. doi: 10.1021/jo701866g. [DOI] [PubMed] [Google Scholar]; (m) Hudon J, Cernak TA, Ashenhurst JA, Gleason JL. Angew Chem, Int Ed. 2008;47:8885–8888. doi: 10.1002/anie.200803344. [DOI] [PubMed] [Google Scholar]; (n) Wang S, Romo D. Angew Chem, Int Ed. 2008;47:1284–1286. doi: 10.1002/anie.200703998. [DOI] [PubMed] [Google Scholar]; (o) Li Q, Hurley P, Ding H, Roberts AG, Akella R, Harran PG. J Org Chem. 2009;74:5909–5919. doi: 10.1021/jo900755r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (p) Namba K, Kaihara Y, Yamamoto H, Imagawa H, Tanino K, Williams RM, Nishizawa M. Chem-Eur J. 2009;15:6560–6563. doi: 10.1002/chem.200900622. [DOI] [PMC free article] [PubMed] [Google Scholar]; (q) Sivappa R, Mukherjee S, Dias HVR, Lovely CJ. Org Biomol Chem. 2009;7:3215–3218. doi: 10.1039/b909482b. [DOI] [PubMed] [Google Scholar]; (r) Doundoulakis T, Lovely CJ. ORGN-508 2010 [Google Scholar]; (s) Namba K, Inai M, Sundermeier U, Greshock TJ, Williams RM. Tetrahedron Lett. 2010;51:6557–6559. doi: 10.1016/j.tetlet.2010.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]; (t) Feldman KS, Nuriye AY, Li J. J Org Chem. 2011;76:5042–5060. doi: 10.1021/jo200740b. [DOI] [PubMed] [Google Scholar]; (u) Ma Z, Lu J, Wang X, Chen C. Chem Commun (Cambridge, U K) 2011;47:427–429. doi: 10.1039/c0cc02214d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.(a) Jacquot DEN, Lindel T. Curr Org Chem. 2005;9:1551–1565. [Google Scholar]; (b) Köck M, Grube A, Seiple IB, Baran PS. Angew Chem, Int Ed. 2007;46:6586–6594. doi: 10.1002/anie.200701798. [DOI] [PubMed] [Google Scholar]; (c) Takao K-i, Tadano K-i. Kagaku (Kyoto, Jpn) 2009;64:70–71. [Google Scholar]; (d) Gaich T, Baran PS. J Org Chem. 2010;75:4657–4673. doi: 10.1021/jo1006812. [DOI] [PubMed] [Google Scholar]; (e) Namba K. Yuki Gosei Kagaku Kyokaishi. 2010;68:1249–1260. [Google Scholar]; (f) Hoffmann H, Lindel T. Synthesis. 2003;1753–1783 [Google Scholar]
- 23.(a) Hoffmann H. Universität Heidelberg; 1999. [Google Scholar]; (b) Trenkle W. University of California; Irvine: 2000. [Google Scholar]; (c) Hochgürtel M. Universität Heidelberg; 2000. [Google Scholar]; (d) Starr J. California Institute of Technology; 2001. [Google Scholar]; (e) Dolaine R. McGill University; 2001. [Google Scholar]; (f) Yu E. University of Texas; Arlington: 2002. [Google Scholar]; (g) Koenig S. Yale University; 2002. [Google Scholar]; (h) Poullennec K. Texas A&M University; 2003. [Google Scholar]; (i) Dilley A. Texas A&M University; 2003. [Google Scholar]; (j) Jacquot D. Ludwig-Maximilians-Universität; München: 2003. [Google Scholar]; (k) Breckle G. Ludwig-Maximilians-Universität; München: 2004. [Google Scholar]; (l) Du H. University of Texas; Arlington: 2004. [Google Scholar]; (m) He Y. University of Texas; Arlington: 2005. [Google Scholar]; (n) Spoering R. Harvard University; 2005. [Google Scholar]; (o) Skoumbourdis A. Pennsylvania State University; 2005. [Google Scholar]; (p) Huang A. University of California; Irvine: 2006. [Google Scholar]; (q) Ashenhurst R. McGill University; 2006. [Google Scholar]; (r) Wang S. Texas A&M University; 2007. [Google Scholar]; (s) Zöllinger M. Ludwig-Maximilians-Universität; München: 2007. [Google Scholar]; (t) Gergely J. University of California; Irvine: 2007. [Google Scholar]; (u) Cernak T. McGill University; 2007. [Google Scholar]; (v) Pöverlein C. Ludwig-Maximilians-Universität; München: 2008. [Google Scholar]; (w) Li Q. University of Texas Southwestern Medical Center; Dallas: 2008. [Google Scholar]; (x) O’Malley DP. The Scripps Research Institute; 2008. [Google Scholar]; (y) White N. University of California; Irvine: 2009. [Google Scholar]; (z) Bultman M. University of Illinois; Urbana-Champaign: 2009. [Google Scholar]; (aa) Hudon J. McGill University; 2010. [Google Scholar]; (bb) Mukherjee S. University of Texas; Arlington: 2011. [Google Scholar]
- 24.(a) Yamaguchi J, Seiple IB, Young IS, O’Malley DP, Maue M, Baran PS. Angew Chem, Int Ed. 2008;47:3578–3580. doi: 10.1002/anie.200705913. [DOI] [PubMed] [Google Scholar]; (b) O’Malley DP, Yamaguchi J, Young IS, Seiple IB, Baran PS. Angew Chem, Int Ed. 2008;47:3581–3583. doi: 10.1002/anie.200801138. [DOI] [PubMed] [Google Scholar]
- 25.Su S, Seiple IB, Young IS, Baran PS. J Am Chem Soc. 2008;130:16490–16491. doi: 10.1021/ja8074852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Seiple IB, Su S, Young IS, Lewis CA, Yamaguchi J, Baran PS. Angew Chem, Int Ed. 2010;49:1095–1098. doi: 10.1002/anie.200907112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Olofson A, Yakushijin K, Horne DA. J Org Chem. 1998;63:1248–1253. doi: 10.1021/jo972295d. [DOI] [PubMed] [Google Scholar]
- 28.(a) Murahashi S, Naota T, Taki H. J Chem Soc, Chem Commun. 1985:613–614. [Google Scholar]; (b) Moriarty RM, Vaid RK, Duncan MP, Ochiai M, Inenaga M, Nagao Y. Tetrahedron Lett. 1988;29:6913–6916. [Google Scholar]; (c) Goti A, Romani M. Tetrahedron Lett. 1994;35:6567–6570. [Google Scholar]; (d) Nicolaou KC, Mathison CJN, Montagnon T. J Am Chem Soc. 2004;126:5192–5201. doi: 10.1021/ja0400382. [DOI] [PubMed] [Google Scholar]; (e) Choi H, Doyle MP. Chem Comm. 2007;745–747 doi: 10.1039/b615805f. [DOI] [PubMed] [Google Scholar]
- 29.(a) Murahashi S, Naota T, Kuwabara T, Saito T, Kumobayashi H, Akutagawa S. J Am Chem Soc. 1990;112:7820–7822. [Google Scholar]; (b) Nagamatsu T, Yamasaki H. J Chem Soc, Chem Commun. 1995:2041–2043. [Google Scholar]; (c) Han G, LaPorte MG, Folmer JJ, Werner KM, Weinreb SM. J Org Chem. 2000;65:6293–6306. doi: 10.1021/jo000260z. [DOI] [PubMed] [Google Scholar]; (d) Qiu XL, Qing FL. J Org Chem. 2005;70:3826–3837. doi: 10.1021/jo050057+. [DOI] [PubMed] [Google Scholar]
- 30.Keifer PA, Schwartz RE, Koker MES, Hughes RG, Jr, Rittschof D, Rinehart KL. J Org Chem. 1991;56:6728. [Google Scholar]
- 31.(a) The often cited, purely theoretical and speculative biogenetic hypothesis of Al Mourabit and Potier is based upon original findings and actual research by the Rinehart group (reference 2d) and the Horne group (published in a poster at the 217th ACS meeting in 1997, shared with Potier and Al Mourabit at this time). The Horne group was the first to demonstrate that 2-aminoimidazoles and related imidazolones can be functionalized on the ring and at the alpha and beta positions. See: Xu Y-z, Yakushijin K, Horne DA. J Org Chem. 1996;61:9569–9571.Olofson A, Yakushijin K, Horne DA. J Org Chem. 1997;62:7918–7919. doi: 10.1021/jo9715682.Sosa ACB, Yakushijin K, Horne DA. Org Lett. 2000;2:3443–3444. doi: 10.1021/ol000233v.Barrios Sosa AC, Yakushijin K, Horne DA. J Org Chem. 2002;67:4498–4500. doi: 10.1021/jo020063v.
- 32.(a) Tiefenbacher K, Arion VB, Mulzer J. Angewandte Chemie International Edition. 2007;46:2690–2693. doi: 10.1002/anie.200604781. [DOI] [PubMed] [Google Scholar]; (b) White JD, Choi Y. Helvetica Chimica Acta. 2002;85:4306–4327. [Google Scholar]; (c) Ward DE, Shen J. Organic Letters. 2007;9:2843–2846. doi: 10.1021/ol070994z. [DOI] [PubMed] [Google Scholar]
- 33.Ryu DH, Corey EJ. J Am Chem Soc. 2003;125:6388–6390. doi: 10.1021/ja035393r. [DOI] [PubMed] [Google Scholar]
- 34.Liu D, Canales E, Corey EJ. J Am Chem Soc. 2007;129:1498–1499. doi: 10.1021/ja068637r. [DOI] [PubMed] [Google Scholar]
- 35.Evans DA, Miller SJ, Lectka T. J Am Chem Soc. 1993;115:6460–6461. [Google Scholar]
- 36.We are extremely greatful to the Shibasaki laboratory for performing these experiments based on their very elegant work with barium-catalyzed Diels-Alder reactions: Yamatsugu K, Yin L, Kamijo S, Kimura Y, Kanai M, Shibasaki M. Angew Chem, Int Ed. 2009;48:1070–1076. doi: 10.1002/anie.200804777.
- 37.We are extremely greatful to the Ishihara laboratory for a gracious donation of ligand 31. These experiments were performed according to the following publication: Sakakura A, Kondo R, Matsumura Y, Akakura M, Ishihara K. J Am Chem Soc. 2009;131:17762–17764. doi: 10.1021/ja906098b.
- 38.Newhouse T, Lewis CA, Baran PS. J Am Chem Soc. 2009;131:6360–6361. doi: 10.1021/ja901573x. [DOI] [PubMed] [Google Scholar]
- 39.(a) Saxon E, Armstrong JI, Bertozzi CR. Organic Letters. 2000;2:2141–2143. doi: 10.1021/ol006054v. [DOI] [PubMed] [Google Scholar]; (b) Nilsson BL, Kiessling LL, Raines RT. Organic Letters. 2000;2:1939–1941. doi: 10.1021/ol0060174. [DOI] [PubMed] [Google Scholar]; (c) Nilsson BL, Kiessling LL, Raines RT. Organic Letters. 2000;3:9–12. doi: 10.1021/ol006739v. [DOI] [PubMed] [Google Scholar]; (d) Soellner MB, Nilsson BL, Raines RT. J Org Chem. 2002;67:4993–4996. doi: 10.1021/jo025631l. [DOI] [PubMed] [Google Scholar]; (e) Soellner MB, Nilsson BL, Raines RT. J Am Chem Soc. 2006;128:8820–8828. doi: 10.1021/ja060484k. [DOI] [PubMed] [Google Scholar]
- 40.David O, Meester WJN, Bieräugel H, Schoemaker HE, Hiemstra H, van Maarseveen JH. Angew Chem, Int Ed. 2003;42:4373–4375. doi: 10.1002/anie.200351930. [DOI] [PubMed] [Google Scholar]
- 41.(a) Clarke TG, Hampson NA, Lee JB, Morley JR, Scanlon B. Can J Chem. 1969;47:1649–1654. [Google Scholar]; (b) Clarke TG, Hampson NA, Lee JB, Morley JR, Scanlon BF. J Chem Soc, C. 1970:815–817. [Google Scholar]; (c) Lee JB, Parkin C, Shaw MJ, Hampson NA, MacDonald KI. Tetrahedron. 1973;29:751–752. [Google Scholar]
- 42.(a) Hartwig JF. Angew Chem, Int Ed. 1998;37:2046–2067. doi: 10.1002/(SICI)1521-3773(19980817)37:15<2046::AID-ANIE2046>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]; (b) Muci AR, Buchwald SL. Top Curr Chem. 2002;219:131–209. [Google Scholar]
- 43.Monnier F, Taillefer M. Angew Chem, Int Ed. 2009;48:6954–6971. doi: 10.1002/anie.200804497. [DOI] [PubMed] [Google Scholar]
- 44.After months of work and two very costly scale-ups, a manuscript titled “A Macrocyclic Isomer of the Palau’amine Core” was drawn up and ready to submit, just when the successful conditions for the conversion of 65 to 64 were discovered.
- 45.(a) Köck M, Junker J. J Org Chem. 1997;62:8614–8615. [Google Scholar]; (b) Kaptein R, Boelens R, Scheek RM, Van GWF. Biochemistry. 1988;27:5389–5395. doi: 10.1021/bi00415a001. [DOI] [PubMed] [Google Scholar]; (c) Weber PL, Morrison R, Hare D. J Mol Biol. 1988;204:483–487. doi: 10.1016/0022-2836(88)90589-x. [DOI] [PubMed] [Google Scholar]; (d) Holak TA, Gondol D, Otlewski J, Wilusz T. J Mol Biol. 1989;210:635–648. doi: 10.1016/0022-2836(89)90137-x. [DOI] [PubMed] [Google Scholar]; (e) Scheek RM, Van GWF, Kaptein R. Methods Enzymol. 1989;177:204–218. doi: 10.1016/0076-6879(89)77012-9. [DOI] [PubMed] [Google Scholar]; (f) Mierke DF, Reggelin M. J Org Chem. 1992;57:6365–6367. [Google Scholar]; (g) Kock M, Junker J. J Mol Model. 1997;3:403–407. [Google Scholar]; (h) Reggelin M, Köck M, Conde-Frieboes K, Mierke D. Angew Chem, Int Ed. 1994;33:753–755. [Google Scholar]
- 46.Albeiicio FC, Rafael, Dodsworth David J, Nájera, Carmen Organic Preparations and Procedures International. 2001;33:203–303. [Google Scholar]
- 47.Seiple IB, Su S, Rodriguez RA, Gianatassio R, Fujiwara Y, Sobel AL, Baran PS. J Am Chem Soc. 2010;132:13194–13196. doi: 10.1021/ja1066459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fujiwara Y, Domingo V, Seiple IB, Gianatassio R, Del Bel M, Baran PS. J Am Chem Soc. 2011;133:3292–3295. doi: 10.1021/ja111152z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.(a) Baran PS, Maimone TJ, Richter JM. Nature (London, U K) 2007;446:404–408. doi: 10.1038/nature05569. [DOI] [PubMed] [Google Scholar]; (b) Young IS, Baran PS. Nat Chem. 2009;1:193–205. doi: 10.1038/nchem.216. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.