

Abstract
Background
Examining the distribution of variation has proven an extremely profitable technique in the effort to identify sequences of biological significance. Most approaches in the field, however, evaluate only the conserved portions of sequences – ignoring the biological significance of sequence differences. A suite of sophisticated likelihood based statistical models from the field of molecular evolution provides the basis for extracting the information from the full distribution of sequence variation. The number of different problems to which phylogeny-based maximum likelihood calculations can be applied is extensive. Available software packages that can perform likelihood calculations suffer from a lack of flexibility and scalability, or employ error-prone approaches to model parameterisation.
Results
Here we describe the implementation of PyEvolve, a toolkit for the application of existing, and development of new, statistical methods for molecular evolution. We present the object architecture and design schema of PyEvolve, which includes an adaptable multi-level parallelisation schema. The approach for defining new methods is illustrated by implementing a novel dinucleotide model of substitution that includes a parameter for mutation of methylated CpG's, which required 8 lines of standard Python code to define. Benchmarking was performed using either a dinucleotide or codon substitution model applied to an alignment of BRCA1 sequences from 20 mammals, or a 10 species subset. Up to five-fold parallel performance gains over serial were recorded. Compared to leading alternative software, PyEvolve exhibited significantly better real world performance for parameter rich models with a large data set, reducing the time required for optimisation from ~10 days to ~6 hours.
Conclusion
PyEvolve provides flexible functionality that can be used either for statistical modelling of molecular evolution, or the development of new methods in the field. The toolkit can be used interactively or by writing and executing scripts. The toolkit uses efficient processes for specifying the parameterisation of statistical models, and implements numerous optimisations that make highly parameter rich likelihood functions solvable within hours on multi-cpu hardware. PyEvolve can be readily adapted in response to changing computational demands and hardware configurations to maximise performance. PyEvolve is released under the GPL and can be downloaded from http://cbis.anu.edu.au/software.
Background
Examining the distribution of variation between related biological sequences has proven an extremely profitable technique in the effort to identify sequences of biological significance. The motivating rationale has been that sequence conservation implies biological significance. Yet, as all biologists know, evolution by natural selection proceeds by sequence change and therefore it is the differences in genome sequence that are responsible for different organismal phenotypes. Analytical techniques that consider the full distribution of variation will therefore have greater power to identify sequences of biological significance.
A suite of sophisticated statistical models from the field of molecular evolution provides the basis for extracting the information from the full distribution of sequence variation [for a review see [1]]. The two key mathematical aspects of these statistical models are the representation of sequence change over some time period as a Markov process and the formulation for calculating the likelihood of a multiple-sequence alignment given a phylogenetic tree [2]. We restrict ourselves here to a simple description of these two procedures, and refer the interested reader to a review by Lio and Whelan [3] and the original paper of Felsenstein [2].
Probabilistic models of substitution are central to our ability to dissect the forces responsible for the molecular changes distinguishing different sequences. The probability a sequence motif changes (or remains the same) can be parameterized according to biochemical attributes of the motifs involved. This Markov process is represented as a matrix of average relative rates of instantaneous change and the matrix of substitution probabilities for a given time period is determined by a matrix exponentiation procedure. For the more complex substitution models, explicit solutions to this matrix exponentiation are not possible and approximation techniques [4] are used instead.
The likelihood framework provides the basis for statistical hypothesis testing using molecular sequence data. The likelihood of an alignment is the probability of observing that alignment given a model of sequence evolution and a phylogenetic tree relating the sequences in the alignment. This likelihood is calculated using the "pruning" algorithm [2]. The explanatory power of different hypotheses, which differ in terms of their parameterisation of the process of molecular evolution, can be formally compared using a likelihood-ratio (LR) test. In many cases, the LR test statistic can be evaluated using the χ2 distribution [for examples of such models see [5,6]]. In other cases, such as for non-nested hypotheses or for models that violate assumptions of the likelihood model, parametric bootstrapping is the preferred technique [7,8]. Parametric bootstrapping can also be used for estimating parameter confidence intervals.
There is a diverse array of problems to which phylogeny-based maximum-likelihood software can be applied. Even within the scope of conventional hypothesis testing, numerous model parameterisations can be developed to dissect biological processes ranging from detecting natural selection [9], testing the molecular clock [10], detecting gene recombination events [11], or phylogenetic reconstruction [12]. Each of the different use cases presents distinct challenges to the end-user in terms of interaction, and distinct challenges to the developer in terms of maximising speed.
As the number and taxonomic diversity of DNA sequences increases, propelled by genomics technology, so too does the imperative for developing software that can both scale with the problem and maximise the information gained from the data. The groundbreaking package in statistical modelling of molecular evolution was PAML [13]. While the suite of models calculable by PAML is extensive, it doesn't scale well as it only runs on single processors. Additionally, the software is sufficiently complex in design as to inhibit its extension by other developers. Control of model parameterisations is achieved by a settings file that uses integers to differentiate between alternative model configurations, and in some instances, by manual editing of the input phylogeny. Both of these approaches are particularly prone to errors, even for experienced users. Defining different statistical models by numbers requires frequent referral to the manual and example files, while it is extremely easy to make mistakes editing large phylogenies in the Newick format. Additionally, changing the bounds for a parameter requires modifying the source code and recompiling, which has obvious disadvantages.
The HYPHY package http://www.hyphy.org has many features, including a GUI, and a batch language for implementing non-standard analyses. The latter approach is intriguing but aspects of its' implementation prove limiting. Since the batch language is unique to the package, users must learn a highly specialised programming language, with very restricted utility. Hence, such simple matters as unsupported data formats quickly become bottlenecks. The ability to integrate HYPHY into automated analysis pipelines is not trivial as it involves moving between languages. Other limitations of HYPHY's batch file language are the mechanism for defining novel models of substitution, the restriction that the models must be reversible, and a cumbersome approach to identifying sub-tree's for modelling purposes. Moreover, while components of this program have been parallelised, the parallelisation is "fixed" and can therefore not be tailored by the user to suite a particular problem.
PyEvolve should prove useful to both modellers and developers. It has been designed to be adaptable to an extensive array of use cases and to overcome the perceived shortcomings of other packages. Here we present a summary of the toolkit's features and illustrate it's utility by applying it to develop a novel model of dinucleotide substitution, and perform a hypothesis test to evaluate the new model. We further demonstrate the usage and performance of PyEvolve's multi-level parallelisation schema.
Implementation
We aimed to develop a toolkit for phylogeny-based maximum-likelihood modelling of molecular evolution that is:
• Extensible, fast and scalable
• Straightforward to implement most existing methods, and simplifies the development of novel methods
• Straightforward to learn by novice biologists or by developers
• Free and can be modified without restriction
To meet these objectives, PyEvolve is implemented as a Python module with the most computationally intensive algorithms written in C/C++ and is released under the GPL.
There are several reasons behind the choice of Python. Principal among them is Python's utility as a programmable scripting language. By selecting a popular language as the principal interface to the toolkit, learning the toolkit for those familiar with Python will be trivial, and for others the general utility of the language means it can be applied to many other problems. Development related factors also affected our choice. Python is designed to be able to readily connect with external modules written in other languages such as C/C++ or Fortran with little fuss. This ability to glue pieces of code from different languages increases choice for selecting from existing code for critical numerical routines (e.g. optimisation). The syntactical clarity of Python, the ready availability of good documentation suitable for novices [such as the open book project, [14]], and the self documenting capabilities of Python code all contribute to lowering the barrier to use of the toolkit. Together these attributes significantly reduced the amount of work required by us to generate a toolkit useful to an end-user group with diverse interests and expertise.
Object architecture
The top-level objects of PyEvolve and their relationships are illustrated in Figure 1. Briefly, these objects and their functions are:
Figure 1.
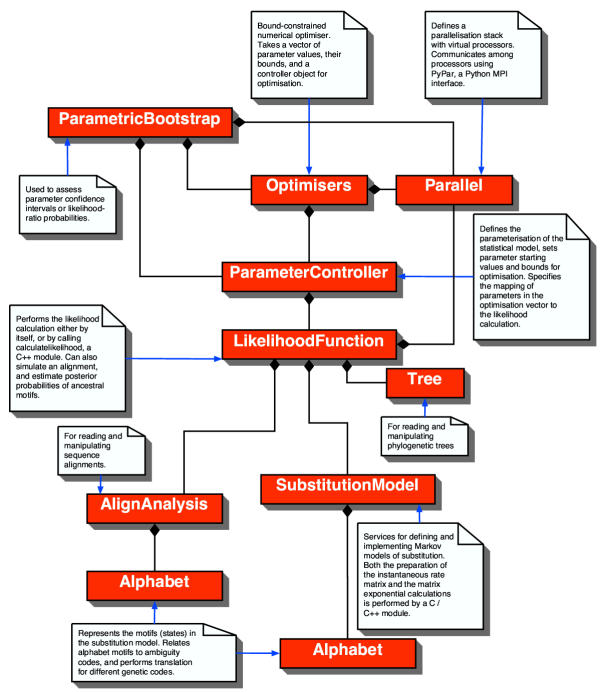
Relationship among the PyEvolve components The boxes represent the objects (classes) that make up PyEvolve. The filled diamonds, as specified by the Unified Modelling language specification, indicate the object at which the diamonds end has the connected objects as essential components.
Parallel
Defines a parallelisation stack with virtual processors. Communicates among processors using PyPar, a Python MPI toolkit.
Bootstrapping
A parametric bootstrapping module. Can be used to assess parameter confidence intervals or probabilities. In either case, it requires users to provide a reference to function(s) that construct controller object(s). Runs in parallel when multiple cpu's are available.
Optimisers
A generic interface to bound-constrained numerical optimisers. Takes a vector of floating point values for optimisation, corresponding vectors of bounds defining the range of acceptable values, and a ParameterController object. Presently, a simulated annealing optimiser is provided.
ParameterController
Defines the parameterisation of the statistical model, sets parameter starting values and bounds for optimisation. Specifies the mapping of parameters in the optimisation vector to the likelihood calculation.
LikelihoodFunction
Performs the likelihood calculation by calling the calculatelikelihood module (which is written in C++). The calculatelikelihood module also calculates the matrix of substitution probabilities, unless overridden. Can also simulate an alignment, and estimate posterior probabilities of ancestral motifs [15].
Tree
For reading and manipulating phylogenetic trees. Tree also provides methods for extracting sub-tree's, and for identifying portions of a tree using the names of tips.
AlignAnalysis
For reading and manipulating sequence alignments.
SubstitutionModel
Provides services for defining and implementing Markov process models of substitution. Both the preparation of the instantaneous average relative rate matrix and the matrix exponential calculations can be performed by calc_psubs, a module written in C / C++. In the fastest implementation, the instantaneous matrix and matrix exponentiation routines are in-lined with the calculatelikelihood C++ module. In this case, SubstitutionModel is relegated to defining the configuration of the substitution model.
Alphabet
Represents the motifs (states) in the substitution model. Relates alphabet motifs to IUPAC ambiguity codes, and performs translation for different genetic codes.
Serial performance innovations
At the heart of the computational challenge is the poor scalability of the pruning and matrix exponentiation algorithms. The order of Felsenstein's pruning algorithm is ~O(NA) where A is the number of motifs in the sequence alphabet (e.g. 4 for DNA, 20 for amino-acids) and N is the number of sequences. The matrix exponentiation, which is done by eigenvalue decomposition using the same code as that used by PAML [13], also scales poorly (> ~ O(N2)). Given the computationally intensive nature of these algorithms they have been implemented in C / C++.
One way efficiencies can be gained is by reducing the number of times the algorithm is used. When the sequences descended from a node are identical at several sites in the alignment, the partial likelihoods for that node of the tree will also be identical for those sites. This has commonly been exploited for the special case in which the node is the root node, in other words all sequences are identical between the sites. In this conventional case the likelihood is calculated only for one instance of a site-pattern and the site-patterns' log-likelihood is then multiplied by its' frequency of occurrence. PyEvolve implements an advanced site-pattern algorithm whereby the data are pre-processed to identify node-specific site-patterns that don't fall within the site-pattern of a higher node (such as the root). The partial likelihoods calculated for the first instance of a node specific site-pattern are reused for all other cases of that site-pattern.
Another optimisation strategy involves storing the results of previous calculations. Many numerical optimisers change one parameter at a time in the vector to be optimised for a function. For a parameter that has a local (single branch) scope, then only the matrix exponentiation for that branch need be recomputed. PyEvolve takes advantage of such optimisers by storing the results of each exponentiation. If the optimiser returns a parameter vector with parameters relevant to a subset of branches being unchanged, the stored probability of substitution matrices are used instead. If parameters for a branch remain unchanged twice in a row, a second level backup of that matrix is created. This second-level becomes most valuable in the later stages of optimisation when most changes to the parameter set are sub-optimal. The performance improvement conferred by this approach is striking for a model in which most parameters are local (e.g. a model with branch lengths and local substitution model parameters). In this instance, the majority of likelihood function evaluations involve recalculating the probabilities of substitution for a single branch.
PyEvolve has also been modified to store previous partial likelihoods for reuse, taking advantage again of the optimiser behaviour. The effect of this approach is that the partial likelihoods need only be determined for nodes affected by a parameter change, and their parents. The optimisation vector has been ordered so as to ensure that the minimum number of nodes need recalculating. The vector is in a natural tree traverse order, where the descendants of each node are added before the node.
The memory impact of storing previous results is minimal for both strategies. Both the partial likelihood and probability of substitution matrix reuse algorithms minimise the overhead for backing up by using the same memory space. The partial likelihood reuse algorithm has also been implemented with two versions that differ in memory usage. The default version keeps all the tree tips and nodes for the fastest and simplest reuse of previous partial likelihoods. The second version only keeps the nodes, thus requiring only half the memory. The performance hit is low because the partial likelihoods are considerably quicker to recalculate for tips than nodes. This algorithm may be of greatest value with bigger trees as the memory saving becomes more important. The reuse algorithm can be specified using an argument of the LikelihoodFunction constructor. The higher memory algorithm is on by default.
An optional efficiency related to pruning in PyEvolve concerns the number of motifs in the alphabet. As the pruning algorithm is most sensitive to the number of motifs in the alphabet, reducing these can provide enormous benefits. This efficiency can be trivially achieved in PyEvolve by reducing the number of motifs in the alphabet to only those observed in the data, and constructing the SubstitutionModel with the revised alphabet.
Parallel performance innovations
With the availability of multi-processor clusters of computers becoming commonplace, performance gains can be achieved by parallelising portions of the computations. The limits to the maximum gain in efficiency are governed by the proportion of the algorithm that is strictly serial (as suggested by Amdahl's Law). Accordingly, the general principle for parallelisation is to minimise the amount of serial computation. A secondary, but similarly important, impact on the performance of a parallelised program is the time taken up by communication between nodes in the compute cluster. The combined impact of the diversity of possible use cases and the heterogeneity in hardware performance precludes a simple fixed parallelisation scheme.
Within a single model there are three obviously parallelisable stages. At the highest level, portions of the numerical optimisation procedure may be parallelised. The details of this parallelisation effort differ between procedures. For instance, optimisers that use a finite difference method for estimating the gradient of the likelihood function can be trivially parallelised at the finite differences step. While this provides a significant performance boost, these optimisers can suffer from a tendency to find local maxima rather than the global maxima being sought. This may require the optimiser to be started multiple times, each from a different position in the parameter space, significantly reducing real world performance. Although global optimisation techniques tend to be slower and, at least for the simulated annealing procedure implemented in PyEvolve, benefit less from parallelisation, global procedures can be more efficient [16]. For these reasons we have developed a parallelised version of the bound-constrained simulated annealing algorithm [16]. This level of parallelisation will be referred to as the SA level.
The two other obvious opportunities for parallelisation are the calculation of the probabilities of substitution by matrix exponentiation and the calculation of the log-likelihood for columns in the alignment. We have not implemented the former because the overhead for communicating an array of 3721 double precision floating point values, as would be required for a codon substitution model, would negate the benefit of parallelising the matrix exponentiation on most hardware. The likelihood calculation has been parallelised at the level of the alignment, which will be referred to as the LF level parallelisation.
In order to permit adaptation of the mixture of parallelised routines to different tasks we have implemented a flexible multi-level parallelisation schema in PyEvolve. The Parallel component of PyEvolve uses the PyPar interface to MPI [17]. The basic principle of MPI/PyPar parallelisation is that a copy of the same program runs on all processors, but that each process uses knowledge that the other processes exist to selectively do less work. This works because each process knows how many processors there are in total, and their ID (or rank) within this number. Usually the final result is collected onto processor 0 and then at the beginning of the next calculation the starting state is sent out to all the other processes.
PyEvolve's schema allows processors to be grouped into a virtual processor, and provides methods for stacking these virtual processors on top of one another. The parallelisation stack created to perform the hypothesis test described below is illustrated in Figure 2. Each level of the parallelisation stack is made up of a number of virtual processors that in turn correspond to the same, or a greater, number of actual cpu's. The following rules must be satisfied in order to define a multi-level parallelisation stack – at each level, the maximum number of processors that make up the virtual cpu (the subgroup size) must be a divisor of the number of cpu's in the level above. At the top level, the subgroup size must be a divisor of the total number of cpu's assigned to the program.
Figure 2.
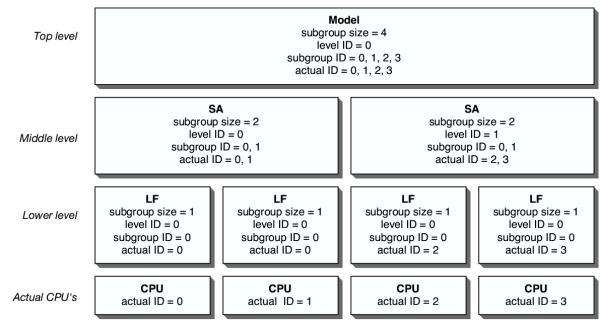
The 3-level parallelisation stack for a model Model – the null or alternative hypothesis parameterisations; SA – the simulated annealing parallelisation level; LF – the likelihood function parallelisation level; CPU – actual hardware cpu's; level ID – an identifier relative to the virtual cpu's at the specified level; subgroup ID – an identifier relative to the processors within a specified virtual processor at the specified level; actual ID – the standard MPI identifier for each real cpu.
Results
Defining a new substitution model
We illustrate how PyEvolve can be used to develop and implement new models of substitution by presenting a dinucleotide model of substitution that incorporates a term for mutation of the commonly methylated dinucleotide CpG. Defining a Markov model of substitution in PyEvolve involves defining a set of rules that govern the assignment of parameter values into the matrix of instantaneous change [for a more detailed explanation of the nature of Markov models in molecular evolution see [1,3]]. These rules correspond to Python functions that return True or False when comparing two sequence alphabet motifs.
The biochemical phenomenon we are interested in is that a methylated C (which occur predominantly at CpG dinucleotides) mutates to a T at a higher rate than an unmethylated C. If the plus strand C is methylated, the CpG mutates to TpG, if it is on the negative strand CpG mutates to CpA. In Figure 3(a) is an 8 line Python function that returns True if a pair of dinucleotide motifs is either (CG and TG) or (CG and CA), False otherwise. To satisfy the reversibility assumption of the likelihood model, the function considers both possible orders in which the dinucleotides could be received. Having written this rule function, implementing the novel dinucleotide methylation model requires passing a reference to the function indexed by the parameter name that we wish to refer to it by. In the example in Figure 3(b), the SubstitutionModel object will then iterate over all possible combinations of dinucleotides, passing each dinucleotide pair to the two assigned rules – submodelobj. istransition and ismethylatedmutation. When either order of the CG/TG, CG/CA dinucleotide pairs is passed to the ismethylatedmutation function, it returns True resulting in the assignment of the parameter 'meth' to that position of the matrix of instantaneous change.
Figure 3.

Python code required to define a novel model of substitution (a) A function to specify a new substitution model rule. This function identifies dinucleotides that differ from each other by mutation of a methylated C. (b) The step to define a dinucleotide substitution model with terms for both transition and methylation induced changes.
Testing a hypothesis with parallelisation
The contribution of the additional methylation parameter to explaining the pattern of substitution observed in a data set is tested using a conventional LR test. This process involves constructing near identical ParameterController objects that differ only by their substitution models. The alternative hypothesis substitution model parameter rules include the ismethylatedmutation reference. Each controller object is then passed to an optimiser object that is run to determine the maximum-likelihood parameter estimates and maximum log-likelihoods, the latter statistics are then used to perform a conventional LR test using the χ2 approximation with 1 degree-of-freedom.
We seek to test the hypothesis that a parameter for methylated CpG mutations does not significantly improve the fit of our model. For this example we use a 38.6 kb non-coding region from Chimpanzee (AF190865) and Gorilla (AF190871) [18]. To remove the potential effect of repeat expansions, which derive from a different mutational process, on the results we masked all dinucleotide repeats greater than five prior to alignment with ClustalW 1.8 [19]. More detailed analyses of these data are reported elsewhere (Wakefield, Isaev and Huttley, in preparation). We parallelise the script at three levels: (1) the model; (2) the SA level; and (3) the LF level. The algorithm for setting up the parallelisation stack for a single model is illustrated in Figure 2. A python script that implements this example is available in the standard distribution of PyEvolve and also as supplementary materials [see Additional file 1]. In the example the top-level subgroup size is 4, the mid-level subgroup size is 2, and the low-level subgroup size is 1. This structure defines two virtual cpu's at the top (each made up of 4 actual cpu's) which run the different models. The script descends to the mid-level because the simulated annealing optimiser assumes it has been initialised at the level at which it is to be run. Note that taking advantage of the likelihood function parallelisation does not require explicit stack traversal statements by the user as the presence of a lower-level is automatically detected. In order to perform the LR test the stack must be ascended to the top level and the results communicated between the two processors. The result of the LR test is highly significant (P < 10-53).
Benchmarking
To illustrate the performance improvements arising from parallelisation we consider different parallelisation stack configurations and numbers of total cpu's for two different substitution models. The parallelisation levels were SA, LF or both SA and LF (referred to as BOTH). We use a dinucleotide model with only a transition transversion parameter, and the codon model of Goldman and Yang [20] applied to an alignment of the gene BRCA1 for 20 mammals [6]. Species abbreviations, scientific names and accession numbers are: Anteater Tamandua tetradactyla AF284001; Chimpanzee Pan troglodytes AF207822; FlyingLem Cynocephalus variegatus AF019081; FlyingSqu Glaucomys volans AF284003; Galago Otolemur crassicaudatus AF019080; Gorilla Gorilla gorilla AF019076; HairyArma Chaetophractus villosus AF284000; Hedgehog Erinaceus europaeus AF284008; HowlerMon Alouatta seniculus AF019079; Human Homo sapiens NM_007306; Jackrabbit Lepus capensis AF284005; Mole Scalopus aquaticus AF284007; Mouse Mus musculus MMU36475; NineBande Dasypus novemcinctus AF283999; OldWorld Hystrix africaeaustralis AF284004; Orangutan Pongo pygmaeus AF019077; Rat Rattus norvegicus NM_012514.1; Rhesus Macaca mulatta AF019078; Sloth Bradypus tridactylus AF284002; TreeShrew Tupaia tana AF284006. Either 10 or 20 sequences are used. The 10 species data set consisted of FlyingSqu, Galago, Gorilla, Hedgehog, HowlerMon, Jackrabbit, Mole, Mouse, Oldworld, and Rat. The alignment was 2883 nucleotides (961 codons) long and is available as part of PyEvolve's distribution. All parameters, aside from branch lengths, were treated as global across the tree (i.e. identical for all branches). We examine the parallel performance of the SA level, the LF level and the two levels jointly. We used the Australian Partnership for Advanced Computing linux cluster. The cluster consisted of single cpu Dell Precision 350's, connected by a gigabit ethernet switch. More details of the hardware and cluster configuration are available at http://nf.apac.edu.au.
The results of performance testing (Table 1) indicate that both the serial optimisations and parallelisation efforts had significant impacts. The difference between time per lfe (likelihood function evaluation) for the 10 and 20 species trees ranged from -7% to 12% of the 10 species time. The decrease in time for 20 taxa probably reflects increased identity in portions of the tree by inclusion of additional taxa. The differences between the dinucleotide and codon models reflect the increasing proportion of time taken for matrix exponentiation by the codon model. These differences indicate the considerable savings achieved by the serial optimisations. Parallelisation gains for 16 cpu's ranged from a 3.1 to 4.5 fold increase over the single cpu performance for the codon model and 3.8 to 5.2 for the dinucleotide model. For both substitution models the typical order of level gains achieved were BOTH > LF > SA for a given number of total cpu's. Parallelisation gains were consistently less pronounced for the larger data set with the codon substitution model, while the dinucleotide model showed more exceptions to this relationship. For the codon model the benefits from the SA vs. LF level depended on the number of taxa, with SA being marginally better for the larger number of sequences. The LF level showed a more consistent advantage for the dinucleotide model. The multi-level parallelisation gave the best performance for both substitution models, providing another 1 × serial fold improved performance over either level alone for the codon model and 1/2 × serial fold improvement for the dinucleotide model.
Table 1.
PyEvolve benchmarking. Time taken was estimated as time for optimisation. Number of runs per condition ranged from 1 to 5. 1Model – See text for details of the codon and dinuc substitution models; 2Levels – indicates whether Simulated Annealing (SA), Likelihood Function (LF) or BOTH parallelisation levels were used; 3Parallel degree refers to the number of virtual cpu's at the 4SA or LF levels (for the LF level, this is defined per SA virtual cpu); 5the number of likelihood function evaluations made during the optimisation for 610 or 20 sequences, expressed in thousands. See text for details of the data and hardware used.
| Model 1 | Levels 2 | Total cpus | Parallel degree 3 | lfe (1000's) 5 | Total Time (minutes) | Time (seconds) per 1000 lfe | ||||
| SA 4 | LF 4 | 10 6 | 20 6 | 10 | 20 | 10 | 20 | |||
| codon | Serial | 1 | 1 | 1 | 56 | 121 | 124 | 269 | 133.06 | 133.65 |
| LF | 2 | 1 | 2 | 56 | 121 | 81 | 182 | 86.49 | 90.16 | |
| 4 | 1 | 4 | 56 | 122 | 55 | 130 | 58.69 | 63.78 | ||
| 8 | 1 | 8 | 57 | 122 | 41 | 100 | 43.99 | 49.02 | ||
| 16 | 1 | 16 | 57 | 120 | 35 | 82 | 36.52 | 41.02 | ||
| SA | 2 | 2 | 1 | 57 | 122 | 85 | 178 | 89.40 | 87.22 | |
| 4 | 4 | 1 | 57 | 121 | 58 | 121 | 60.19 | 60.15 | ||
| 8 | 8 | 1 | 57 | 122 | 44 | 99 | 46.10 | 48.83 | ||
| 16 | 16 | 1 | 58 | 122 | 38 | 88 | 39.48 | 43.16 | ||
| BOTH | 4 | 2 | 2 | 57 | 125 | 56 | 125 | 59.39 | 60.03 | |
| 8 | 2 | 4 | 58 | 122 | 40 | 89 | 41.57 | 43.74 | ||
| 8 | 4 | 2 | 57 | 121 | 39 | 85 | 40.92 | 42.37 | ||
| 16 | 2 | 8 | 56 | 121 | 30 | 69 | 32.28 | 34.47 | ||
| 16 | 4 | 4 | 58 | 121 | 28 | 63 | 29.31 | 31.27 | ||
| 16 | 8 | 2 | 57 | 121 | 31 | 71 | 32.70 | 35.30 | ||
| dinuc | Serial | 1 | 1 | 1 | 54 | 119 | 17 | 37 | 19.22 | 18.47 |
| LF | 2 | 1 | 2 | 54 | 119 | 11 | 24 | 12.59 | 12.29 | |
| 4 | 1 | 4 | 54 | 119 | 7 | 16 | 7.80 | 7.82 | ||
| 8 | 1 | 8 | 54 | 117 | 5 | 11 | 5.30 | 5.55 | ||
| 16 | 1 | 16 | 55 | 119 | 4 | 9 | 4.04 | 4.41 | ||
| SA | 2 | 2 | 1 | 53 | 118 | 11 | 23 | 12.19 | 11.51 | |
| 4 | 4 | 1 | 54 | 118 | 7 | 15 | 8.32 | 7.77 | ||
| 8 | 8 | 1 | 54 | 118 | 5 | 12 | 5.91 | 5.89 | ||
| 16 | 16 | 1 | 53 | 118 | 4 | 10 | 4.73 | 4.86 | ||
| BOTH | 4 | 2 | 2 | 54 | 118 | 7 | 15 | 8.07 | 7.69 | |
| 8 | 2 | 4 | 54 | 118 | 5 | 10 | 5.14 | 5.04 | ||
| 8 | 4 | 2 | 54 | 117 | 5 | 10 | 5.61 | 5.28 | ||
| 16 | 2 | 8 | 54 | 118 | 3 | 7 | 3.76 | 3.74 | ||
| 16 | 4 | 4 | 54 | 119 | 3 | 7 | 3.76 | 3.57 | ||
| 16 | 8 | 2 | 54 | 119 | 4 | 8 | 4.13 | 4.10 | ||
Discussion
PyEvolve's technique for constructing substitution models confers several advantages. Firstly, with very little coding it is possible to implement existing or generate entirely novel models of substitution. This approach reduces the opportunity for errors in defining existing models. By lowering the barrier for developing novel models of substitution, more biologists should be able to develop models that reflect their own knowledge. As illustrated by the methylation model presented here, such knowledge can lead to striking improvements in model fit. The overall performance of the software is largely independent of the code efficiency used to define these model rules.
There are two measures that we have used to evaluate PyEvolve's performance: (1) the time taken to calculate the likelihood of an alignment measured in terms of 1000 lfe; (2) the real world performance measured as the time taken to maximise the likelihood. For the first of these measures, PyEvolve was faster than PAML. For instance, the time taken per 1000 lfe by PAML's codeml application implementing the Goldman and Yang [20] substitution model for the 20 sequence data set was ~326.36 seconds on the same hardware, which is ~2.4 to ~10.4 fold slower than the fastest single and multi- cpu performance of PyEvolve respectively. The real world performance of PyEvolve, however, was sometimes slower than that of PAML. codeml was able to optimise the likelihood function for these tests with as few as ~3500 lfe (using the original slower codeml optimiser) compared with ~125000 lfe required by PyEvolve's simulated annealing optimiser. Yet the real world performance edge of codeml implied by these test cases did not translate to a more parameter rich model and larger data set. For a 55 mammal species BRCA1 data set and a codon substitution model, PyEvolve running on 16 fast Pentium cpu's of the test hardware required ~6 hours to optimise the function (~30.5 seconds per 1000 lfe). In contrast, on the same data set codeml (using the fast optimiser option) has taken ~240 hours to optimise the same likelihood on faster hardware (Alpha 21264C cpu). A codeml run on the fast Pentium hardware was terminated at 12 hours.
The potential for attaining performance benefits with PyEvolve depends on the data and the model complexity. Although parallelisation benefits were typically largest for the smaller data set, the results are likely to be different for a data set with a different tree and levels of divergence. For instance, LF level parallel performance gains will be possible for a highly diverged set of sequences if the alignment is long. Users will therefore need to experiment with their own data, and can use the scripts we provide in the PyEvolve distribution [see Additional file 1] and the structure of Table 1 as the basis for establishing the fastest configuration for their analysis problems. There are some general rules, however, that can point to the most likely configuration to implement. Parameterisations that increase the number of global parameters will benefit most from the SA level parallelisation. Factors that increase the proportion of time spent in the pruning algorithm will benefit most from the LF level. Alphabets with a smaller numbers of motifs, such as a nucleotide alphabet, will benefit because the proportion of time taken by the matrix exponentiation algorithm is small. A less balanced tree, which requires more partial likelihood recalculations for a local parameter change since the average number of nodes to the root increases, will also benefit from the LF level.
Clearly there is still scope for improving PyEvolve's performance. Given the dramatic effect of the optimisation algorithm on real world performance, implementing an optimiser similar to that used by PAML will have a major impact on time taken and should increase the number of problems for which PyEvolve is faster. Matrix exponentiation, although being done by a C module (using the same source code as that used by PAML), is a costly algorithm. Other techniques for approximating exponentials are possible, some of which have good performance for sparse matrices [4] suggesting they may be candidates to improve the performance of codon substitution models. Effort's to better integrate the advanced site-pattern algorithm with alignment level parallelisation should also reduce the cost of the latter, conferring benefits of this level to shorter and less-diverged alignments. Additionally, the vector processing units available on most modern cpu's can be exploited to speed up the considerable vector multiplication and addition operations performed in the pruning algorithm.
PyEvolve has numerous potential uses beyond the straightforward statistical modelling we have applied it to here. As we have shown, the toolkit can be readily applied to the development of new models of substitution. Another potential use is as the computational centrepiece for phylogenetic reconstruction methods. For instance, only a topology space search procedure, such as the step-wise or advanced step-wise addition algorithms [2,12], is required to deploy PyEvolve in a likelihood based phylogenetic reconstruction method. Given PyEvolve's parallelisation schema, implementing parallelised versions of such topology space search algorithms becomes straightforward.
Conclusion
PyEvolve provides flexible functionality that can be used either for statistical analysis of sequence data, or the development of new methods in molecular evolution. Here we demonstrated the ease with which a novel model of dinucleotide substitution can be developed and tested. The toolkit implements a novel parallelisation schema, and objects within the toolkit that can take advantage of this schema, that allows the program to be adapted to suit a broad range of problems. PyEvolve performance scales well with increasingly complex data sets and models, with significantly faster performance than codeml for parameter rich models. The modular design ensures that modifications, such as inclusion of more efficient numerical optimisation techniques, should be straightforward. The toolkit also provides sequence alignment and phylogenetic tree manipulation tools that are of general utility.
Availability and requirements
Project name: PyEvolve
Project home page: http://cbis.anu.edu.au/software
Operating system(s): Platform independent
Programming language: Python, C, C++
Other requirements: Python 2.3 or higher
License: GPL
Any restrictions to use by non-academics: None
List of abbreviations used
BOTH – parallelised at both the LF and SA levels
LF – likelihood function parallelised at the alignment column level
lfe – likelihood function evaluation
LR – likelihood ratio
SA – parallelised simulated annealing numerical optimiser
Authors' contributions
AB was responsible for most of the software design and implementation. VV translated the Fortran implementation of the simulated annealing optimiser into Python. EL assisted with establishing the utility of PyPar for parallelisation, and wrote the disutils component for distribution. CL made contributions to the data and alphabet components. MJW contributed to the bootstrapping component. AI assisted with the mathematical aspects of the likelihood calculations. GAH wrote an initial prototype, contributed to aspects of design, oversaw and managed contributions to the project, designed and performed the benchmarking and wrote the manuscript. All authors read and approved the final manuscript.
Supplementary Material
PyEvolve distribution. Includes source code of the benchmarked version (0.8), installation script, documentation, example scripts and the code used to perform benchmarking. The latest version of the source code can be obtained either from the project home page or the corresponding author.
Acknowledgments
Acknowledgements
We thank Ziheng Yang who has given permission to include his source code for performing matrix exponentials. We acknowledge support provided by the Australian Partnership for Advanced Computing whose facilities we used for the benchmarking.
Contributor Information
Andrew Butterfield, Email: andrew.butterfield@anu.edu.au.
Vivek Vedagiri, Email: vvedagiri@helixense.com.
Edward Lang, Email: edward.lang@anu.edu.au.
Cath Lawrence, Email: cath.lawrence@anu.edu.au.
Matthew J Wakefield, Email: matthew.wakefield@anu.edu.au.
Alexander Isaev, Email: alexander.isaev@anu.edu.au.
Gavin A Huttley, Email: gavin.huttley@anu.edu.au.
References
- Whelan S, Lio P, Goldman N. Molecular phylogenetics: state-of-the-art methods for looking into the past. Trends Genet. 2001;17:262–272. doi: 10.1016/S0168-9525(01)02272-7. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. Evolutionary trees from DNA sequences: a maximum likelihood approach. J Mol Evol. 1981;17:368–376. doi: 10.1007/BF01734359. [DOI] [PubMed] [Google Scholar]
- Lio P, Goldman N. Models of molecular evolution and phylogeny. Genome Res. 1998;8:1233–1244. doi: 10.1101/gr.8.12.1233. [DOI] [PubMed] [Google Scholar]
- Moler C, Van Loan C. Dubious Ways to Compute the Exponential of a Matrix, Twenty-Five Years Later. SIAM Review. 2003;45:3–49. [Google Scholar]
- Messier W, Stewart CB. Episodic adaptive evolution of primate lysozymes. Nature. 1997;385:151–154. doi: 10.1038/385151a0. [DOI] [PubMed] [Google Scholar]
- Huttley GA, Easteal S, Southey MC, Giles GG, McCredie MRE, Hopper JL, Venter DJ. Adaptive evolution of the tumor suppressor BRCA1 in humans and chimpanzees. Nat Genet. 2000;24:410–413. doi: 10.1038/78092. [DOI] [PubMed] [Google Scholar]
- Hall P, Wilson SR. Two guidelines for bootstrap hypothesis testing. Biometrics. 1991;47:757–762. [Google Scholar]
- Goldman N. Statistical tests of models of DNA substitution. J Mol Evol. 1993;36:182–198. doi: 10.1007/BF00166252. [DOI] [PubMed] [Google Scholar]
- Yang Z. Likelihood ratio tests for detecting positive selection and application to primate lysozyme evolution. Mol Biol Evol. 1998;15:568–573. doi: 10.1093/oxfordjournals.molbev.a025957. [DOI] [PubMed] [Google Scholar]
- Muse SV, Gaut BS. A likelihood approach for comparing synonymous and nonsynonymous nucleotide substitution rates, with application to the chloroplast genome. Mol Biol Evol. 1994;11:715–724. doi: 10.1093/oxfordjournals.molbev.a040152. [DOI] [PubMed] [Google Scholar]
- McGuire G, Wright F. TOPAL: recombination detection in DNA and protein sequences. Bioinformatics. 1998;14:219–220. doi: 10.1093/bioinformatics/14.2.219. [DOI] [PubMed] [Google Scholar]
- Wolf MJ, Easteal S, Kahn M, McKay BD, Jermiin LS. TrExML: a maximum-likelihood approach for extensive tree-space exploration. Bioinformatics. 2000;16:383–394. doi: 10.1093/bioinformatics/16.4.383. [DOI] [PubMed] [Google Scholar]
- Yang Z. PAML: a program package for phylogenetic analysis by maximum likelihood. Comput Appl Biosci. 1997;13:555–556. doi: 10.1093/bioinformatics/13.5.555. [DOI] [PubMed] [Google Scholar]
- Downey A, Elkner J, Meyers C. How to think like a computer scientist http://ibiblio.org/obp/thinkCSpy
- Yang Z, Kumar S, Nei M. A new method of inference of ancestral nucleotide and amino acid sequences. Genetics. 1995;141:1641–1650. doi: 10.1093/genetics/141.4.1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goffe WL, Ferrier GD, Rogers J. Global Optimization of Statistical Functions with Simulated Annealing. Journal of Econometrics. 1994;60:65–100. doi: 10.1016/0304-4076(94)90038-8. [DOI] [Google Scholar]
- Nielsen O. PyPAR – Parallel Python, efficient and scalable parallelism using the message passing interface (MPI) Version 164 edn. 2001. http://datamining.anu.edu.au/~ole/pypar
- Bohossian HB, Skaletsky H, Page DC. Unexpectedly similar rates of nucleotide substitution found in male and female hominids. Nature. 2000;406:622–625. doi: 10.1038/35020557. [DOI] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman N, Yang Z. A codon-based model of nucleotide substitution for protein-coding DNA sequences. Mol Biol Evol. 1994;11:725–736. doi: 10.1093/oxfordjournals.molbev.a040153. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PyEvolve distribution. Includes source code of the benchmarked version (0.8), installation script, documentation, example scripts and the code used to perform benchmarking. The latest version of the source code can be obtained either from the project home page or the corresponding author.