

Abstract
Intracellular bacterial pathogens Francisella novicida and the Live Vaccine Strain (LVS) are recognized in the macrophage cytosol by the AIM2 inflammasome, which leads to the activation of caspase-1 and the processing and secretion of active IL-1β, IL-18 and pyroptosis. Previous studies have reported that F. novicida and LVS mutants in specific genes (e.g., FTT0584, mviN and ripA) induce elevated inflammasome activation and hypercytotoxicity in host cells, leading to the proposal that F. novicida and LVS may have proteins that actively modulate inflammasome activation. However, there has been no direct evidence of such inflammasome evasion mechanisms. Here, we demonstrate for the first time that the above mutants, along with a wide range of F. novicida hypercytotoxic mutants that are deficient for membrane-associated proteins (ΔFTT0584, ΔmviN, ΔripA, ΔfopA and ΔFTN1217) or deficient for genes involved in O-antigen or LPS biosynthesis (ΔwbtA and ΔlpxH) lyse more intracellularly, thus activating increased levels of AIM2-dependent pyroptosis and other innate immune signaling pathways. This suggests that an inflammasome-specific evasion mechanism may not be present in F. novicida and LVS. Furthermore, future studies may need to consider increased bacterial lysis as a possible cause of elevated stimulation of multiple innate immune pathways when the protein composition or surface carbohydrates of the bacterial membrane is altered.
Keywords: AIM2, bacteriolysis, Francisella, hypercytotoxic, pyroptosis
Introduction
Innate immunity against microbial pathogens is initiated very early during the course of infection by germline-encoded pattern recognition receptors (PRRs), specialized receptors that recognize and respond to highly conserved microbial components collectively known as pathogen associated molecular patterns (PAMPs) (Akira et. al., 2006). An array of PRRs recognizes a wide spectrum of microbial molecules in the extracellular compartment, endosomes or in the cytosol (Akira et. al., 2009). Sensing of the PAMPs by these receptors in turn activates intracellular signaling cascades that induce the expression of a wide range of genes involved in the inflammatory and immune response (Akira et. al., 2006). For instance, the toll-like receptors (TLRs) located in the cytoplasmic or endosomal membranes sense microbial molecules such as lipopolysaccharide, flagellin and nucleic acids, and trigger NF-κB and interferon regulatory factor (IRF) signaling by pathways that involve the signaling adaptors MyD88 and Trif, which induce the expression of multiple antimicrobial molecules, including proinflammatory cytokines (Akira et. al., 2006). In addition to TLRs, some innate immune cells detect signatures of microbial pathogens in the host cell cytosol. Macrophages and dendritic cells utilize cytosolic PRRs, such as the NOD-like receptor (NLR) family, to detect microbial products introduced into the cytosol by multiple mechanisms, including by bacterial secretion systems or by virulent bacteria that escape to the host cytosol (Franchi et. al., 2009, Takeuchi et. al., 2010). The NLRs, NOD1 and NOD2 sense bacterial peptidoglycan in the cytosol and trigger NFκB and MAPK signaling by a pathway that involves the signaling adaptor RIP2 (Akira et. al., 2006) and CARD9 (Hsu et. al., 2007). In addition, several NLRs participate in the assembly of a multi-protein caspase-1 activating complex known as the inflammasome, resulting in the processing and release of mature IL-1β, IL-18 and host cell death via pyroptosis (Schroder et. al., 2010).
Many bacterial pathogens utilize virulence mechanisms in order to modulate host cell signaling and avoid, manipulate or silence immune responses (Bhavsar et. al., 2007). Indeed, some bacterial pathogens seem to block caspase-1 activation (Brodsky et. al., 2009). In this study, Francisella tularensis was used as a model organism to study caspase-1 activation in response to a bacterial pathogen that resides in the host cell cytosol. Francisella tularensis is a facultative intracellular Gram-negative pathogen that causes the highly fulminating disease, tularemia (Oyston et. al., 2004). There are currently 4 known subspecies of F. tularensis that cause disease of different severities, namely F. tularensis subspecies tularensis, subspecies holarctica, subspecies mediasiatica and subspecies novicida. One of Francisella's replicative niches is the macrophage (Fortier et. al., 1994). Francisella is sensed by TLR2 at the surface and in the phagosome, leading to the production of proinflammatory cytokines like TNF-α and IL-6. After being taken up by the macrophage, the bacterium escapes the phagosome where it replicates to high numbers within the host cell cytosol (Checroun et. al., 2006, Santic et. al., 2007). Cytosolic F. tularensis trigger a proinflammatory host innate immune program characterized by initial type 1 interferon (type 1 IFN) production (Henry et. al., 2007). Although the Francisella ligand and PRR responsible for type 1 IFN induction is still unknown, the adaptor stimulator of interferon genes (STING) is required (Jones et. al., 2010). Type 1 IFN in turn increases the expression of the cytosolic DNA sensor called absent in melanoma 2 (AIM2) (Fernandes-Alnemri et. al., 2010, Henry et. al., 2007, Jones et. al., 2010, Rathinam et. al., 2010), a protein containing both pyrin (PYD) and hematopoietic interferon-inducible nuclear antigens with 200 amino acids repeats (HIN200) domains. AIM2 binds transfected DNA at the HIN200 domain and then associates with ASC via the PYD domain to form an active inflammasome complex (Burckstummer et. al., 2009, Fernandes-Alnemri et. al., 2009, Hornung et. al., 2009, Landolfo et. al., 1998). It has also recently been demonstrated that cytosolic F. tularensis DNA is recognized by the AIM2 inflammasome, leading to processing and release of mature IL-1β, IL-18 and a pro-inflammatory host cell death called pyroptosis (Fernandes-Alnemri et. al., 2010, Jones et. al., 2010, Rathinam et. al., 2010).
We, and others have previously reported that various Francisella mutants induce higher levels of host cell death during infections of macrophages compared to wild-type strains. The host cell death pathway triggered by both the wild-type and hypercytotoxic mutants requires caspase-1, which mediates pyroptosis (Mariathasan et. al., 2005). For example, a F. novicida strain that contains a deletion in a gene that appears to be essential for virulence, FTT0584, triggers higher levels of pyroptosis compared to the wild-type strain (Weiss et. al., 2007b). Similarly, F. tularensis live vaccine strain (LVS) mutants that lack mviN, which encodes a putative lipid II flippase, or ripA, which encodes a cytoplasmic membrane protein of unknown function, have been reported to trigger increased levels of pyroptosis (Huang et. al., 2010, Ulland et. al., 2010). In addition, we and others conducted a genome-wide negative selection screen using the Francisella tularensis SchuS4 strain in human-derived macrophages (huMDMs) to identify bacterial genes that are important for bacterial survival and/ or replication in the macrophage intracellular replicative niche (Lindemann et. al., 2010). Francisella mutant strains that lack one or more of the genes that play a role in O-antigen and capsule biosynthesis, FTT1236, FTT1237 and FTT1238, were more sensitive to complement-dependent killing mechanisms and induced higher levels of macrophage cytotoxicity compared to wild type bacteria (Lindemann et. al., 2010). Taken together, these results indicate that many Francisella genes can have an impact on the cell death kinetics of infected macrophages.
We initially hypothesized that the gene product encoded by FTT0584 was somehow involved in modulating the activation of the inflammasome. However, given that the numerous previously described hypercytotoxic bacterial strains are deficient for genes that encode proteins with diverse biological functions and yet exhibit a similar phenotype in macrophages, we instead postulated that the mechanism for inducing elevated macrophage cytotoxicity involves increased bacterial lysis, and therefore increased release of bacterial DNA as well as other PAMPs into the intracellular milieu. Now that we have a better understanding of the mechanisms underlying inflammasome activation, we have set out here to test this hypothesis.
In this study, we demonstrate that a wide range of F. novicida mutants including the previously reported ΔFTT0584, ΔmviN and ΔripA mutants trigger elevated levels of AIM2 inflammasome activation compared to wild-type bacteria, leading to higher levels of active caspase-1 and IL-1β production and macrophage pyroptosis. In addition, we show that F. novicida mutants that are deficient for a membrane-associated protein ΔfopA (Fulop et. al., 1995) or a putative membrane-associated transporter protein, ΔFTN1217 as well as mutants deficient in O antigen and lipopolysaccharide biosynthesis (ΔwbtA and ΔlpxH) also trigger higher levels of AIM2 inflammasome activation. Infection of macrophages with these hypercytotoxic mutants also triggered higher levels of type 1 interferon production and pro-inflammatory cytokine signaling compared to wild type bacteria as evidenced by elevated TNF-α and IL-6 secretion, further supporting the idea that these mutants may exhibit increased lysis in the host cell. In addition, these mutants displayed aberrant colony morphology by scanning electron microscopy. Perhaps most importantly, we further demonstrate that these hypercytotoxic mutants exhibit higher levels of bacterial lysis within the host cell compared to wild type bacteria, as visualized via confocal miscroscopy and measured quantitatively by delivery of a luciferase reporter plasmid. Our results indicate that the Francisella genes that we report on in this study are important for minimizing bacterial susceptibility to intracellular lysis and are not actively suppressing or modulating the host innate immune sensing pathways, further suggesting that contrary to common belief, an inflammasome-specific evasion mechanism may not exist in Francisella.
Results
F. novicida ΔFTT0584, ΔmviN, ΔripA, ΔfopA, ΔFTN1217, ΔwbtA and ΔlpxH mutants are hypercytotoxic in macrophages
Upon infection of macrophages, F. novicida and LVS activate pyroptosis by a mechanism that requires the inflammasome receptor AIM2, the adaptor protein ASC and the effector cysteine protease caspase-1. To gain more insight into the interactions between Francisella and the cell death pathway that leads to pyroptosis, we characterized three F. novicida mutants that had been previously described as having a hypercytotoxic phenotype in either F. novicida (ΔFTT0584; (Weiss et. al., 2007b)) or LVS (ΔmviN or ΔripA; (Huang et. al., 2010, Ulland et. al., 2010)). FTT0584 encodes a putative membrane protein of unknown function and is important for virulence in mice (Weiss et. al., 2007b). The gene product encoded by mviN is a putative multi-drug/oligosaccharidyl-lipid/ polysaccharide transporter that also shows homology to a lipid II flippase in Escherichia coli. Although the mviN homolog in E. coli is involved in peptidoglycan synthesis (Inoue et. al., 2008, Ruiz et. al., 2008), the function of its homolog in other bacterial species has yet to be defined. ripA encodes a cytoplasmic membrane protein that is required for intracellular survival (Fuller et. al., 2008), however the function of ripA is unknown. In addition, we also characterized F. novicida mutants that were identified in genetic screens of a comprehensive transposon library of F. novicida for mutant strains that increased the rate of cell death following infection in J774 macrophages (Lai et. al., 2010) or in murine bone marrow-derived macrophages (BMDMs) (our unpublished results). Briefly, both screens were conducted by indirectly monitoring host cell death by measuring the amount of macrophage lactate dehydrogenase released into the supernatant. Both screens identified mutants that caused increased cytotoxicity compared to the parental strain. We focused our attention on mutants that were identified in both screens and are deficient for membrane-associated proteins (fopA and FTN1217; Table 1) or are involved in O-antigen and LPS biosynthesis (wbtA and lpxH; Table 1). For example, a transposon insertion in fopA, a gene that encodes an outer membrane protein, was identified in both screens as having a hypercytotoxic phenotype ((Lai et. al., 2010); our unpublished results). fopA is highly homologous to ompA in Escherichia coli (∼60% similarity) (Nano et. al., 1988), which is a major outer membrane protein of E. coli and which plays a structural role in the integrity of the bacterial cell surface presumably because it provides a physical linkage between the outer membrane and the underlying peptidoglycan layer (Koebnik et. al., 2000). The other outer membrane mutant that caused increased cytotoxicity in our screen harbored a transposon insertion in FTN1217, which encodes an ATP-binding cassette superfamily protein, a group of transporters that hydrolyze ATP during translocation of substrates across bacterial membranes (Linton et. al., 1998). Finally, we identified mutants in wbtA and lpxH, which are involved in O-antigen and lipopolysaccharide (LPS) biosynthesis respectively (Raetz et. al., 2007, Raynaud et. al., 2007). Other mutants defective in LPS and capsule biosynthesis have also been recently described to have a hypercytotoxic effect in infected macrophages (Lai et. al., 2010, Lindemann et. al., 2010).
Table I. List of Francisella bacterial mutant strains used in this study.
| Mutant strain | Gene description | References |
|---|---|---|
| ΔFTT0584 | Putative membrane protein of unknown function | Weiss et al, 2007a |
| ΔmviN | multidrug, oligosaccharidyl-lipid/polysaccharide (MOP) transporter family protein/ putative lipid II flippase | Ruiz 2008, Ulland et al, 2010 |
| ΔripA | Cytoplasmic membrane protein of unknown function | Huang et al, 2010 |
| ΔfopA | Major outer membrane protein of OmpA family | Nano 1988 |
| ΔFTN1217 | Membrane transporter protein; ATP-binding cassette (ABC) superfamily protein | |
| ΔwbtA | Involved in O-antigen biosynthesis | Raynaud et al, 2007 |
| ΔIpxH | Involved in lipopolysaccharide biosynthesis | Raetz et al, 2007 |
To determine the contribution of all the aforementioned genes to the hypercytotoxic phenotype in macrophages, we constructed in-frame deletions of these genes in F. novicida (Table 1) and measured the kinetics of macrophage death induced by these mutant bacterial strains compared to that induced by the parental F. novicida wild-type strain, U112.
Since it had been previously reported that some Francisella LPS mutants may be internalized by macrophages at higher numbers compared to the wild type strain (Lindemann et. al., 2010, Lai et. al., 2010), we first determined the rates at which the wild-type and mutant bacterial strains were invading macrophages. Indeed, some of the hypercytotoxic bacterial mutant strains were associating with or invading macrophages at different rates compared to the wild type parental strain at 30 min post infection (Figure S1A). This difference in the invasion rates of the mutant strains would result in different numbers of intracellular bacteria and subsequently different levels of cell death. Therefore, the MOIs were adjusted accordingly for the following experiments (subsequently referred to as effective MOI) in order to ensure that for each of the different bacterial strains, similar numbers of viable bacteria were associating with and invaded BMDMs (details in materials and methods). The validity of these adjusted infection conditions was verified using standard gentamicin-protection assays demonstrating that comparable bacterial numbers of each strain were consistently recovered from infected BMDMs at both 30 min (data not shown) and 2 hrs post infection (Figure S1B).
In agreement with previous reports (Ulland et. al., 2010, Huang et. al., 2010, Weiss et. al., 2007b), we confirmed that the F. novicida ΔFTT0584, ΔmviN and ΔripA mutants induced increased levels of macrophage death and with faster kinetics compared to the parental strain (Figure 1A). The ΔfopA, ΔFTN1217, ΔwbtA or ΔlpxH mutants also induced BMDM death sooner than wild-type bacteria. Indeed, at 6 h post-infection, the mutant bacterial strains induced 2- to 10-fold higher levels of cell death than the wild-type bacterial strain, which had not yet induced significant levels of death (Figure 1A). At 10 h, the levels of cell death induced by the mutant bacterial strains were significantly higher (40-90%) than the level of death induced by the wild type strain (∼18%) (Figure 1B). As previously published, the Francisella pathogenicity island deletion mutant, ΔFPI, did not induce any macrophage death (Figure 1A). Furthermore, the hypercytotoxic phenotypes of the ΔmviN and ΔfopA mutants were restored to the parental strain levels by adding back a wild-type copy of each of these genes (Figure 1B). Taken together, our findings suggested that the hypercytotoxic mutants might stimulate a pyroptotic cell death pathway sooner than the wild-type strain.
Figure 1. F. novicida ΔFTT0584, ΔmviN, ΔripA, ΔfopA, ΔFTN1217, ΔwbtA and ΔlpxH mutants are hypercytotoxic in macrophages.

Cytotoxicity levels by infected wild type BMDMs at (A) indicated times post infection or (B) 10 hrs post infection. BMDMs were infected with the indicated bacterial strains at an effective MOI of 20. Data is representative of 3 independent experiments. * p value < 0.05 using the Student's t test.
Francisella hypercytotoxic mutants trigger elevated AIM2 inflammasome activation in macrophages
To determine if our mutants had any effect on activation of the inflammasome in macrophages, BMDMs were infected with the wild type U112 strain or each of the hypercytotoxic mutant bacterial strains. We first monitored the release of IL-1β into the supernatant at 2, 4, 6, 8 and 10 h post infection. The hypercytotoxic mutants induced higher levels of IL-1β secretion sooner and to higher levels than the parental strain, beginning at about 5 h post-infection (Figure 2A). By 10 h, all of the hypercytotoxic mutants had induced significantly more IL-1β secretion compared to the parental strain (Figure 2B). Furthermore, there was a correlation between the levels of cell death and IL-1β secretion induced by the mutants. For example, the ΔfopA and ΔwbtA mutants consistently induced the highest level of macrophage death and similarly induced the highest levels of IL-1β secretion (Figure 1B and 2B). Furthermore, the elevated levels of IL-1β secretion induced by the ΔmviN and ΔfopA mutants was restored to the parental strain levels by adding back a wild type copy of each of these genes (Figure 2B). In addition, we wanted to test whether the release of IL-1β correlated with caspase-1 activation by immunoblotting for the caspase-1 p10 subunit in the supernatants of infected BMDMs. Indeed, infections of BMDMs with the hypercytotoxic mutants resulted in the release of processed caspase-1 p10 subunit into the supernatants, suggesting that the release of IL-1β was dependent on caspase-1 (Figure S2).
Figure 2. Francisella hypercytotoxic mutants trigger elevated AIM2 inflammasome activation in macrophages.

IL-1β secretion by infected wild type BMDMs at (A) indicated times post infection or (B) 10 h post infection. (C) AIM2 transcript levels (D) cytotoxicity levels and (E) IL-1β secretion by the indicated strains of infected BMDMs at 8 hr post infection. BMDMs were infected with the indicated bacterial strains at an effective MOI of 20. Data is representative of 3 independent experiments. * p value < 0.05 using the Student's t test.
To ensure that the elevated levels of cytotoxicity and IL-1β secretion observed was not due to higher amounts of AIM2 present in macrophages infected with the hypercytotoxic mutants, levels of AIM2 transcript in the cell lysates of BMDMs infected with either the hypercytotoxic mutants or wild type U112 strain were determined via quantitative RT-PCR. There was no significant difference in the levels of AIM2 transcript in macrophages infected with the hypercytotoxic mutant strains compared to those infected with the wild type parental strain (Figure 2C). The AIM2 protein levels were the same for macrophages infected with either the wild type or hypercytotoxic mutant strains, as confirmed by Western blotting (data not shown).
Bacterial numbers within the cell can directly impact macrophage cytotoxicity levels. To ensure that the increased macrophage hypercytotoxicity observed in BMDMs infected with the hypercytotoxic mutants was not an effect of increased bacterial replication post-infection, the levels of intracellular bacteria were determined for each of the hypercytotoxic bacterial mutant strains in BMDMs and RAW macrophages and compared to the wild-type, parental strain. The levels of intracellular bacteria were determined during the course of infection and were not significantly different compared to the wild type U112 strain with the exception of the ripA mutant, which has been previously reported to have a replication defect in macrophages (Figure S3A and B; (Fuller et. al., 2008)). Taken together, our data indicate that the hypercytotoxic mutants trigger a more rapid and robust inflammasome response in BMDMs.
As discussed earlier, assembly of the AIM2 inflammasome complex is critical for caspase-1 activation in response to Francisella infection. To determine if the hypercytotoxic response observed in BMDMs infected with our mutants was dependent upon the assembly of an AIM2/ASC inflammasome, similar to the wild-type, parental F. novicida strain, WT, Aim2-/-, ASC-/- and caspase-1-/- macrophages were infected with each of the bacterial strains and the levels of IL-1β release and cytotoxicity were determined. As described above, the hypercytotoxic bacterial mutant strains induced significantly higher levels of host cell death in wild-type BMDMs compared to the parental strain and similar results were seen for IL-1β release (Figure 2D and E). In contrast, the hypercytotoxic mutants did not induce cell death and IL-1β release in Aim2-/-, ASC-/- and caspase-1-/- macrophages (Figure 2D and E). The low levels of cell death observed in caspase-1-/- macrophages is most likely due to the previously described ASC dependent, caspase-1 independent cell death pathway (Henry et. al., 2007). Previous reports indicate that wild-type F. novicida and LVS-induced pyroptosis does not depend on the NLRs NLRP3 and NLRC4 (Mariathasan et. al., 2006, Weiss et. al., 2007a). To determine whether cell death induced by the hypercytotoxic mutants was dependent on NLRP3 and/or NLRC4, we infected nlrp3-/-nlrc4-/- BMDMs with each of the bacterial mutant strains. In contrast to the results with Aim2-/-, ASC-/- and caspase-1-/- macrophages, no significant differences in cytotoxicity levels and IL-1β release were observed between WT and nlrp3-/-nlrc4-/- BMDMs (Figure 2D and E).
Taken together, these results indicate that the F. novicida hypercytotoxic mutants induce elevated levels of AIM2/ASC inflammasome activation compared to the wild type parental strain.
Francisella hypercytotoxic mutants hyperstimulate type 1 interferon and proinflammatory cytokine signaling
As described earlier, apart from AIM2 inflammasome activation, macrophage infections with F. novicida or LVS triggers a proinflammatory host innate immune program characterized by an initial type 1 IFN production (Henry et. al., 2007) as well as secretion of pro-inflammatory cytokines such as TNF-α and IL-6 (Katz et. al., 2006, Li et. al., 2006, Malik et. al., 2006). To determine if the hypercytotoxic F. novicida mutants also stimulated other proinflammatory responses apart from AIM2 inflammasome activation, we infected ASC-/- macrophages with the wild-type U112 strain or each of the hypercytotoxic mutant strains and measured type 1 IFN, TNF-α and IL-6 secretion into the supernatants at 8 h post-infection. ASC-/- macrophages were used to eliminate host cell death.
Macrophages infected with the hypercytotoxic mutant bacterial strains consistently released more type 1 IFN (Figure 3A), TNF-α (Figure 3B) and IL-6 (Figure 3C) into the culture supernatants compared to macrophages infected with the wild-type U112 strain. These results suggest that the hypercytotoxic mutants release more PAMPs and/ or have more PAMPs exposed on their surfaces that lead to the stimulation of innate immune signaling pathways that are upstream of the AIM2 inflammasome and TNF-α and IL-6 secretion (e.g. TLR2 and Nod1/2) and are not just hyperstimulating the AIM2 inflammasome.
Figure 3. Francisella hypercytotoxic mutants hyperstimulate type 1 interferon and proinflammatory cytokine signaling.

(A) Type 1 IFN, (B) TNF-α and (B) IL-6 secretion by infected ASC BMDMs at 8 hours post infection. BMDMs were infected with the indicated bacterial strains at an effective MOI of 20. Data is representative of 3 independent experiments. * p value < 0.05 using the Student's t test.
F. novicida hypercytotoxic mutants require phagosomal escape to induce AIM2-mediated pyroptosis
Although it is currently unknown if bacterial DNA is released while Francisella is still within the phagosome or when the bacterium is within the cytosol or both, F. novicida and LVS mutants that fail to escape the phagosome, such as the ΔFPI mutant, do not induce IL-1β production or macrophage cytotoxicity (Nano et. al., 2004, Weiss et. al., 2007b). These observations indicate that regardless of the subcellular location at which bacteriolysis occurs, AIM2-mediated pyroptosis occurs only upon detection of Francisella DNA within the cytosolic compartment. This can occur either if the bacterium lyses within the cytosol or if the bacterium lyses within the phagosome, breakage of the phagosomal membrane is needed for bacterial DNA to be released into the cytosol. To determine whether the increased activation of the AIM2 inflammasome by the hypercytotoxic mutants also required phagosomal escape, we deleted the FPI in each of the mutant bacterial strains. Following infection of BMDMs with these double deletion mutant strains, the levels of cell death and IL-1β production was determined. The hypercytotoxic, ΔFPI double mutants did not induce IL-1β secretion (Figure 4A) or host cell death (Figure 4B), similar to the ΔFPI mutant strain (Figure 4A and B). Our observations that the F. novicida hypercytotoxic mutants require phagosomal escape in order to hyperinduce the inflammasome strongly suggests that like the wild type U112 strain, the ligand responsible for hyper-stimulating AIM2-mediated pyroptosis in these hypercytotoxic mutants is bacterial DNA, further lending support to our hypothesis that these hypercytotoxic mutants are likely to be lysing more within the macrophage.
Figure 4. Francisella hypercytotoxic mutants require phagosomal escape to induce AIM2-mediated pyroptosis.

(A) Cell death and (B) IL-1β secretion by infected wild type BMDMs at 10 hrs post infection. BMDMs were infected with the indicated bacterial strains at an effective MOI of 20. Data is representative of 3 independent experiments. * p value < 0.05 using the Student's t test.
Francisella hypercytotoxic mutants exhibit increased lysis in the intracellular milieu as visualized via confocal microscopy
We recently demonstrated that AIM2 binding to bacterial DNA in the macrophage cytosol can be visualized by confocal microscopy as AIM2 specks colocalizing with bacterial DNA (Jones et. al., 2010). We have also seen that multiple AIM2 specks can form next to bacterial remnants in an infected cell (Jones et. al., 2010). Thus, we hypothesized that the hypercytotoxic mutants may be lysing more frequently in the macrophage cytosol and that we might be able to visualize a higher number of AIM2 specks colocalizing with bacterial DNA in host cells infected with the hypercytotoxic mutants compared to the wild type strain. To test this, BMDMs were infected with the parental strain U112, ΔFPI, ΔfopA or ΔwbtA mutant bacterial strains and stained for bacteria with an anti-F. novicida antibody (Monack lab), AIM2 protein with an anti-AIM2 antibody (Jones et. al., 2010) and for DNA with DAPI. BMDMs infected with either the ΔfopA or ΔwbtA hypercytotoxic mutant bacterial strains contained significantly higher numbers of AIM2 specks that colocalized with DNA and bacteria compared to cells infected with a similar number of wild-type bacteria (Figure 5A). When we counted the number of AIM2 specks, we saw significantly more specks in the cells infected with the hypercytotoxic mutants compared to the wild-type stain (Figure 5B). In contrast, we did not detect any AIM2 specks in macrophages infected with the ΔFPI mutant (Figure 5A). Consistent with previous reports, only one ASC focus was observed in each infected cell. However, we detected that a higher percentage of cells infected with the hypercytotoxic mutants had an ASC focus compared to cells infected with the wild-type U112 strain (data not shown). Taken together, these results suggest that the hypercytotoxic mutants exhibit increased lysis compared to the wild-type U112 strain in macrophages.
Figure 5. Francisella hypercytotoxic mutants exhibit increased lysis in the intracellular milieu as visualized via confocal microscopy.
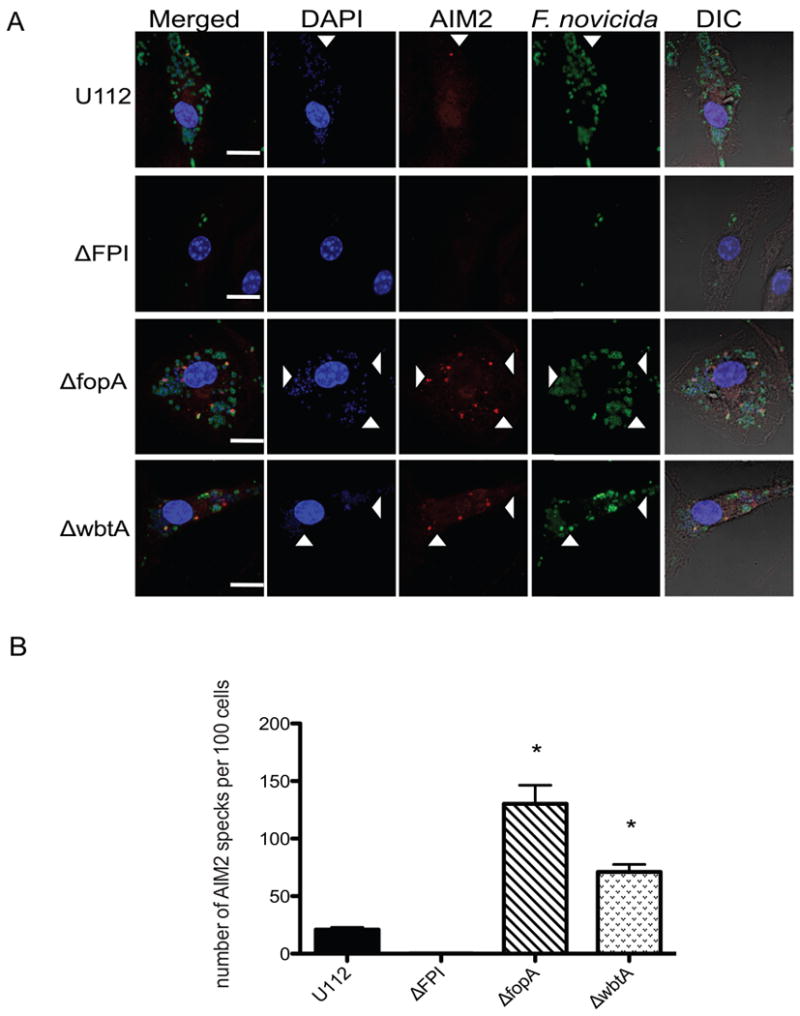
(A) Immunofluorescence microscopy of BMDMs infected with the indicated bacterial strains at 7 hours post infection. DIC, differential interference contrast. Arrowheads indicate examples of colocalization of bacterial DNA, AIM2 and bacteria. (B) Graph showing the number of AIM2 specks in 100 BMDMs from Figure 5A. Scale bars; 10 μm. Data is representative of 2 independent experiments. * p value < 0.05 using a student's t test.
Francisella hypercytotoxic mutants exhibit aberrant morphologies during growth in minimal media
Our immunofluorescence results demonstrating that BMDMs infected with hypercytotoxic mutants contained more AIM2 specks that colocalized with bacterial DNA in the cytosol indicated that the hypercytotoxic mutants might have defects in structural integrity especially when they enter stressful conditions such as the host cell. This led us to investigate the morphology of the hypercytotoxic bacterial mutant strains during growth in a rich media such as tryptic soy broth (TSB) versus a minimal media such as Chamberlain's defined media (CDM). All of the strains, including the hypercytotoxic mutant strains, wild-type U112 and ΔFPI mutant strains had typical polymorphic morphologies with a smooth surface when grown in TSB to the late exponential growth phase (Figure 6A). When the bacteria were grown in CDM to the late exponential growth phase, the wild-type and ΔFPI strains looked very similar in size and shape to the TSB grown bacteria (Figure 6B). In contrast, the hypercytotoxic mutants grown in CDM exhibited aberrant coccoid morphologies and significant dimpling was observed on the bacterial surface, albeit to different extents in the different mutants (Figure 6B). In addition, significantly fewer CFUs were consistently recovered for the hypercytotoxic mutant bacterial strains grown in vitro in CDM compared to the wild type U112 and ΔFPI strains (data not shown). These data strongly suggested that the F. novicida hypercytotoxic mutants are attenuated in stressful conditions and either don't replicate as well as wild-type bacteria in CDM or lyse more than the wild-type strain. These data support our hypothesis that the hypercytotoxic F. novicida mutants are stimulating more AIM2-dependent caspase-1 activation because they are lysing more than the parental strain in the macrophage.
Figure 6. Francisella hypercytotoxic mutants exhibit aberrant morphologies during growth in minimal media.

Bacterial morphology as visualized via scanning electron micropscopy of bacterial strains grown to late exponential phase in (A) tryptic soy broth supplemented with cysteine or (B) Chamberlain's defined media. Scale bars, 2μm. Images are representative of 2 independent experiments.
Francisella hypercytotoxic mutants exhibit increased lysis in the intracellular milieu as determined quantitatively via luminescence production
To test our hypothesis and to demonstrate quantitatively that the hypercytotoxic mutants are lysing and releasing more DNA into the macrophage compared to wild-type F. novicida, we utilized a luciferase assay previously described by Sauer et al. to quantify bacterial lysis within the host cell. In this assay, the firefly luciferase gene and cytomegalovirus (CMV) promoter was cloned into a Francisella shuttle vector, pFNLTP6 (Maier et. al., 2004) and the resulting plasmid was transformed into each of the bacterial strains. Since expression of the firefly luciferase gene was placed under the control of the CMV promoter, functional luciferase protein would only be expressed when the bacterium lyses and plasmid DNA is released into the host cell cytosol. BMDMs were infected with the various bacterial strains and at 7 h post-infection luminescence levels were determined. BMDMs infected with the FPI mutant strain displayed basal luminescence levels while cells infected with the hypercytotoxic mutant bacterial strains consistently exhibited higher luminescence levels compared to those infected with the wild type U112 strain (Figure 7). Taken together, these results clearly demonstrate that the F. novicida hypercytotoxic mutant strains are lysing more than the wild-type U112 parental stain within the intracellular milieu.
Figure 7. Francisella hypercytotoxic mutants exhibit increased lysis in the intracellular milieu as determined quantitatively via luminescence production.

Luminescence levels of BMDMs infected with the indicated bacterial strains containing the luciferase reporter construct at 7 hours post infection. Data is represented as the average of at least 3 independent experiments. * p value < 0.05 using a student's t test.
Discussion
Francisella tularensis is a facultative intracellular pathogen known to trigger a cytosolic innate immune response upon phagosomal escape. One of the key innate immune responses triggered is the sensing of Francisella DNA by the cytosolic DNA sensor AIM2, leading to the assembly of a functional inflammasome complex. This in turn leads to the processing and release of mature IL-1β, IL-18 and pyroptosis (Fernandes-Alnemri et. al., 2010, Jones et. al., 2010, Rathinam et. al., 2010). With this new knowledge, many questions remain. Although bacterial DNA colocalization with AIM2, in close proximity to what is proposed to be bacterial remnants has been visualized in Francisella infection of macrophages via confocal microscopy (Jones et. al., 2010), a quantitative way to demonstrate Francisella lysis within the intracellular milieu had yet to be described. Furthermore, the bacterial factors, if any, that affect susceptibility to these lysis events are also largely undefined. It is also unknown if Francisella encodes any proteins that are involved in modulation of the inflammasome.
Recently, Huang et al. and Ulland et al. described LVS mutants that exhibit a hypercytotoxic and increased AIM2 activation phenotype in macrophages (Huang et. al., 2010, Ulland et. al., 2010). These observations led these groups to propose that Francisella possesses proteins that are involved in active modulation or suppression of the host AIM2 inflammasome complex. Several groups have also recently described mutant strains of F. novicida as well as F. tularensis Type A Schu S4 that are deficient for LPS and capsule biosynthesis which induce a similar hypercytotoxic phenotype in macrophages (Lai et. al., 2010, Lindemann et. al., 2010). In this study, we demonstrated that a range of F. novicida hypercytotoxic bacterial mutant strains; irrespective of their innate biological function hyper-stimulate the AIM2 inflammasome (Figure 2). These hypercytotoxic mutants also stimulated increased levels of type 1 IFN, TNF-α and IL-6 secretion in infected macrophages compared to the wild-type U112 strain (Figure 3). In contrast, AIM2 transcript levels in macrophages infected with the hypercytotoxic bacterial mutant strains were not significantly different than the levels in macrophages infected with the wild type U112 strain (Figure 2). One possible explanation for this result is that there are known differences in signaling pathways and transcription factors involved in AIM2 transcription compared to that of TNF-α and IL-6. For instance, expression of AIM2 has been shown to be affected by type 1 IFN signaling while that of TNF-α and IL-6 is affected by signaling via NF-kB (Jones et. al., 2010, Libermann et. al., 1990, Craig et. al., 2000, Shakhov et. al., 1990). Furthermore, other signaling pathways or transcription factors that can affect AIM2 expression may exist, which we currently still do not fully understand. It is also unknown if type 1 IFN dependent induction of AIM2 is dose-dependent.
In addition, the F. novicida hypercytotoxic mutants described in this study exhibit aberrant morphologies (Figure 6) and we definitively demonstrated for the first time that these hypercytotoxic Francisella mutants displayed increased bacterial lysis within the intracellular milieu (Figure 5 and 7). It is interesting to note that although the hypercytotoxic mutants exhibited increased bacterial lysis in the macrophage cytosol, we did not detect significantly different levels of intracellular replication compared to the wild type U112 strain. These results suggest that the differences in recoverable viable bacteria between the hypercytotoxic mutant strains and the wild type strain are not large enough to be detected by the standard gentamicin-protection assay. In addition, these results indicate that the immune surveillance system in the cytosol of macrophages is extremely sensitive and that the mammalian innate immune system has evolved to react very rapidly to cytosolic danger signals.
The cellular structure of Gram-negative bacteria consists of a cytoplasmic membrane and an outer membrane separated by peptidoglycan and the periplasmic space. These membranes are lipid bilayers interspersed with a variety of carbohydrates and proteins that allow the membranes to carry out their different biological functions. For instance, the bacterial LPS forms a picket fence structure to keep antibodies and complement at a defined distance from the susceptible outer membrane (Costerton et. al., 1974) while matrix proteins are critical for maintaining the integrity of the outer membrane (DiRienzo et. al., 1978). Cell wall associated degradative enzymes also provide a digestive function to break down complex food molecules into simple monomeric molecules in a zone immediately surrounding the bacterial cell where they can be subsequently translocated into the cell via porins, permeases and other transporters found in the membranes (Benz et. al., 1988, Costerton et. al., 1974, DiRienzo et. al., 1978). Although the structural integrity of the bacterial cell is usually attributed to the peptidoglycan, many studies have demonstrated strong interactions between membrane proteins and the peptidoglycan layer (DiRienzo et. al., 1978). Extensive protein-protein interactions also occur over the entire surface of the membrane (DiRienzo et. al., 1978). Thus, it is highly likely that when a membrane-associated protein is deleted, this leads to altered protein composition of the membrane. These alterations may affect the stability or integrity of the bacterial membrane and increase susceptibility of the bacterium to lysis events under stress (Costerton et. al., 1974, DiRienzo et. al., 1978, Wang et. al., 2002). In addition, mutational defects in LPS structure can also affect the organization and proper assembly of the outer membrane proteins (DiRienzo et. al., 1978). Since all of the F. novicida and LVS hypercytotoxic mutants described previously by others as well as in this study all encode either membrane-associated proteins or surface structures, it is likely that the deletion of these proteins led to destabilization of the bacterial cell structures, thus increasing susceptibility to bacterial lysis in the intracellular milieu, a presumably stressful environment with the myriad of antimicrobial peptides and host innate immune defense molecules (Edwards et. al., 2010).
In addition, Shimada et al. also previously reported that during Staphylococcus aureus infection of macrophages, lysozyme-based bacterial cell wall degradation was needed for inflammasome activation and IL-1β production (Shimada et. al., 2010). They further reported that presence of PGN-O-acetyltransferase A, a S. aureus enzyme that is required for making the bacterial cell wall peptidoglycan resistant to lysozyme, plays a role in preventing inflammasome activation (Shimada et. al., 2010). These findings, together with ours, appear to highlight a common theme where the structural integrity of a bacterium can in turn affect accessibility of various microbial PAMPs to the myriad of host innate immune sensing molecules present, such as inflammasome complexes.
It has also been recently reported that the overexpression of a bacterial transporter in L. monocytogenes led to the transport of cyclic diadenosine monophosphate (c-di-AMP), a molecule that the authors proposed to be responsible for triggering type 1 IFN production (Woodward et. al., 2010). However, in that study, the authors did not verify if overexpression of the bacterial transporter in question, also a membrane-associated protein, affects bacterial susceptibility to lysis in the host cell. In light of our new data, it is imperative to keep in mind that when the protein composition of the bacterial membrane is altered, that this could lead to increased bacterial lysis and stimulation of multiple innate immune pathways.
In this study, contrary to what had been previously proposed by other Francisella groups, we definitively demonstrate for the first time that hypercytotoxic F. novicida mutants stimulate increased AIM2 inflammasome activation because they exhibit increased bacterial lysis in macrophages. Thus, there is not currently any direct evidence demonstrating that Francisella possesses bacterial factors involved in active modulation of the host inflammasome complex. However, the role of the inflammasome in macrophages infected with the more virulent F. tularensis Type A strain has not been investigated. It is possible that the Type A strains possess a factor that directly modulates the inflammasome and this may contribute to its increased virulence. However, there is also a possibility that the bacterial physiology and surface structures such as the capsule and LPS of the Type A strains may differ from that of the F. novicida and LVS strains and this could directly affect the levels of intracellular bacteriolysis and inflammasome activation. F. novicida, LVS and Listeria monocytogenes are intracellular pathogens that replicate within the cytosol and have been demonstrated to be sensed by AIM2. This suggests that perhaps AIM2 has evolved to be a very sensitive detector of DNA mislocalization, capable of detecting even low levels of bacterial DNA in the cytosolic compartment. In future, it will be interesting to determine whether other bacterial pathogens that reside within the cytosol such as Shigella, Burkholderia and Rickettsia are also detected by AIM2. If not, our findings suggest that the differential ability of various cytosolic bacterial pathogens to by sensed by AIM2 could possibly be attributed, at least in part, to their differential resistance to bacteriolysis in the intracellular milieu. This may be influenced by inherent differences in bacterial physiology such as membrane protein composition and surface structures. The interplay between intracellular bacteria and the host cell, especially in the context of the cytosolic milieu is an area that warrants further investigation and is likely to greatly extend our understanding of host-pathogen interactions.
Materials and Methods
Bacteria
Wild-type F. tularensis subspecies novicida strain U112 was described (Gill et. al., 1997). All bacterial strains were grown overnight at 37°C with aeration in modified tryptic soy broth (TSB; Difco/BD, Sparks, MD) and supplemented with 0.2% L-cysteine.
Targeted Mutagenesis
Mutant and complemented strains were constructed as previously described. Briefly, primers were used to amplify sequences flanking genes of interest as well as a kanamycin resistance cassette. The PCR products were then put together using a sewing-PCR approach. Resulting constructs were chemically transformed into U112 and Kan-resistant colonies representing strains with double crossover events were selected. Integration of the Kan cassette into the correct region of the genome was confirmed by sequencing. The generation and verification of the FPI mutant has been previously described (Weiss et. al., 2007b). Constructs for complemented strains were engineered similarly but with a chloramphenicol cassette and resulting constructs were chemically transformed into the corresponding mutant strain of interest. Allelic replacement was confirmed by chloramphenicol resistance and Kan sensitivity as well as by sequencing.
Macrophage Infections
Macrophages were seeded at the appropriate density as described for each of the different assays and allowed to adhere overnight at 37°C, 5% CO2. The following day, bacteria were added to the cells and plates were centrifuged at 19000 rpm for 15 minutes at room temperature to spin the bacteria onto the cells and synchronize the infection. 30 minutes post infection, the cell culture medium was replaced with culture medium containing 10 μg/ml of gentamicin and the cells were then cultured in the gentamicin containing media till the appropriate time points post infection.
Cytotoxicity and Cytokine Measurement
For cytotoxicity and cytokine measurement, macrophages were seeded at a density of 5 × 104 macrophages per well in 96-well plates and infected as described above at an effective MOI of 20. Cytotoxicity was measured via lactate dehydrogenase release (Promega, Madison, WI) and levels of IL-1β, TNF-α and IL-6 in cell culture supernatants were measured via ELISA (R & D systems, Minneapolis, MN).
Type 1 Interferon Measurement
For measurement of type 1 IFN levels, macrophages were seeded at a density of 5 × 104 macrophages per well in 96-well plates and infected as described above at an effective MOI of 20. At 8h post infection, culture supernatants were collected. ISRE-L929 reporter cells were used to measure type 1 IFN levels in the supernatants as previously described (Jiang et. al., 2005). Briefly, ISRE-L929 cells were seeded at a density of 1 × 105 cells per well in 96 well viewplate-96 TC plates (Perkin Elmer, Waltham, MA) and allowed to adhere overnight at 37°C, 5% CO2. ISRE-L929 cells were incubated with 60 μl of the collected macrophage supernatants for 4 hrs at 37°C, 5% CO2. After 4 hrs, the supernatant was removed and the ISRE-L929 cells were lysed with 40 μl per well of Glo lysis buffer (Promega, Madison, WI) for 7 min with agitation. 40 μl per well of bright-glo luciferase substrate (Promega, Madison, WI) was added and luminescence levels were measured using a luminometer (Veritas, Turner Biosystems, Sunnyvale, CA).
RNA Isolation and qRT-PCR analysis
For measurement of AIM2 transcript levels, macrophages were seeded at a density of 1 × 106 macrophages per well in 6-well plates and infected as described above at an effective MOI of 20. At 8h post infection, 1 ml of Trizol reagent (Invitrogen) was added to the BMDMs and RNA was isolated using the RNeasy minikit (Qiagen). Quantitative real-time RT-PCR (qRT-PCR) was performed on a real-time detection system (iCycler; Bio-Rad Laboratories) using the rTth enzyme (Applied Biosystems), SYBR green, and the respective primers (see Table S1 in the supplemental material) were used. AIM2 transcripts were normalized to the amount of β-actin mRNA to obtain relative quantities of message and compared to uninfected controls.
Immunofluorescence Microscopy
BMDMs were seeded at a density of 1.5×105 cells/ well onto glass coverslips in 24-well plates and infected as described above at an effective MOI of 10. At the appropriate time point post infection, cells were washed three times with PBS, fixed for 15 minutes at 37°C with 4% paraformaldehyde in PBS, washed three times with PBS and stained with rabbit anti-mouse AIM2 at 1/200 (Genentech) and chicken anti-Francisella at 1/2000 (Monack Laboratory) for 30 minutes in blocking buffer (3% BSA and 0.1% saponin in PBS). Cells were washed three times with PBS and incubated for 30 minutes with Alexa488 anti-chicken and Alexa594 anti-rabbit antibodies (Invitrogen). Cells were washed four times with PBS, mounted onto microscope slides and imaged using a Zeiss LSM700 confocal microscope.
Scanning Electron Microscopy
Overnight cultures of the bacterial strains were subcultured at 1:50 in Chamberlain's defined media and grown till the late exponential growth phase. The bacteria was spun down at 8000g for 5 min and processed for scanning electron microscopy. Bacteria were fixed overnight at 4°C in 4% paraformaldehyde with 2% glutaraldehyde in 0.1 M Na cacodylate buffer (pH 7.3, electron microscopy grade, EMS, Hatfield, PA). After primary fixation, samples were rinsed in the same buffer, postfixed in 1% aqueous OsO4 for 1 hr and dehydrated in an ascending ethanol series (30, 50, 70, 80, 90 and 100% for 20 min each), followed by critical-point drying with liquid CO2 using a Tousimis SAMDRI-VT-3B apparatus (Tousimis, Rockville, MD). Samples were mounted on adhesive carbon film on 15mm aluinium stubs and sputter coated with 100A gold/palladium using a Denton Desk 11 TSC sputter coater. Visualization was carried out with a Hitachi S-3400N VP scanning electron microscope (Hitachi Ltd, Pleasanton, CA) operated at 10-15 kV at a working distance of 8 to 10 mm with a secondary electron detector. Images were captured in TIFF format.
Luciferase Reporter Delivery Assay
The luciferase reporter plasmid was constructed via the cloning of the CMV enhancer, immediate early promoter, chimeric intron and the modified firefly luciferase gene from the previously described pBHE573 (Sauer et. al., 2010) into a Francisella shuttle vector pFNLTP6 to create the Francisella luciferase reporter plasmid pFNLTP6-CMV-Luc. Nonstimulated bone marrow-derived IFNAR-/- macrophages were seeded at a density of 2×105 macrophages per well of an opaque 96 well plate and allowed to adhere overnight at 37°C, 5% CO2. The following day, the macrophages were infected with F. tularensis strains carrying the pFNLTP6-CMV-Luc luciferase reporter plasmid at an effective MOI of 100 for 1 hr. At 1 hr post infection, the cell culture media was removed and replaced with media containing 10 ug/ml of gentamicin. 7 hours post infection, bright-glo luciferase substrate (Promega, Madison, WI) was added and luminescence levels were measured using a luminometer (Veritas, Turner Biosystems, Sunnyvale, CA).
Statistical Analysis
All statistical data analysis was done as indicated using Prism 5 (GraphPad Software, Inc).
Supplementary Material
Acknowledgments
We thank John-Damien Sauer and Daniel Portnoy for the kind gift of pBHE573; Stanley Falkow and the Monack lab for helpful discussions and Sara Fisher for administrative assistance. Kaitian Peng is supported by a graduate fellowship from the Agency for Science, Technology and Research, Singapore. Petr Broz is supported by a long-term post-doctoral fellowship from the Human Frontiers in Science Program (HFSP). This work was supported by grants AI063302 and AI065359 from the NIH-NIAID to Denise Monack.
Footnotes
Author contributions: K.P and D.M.M designed the experiments. K.P., J.W.J., P.B., L.M.J performed the experiments. K.P. and D.M.M. wrote the paper.
The authors declare no conflict of interests.
References
- Akira S. Innate immunity to pathogens: diversity in receptors for microbial recognition. Immunol Rev. 2009;227:5–8. doi: 10.1111/j.1600-065X.2008.00739.x. [DOI] [PubMed] [Google Scholar]
- Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- Benz R. Structure and function of porins from gram-negative bacteria. Ann Rev Microbiol. 1988;42:359–393. doi: 10.1146/annurev.mi.42.100188.002043. [DOI] [PubMed] [Google Scholar]
- Bhavsar AP, Guttman JA, Finlay BB. Manipulation of host-cell pathways by bacterial pathogens. Nature. 2007;449:827–834. doi: 10.1038/nature06247. [DOI] [PubMed] [Google Scholar]
- Brodsky IE, Monack D. NLR-mediated control of inflammasome assembly in the host response against bacterial pathogens. Semin Immunol. 2009;21:199–207. doi: 10.1016/j.smim.2009.05.007. [DOI] [PubMed] [Google Scholar]
- Burckstummer T, Caumann C, Blumi S, Dixit E, Durnberger G, Jahn H, Planyavsky M, Bilban M, Colinge J, Bennett KL, Superti-Furga G. An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat Immunol. 2009;10:266–272. doi: 10.1038/ni.1702. [DOI] [PubMed] [Google Scholar]
- Checroun C, Wehrly TD, Fischer ER, Hayes SF, Celli J. Autophagy-mediated reentry of Francisella tularensis into the endocytic compartment after cytoplasmic replication. PNAS. 2006;103:14578–14583. doi: 10.1073/pnas.0601838103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costerton JW, Ingram JM, Cheng KJ. Structure and function of the cell envelope of gram-negative bacteria. Bacteriological Reviews. 1974;38:87–110. doi: 10.1128/br.38.1.87-110.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig R, Larkin A, Mingo AM, Thuerauf DJ, Andrews C, McDonough PM, Glembotski CC. p38 MAPK and NF-kappa B collaborate to induce interleukin-6 gene expression and release. Evidence for a cytoprotective autocrine signaling pathway in a cardiac monocyte model system. J Biol Chem. 2000;275:23814–23824. doi: 10.1074/jbc.M909695199. [DOI] [PubMed] [Google Scholar]
- DiRienzo JM, Nakamura K, Inouye M. The outer membrane proteins of gram-negative bacteria: biosynthesis, assembly and functions. Ann Rev Biochem. 1978;47:481–532. doi: 10.1146/annurev.bi.47.070178.002405. [DOI] [PubMed] [Google Scholar]
- Edwards JA, Rockx-Brouwer D, Nair V, Celli J. Restricted cytosolic growth of Francisella tularensis subsp. tularensis by IFN-gamma activation of macrophages. Microbiology. 2010:327–339. doi: 10.1099/mic.0.031716-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes-Alnemri T, Yu JW, Datta P, Wu J, Alnemri ES. Aim2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature. 2009;458:509–513. doi: 10.1038/nature07710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes-Alnemri T, Yu JW, Juliana C, Solorzano L, Kang S, Wu J, Datta P, McCormick M, Huang L, McDermott E, Eisenlohr L, Landel CP, Alnemri ES. The Aim2 inflammasome is critical for innate immunity to Francisella tularensis. Nat Immunol. 2010;11:385–393. doi: 10.1038/ni.1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortier AH, Green SJ, Polsinelli T, Jones TR, Crawford RM, Leiby DA, Elkins KL, Meltzer MS, Nacy CA. Life and death of an intracellular pathogen: Francisella tularensis and the macrophage. Immunol Ser. 1994;60:349–361. [PubMed] [Google Scholar]
- Franchi L, Warner N, Viani K, Nunez G. Function of Nod-like receptors in microbial recognition and host defense. Immunol Rev. 2009;227:106–128. doi: 10.1111/j.1600-065X.2008.00734.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller JR, Craven RR, Hall JD, Kijek TM, Taft-Benz S, Kawula TH. RipA, a cytoplasmic membrane protein conserved among Francisella species, is required for intracellular survival. Infect Immun. 2008;76:4934–4943. doi: 10.1128/IAI.00475-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulop M, Manchee R, Titball R. Role of lipopolysaccharide and a major outer membrane protein from Francisella tularensis in the induction of immunity against tularemia. Vaccine. 1995;13:1220–1225. doi: 10.1016/0264-410x(95)00062-6. [DOI] [PubMed] [Google Scholar]
- Gill V, Cunha BA. Tularemia pneumonia. Semin Respir Infect. 1997;12:61–67. [PubMed] [Google Scholar]
- Henry T, Brotcke A, Weiss DS, Thompson LJ, Monack DM. Type 1 interferon signaling is required for activation of the inflammasome during Francisella infection. J Exp Med. 2007;204:987–994. doi: 10.1084/jem.20062665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, Latz E, Fitzgerald KA. Aim2 recognizes cytosolic dsDNA and forms a caspase-1- activating inflammasome with ASC. Nature. 2009;458:514–518. doi: 10.1038/nature07725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu YM, Zhang Y, You Y, Wang D, Li H, Duramad O, Qin XF, Dong C, Lin X. The adaptor protein CARD9 is required for innate immune responses to intracellular pathogens. Nat Immunol. 2007;8:198–205. doi: 10.1038/ni1426. [DOI] [PubMed] [Google Scholar]
- Huang MT, Mortensen BL, Taxman DJ, Craven RR, Taft-Benz S, Kijek TM, Fuller JR, Davis BK, Allen IC, Brickey WJ, Gris D, Wen H, Kawula TH, Ting JP. Deletion of ripA alleviates suppression of the inflammasome and MAPK by Francisella tularensis. J Immunol. 2010;185:5476–5485. doi: 10.4049/jimmunol.1002154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue A, Murata Y, Takahashi H, Tsuji N, Fujisaki S, Kato J. Involvement of an essential gene, mviN, in murein synthesis in Escherichia coli. J Bacteriol. 2008;190:7298–7301. doi: 10.1128/JB.00551-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Z, Georgel P, Du X, Shamel L, Sovath S, Mudd S, Huber M, Kalis C, Keck S, Galanos C, Freudenberg M, Beutler B. CD14 is required for MyD88-independent LPS signaling. Nat Immunol. 2005;6:565–570. doi: 10.1038/ni1207. [DOI] [PubMed] [Google Scholar]
- Jones JW, Kayagaki N, Broz P, Henry T, Newton K, O' Rourke K, Chan S, Dong J, Qu Y, Roose-Girma M, Dixit VM, Monack DM. Absent in melanoma 2 is required for innate immune recognition of Francisella tularensis. PNAS. 2010;107:9771–9776. doi: 10.1073/pnas.1003738107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz J, Zhang P, Martin M, Vogel SN, Michalek SM. Toll-like receptor 2 is required for inflammatory responses to Francisella tularensis LVS. Infect Immun. 2006;74:2809–2816. doi: 10.1128/IAI.74.5.2809-2816.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koebnik R, Locher KP, Van Gelder P. Structure and function of a bacterial outer membrane proteins: barrels in a nutshell. Molecular Microbiology. 2000;37:239–253. doi: 10.1046/j.1365-2958.2000.01983.x. [DOI] [PubMed] [Google Scholar]
- Lai XH, Shirley RL, Crosa L, Kanistanon D, Tempel R, Ernst RK, Gallagher LA, Manoil C, Heffron F. Mutations of Francisella novicida that alter the mechanism of its phagocytosis by murine macrophages. PLoS One. 2010;5:e11857. doi: 10.1371/journal.pone.0011857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landolfo S, Gariglio M, Bribaudo G, Lembo D. The Ifi 200 genes: An emerging family of IFN-inducible genes. Biochimie. 1998;80:721–728. doi: 10.1016/s0300-9084(99)80025-x. [DOI] [PubMed] [Google Scholar]
- Li H, Nookala S, Bina XR, Bina JE, Re F. Innate immune response to Francisella tularensis is mediated by TLR2 and caspase-1 activation. J Leukoc Biol. 2006;80:766–773. doi: 10.1189/jlb.0406294. [DOI] [PubMed] [Google Scholar]
- Libermann TA, Baltimore D. Activation of interleukin-6 gene expression through the NF-kB transcription factor. Molecular and Cellular Biology. 1990;10:2327–2334. doi: 10.1128/mcb.10.5.2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindemann SR, Peng K, Long ME, Hunt JR, Apicella MA, Monack DM, Allen LA, Jones BD. F. tularensis Schu S4 mutants in O-antigen and capsule biosynthesis genes induce early cell death in human macrophages. Infect Immun. 2010 doi: 10.1128/IAI.00863-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linton KJ, Higgins CF. The Escherichia coli ATP binding cassette (ABC) proteins. Mol Microbiol. 1998;28:5–13. doi: 10.1046/j.1365-2958.1998.00764.x. [DOI] [PubMed] [Google Scholar]
- Maier TM, Havig A, Casey M, Nano FE, Frank DW, Zahrt TC. Construction and characterization of a highly efficient Francisella shuttle plasmid. Appl Environ Microbiol. 2004;70:7511–7519. doi: 10.1128/AEM.70.12.7511-7519.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik M, Bakshi CS, Sahay B, Shah A, Lotz SA, Sellati TJ. Toll-like receptor 2 is required for control of pulmonary infection with Francisella tularensis. Infect Immun. 2006;74:3657–3662. doi: 10.1128/IAI.02030-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariathasan S, Weiss DS, Dixit VM, Monack DM. Innate immunity against Francisella tularensis is dependent on the ASC/ caspase 1 axis. J Exp Med. 2005:1043–1049. doi: 10.1084/jem.20050977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariathasan S, Weiss DS, Newton K, McBride J, O' Rourke K, Roose-Girma M, Lee WP, Weinrauch Y, Monack DM, Dixit VM. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440:228–232. doi: 10.1038/nature04515. [DOI] [PubMed] [Google Scholar]
- Nano FE. Identification of a heat-modifiable protein of Francisella tularensis and molecular cloning of the encoding gene. Microb Pathog. 1988;5:109–119. doi: 10.1016/0882-4010(88)90013-7. [DOI] [PubMed] [Google Scholar]
- Nano FE, Zhang N, Cowley SC, Klose KE, Cheung KK, Roberts MJ, Ludu JS, Letendre GW, Meierovics Al, Stephens G, Elkins KL. A Francisella tularensis pathogenicity island required for intramacrophage growth. J Bacteriol. 2004;186:6430–6346. doi: 10.1128/JB.186.19.6430-6436.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyston PC, Sjostedt A, Titball RW. Tularemia: bioterrorism defence renews interest in Francisella tularensis. Nat Rev Microbiol. 2004;12:967–978. doi: 10.1038/nrmicro1045. [DOI] [PubMed] [Google Scholar]
- Raetz C, Reynolds CM, Trent MS, Bishop RE. Lipid A modification systems in gram-negative bacteria. Annu Rev Biochem. 2007;76:295–329. doi: 10.1146/annurev.biochem.76.010307.145803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathinam VA, Jiang Z, Waggoner SN, Sharma S, Cole LE, Waggoner L, Vanaja SK, Monks BG, Ganesan S, Latz E, Hornung V, Vogel SN, Szomolanyi-Tsuda E, Fitzgerald KA. The Aim2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat Immunol. 2010;11:395–402. doi: 10.1038/ni.1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raynaud C, Meibom KL, Lety MA, Dubail I, Candela T, Frapy E, Charbit A. Role of the wbt locus of Francisella tularensis in lipopolysaccharide O-antigen biogenesis and pathogenicity. Infect Immun. 2007;75:536–541. doi: 10.1128/IAI.01429-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz N. Bioinformatics identification of MurJ (MviN) as the peptidoglycan lipid II flippase in Escherichia coli. PNAS. 2008;105:15553–15557. doi: 10.1073/pnas.0808352105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santic M, Molmeret M, Barker JR, Klose KE, Dekanic A, Doric M, Abu Kwaik Y. A Francisella tularensis pathogenicity island protein essential for bacterial proliferation within the host cell cytosol. Cell Microbiol. 2007;9:2391–2403. doi: 10.1111/j.1462-5822.2007.00968.x. [DOI] [PubMed] [Google Scholar]
- Sauer JD, Witte CE, Zemansky J, Hanson B, Lauer P, Portnoy DA. Listeria monocytogenes triggers Aim2-mediated pyroptosis upon infrequent bacteriolysis in the macrophage cytosol. Cell Host Microbe. 2010;7:412–419. doi: 10.1016/j.chom.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140:821–832. doi: 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- Shakhov AN, Collart MA, Vassalli P, Nedospasov SA, Jongeneel CV. Kappa B-type enhancers are involved in lipopolysaccharide-mediated transcriptional activation of the tumor necrosis factor alpha gene in primary macrophages. J Exp Med. 1990;171:35–47. doi: 10.1084/jem.171.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada T, Park BG, Wolf AJ, Brikos C, Goodridge HS, Becker CA, Reyes CN, Miao EA, Aderem A, Gotz F, Liu GY, Underhill DM. Staphylococcus aureus evades lysozyme-based peptidoglycan digestion that links phagocytosis, inflammasome activation and IL-1beta secretion. Cell Host Microbe. 2010;7:38–49. doi: 10.1016/j.chom.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- Ulland TK, Buchan BW, Ketterer MR, Fernandes-Alnemri T, Meyerholz DK, Apicella MA, Alnemri ES, Jones BD, Nauseef WM, Sutterwala FS. Cutting edge: mutation of Francisella tularensis mviN leads to increased macrophage absent in melanoma 2 inflammasome activation and a loss of virulence. J Immunol. 2010;185:2670–2674. doi: 10.4049/jimmunol.1001610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y. The function of OmpA in Escherichia coli. Biochemical and Biophysical Research Communications. 2002;292:396–401. doi: 10.1006/bbrc.2002.6657. [DOI] [PubMed] [Google Scholar]
- Weiss D, Henry T, Monack DM. Francisella tularensis: activation of the inflammasome. Ann N Y Acad Sci. 2007a;1105:219–237. doi: 10.1196/annals.1409.005. [DOI] [PubMed] [Google Scholar]
- Weiss DS, Brotcke A, Henry T, Margolis JJ, Chan K, Monack DM. In vivo negative selection screen identifies genes required for Francisella virulence. PNAS. 2007b;104:6037–6042. doi: 10.1073/pnas.0609675104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodward JJ, Lavarone AT, Portnoy DA. c-di-AMP secreted by intracellular Listeria monocytogenes activates a host type 1 interferon response. Science. 2010;328:1703–1705. doi: 10.1126/science.1189801. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.