

Abstract
Systemic lupus erythematosus (SLE) is a multi-system disorder resulting from interaction of susceptibility genes and environmental factors. SLE has protean clinical presentations at the initial diagnosis and relapses. SLE-related autoantibodies have unique patterns of diversification to linked proteins such as the snRNP particle and the diversification takes years before clinical diagnosis. There are both clinical and experimental evidence to indicate that separate genes contribute to autoimmunity and end organ damage and these genes are independent and interactive. Among the numerous susceptibility genes, HLA-D complex is dominant. Results from the authors’ laboratories led us to postulate a unified hypothesis for SLE pathogenesis. This hypothesis states that SLE-autoantibodies are initiated by environmental T cell epitope mimics of the SLE-related autoantigens in hosts with susceptible HLA-D alleles. These autoantibodies diversify over a period of years due the accumulation of cross-reactive T cells. This process ultimately leads to the generation of organ specific autoantibodies and autoreactive effector T cells due to the polyreactive nature of T and B cell receptors from hosts with susceptibility genes to end organ damage, resulting in protean clinical presentations. This hypothesis accounts for most of the features unique to SLE and has clinical implications as to how patients should be treated.
Keywords: Systemic lupus erythematosus Pathogenesis, Epitope spreading, Cross-reactive T and B cell epitopes, HLA-D and end organ damage
Systemic lupus erythematosus (SLE) is a multi-system autoimmune disorder with protean clinical picture in its initial presentation and relapses. It is considered a prototype of autoimmune disorder. SLE has been a subject of intense investigation during the past five decades. Despite significant progress regarding its pathogenesis, many questions remain to be answered. In this review, we will mainly draw on the studies carried out in our laboratory and provide a hypothesis that may be useful as a framework for future investigations.
Historical Perspectives
The history of SLE has been well summarized by Dr. Robert Lahita (1) and Thomas Benedek (2). The term lupus was first applied to described the erosive facial lesion that resembled a “wolf’s bite” in the 13 century. In the mid 1800s, interest in this skin disorder was rekindled. With the characteristic skin disease as a marker, it was soon realized that the disease also affects multiple internal organs. In the early 1900s, by postmortem studies, it was soon realized that SLE could affect internal organs without the skin disorder. Thus the wide spectrum of clinical presentations was well established by the 1970s and 1980s. With the advent of laboratory tests in the last half of the 20th century such as false positive syphilis test, LE cell test, indirect immunofluorescence test for antinuclear antibodies (ANA), anti-dsDNA, anti-Sm, anti-RNP, anti-Ro and anti-La autoantibodies, the diagnosis of SLE was made easier. However, it was soon discovered that the majority of these autoantibodies are not specific for SLE and that these antibodies (Abs) are often found in some healthy individuals and in patients with other diseases. It was also evident that there are autoantibodies with numerous specificities other than those listed above (3). Nevertheless the aforementioned autoantibodies dominate the diagnostic and research arenas for historical reasons.
End Organ Damage vs Autoimmunity
It is apparent from the above brief summary of the history of SLE, the diagnosis of SLE remains problematic because of its protean clinical presentation and the non-specific nature of many of the diagnostic tests. This led to the formulation of the 1982 American College of Rheumatology criteria for the classification of SLE by a committee led by Dr. Eng Tan (4) and these criteria have been updated by Dr. Mark. Hochberg (5). In the 1977 updated criteria, there are 11 criteria (table 1) and for the purpose of identifying patients in clinical studies, a person has SLE if any 4 or more of the 11 criteria are present either simultaneously or serially during the interval of observation. Criteria 1 to 9 deal with clinical manifestations and these manifestations should be considered as end organ damage. Criteria 10 and 11 deal with positive serological tests for lupus-related antigens. Thus in this classification, autoimmunity as measured by the established tests and end organ damage are not linked. The dissociation of end organ damage and autoimmunity is important in both basic consideration of the pathogenesis of SLE and clinical care. Implicitly, there are other autoantibodies not currently measured by clinical laboratories in patients without positive ANA and Abs to the SLE-related autoantigens. In addition, the reality that the autoimmunity as manifested by the presence of ANA and Abs to the SLE-related antigens are present in asymptomatic or healthy individuals was acknowledged by the committee in 1982 (4).
Table 1.
The Revised Criteria for the Classification of Systemic Lupus Erythematosus
| End Organ Damage | Autoimmunity |
|---|---|
|
|
Long Lag Period between the Detection of First Autoantibodies and Diagnosis of SLE with Diversification of Autoantibody Specificities
Shortly after the description of false positive test for syphilis being detected in SLE, Haserick and Long (6) reported in 1952, five cases of SLE in whom the appearance of these false positive biological reactions preceded the clinical signs of SLE by as long as eight years. In a recent publication, Arbukle et al (7) reaffirmed this earlier finding showing that lupus related autoantibodies are present many years before the diagnosis of SLE. The lag period was up to more than 9 years. It was also documented that patients accumulated additional autoantibodies with diverse specificities before the diagnosis was made. In a more recent detailed study (8) this group of investigators provided evidence to support the conclusion that there are unique patterns of responses to linked particles such as snRNP and Ro60/La. This diversification of autoantibody specificities has been termed epitope spreading.
HLA-D Region Identified as a Dominant Lupus Susceptibility Locus
The familial occurrence of SLE has been well recognized. This was well documented by the classical study of Arnett and Shulman (9). Studies on identical twins have documented the high to moderate rate of concordance in monozygotic twins (10, 11). These studies support the thesis that genetics plays a significant role of SLE. Shortly after the description of human HLA-D related serology, it was demonstrated that the HLA-D region contained lupus susceptibility gene(s) (12, 13). Recent genome wide association studies have identified many candidate genes for lupus susceptibility (14). However, HLA-D region was repeatedly identified to be a dominant genetic segment for lupus susceptibility (15, 16). In view of the function of MHC class II genes in antigen presentation, the association of specific haplotypes with SLE (17) must be considered and accounted for in any hypothesis of pathogenesis of SLE.
Major Features of SLE Accommodated by Hypotheses of SLE Pathogenesis
From the above discussion, there are four major features in SLE to be considered in postulating a comprehensive hypothesis. See table 2. These are 1) protean clinical presentations at the initial diagnosis and in relapses with each patient having a unique clinical course; 2) interactive and independent nature of autoimmunity and end organ damage; 3) unique patterns of autoantibody diversification to linked autoantigen sets such as SmD, SmB, and RNP proteins and this diversification process takes years; and 4) the dominant role of HLA-D regions in SLE susceptibility.
Table 2.
Major Features of SLE that should be Accommodated by Hypotheses of SLE Pathogenesis
|
During the past decade, our laboratory has been interested in developing models to address the different aspects of SLE in order to formulate a coherent hypothesis for the pathogenesis of SLE that may account for the aforementioned four major features of human SLE.
NZM2328 and its Congenics: Dissociation of Autoimmunity with End Organ Damage
(NZBXNZW)F1 has been a very useful model for SLE (18). Recently, the New Zealand Mixed (19) strains have been generated and further characterized (20). NZM strains were derived from (NZWXNZB)F1 with multiple backcrosses to NZW. They were selected by coat colors. Multiple strains were established as inbred strains by brother-sister mating. They have various contributions from NZB and have varied phenotypes. They have ANA and varying diseases affecting the kidneys and the neurological system. For example, NZM2410 has severe nephritis and early mortality in both males and females and mild neurological symptoms. NZM2328 females have severe nephritis and early mortality without neurological symptoms. NZM2328 males have mild nephritis without early mortality. NZM88 males have severe neurological symptoms while its females have moderate nephritis. NZM64 have little nephritis and no renal disease.
NZM2410 was utilized by Morel et al to map the lupus susceptibility genes (21). In linkage studies involving the analysis of (NZM2410XB6) X NZM2410, Sle1, 2, 3 were identified as SLE susceptibility genes linked to glomerulonephritis (GN). In this initial study, no genetic loci were identified to be linked to the production of ANA and anti-DNA Abs. These three loci have been bred to B6 to generate congenic strains, B6.NZMSle1, B6.NZMSle2 and B6.NZMSle3. Unfortunately, none of these congenic strains have GN. The characteristics of these congenic strains have been summarized in a review by Morel and Wakeland (22). Sle1, Sle2, and Sle3 apparently interact with various B6 genes, resulting in various autoimmune phenotypes. It is of interest to note that bicongenic and tricongenic strains containing Sle1 with Sle2 or/and Sle3 show varying degrees of severe GN development, indicating the dominance of Sle1 in the development of GN on B6 background (23). In addition, the Sle1 locus has been shown to be a cluster of functionally related genes (24). This cluster contains Sle1a, Sle1b and Sle1c. Because Sle1 was shown to be a lupus susceptibility gene linked to GN, it was postulated that an additional gene, Sle1d within the Sle1 locus contributes to GN. Thus far, the location of Sle1d remains to be determined. Although considerable information has been generated in the characterization of the B6.NZM congenic lines, the relationship of these observations as related to the original mapping data, i.e. the pathogenesis of lupus GN remains to be clarified.
Our laboratory has taken another approach to study the genetics of SLE. NZM2328 was chosen as a model because the clinical picture of this strain resembles that in human lupus nephritis (25). Cognizant of the fact that GN can be readily classified as acute GN (aGN) and chronic GN (cGN) and ANA/anti-DNA Abs, aGN and cGN are the three phenotypes, linkage analysis of a (NZM2328 X C57L/J)F1 X NZM2328 cohort identified distinct loci associated with three phenotypes. Cgnz1 on telomeric chromosome 1 displayed significant linkage to cGN, severe proteinuria, and early mortality in female NZM2328 mice. Adaz1 on chromosome 4 was suggestively linked to elevated levels of ANA and anti-dsDNA Ab. Three loci were suggestively linked to aGN: Agnz1 on distal chromosome 1, Agnz2 on distal chromosome 17, and the H-2-Tnf complex. Two congenic strains, NZM2328.C57L/Jc1 (NZM.Lc1) and NZM2328.C57L/Jc4 (NZM.Lc4) were generated by replacing the respective genetic intervals containing Cgnz1/Agnz1 or Adaz1 with those from C57L/J as shown in figure 1 (26). NZM2328.Lc1 (Lc1) females had markedly reduced incidence of acute GN, chronic GN, severe proteinuria, and autoantibody production. NZM2328.Lc4 (Lc4) females developed acute GN, chronic GN and severe proteinuria without circulating ANA, anti-dsDNA, and anti-nucleosome Ab. The phenotypes of the two congenic lines confirm the genetic analysis results. The Lc4 congenic females show that anti-dsDNA Abs, ANA and anti-nucleosome Abs are not needed for lupus nephritis, and the Lc1 congenic line provided the opportunity to show that aGN and cGN are under separate genetic control.
Figure 1.

NZM.C57/Lc1 (Lc1) and NZM.C57Lc4 (Lc4) congenic lines were derived by replacing the genetic intervals in NZM2328 with those from C57L/J (hatched bar). The genetic intervals with SLE susceptibility genes in NZM2328 delineated by informative microsatellite markers are shown (open bars). Chromosome intervals are drawn to scale. The genes involved in Lc1 and Lc4 congenic lines are Cgnz1 and Agnz1, and Adaz1 respectively. The characteristics of these two congenic lines are included (modified from 26).
These data have allowed us to propose a cooperative model for the pathogenesis of lupus nephritis (figure 2). In this model (26), the production of classical lupus-related autoantibody production is dissociated from end organ damage. The model predicts that autoimmunity may not cause end organ damage in individuals with genes that render their end organ resistant to damage either by immune complex mediated or cell mediated mechanisms. The model also suggests that the interaction between the two pathways may induce organ specific immunity that, in turn, can cause more inflammation and further enhance the autoimmune response.
Figure 2.
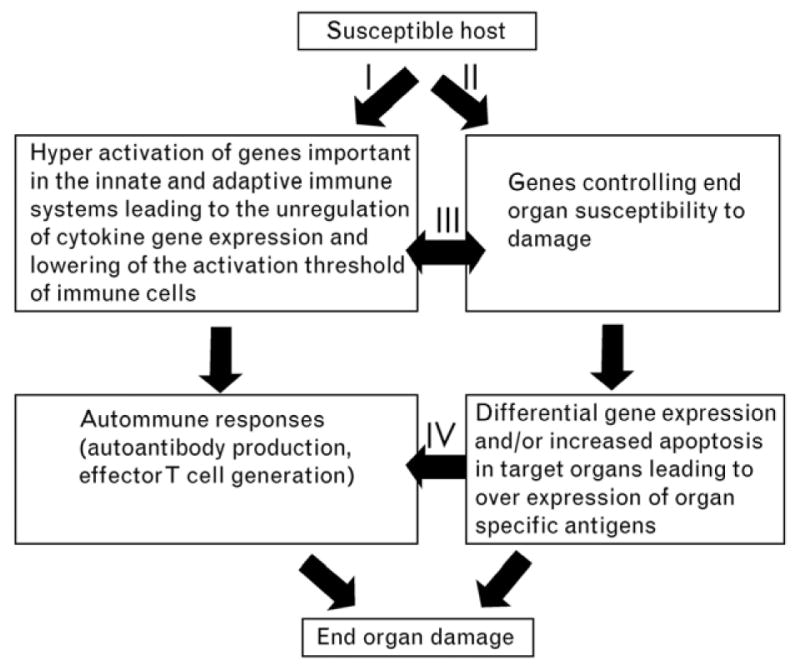
Proposed model for the pathogenesis of SLE describes the independent and yet interactive nature of the genes contributes to autoimmunity and end organ damage. I, Autoantibody production and activation of effector T cells and II, activation of susceptibility for end organ damage, can be initiated independently while they interact at different levels as indicated by pathways III and IV. Interaction of these pathways leads to end organ damage (26).
As stated above, both Cgnz1 and Agnz1 are located on chromosome 1 and these loci are within the replaced chromosome segment in NZM.Lc1. Several intra-chromosomal recombinant lines have been generated. One of them NZM.Lc1R27 (R27) was extensively characterized. R27 has an 8Mb segment from C57L/J. The Cgnz1 was replaced by the allele from C57L/J without the replacement of Agnz1 locus. The females of R27 have aGN by morphology and mild proteinuria without early mortality. Thus aGN does not need to progress to cGN and immune complex deposition with complement activation may not be sufficient to cause end stage renal disease in lupus nephritis. Preliminary data indicates that Cgnz1 locus is located within an 1.34 Mb segment that is identical to the Sle1b segment (27, Ge et al manuscript in preparation). Thus it is likely that the original identified Sle1 locus and the later postulated Sle1d with GN as the phenotype is located in the Sle1b region.
Role of T Cells in Autoantibody Diversification in SLE
Mechanism of Intramolecular Epitope Spreading and the Presence of Similar B Epitopes in SLE-Related Autoantigens
During the past decade, Ro60 and SmD have been used by us to study epitope spreading, a mechanism by which autoantibody diversity is generated (28). The immune response to both mouse and human recombinant Ro60 proteins induced autoantibodies in SJL but not in B6 with reactivity to La, Ro52, and proteins of the snRNP particle such as SmD, SmB, A-RNP and 70kD U1-RNP (29). The Abs against Ro52, La and snRNP proteins were shown to be reactive with B cell epitopes shared by Ro60 and these autoantigens because the immunogen Ro60 absorbed these reactive Abs completely. In addition, a dominant T cell epitope in both mouse and human Ro60 proteins was shown to be located on the peptide, Ro60316–335. Similar to the Ro60 protein, this peptide induced multiple autoantibodies cross-reactive with a panel of SLE-related antigens. In addition this peptide induced autoantibodies to multiple B cell epitopes within the Ro60 peptides (30). Some of these Abs were able to be absorbed by the immunizing peptides while other Abs were not cross-reactive with the immunogen. These data indicate that intramolecular epitope spreading is responsible for the generation of anti-Ro60 Abs with complex specificities. In addition, autoantibodies cross-reactive with multiple SLE-related autoantigens are generated. The presence of multiple cross reactive B cell epitopes among SLE-related of autoantigens has also been confirmed by human autoantibodies (31). Our data extended those by other investigators (8, 32). The cross-reactive epitopes are much more numerous than those analogous to PPPGRRP and related linear determinants expressed by SmD, A-RNP protein, SmB and EBV (8).
Intermolecular B Epitope Spreading within SnRNP
In 1986, Dr. John Hardin (33) proposed the “Particle Hypothesis” for the initiation of antibody responses to SLE-related antigens. Subsequently, Craft and colleagues have specifically sought evidence for this hypothesis (34). Data from our laboratory showed that in A/J mice immunized with recombinant mouse SmD, SmB or A-RNP producd Abs to these proteins in specific patterns as summarized in table 3 (35). In SmD-immunized mice, specific Abs to A-RNP and SmB were generated by 2 mo postimmunization, in addition to the detection of cross-reactive Abs between the immunogen and other snRNPs. Whereas Abs reactive with the immunogen decreased by 5 mo, Abs capable of immunoprecipitating A-RNP and SmB increased. In SmB-immunized mice, specific Abs to A-RNP were readily detectable, in addition to cross-reactive Abs. In contrast, A-RNP-immunized mice had only cross-reactive Abs to SmB without detectable Abs to SmD. However, in these mice, specific Abs to the 70-kDa protein were generated. Abs, which precipitated the native snRNP particle, were generated in all three groups of the immunized mice. These patterns of epitope spreading are very similar to those observed in patients with SLE and in MRL/lpr mice (8, 36). These results suggest that whole snRNP particles need not be the initiating antigens as previously suggested (33, 34, and 36).
Table 3.
Summary of intermolecular epitope spreading in A/J mice immunized with recombinant SmD, SmB, and A-RNP
| Immunogen | Ab Response Spread |
|---|---|
| SmD | A-RNP, SmB |
| SmB | A-RNP, SmDa |
| A-RNP | 70 kDab, cross-reactive Abs to SmB |
Only by immunoprecipitation in one mouse.
Detected by Western blot analysis only. (35)
In further experiments, three T cell epitopes of SmD were mapped in A/J mice (37). One of them, SmD52–66 induced a better response to A-RNP and perhaps SmB than to SmD. This result has significant implications in postulating initiating antigen from analyses of serological activities of serial SLE sera (8). Relevant to this discussion is the observation by Fisher et al (38) that that there were temporal shifts from reactivity to Sm to RNP in SLE patients. In one instance, the shift occurred within an eight week period. Therefore the conclusion that initiating antigen of anti-snRNP response in SLE is the A-RNP may be premature. This is complicated by the observation that different assays may yield differing conclusions regarding the initiating antigens (8).
It has been postulated that B cell mimics can initiate multiple SLE-related autoantibodies (8). This conclusion is based on the observations that two octopeptides, OOOGMRPP and PPPGIRGP that linked to a lysine backbone (MAP™) were able to induce spliceosome autoimmunity (39) and that B cell epitope of EBNA-1 cross-reactive with Ro60 can induce anti-Ro60 Abs (40). In both cases, the immunized rabbits had clinical pictures such as leucopenia, thrombocytopenia and renal dysfunction. These investigators did not consider the role of T cells in these studies. Related to the role of B epitope mimicry in the initiation of SLE-related autoantibodies, the observations by Diamond and colleagues should be considered (reviewed in 41). By a peptide display phage library, DWEES was identified to be a mimotope for dsDNA and DWEYSVWLSN-MAP induction of anti-dsDNA and SLE-like disease is T cell dependent. These observations suggest the revisit of the hypothesis of B cell mimicry as the initiation of SLE-related autoantibodies. At any rate, the B epitope mimicry hypothesis cannot adequately account for the unique patterns of epitope spreading in anti-snRNP responses as discussed above.
It should be apparent that our investigation on the mechanism of B epitope spreading in the anti-snRNP response has been centered on the role of T cells. In SmD immunized mice, using a ELISPOT assay to detect antigen-specific T cells, 52 SmD-specific T cells/1,000,000 splenic cells were detected on day 30. By day 90, this number increased to 176. In contrast, 7 A-RNP-specific T cells/1,000,000 splenic cells were detected on day 30, and this number increased to 23 on day 90 when epitope spreading to A-RNP was evident. With a T-T hybridoma technique, T cells with dual reactivity to SmD and A-RNP were detected (42). Our views on the role of T cells have been summarized in figure 3. Panels I-III depict scenarios that cannot explain the unique patterns of epitope spreading within snRNP when mice were immunized with individual proteins. In these three scenarios, more random patterns of epitope spreading are expected. Thus panel IV represents the most likely hypothesis taking into account that cross reactive T cells are present and pivotal in controlling the patterns of epitope spreading.
Figure 3.
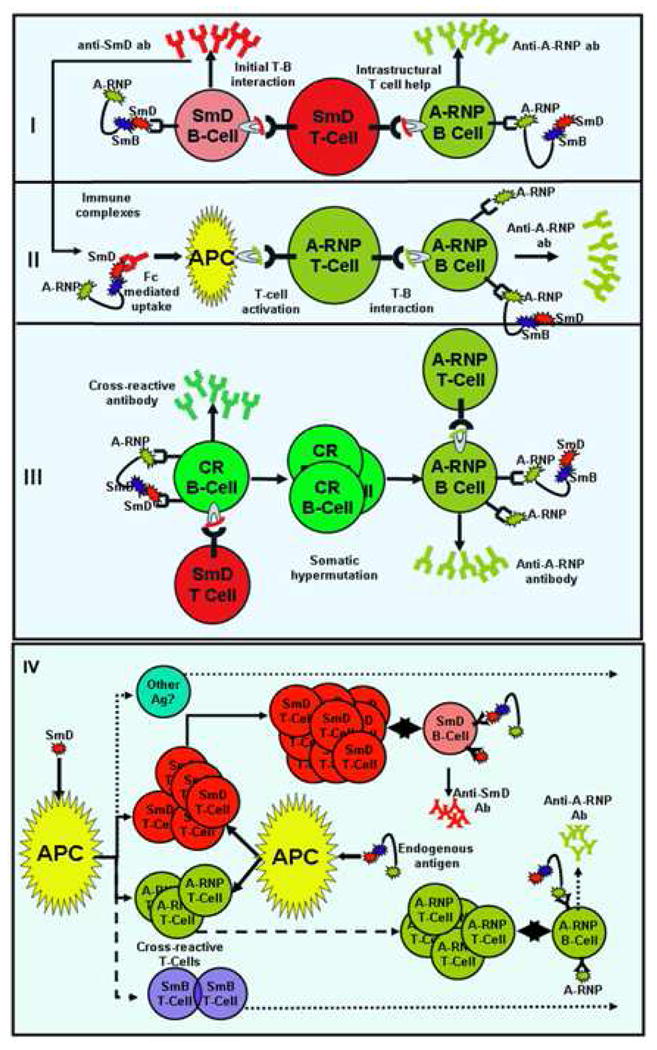
Possible mechanisms for intermolecular epitope spreading within the snRNP complex. For simplicity, only three components of snRNP are included in this illustration. Four possible mechanisms are illustrated: I. Intrastructural T cell help hypothesis. II. Role of immune complexes in epitope spreading. III. Role of cross-reactivity in epitope spreading and IV. Epitope spreading controlled by cross-reactive T cells. Mechanisms I-III cannot account for the specific patterns of epitope spreading. There is no evidence for the generation of specific Abs from cross-reactive Abs. There is evidence to support mechanism IV and further experimental approaches can be designed to test its validity.
Role of HLA-D Region in the Initiation and Diversification of SLE-Related Autoantibodies
In order to elucidate the mechanism by which HLA-D loci exert their dominant role in SLE susceptibility, collaboration was initiated between our laboratory and that of Dr. Chella David to utilize mice with HLA-DR and DQ transgenes to study the HLA-D restricted initiation and diversification of SLE-related autoantibodies. In the initial study (43), mice with HLA-DR2, DR3, DQ6 or DQ8 were immunized with human Ro60 and their immune responses were ascertained over a period of 12 months. Multiple DR- or DQ- restricted T cell epitopes were mapped in human Ro60. Mice with HLA-DR2 and DR3 mounted the most vigorous response when their sera were assayed against mouse Ro60 by immunoprecipitation. By Western blot analysis, epitope spreading was detected to La in immunized DR2 transgenic mice in that the immunogen did not abolish the reactivity to La. The limitation of this investigation is that there are considerable amino acid sequence differences between human and mouse Ro60 molecules and whether these amino acid sequence differences play a role in the observed immune responses remains to be determined. To circumvent this complication, SmD that has an identical amino sequence acid in both human and mice was used as the immunogen (44). The results were remarkable in that by ELISA, mice with DR3, DR4, DQ8, DQ601 or DQ604 transgene responded to immunization with recombinant SmD but only the DR3 and DQ604 transgenic mice made immunoprecipitating Abs to in vitro translated SmD protein. In addition, the immune response was more robust in the DR3 transgenic mice. Among the immunized DR or DQ transgenic mice, only DR3 transgenic mice made Abs to C-RNP and to dsDNA. About half of the anti-dsDNA Abs were absorbed by SmD, the immunogen. It is also remarkable that epitope spreading followed the pattern from SmD to A-RNP and then to SmB. These observations led to the conclusion that HLA-DR3 is a dominant allele in the initiation of anti-snRNP. They also provide insight into the origin of anti-dsDNA Abs.
Initiation of anti-SmD Immune Response by Environmental Antigens with HLA-DR3 Restricted T Cell Epitopes
From the family and twin studies (9–11), it is evident that environmental factors interplay in the pathogenesis of SLE. It is also very instructive that two of the unaffected identical twin pairs in the original report (10) were positive for anti-U1RNP and anti-Sm Abs by ELISA although they did not have positive ANA (45). It is of note that the recently introduced BioRad miltiplex bead assay system, the BioPlex 2200™ for autoantibodies reports semi-quantitative values from 0–8 AI (Ab index). The cut off positive value for each assays was 1 AI, indicating that the autoantibodies of diverse specificities were detected in normals in low AI (0.2–1). Recently, it has been demonstrated in healthy Chinese and Swedish individuals that circulating non cross-reactive IgG anti-pyeloperoxidase, anti-protease 3 and anti-glomerular basement membrane Abs were readily detectable (46). It is of interest to note that the Chinese have more of these Abs. The quantitative differences may be due to environment in that the Chinese may be exposed to more subclinical infections from the environment.
On the T cell level, T cells reactive to snRNP have been reported to be present in both SLE patients and normal individuals (47). Normal individuals have also been shown to have reactivity to Ro60 peptides (S-J Sung, unpublished observation). In other autoimmune diseases such as multiple sclerosis (MS), T cells reactive to MS-related autoantigens have been shown in normal individuals (48). Since CD4+ T cells are positively selected on Class II restriction, it is reasonable to expect that all circulating T cells have some autoreactivity. The summary report by Wucherpfennig et al (49) states clearly the polyspecificity of T and B cell receptor recognition. This information forms the rationale for us to seek bacterial mimics that can induce SLE-related autoantibodies.
With DR3 transgenic mice, a dominant DR3 restricted T cell epitope was identified in the SmD79-93 peptide. A T-T, hybridoma, C1P2 that recognized this peptide and SmD was generated from a DR3 transgenic mouse immunized with SmD79–93 and used to identify contact residues either with HLA-DR3 or with TCR by alanine substitutions. Conservative substitutions were made. Pattern recognition search (http://pir.georgetown.edu) using above structural criteria yielded 430 mimics. This number was reduced to 121 human pathogen or self antigen related mimics. Twenty of these have DR3 binding motif by the software analysis (http://www.imtech.res.in/raghava/propred/). One of these peptides namely P20 was located in galactoside ABC transporter, ATP-binding protein from C. vibrio and was able to stimulate the T-T hybridoma, C1P2 in a DR3 restricted manner. Another T-T hybridoma, P20P1 was generated from a DR3 mouse immunized with P20. This hybridoma was able to respond to SmD, P20, SmD79–93, and P17, a peptide in the putative tetracenomycin polypetide synthesis O-methyltransferase (TcmP) from Streptococcus agalactiae, and P11, a peptide from La-related protein 1 in man. P20, P17 and P11 were used as immunogens to immunize DR3 transgenic mice. They induced anti-SmD Abs with diversification to A-RNP to varying degrees. These autoantibodies are not crossreactive to the immunogens. These results have been presented as an abstract (50). Recently a similar approach has been made with R060 T epitopes and more bacterial mimics have been identified. Immunizations have been initiated with some of the mimics to determine whether the immunized DR3 mice make anti Ro60 Abs. These results indicate that anti-SLE autoantibody response can be initiated by multiple bacterial T epitope mimics in a DR restricted manner, providing definitive evidence to support our hypothesis that autoimmune response to SLE-related antigens are initiated by multiple environmental T epitope mimics that are cross-reactive with the autoantigen of interest and that expansion of the cross-reactive clones may take a long time to accumulate in order to overcome the peripheral regulatory mechanisms to provide sufficient T help to generate autoreactive Abs of sufficient quantity to be of diagnostic value (panel A, figure 4). In this process, effector T cells against various organs may also be generated. The randomness of this process may explain the protean nature of clinical presentations during the initial diagnosis and in relapse (panel B, figure 4).
Figure 4.
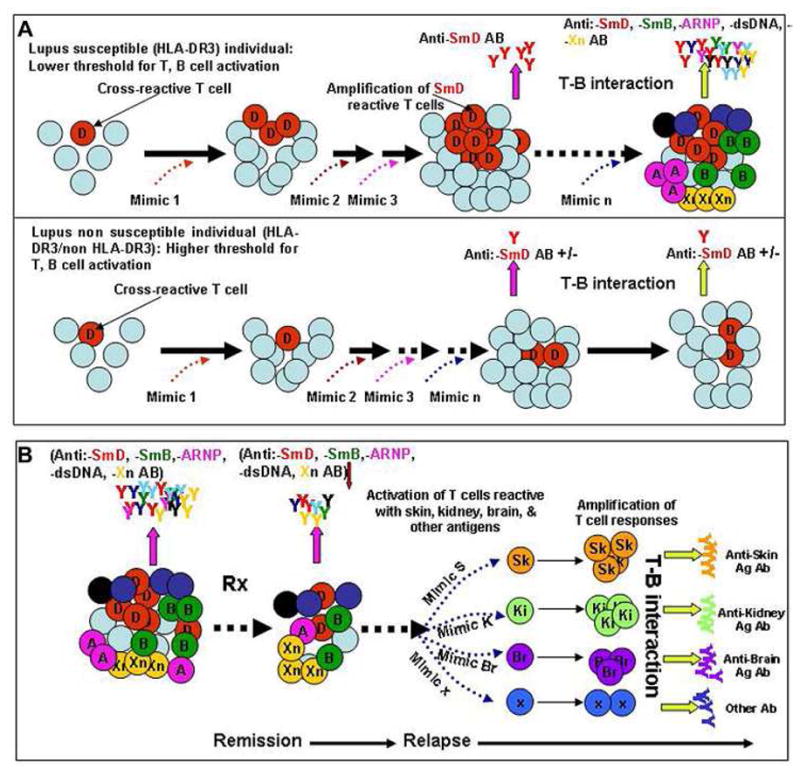
Generation of autoreactive Abs and effector T cells in SLE by environmental T cell epitope mimics. A. Accumulation of cross-reactive T cells as a consequence of response to environmental mimics in hosts (HLA-DR3+) with lupus susceptibility genes but not in hosts (HLA-DR3+) without these genes. B. The accumulation of diverse autoantibodies as a response to these mimics generates pathogenic autoreactive Abs and effector T cells. After therapy, the complexity of these autoantibodies and autoreactive T cells are reduced, leading to remission. Over a period of time after discontinuing therapy, the complexity of autoantibodies and effect T cells returns leading to a protean clinical presentation in relapses. The mimics reside on a diverse array of environmental antigens and the chances for exposure to these mimics are random, providing a scenario in that SLE is not caused by a single pathogen. This mechanism has the flavor of a stochastic process.
Concluding Remarks
In this review, a coherent hypothesis for the pathogenesis of SLE is put forth to account for major features of the syndrome as outlined in table 2. We have postulated that environmental T cell mimics stimulating autoreactive T cells are responsible for the initiation of autoantibodies to SLE-related autoantigens. These T cell responses are restricted by relevant HLA-DR and/or DQ molecules. Normally the generation of these crossreactive T cells is down-regulated by peripheral mechanisms such as the generation of T regulatory cells. In SLE patients, this process was short-circuited by leading to the accumulation of significant crossreactive T cells so that significant autoantibodies are generated to be of diagnostic value. T cell mediated mechanisms leading to autoantibody diversification over a period of years result in complex autoantibody specificity. Some of these Abs may be tissue specific. Through this process, autoreactive effector cells with organ specificity may be generated. These autoreactive Abs and T effector cells will cause end organ damage, which in turn heighten inflammation in the targeted organ. Implicit in this model is that therapy should be targeted to decrease the complexity of the autoreactive Abs and T cells, suggesting that complexity of autoantibody specificity should be a biomarker as a end point for therapy. Since general inflammation may decrease the threshold of autoreactive T and B cells for activation, general suppression of inflammation should be instituted. This scenario suggests that long term therapy with agents such as hydroxychloroquine and immuno-regulatory agents such as mycophenolate and rapimycin can be used in SLE patients to prevent relapses. To reduce side effects, long term use of hydrocortisones such as prednisone should be discouraged.
This review also identifies targets for future investigation. It is important to continue to elucidate mechanisms that lead to specific patterns of B cell epitope spreading. How to translate our observation to patients remains a major challenge. In this regard, studies on SLE patients’ populations in a different environment and studies on selected patients over a long period of time may be helpful. Our hypothesis does not exclude other competing hypotheses except that ours provides an explanation to account for the dominant role of HLA-D as a disease susceptibility locus.
Acknowledgments
We thank Ms Lena Garrison for her help in preparing this manuscript. This work was supported in part by NIH grants P50-AR04522, R01-AR047988, R01-AR049449 to SMF, R01-AI079621 (MPI: USD and SFM), KO1 AR051391(USD), and grants from Alliance for Lupus Research (SMF).
Footnotes
Authors’ Note: The authors express our gratitude to Dr. Chella David for his friendship and generosity. It is our pleasure to be able to contribute this review to be included in this issue of Journal of Autoimmunity to celebrate Chella’s contributions to the area of autoimmunity. We note this is part of the attempt by the Journal of Autoimmunity/Autoimmunity Reviews to highlight distinguished figures in immunology and Chella certainly falls in that category. These issues have included in the past recognition of Noel Rose, Ian Mackay and Harry Moutsopoulos and special issues dealing with lupus, geoepidemiology and Sjogren’s syndrome, amongst others (51–70).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lahita RG. Introduction. In: Lahita RG, editor. Systemic lupus erythematosus. 3. San Diego CA: Academic Press; 1999. pp. xix–xx. [Google Scholar]
- 2.Benedek TG. Historical background of discoid and systemic lupus erythematosus. In: Wallace DJ, Hahn BH, editors. Dubois’ lupus erythematosus. 5. Baltimore MD: Williams & Wilkins; 1997. pp. 3–16. [Google Scholar]
- 3.Amital H, Shoenfeld Y. Autoimmunity and autoimmune diseases such as systemic lupus erythematosus. In: Lahita RG, editor. Systemic lupus erythematosus. 3. San Diego CA: Academic Press; 1999. pp. 1–16. [Google Scholar]
- 4.Tan EM, Cohen AS, Fries JF, Masi AT, McShane DJ, Rothfield NF, et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis & Rheumatism. 1982;25(11):1271–1277. doi: 10.1002/art.1780251101. [DOI] [PubMed] [Google Scholar]
- 5.Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus (letter) Arthritis & Rheumatism. 1997;40(9):1725–1726. doi: 10.1002/art.1780400928. [DOI] [PubMed] [Google Scholar]
- 6.Haserick JR, Long R. Systemic lupus erythematosus preceded by false-positive serologic tests for syphilis: Presentation of five cases. Annuals of Internal Medicine. 1952;37(3):559–565. doi: 10.7326/0003-4819-37-3-559. [DOI] [PubMed] [Google Scholar]
- 7.Arbuckle MR, McClain MT, Rubertone MV, Scofield RH, Dennis GJ, James JA, et al. Development of autoantibodies before the clinical onset of systemic lupus erythematosus. New England Journal of Medicine. 2003;349(16):1526–1533. doi: 10.1056/NEJMoa021933. Retrieved from http://ovidsp.ovid.com/ovidweb.cgi?T=JS&CSC=Y&NEWS=N&PAGE=fulltext&D=med4&AN=14561795. [DOI] [PubMed]
- 8.Heinlen LD, McClain MT, Ritterhouse LL, Bruner BF, Edgerton CC, Keith MP, et al. 60 kD Ro and nRNP A frequently initiate human lupus autoimmunity. PLoS ONE [Electronic Resource] 2010;5(3):e9599. doi: 10.1371/journal.pone.0009599. Retrieved from http://ovidsp.ovid.com/ovidweb.cgi?T=JS&CSC=Y&NEWS=N&PAGE=fulltext&D=medl&AN=20224770. [DOI] [PMC free article] [PubMed]
- 9.Arnett FC, Shulman LE. Studies in familial systemic lupus erythematosus. Medicine. 1976;55(4):313–322. doi: 10.1097/00005792-197607000-00003. [DOI] [PubMed] [Google Scholar]
- 10.Block SR, Lockshin MD, Winfield JB, Weksler ME, Imamura M, Winchester RJ, et al. Immunologic observations on 9 sets of twins either concordant or discordant for SLE. Arthritis & Rheumatism. 1976;19(3):545–554. doi: 10.1002/art.1780190306. [DOI] [PubMed] [Google Scholar]
- 11.Deapen D, Escalante A, Weinrib L, Horwitz D, Bachman B, Roy-Burman P, et al. A revised estimate of twin concordance in systemic lupus erythematosus. Arthritis & Rheumatism. 1992;35(3):311–318. doi: 10.1002/art.1780350310. [DOI] [PubMed] [Google Scholar]
- 12.Gibofsky AM, Winchester RJ, Paterroyo M, Fotino M, Kunkel HG. Disease association of the Ia-like human alloantigens: contrasting patterns in rheumatoid arthritis and systemic lupus erythematosus. Journal of Experimental Medicine. 1978;146(6):1728–1732. doi: 10.1084/jem.148.6.1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reinertsen JL, Klippel JH, Steinberg AD, Decker JL, Mann DL. B-lymphocyte alloantigens associated with systemic erythematosus. New England Journal of Medicine. 1978;299(10):515–518. doi: 10.1056/NEJM197809072991004. [DOI] [PubMed] [Google Scholar]
- 14.Graham RR, Hom G, Ortmann W, Behrens TW. Review of recent genome-wide association scans in lupus. Journal of Internal Medicine. 2009;265(6):680–688. doi: 10.1111/j.1365-2796.2009.02096.x. Retrieved from http://ovidsp.ovid.com/ovidweb.cgi?T=JS&CSC=Y&NEWS=N&PAGE=fulltext&D=medl&AN=19493061. [DOI] [PubMed]
- 15.Harley JB, Alarcón-Riquelme ME, Criswell LA, Jacob CO, Kimberly RP, Moser KL, et al. Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nature Genetics. 2008;40(2):204–210. doi: 10.1038/ng.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hom G, Graham RR, Modrek B, Taylor KE, Ortmann W, Garnier S, et al. Association of systemic lupus erythematosus with C8orf13-BLK and ITGAM-ITGAX. New England Journal of Medicine. 2008;358(9):900–909. doi: 10.1056/NEJMoa0707865. Retrieved from http://ovidsp.ovid.com/ovidweb.cgi?T=JS&CSC=Y&NEWS=N&PAGE=fulltext&D=medl&AN=18204098. [DOI] [PubMed]
- 17.Graham RR, Ortmann WA, Langefeld CD, Jawaheer D, Selby SA, Rodine PR, et al. Visualized human leukocyte antigen class II risk haplotypes in human systemic erythematosus. American Journal of Human Genetics. 2002;71(3):543–553. doi: 10.1086/342290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Theofilopoulos AN, Dixon FJ. Murine models of systemic lupus erythematosus. Advances in Immunology. 1985;37:269–390. doi: 10.1016/s0065-2776(08)60342-9. [DOI] [PubMed] [Google Scholar]
- 19.Rudofsky UH, Evans BD, Balaban SL, Mottironi VD, Gabrielsen AE. Differences in expression of lupus nephritis in New Zealand mixed H-2z homozygous inbred strains of mice derived from New Zealand black and New Zealand white mice. Origins and initial characterization. Laboratory Investigation. 1993;68(4):419–426. [PubMed] [Google Scholar]
- 20.Rudofsky UH, Lawrence DA. New Zealand mixed mice: A genetic systemic lupus erythematosus model for assessing environmental effects. Environmental Health Perspectives. 1999;107(Suppl 5):713–721. doi: 10.1289/ehp.99107s5713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morel L, Rudofsky UH, Longmate JA, Dchiffenbauer J, Walkland EK. Polygenic control of susceptibility to mouse systemic lupus erythematosus. Immunity. 1993;1(3):21229. doi: 10.1016/1074-7613(94)90100-7. [DOI] [PubMed] [Google Scholar]
- 22.Morel L, Wakeland EK. Susceptibility to lupus nephritis in the NZB/W model system. Current Opinion in Immunology. 1998;10(6):718–725. doi: 10.1016/s0952-7915(98)80094-0. [DOI] [PubMed] [Google Scholar]
- 23.Morel L, Croker BP, Blenman KR, Mohan C, Huang G, Gilkeson G, et al. Genetic reconstitution of systemic lupus erythematosus immunopathology with polycongenic murine strains. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(12):6670–6675. doi: 10.1073/pnas.97.12.6670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morel L, Blenman KR, Croker BP, Wakeland EK. The major murine systemic lupus erythematosus susceptibility locus, Sle1, is a cluster of functionally related genes. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(4):1787–1792. doi: 10.1073/pnas.031336098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Waters ST, Fu SM, Gaskin F, Deshmukh US, Sung SS, Kannapell CC, et al. NZM2328: A new mouse model of systemic lupus erythematosus with unique genetic susceptibility loci. Clinical Immunology. 2001;100(3):372–383. doi: 10.1006/clim.2001.5079. [DOI] [PubMed] [Google Scholar]
- 26.Waters ST, McDuffie M, Bagavant H, Deshmukh US, Gaskin F, Jiang C, et al. Breaking tolerance to double stranded DNA, nucleosome, and other nuclear antigens is not required for the pathogenesis of lupus glomerulonephritis. Journal of Experimental Medicine. 2004;199(2):255–264. doi: 10.1084/jem.20031519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ge Y, Jiang C, Gaskin F, Sung SSJ, Bagavant H, Fu SM. Pathogenesis of Proliferative Lupus Nephritis: Different Genetic Control for Acute and Chronic Glomerulonephritis and New Insight Into the Mechanism of Immune Complex Mediated Nephritis. Arthritis & Rheumatism. 2009;60:S755. [Google Scholar]
- 28.Deshmukh US, Bagavant H, Lewis J, Gaskin F, Fu SM. Epitope spreading within lupus-associated ribonucleoprotein antigens. Clinical Immunology. 2005;117(2):112–120. doi: 10.1016/j.clim.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 29.Deshmukh US, Lewis JE, Gaskin F, Kannapell CC, Waters ST, Lou YH, et al. Immune responses to Ro60 and its peptides in mice. I. the nature of the immunogen and endogenous autoantigen determine the specificities of the induced autoantibodies. Journal of Experimental Medicine. 1999;189(3):531–540. doi: 10.1084/jem.189.3.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Deshmukh US, Lewis JE, Gaskin F, Dhakephalkar PK, Kannapell CC, Waters ST, et al. Ro60 peptides induce antibodies to similar epitopes shared among lupus-related autoantigens. Journal of Immunology. 2000;164(12):6655–6661. doi: 10.4049/jimmunol.164.12.6655. [DOI] [PubMed] [Google Scholar]
- 31.Pal R, Deshmukh US, Ohyama Y, Fang Q, Kannapell CC, Gaskin F, et al. Evidence for multiple shared antigenic determinants within Ro60 and other lupus-related ribonucleoprotein autoantigens in human autoimmune responses. Journal of Immunology. 2005;175(11):7669–7677. doi: 10.4049/jimmunol.175.11.7669. [DOI] [PubMed] [Google Scholar]
- 32.Elkon KB, Hines JJ, Chu JL, Parnassa A. Epitope mapping of recombinant HeLa SmB and B′ peptides obtained by the polymerase chain reaction. Journal of Immunology. 1990;145(2):636–643. [PubMed] [Google Scholar]
- 33.Hardin JA. The lupus autoantigens and the pathogenesis of systemic lupus erythematosus. Arthritis & Rheumatism. 1986;29(4):457–460. doi: 10.1002/art.1780290401. [DOI] [PubMed] [Google Scholar]
- 34.Fatenejad S, Brooks W, Schwartz A, Craft J. Pattern of anti-small nuclear ribonucleoprotein antibodies in MRL/Mp-lpr/lpr mice suggests that the intact U1 snRNP particle is their autoimmunogenic target. Journal of Immunology. 1994;152(11):5523–5531. [PubMed] [Google Scholar]
- 35.Deshmukh US, Kannapell CC, Fu SM. Immune responses to small nuclear ribonucleoproteins: Antigen-dependent distinct B cell epitope spreading patterns in mice immunized with recombinant polypeptides of small nuclear ribonucleoproteins. Journal of Immunology. 2002;168(10):5326–5332. doi: 10.4049/jimmunol.168.10.5326. [DOI] [PubMed] [Google Scholar]
- 36.Fatenejad S, Brooks W, Schwartz A, Craft J. Pattern of anti-small nuclear ribonucleoprotein antibodies in MRL/Mp-lpr/lpr mice suggests that the intact U1 snRNP particle is their autoimmunogenic target. Journal of Immunology. 1994;152(11):5523–5531. [PubMed] [Google Scholar]
- 37.Deshmukh US, Bagavant H, Sim D, Pidiyar V, Fu SM. A SmD peptide induces better antibody responses to other proteins within the small nuclear ribonucleoprotein complex than to SmD protein via intermolecular epitope spreading. Journal of Immunology. 2007;178(4):2565–2571. doi: 10.4049/jimmunol.178.4.2565. [DOI] [PubMed] [Google Scholar]
- 38.Fisher DE, Reeves WH, Wisniewolski R, Lahita RG, Chiorazzi N. Temporal shifts from sm to ribonucleoprotein reactivity in systemic lupus erythematosus. Arthritis & Rheumatism. 1985;28(12):1348–1355. doi: 10.1002/art.1780281206. [DOI] [PubMed] [Google Scholar]
- 39.James JA, Gross T, Scofield RH, Harley JB. Immunoglobulin epitope spreading and autoimmune disease after peptide immunization: Sm B/B′-derived PPPGMRPP and PPPGIRGP induce spliceosome autoimmunity. Journal of Experimental Medicine. 1995;181(2):453–461. doi: 10.1084/jem.181.2.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McClain MT, Heinlen LD, Dennis GJ, Roebuck J, Harley JB, James JA. Early events in lupus humoral autoimmunity suggest initiation through molecular mimicry. Nature Medicine. 2005;11(1):85–89. doi: 10.1038/nm1167. [DOI] [PubMed] [Google Scholar]
- 41.Cohen-Solal J, Diamond B. Lessons from an anti-DNA autoantibody. Molecular Immunology. 2011 Jan 6; doi: 10.1016/j.molimm.2010.12.003. (Epub ahead of printing) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Deshmukh US, Gaskin F, Lewis JE, Kannapell CC, Fu SM. Mechanisms of autoantibody diversification to SLE-related autoantigens. Annals of the New York Academy of Sciences. 2003;987:91–98. doi: 10.1111/j.1749-6632.2003.tb06036.x. [DOI] [PubMed] [Google Scholar]
- 43.Paisansinsup T, Deshmukh US, Chowdhary VR, Luthra HS, Fu SM, David CS. HLA class II influences the immune response and antibody diversification to Ro60/Sjögren’s syndrome-A: heightened antibody responses and epitope spreading to Ro52 in mice expressing HLA-DR. molecules. Journal of Immunology. 2002;168(11):5876–5584. doi: 10.4049/jimmunol.168.11.5876. [DOI] [PubMed] [Google Scholar]
- 44.Jiang C, Deshmukh US, Gaskin F, Bagavan H, Hanson J, David CS, Fu SM. Differential responses to Smith D autoantigen by mice with HLA-DR and HLA-DQ transgenes: dominant responses by HLA-DR3 transgenic mice with diversification of autoantibodies to small nuclear ribonucleoprotein, double-stranded DNA, and nuclear antigens. Journal of Immunology. 2010;184(2):1085–1091. 2010. doi: 10.4049/jimmunol.0902670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Reichlin M, Harley JB, Lockshin MD. Serologic studies of monozygotic twins with systemic lupus erythematosus. Arthritis & Rheumatism. 1992;35(4):457–464. doi: 10.1002/art.1780350416. [DOI] [PubMed] [Google Scholar]
- 46.Cui Z, Zhao MH, Segelmark M, Hellmark T. Natural autoantibodies to myeloperoxidase, proteinase 3, and the glomerular basement membrane are present in normal individuals. Kidney International. 2010;78(6):590–597. doi: 10.1038/ki.2010.198. Retrieved from http://ovidsp.ovid.com/ovidweb.cgi?T=JS&CSC=Y&NEWS=N&PAGE=fulltext&D=medl&AN=20592714. [DOI] [PubMed]
- 47.Hoffman RW, Takeda Y, Sharp GC, Lee DR, Hill DL, Kaneoka H, et al. Human T cell clones reactive against U-small nucleoprotein Autoantigens from connective tissue disease patients and healthy individuals. Journal of Immunology. 1993;151(11):6460–6469. [PubMed] [Google Scholar]
- 48.Wucherpfennig KW, Zhang J, Witek C, Matsui M, Modabber Y, Ota K, et al. Clonal expansion and persistence of human T cells specific for an immunodominant myelin basic protein peptide. Journal of Immunology. 1994;152(11):5581–5592. [PubMed] [Google Scholar]
- 49.Wucherpfennig KW, Allen PM, Celada F, Cohen IR, De Boer R, Garcia KC, et al. Polyspecificity of T cell and B cell receptor recognition. Seminars in Immunology. 2007;19(4):216–224. doi: 10.1016/j.smim.2007.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Deshmukh US, Sim D, Rajagopalan G, David C, Gaskin F, Fu SM. HLA-DR3 Restricted T Cell Epitope Mimics of a Lupus-associated autoantigen can Initiate Autoimmune Responses. Arthritis & Rheumatism 58. 2008;1926:S872. [Google Scholar]
- 51.Meroni PL, Shoenfeld Y. Systemic lupus erythematosus and the SLE galaxy. Autoimmunity Reviews. 2010;10:1–2. doi: 10.1016/j.autrev.2010.09.006. [DOI] [PubMed] [Google Scholar]
- 52.Gualtierotti R, Biggioggero M, Penatti AE, Meroni PL. Updating on the pathogenesis of systemic lupus erythematosus. Autoimmunity Reviews. 2010;10:3–7. doi: 10.1016/j.autrev.2010.09.007. [DOI] [PubMed] [Google Scholar]
- 53.Pisetsky DS, Ullal AJ. The blood nucleome in the pathogenesis of SLE. Autoimmunity Reviews. 2010;10:35–37. doi: 10.1016/j.autrev.2010.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Munoz LE, Janko C, Schulze C, Schorn C, Sarter K, Schett G, Herrmann M. Autoimmunity and chronic inflammation - Two clearance-related steps in the etiopathogenesis of SLE. Autoimmunity Reviews. 2010;10:38–42. doi: 10.1016/j.autrev.2010.08.015. [DOI] [PubMed] [Google Scholar]
- 55.Youinou P. Haralampos M. Moutsopoulos: A lifetime in autoimmunity. Journal of Autoimmunity. 2010;35:171–175. doi: 10.1016/j.jaut.2010.06.003. [DOI] [PubMed] [Google Scholar]
- 56.Kontaki E, Boumpas DT. Innate immunity in systemic lupus erythematosus: Sensing endogenous nucleic acids. Journal of Autoimmunity. 2010;35:206–211. doi: 10.1016/j.jaut.2010.06.009. [DOI] [PubMed] [Google Scholar]
- 57.Manoussakis MN, Spachidou MP, Maratheftis CI. Salivary epithelial cells from Sjogren’s syndrome patients are highly sensitive to anoikis induced by TLR-3 ligation. Journal of Autoimmunity. 2010;35:212–218. doi: 10.1016/j.jaut.2010.06.010. [DOI] [PubMed] [Google Scholar]
- 58.Vlachoyiannopoulos PG, Routsias JG. A novel mechanism of thrombosis in antiphospholipid antibody syndrome. Journal of Autoimmunity. 2010;35:248–255. doi: 10.1016/j.jaut.2010.06.015. [DOI] [PubMed] [Google Scholar]
- 59.Scheinecker C, Bonelli M, Smolen JS. Pathogenetic aspects of systemic lupus erythematosus with an emphasis on regulatory T cells. Journal of Autoimmunity. 2010;35:269–275. doi: 10.1016/j.jaut.2010.06.018. [DOI] [PubMed] [Google Scholar]
- 60.Youinou P, Pers JO. The international symposium on Sjogren’s syndrome in Brest: The “top of the tops” at the “tip of the tips”. Autoimmunity Reviews. 2010;9:589–590. doi: 10.1016/j.autrev.2010.05.014. [DOI] [PubMed] [Google Scholar]
- 61.Varin MM, Le Pottier L, Youinou P, Saulep D, Mackay F, Pers JO. B-cell tolerance breakdown in Sjogren’s syndrome: Focus on BAFF. Autoimmunity Reviews. 2010;9:604–608. doi: 10.1016/j.autrev.2010.05.006. [DOI] [PubMed] [Google Scholar]
- 62.Lu Q, Renaudineau Y, Cha S, Ilei G, Brooks WH, Selmi C, Tzioufas A, Pers JO, Bombardieri S, Gershwin ME, Gay S, Youinou P. Epigenetics in autoimmune disorders: Highlights of the 10th Sjogren’s syndrome symposium. Autoimmunity Reviews. 2010;9:627–630. doi: 10.1016/j.autrev.2010.05.011. [DOI] [PubMed] [Google Scholar]
- 63.Youinou P, Pers JO, Gershwin ME, Shoenfeld Y. Geo-epidemiology and autoimmunity. Journal of Autoimmunity. 2010;34:J163–J167. doi: 10.1016/j.jaut.2009.12.005. [DOI] [PubMed] [Google Scholar]
- 64.Shapira Y, Agmon-Levin N, Shoenfeld Y. Defining and analyzing geoepidemiology and human autoimmunity. Journal of Autoimmunity. 2010;34:J168–J177. doi: 10.1016/j.jaut.2009.11.018. [DOI] [PubMed] [Google Scholar]
- 65.Hoffmann MH, trembleau S, Muller S, Steiner G. Nucleic acid-associated autoantigens: Pathogenic involvement and therapeutic potential. Journal of Autoimmunity. 2010;34:J178–J206. doi: 10.1016/j.jaut.2009.11.013. [DOI] [PubMed] [Google Scholar]
- 66.Chang C, Gershwin ME. Drugs and autoimmunity - A contemporary review and mechanistic approach. Journal of Autoimmunity. 2010;34:J266–275. doi: 10.1016/j.jaut.2009.11.012. [DOI] [PubMed] [Google Scholar]
- 67.Borchers AT, Naguwa SM, Keen CL, Gershwin ME. The implications of autoimmunity and pregnancy. Journal of Autoimmunity. 2010;34:J287–J299. doi: 10.1016/j.jaut.2009.11.015. [DOI] [PubMed] [Google Scholar]
- 68.Rubstov AV, Rubstova K, Kappler JW, Marrack P. Genetic and hormonal factors in female-biased autoimmunity. Autoimmunity Reviews. 2010;9:494–498. doi: 10.1016/j.autrev.2010.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ansari AA, Gershwin ME. Navigating the passage between Charybdis and Scylla: Recognizing the achievements of Noel Rose. Journal of Autoimmunity. 2009;33:165–169. doi: 10.1016/j.jaut.2009.07.007. [DOI] [PubMed] [Google Scholar]
- 70.Mackay IR. Clustering and commonalities among autoimmune diseases. Journal of Autoimmunity. 2009;33:170–177. doi: 10.1016/j.jaut.2009.09.006. [DOI] [PubMed] [Google Scholar]