

Abstract
The HIV-1 accessory protein Nef is well known for its manipulation of host cell endosomal trafficking. By linking transmembrane proteins to endosomal coats, Nef removes them from the surface of infected cells. Modulation of MHC proteins leads to viral evasion of cellular adaptive immunity, whereas modulation of receptors for the HIV envelope glycoprotein, including CD4, enhances viral infectivity. The other HIV-1 accessory proteins, Vif, Vpr and Vpu, share a mechanism of action distinct from Nef in that each interacts with a multi-subunit ubiquitin ligase complex to target cellular proteins for proteosomal degradation. However, newly uncovered functions and mechanistic aspects of Vpu likely involve endosomal trafficking: these include counteraction of the innate antiviral activity of the cellular transmembrane protein BST-2 (tetherin), as well as the removal of the lipid-antigen presenting protein CD1d and the natural killer cell ligand NTB-A from the cell surface. This review focuses on how Nef and Vpu interfere with normal intracellular membrane trafficking to facilitate the spread and virulence of HIV-1.
Key words: HIV-1 accessory proteins, Vpu, Nef, host cell trafficking, modulation of surface expression of host cell receptors
Introduction
The chronic, persistent viral replication that characterizes untreated HIV-1 infection reflects the interplay of host defenses and viral countermeasures. To infect a new host, persistently replicate in that host for years, and spread to new individuals, HIV-1 must evade both innate defenses, including the so-called antiviral restriction factors and adaptive defenses, including antigen-specific cell-mediated immunity. To date, three restriction factors active against HIV-1 have been identified: APOBEC3 family proteins, TRIM5α and BST-2/tetherin. HIV-1 has evolved mechanisms to evade these factors, either by mutation of the targeted viral protein or by the acquisition of genes encoding specific proteins that antagonize them. Old world monkey TRIM5α causes premature uncoating of HIV-1 virion cores and consequently blocks the reverse transcription of the viral RNA genome in newly infected target cells. However, HIV-1 can replicate in human cells, because the viral capsid protein is not recognized efficiently by human TRIM5α.1,2 The cytosine deaminases APOBEC3G and F cause hypermutation of viral DNA, but the HIV-1 accessory protein Vif links these proteins to the ubiquitin-proteasome system, thus degrading and antagonizing them.3,4 The interferon-inducible protein BST-2/tetherin retains mature HIV-1 virions at the cell surface, restricting their release.5,6 The viral accessory protein Vpu antagonizes BST-2 by removing it from its site of action on the cell surface; this enables the efficient release of progeny virions from the infected cell.4,7–10 The HIV-1 accessory protein Nef is not yet known to antagonize a host cell restriction factor, although the Nef proteins of several SIVs, including SIVcpz, do counteract simian BST-2.11–13 HIV-1 Nef nevertheless modulates several cellular transmembrane proteins to the advantage of the virus. Nef contributes to viral evasion of acquired cellular immunity by modulating major histocompatibility complex (MHC) proteins.14–16 Nef also prevents cellular receptors for the viral envelope glycoprotein such as CD4 and CCR5 from interfering with viral assembly and release.17–19
The formation of a ternary complex between the viral accessory protein, the host restriction factor or other protein target, and a host co-factor (or several co-factors) is a common mechanism by which viral accessory genes neutralize host intrinsic and adaptive immunity. For example, counteraction of APOBEC3 proteins by Vif relies on the formation of a complex between Vif, APOBEC3, and a Cullin-5-containing ubiquitin ligase complex, with subsequent degradation of APOBEC3G by the proteosome. Similarly, the interaction between the viral accessory protein Vpr and the cullin 4A-DDB1 complex is presumably required for ubiquitination and degradation of an unidentified restriction factor. Like Vif and Vpr, Vpu and Nef can be considered as adaptors that link cellular targets to degradative pathways or to alternative trafficking pathways in the cell: Vpu by the induction of ubiquitination of its targets and Nef by linking its targets to endosomal coat proteins. However, unlike the other viral accessory genes, both Vpu and Nef are membrane-associated proteins. Moreover, unlike the known targets of the other viral accessory genes, the targets of both Vpu and Nef are transmembrane proteins. Here, we will focus on how Vpu and Nef manipulate intracellular trafficking of host cell transmembrane proteins to benefit viral spread and persistence.
Functions
Nef.
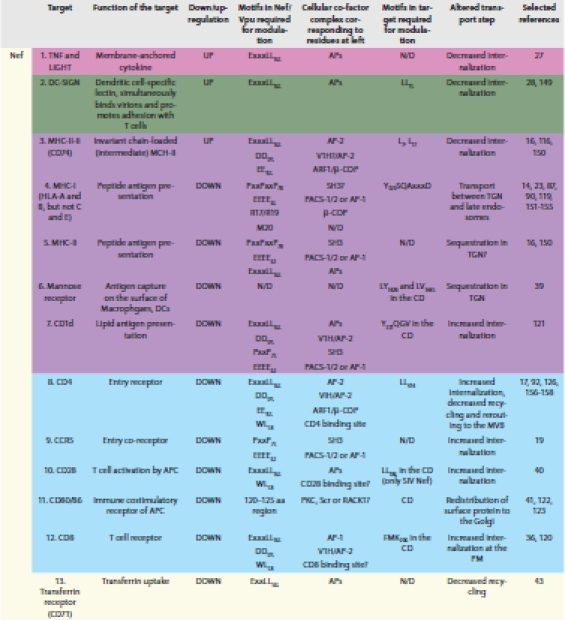
Nef is required for the rapid progression of primate lentiviral infections towards acquired immunodeficiency or AIDS.20,21 Nef is expressed early during the viral replication cycle with the general purpose of converting the host cell into an efficient virus production and dissemination factory. Nef achieves this in part by reorganizing cellular signaling and trafficking machineries, while protecting the infected cell from immune surveillance long enough to ensure robust production of progeny virions. Although it has no enzymatic activity, Nef is involved in an apparent plethora of interactions with host cell proteins and acts as a molecular adaptor, establishing linkages between cellular proteins. The multiple functions of HIV-1 Nef have been extensively reviewed elsewhere.22–26 Here, we will focus on functions that are related to the manipulation of intracellular trafficking. These functions involve modulation of the cell surface expression of specific receptors and the co-option of endosomal coat proteins and are listed in Table 1. In a few cases these receptors are upregulated to the cell surface, but in most cases they are downregulated from the cell surface by Nef.
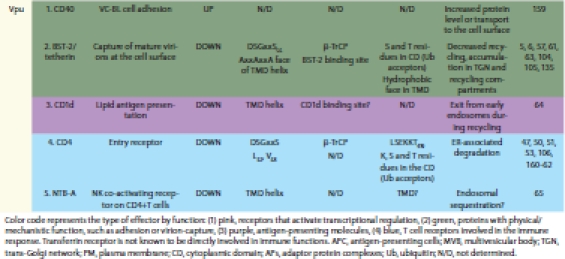
Table 1.
Effectors of Nef and Vpu that are modulated at the cell surface by altered intracellular membrane trafficking
 |
 |
Proteins upregulated by Nef include the membrane-anchored cytokines TNF and LIGHT, the dendritic cell-specific lectin DC-SIGN, and immature MHC-II bound to the invariant chain (MHC-II-Ii).16,27,28 TNF and LIGHT are members of the TNF family that activate NF-κB and c-Jun N-terminal kinase pathways. The immediate advantage of upregulating TNF and LIGHT at the surface of the infected cell for the virus is not clear, but because TNF is a potent activator of HIV transcription, this might accelerate virus spread.29 The lectin DC-SIGN binds HIV virions and promotes adhesion between dendritic cells and T cells. By increasing the cell surface expression of DC-SIGN, Nef facilitates the so-called trans-infection of T lymphocytes, in which viruses bound to dendritic cells are transferred to T cells.28,30 Upregulation of MHC-II-Ii and downregulation of mature MHC-II lead to defective MHC-II-mediated peptide presentation and consequently impaired stimulation of CD4+ T helper cells by HIV-infected cells.16,31
Downregulation of MHC-I (HLA-A and -B, but not -C and -E) protects HIV-infected cells from lysis by cytotoxic T lymphocytes and natural killer cells.14,32 The CD1d molecule presents lipid antigens to CD1d-restricted natural killer T cells (NKTs). Whether any lipid antigens are specific to HIV-1 is unclear, but the downregulation of CD1d from the surface of dendritic cells could uncouple the interaction between these cells and NKTs and affect the subsequent initiation of cellular immune responses. The downregulation of the virus attachment and entry receptors CD4, CCR5, and CXCR4 protects the infected cell from superinfection, which would lead to premature cell death and inefficient virus replication.19,33,34 These receptors can also interfere with the release of virus from the cell surface and impair the infectivity of released viruses due to their high binding affinity to Env.18,35 In addition to CD4, another T cell co-receptor, CD8, is downregulated by Nef, presumably to avoid immune activation.36 Macrophage mannose receptor can aid CD4-independent virus entry37 and is required for efficient antigen internalization and MHC-II loading.38 The removal of this receptor from the cell surface by Nef might impair superinfection, enhance virion release, and impair MHC-II-mediated antigen presentation.39 Downregulation of the T cell receptor CD28 and its ligands in antigen-presenting cells, CD80 and CD86, may cause impaired activation of naïve T cells.40,41 Finally, and perhaps not surprisingly given its many effects on trafficking, Nef alters the morphology of the endocytic compartment and the distribution of markers for membrane recycling compartments: Nef impairs recycling of transferrin receptor (TfR) and causes redistribution of the recycling center marker and regulatory GTPase Rab11.42,43 Thus, trafficking defects caused by Nef lead to a broad range of benefits for the virus: from facilitated virus replication to immune evasion.
Vpu.
Unlike Nef, HIV-1 Vpu is expressed late during the viral replication cycle, but like Nef, it plays an important role in viral virulence. The role of Vpu in vivo has been demonstrated using pigtailed macaques infected with SIV-HIV chimeric viruses (SHIVs) that express Vpu from HIV-1, since most SIVs lack a vpu gene. Infection of monkeys with SHIVs expressing no Vpu (or Vpu proteins with a scrambled transmembrane sequence or with inability to recruit a cellular β-TrCP-containing E3 ubiquitin ligase complex) yields virus levels in the blood that are at least 10-fold lower than infection with a SHIV encoding wild type Vpu; moreover, the animals infected with the vpu mutants have little or no loss of CD4-positive T lymphocytes, the hallmark of immunodeficiency in AIDS.44–46 These effects are presumably due to the ability of Vpu to facilitate virion release and to contribute to immune evasion. On the molecular level, these activities have been linked to the downregulation of the cell surface levels of antigen-presenting complexes, T cell receptors and BST-2/tetherin as discussed below and summarized in Table 1.
Similarly to Nef, Vpu reduces CD4 levels at the cell surface, in this case not by affecting endosomal trafficking but by targeting newly synthesized CD4 for ubiquitination and an ERAD-related pathway at the ER;47–51 this disables the delivery of CD4 to downstream compartments including the plasma membrane. The extraction of CD4 from the ER membrane by Vpu also ensures release of the viral envelope glycoprotein (Env) precursor from the ER, where it would otherwise be retained by CD4 due to the high binding affinity of these proteins for each other.52 The targeting of CD4 to the proteasome depends on the interaction of Vpu with the SCF-E3 ubiquitin ligase complex subunit β-TrCP, which is mediated by a canonical DSGxxS motif in the cytoplasmic domain of Vpu.53 Of the two isoforms of β-TrCP, β-TrCP2 is the one required for Vpu-dependent degradation of CD4.54 The interaction of Vpu with β-TrCP also causes the sequestration of this ubiquitin ligase complex from its natural substrates, resulting in the stabilization of certain cellular proteins. Such substrates include the regulators of transcription IκB and β-catenin.55,56 While IκB inhibits the transcription factor NFκB and limits the expression of anti-apoptotic proteins, β-catenin is involved in cell adhesion and interactions with nuclear transcription factors that initiate the expression of various oncogenes. The immediate advantage of such effects to the virus is not clear, especially since Nef upregulates NFκB while Vpu seems to do the opposite. These effects on protein stabilization by Vpu could be incidental byproducts of the interaction between Vpu and β-TrCP.
Vpu downregulates the interferon-inducible host restriction factor BST-2/tetherin from the cell surface by an incompletely described mechanism, which partially relies on Vpu-dependent ubiquitination.6,7,57–59 However, the degradation of BST-2 is not directly correlated with downregulation from the cell surface or with enhanced virion release in some experimental systems.57,60–63 This raises the possibility that rather than simply degrading BST-2 via the ubiquitin-proteasome system, Vpu might modulate the intracellular trafficking of BST-2 as discussed below. Two recent studies identified novel targets of Vpu: CD1d and NTB-A, both of which are downregulated from the cell surface.64,65 CD1d downregulation appears to be a function shared by Vpu and Nef.64 Vpu alone downregulates the NTB-A receptor from the surface of HIV-1-infected T cells; this inhibits the lysis of infected cells by natural killer cells.65 Neither the downregulation of CD1d nor of NTB-A involves degradation but seems instead to involve altered intracellular trafficking of these proteins.
Interestingly, whereas the effect of Nef on TfR suggests a generalized perturbation of traffic within endosomal pathways, Vpu does not affect TfR and thus alters endosomal traffic more specifically.64,66 However, Vpu does delay secretion along the biosynthetic pathway, and this effect depends on the DSGxxS motif.66 This mechanism might be involved in the Vpu-mediated downregulation of MHC-I from the cell surface,67 as well as in the retention of BST-2 in the trans-Golgi network (TGN), as discussed below.
Other Vpu-interacting proteins include CAML, MHC-II-Ii, and UBP (all targets identified in yeast two-hybrid screens), and TASK-1 (an ion channel protein), whose transmembrane domain is partly homologous to that of Vpu. The functions of these interactions remain elusive. Although initially proposed as a cellular protein that Vpu antagonizes to enhance virion release, CAML appears to restrict virion release only in simian cells.68,69 The interaction of Vpu with MHC-II-Ii may affect MHC-II-mediated antigen presentation.70 Overexpression of UPB and the interaction of this protein with Gag results in limited virion release, thus the interaction of Vpu with UPB may disrupt the UBP-Gag association and facilitate virion release.71 The interaction of Vpu with TASK-1 results in the disruption of the ion channel activities of both proteins and was proposed to mediate Vpu-dependent enhancement of virion release.72,73 However, the ability of Vpu to enhance virion release is now ascribed to its interaction with BST-2/tetherin, an interaction that could in principle be inhibited by the overexpression of TASK-1.
Structure and Motifs Required for Trafficking Effects
Nef.
HIV-1 Nef is typically 27 kDa in apparent mass and 206 amino acids in size. It is membrane-associated via N-terminal myristoylation and forms dimers. Myristoylation at Gly2 in at the N-terminal MGxxxS motif, as well as Asp123-dependent dimerization, are apparently essential for most Nef functions, although whether dimerization is the key functional attribute of Asp123 is not clear.74,75 NMR spectroscopy and X-ray crystallography have revealed three structural parts of Nef: a flexible N-terminal region (residues 1–57), a C-terminal folded core (residues 58–149 and 181–206) and a flexible C-terminal loop (residues 150–180). A surface interacting with CD4 was determined by solution NMR and includes Trp57, Leu58, Glu59 and Arg106.76 The same motif presumably interacts with CD8 and CD28, as mutating these residues, specifically Trp57 and Leu58, disrupts downregulation of these receptors.36,40 The sites in Nef for interaction with other targets such as MHC-I and MHC-II are not known. However, because Nef functions related to CD4 and MHC-I are genetically separable, the site for interaction with MHC-I is presumably elsewhere.
The motifs in Nef required for interaction with several cellular co-factors have also been characterized, and some of these are directly involved in endosomal trafficking (Fig. 1). An acidic motif (or cluster) EEEE65 just N-terminal of the folded core binds to the phosphofurin acid cluster sorting protein 1 (PACS-1), which may mediate retrograde endosome-to-TGN transport.77,78 Alternatively or in addition, an interaction with PACS-2 may trigger a multi-kinase cascade leading to the downregulation of MHC-I.79 A contrasting model proposes that the EEEE65 acidic cluster instead participates directly in the interaction of Nef with the µ subunit of the adaptor protein complex 1 (AP-1).80,81 The polyproline domain including the PxxP75 sequence of HIV-1 Nef binds the SH3 domains of Src kinases, including the monocyte-specific kinase Hck and the lymphocyte-specific kinase Lck.82,83 The importance of the interactions between Src kinases and Nef for its trafficking functions is not clear, although signaling cascades involving an unidentified Src-family kinase have been proposed as important for the Nef-mediated modulation of MHC-I.83 Moreover, a recent study suggests that Nef-induced activation and recruitment of Hck results in the perturbation of Golgi structure and Golgi-associated glycosylation, resulting in trafficking defects such as the reduced expression of the receptor for macrophage colony stimulating factor.84 On the other hand, a nearby residue in the HIV-1 Nef polyproline helix, P78, is required for the downregulation of MHC-I, but it is not required for SH3-binding; this suggests that the polyproline domain of Nef might participate in the modulation of MHC-I by an alternative mechanism.85 An acidic di-leucine motif ExxxLL165 in the flexible C-terminal loop is required for the interaction with AP complexes and for the downregulation of CD4.86,87 It is preceded by a diacidic sequence EE155, which interacts with β-COP, possibly via the regulatory endosomal GTPase ARF1, and is required for optimal CD4 degradation.88,89 An alternative interface for the interaction with β-COP is formed by residues arginine 17 and 19 and is required for the degradation of MHC-I.90 Two aspartic acids succeeding the di-leucine motif, DD175, are required for binding to V1H, the catalytic subunit of an endosomal ATPase involved in acidification.91 However, this motif has also been shown to participate, along with the ExxxLL motif, in the direct binding of Nef to AP-2, the endosomal clathrin adaptor involved in endocytosis.92
Figure 1.

The effects of Nef and Vpu on intracellular trafficking. Top: Nef alters multiple intracellular trafficking steps. The ExxxLL motif in Nef directs interactions with AP complexes. Golgi-endosome-related trafficking and designated residues in Nef are depicted in purple; PM endosome in orange; endosome-MVB in red. SFK indicates unidentified Src-family kinase. Full length Nef molecule with the interaction motifs represents a composite of two solution NMR structures, Protein Data Bank ID's 2NEF and 1QA5,146,147 assembled in PyMOL. Bottom: intracellular trafficking steps likely affected by Vpu. The cytosolic domain of Vpu with designated DpSGxxpS motif represents solution NMR structure 1VPU.148 In both panels, wide green arrows represent normal intracellular trafficking pathways; other-colored narrow arrows represent trafficking stimulated by HIV-1 Nef or Vpu. Cellular structures: CCV, clathrin-coated vesicles; NCV, non-clathrin vesicles; SE, sorting endosome; MVB, multivesicular body; ERC, endosomal recycling center; RE, recycling endosomes; SV, secretory vesicles. Two structures involved in protein-degradation, proteosomes and lysosomes, are depicted in yellow.
Due to its apparent flexibility and large solvent-accessible surface area, Nef can potentially assume various conformations that expose different sets of interaction motifs and thus recruit functionally distinct co-factor complexes as illustrated in Figure 2.26,93,94 Such alternative conformations could be adopted upon binding with specific target proteins.
Figure 2.

Tripartate complex formation between Nef or Vpu, cellular co-factors, and target proteins. Left: Nef binds differently to the AP-complexes depending on its target: when Nef downregulates CD4 it binds AP-2 via its ExxxLL motif and the α/σ2 subunits of AP-2; when Nef downregulates MHC-I it binds via unclear sequences to the µ subunit of AP-1, while directing a key tyrosine residue of MHC-I to the canonical binding site for Yxxφ motifs on the µ subunit. Right: Vpu binds an E3 ubiquitin ligase complex, which results in ubiquitination and downregulation of CD4 and BST-2. “X” refers to unidentified factors that might bind the cytoplasmic domain of Vpu or BST-2; in the case of BST-2 such factors could recognize ubiquitin.
Vpu.
HIV-1 Vpu is typically 17 kDa in apparent mass and 81 amino acids in size. Like Nef, it is associated with membranes but via a single transmembrane domain (TMD) in a type I orientation. NMR studies have revealed a short N-terminal luminal domain (residues 1–3), a transmembrane α-helix (residues 4–27), two cytosolic α-helices (residues 32–49 and 57–72), a flexible connector loop (50–56), and a flexible C-terminal tail.95–99
The downregulation of NTB-A and BST-2 functionally maps to the TMDs of these proteins and the TMD of Vpu. An interaction between the TMDs of Vpu and BST-2 has been demonstrated by immunoprecipitation, FRET, and bi-molecular fluorescence complementation.61,100–104 These data support a model in which the α-helices of the two proteins bind each other in an anti-parallel orientation. Unlike the case of Nef, only one Vpu-interacting cellular co-factor with meaningful function is currently known: β-TrCP. Phosphorylation of the two serines (Ser52 and Ser56) in the DSGxxS motif in the flexible region of the cytosolic domain of Vpu is required for the association of Vpu with the β-TrCP, which occurs via the WD domain of β-TrCP.53 The interaction between Vpu and β-TrCP is required for the downregulation of CD4; it contributes to the downregulation of BST-2; and it is dispensable for the downregulation of NTB-A. These results prompt the continuing search for other Vpu-binding proteins, as well as for additional ways in which Vpu might interfere with the surface expression of cellular proteins independently of β-TrCP-mediated ubiquitination (indicated by “X-factor” in Fig. 2).
Additional sites with the potential to interact with cellular co-factors have been noted in the cytosolic domain of Vpu: these include a potential tyrosine-based AP-binding motif (Yxxφ) in the membrane-proximal hinge region upstream of helix 1, which overlaps with a potential leucine-based motif (E/D)xxxLφ, as well as residues L63 and V64 in helix 2, which could also comprise a leucine-based AP-binding motif. Whereas L63 is required for CD4 downregulation, the substitution R30A,K31A within the Yxxφ sequence and the truncation of helix 2 affect Vpu's localization to the TGN and its ability to enhance virion release.105,106 However, whether the aforementioned residues or regions are involved in the binding of Vpu to specific cellular co-factors is unclear.
Traffic Control
Major trafficking routes within the membrane systems of eukarytic cells are depicted in Figure 1 and represent potential sites of interference by viral proteins. In brief, cargo (i.e., transmembrane proteins) within the secretory pathway is synthesized in the endoplasmic reticulum (ER) and transported to the plasma membrane (PM) through successive compartments: first the Golgi, then the TGN and finally secretory vesicles. Once at the PM, certain receptors are subject to endocytosis, then transport through early/sorting endosomes and recycling endosomes. From both sorting and recycling endosomes, cargo can be sent directly back to the PM or be retrieved to the TGN. A distinct compartment, the endocytic recycling center, is identified by the presence of the regulatory GTPase Rab11 and is localized in the perinuclear region. Direct transport from sorting endosomes to the PM is referred to as fast recycling and takes 1–2 min; in contrast, recycling from the perinuclear recycling endosomes to the PM takes about 12 min.107 From sorting endosomes, cargo can also be delivered to late endosomes or multivesicular bodies (MVB) and subsequently, to the lysosome.108 Lysosomes, like proteosomes, are a major site of protein degradation. While lysosomes are a terminal branch of the endocytic pathway, proteosomal degradation of membrane proteins generally occurs after the protein is extracted from the ER and is thus linked to ER-associated degradation, a process that includes the degradation of misfolded proteins. Initial internalization of transmembrane proteins at the PM can be clathrindependent or -independent. These two pathways merge in the sorting endosome.109 In the endosomal system, each cargo destination is defined by a specific set of factors that interact with the cargo either directly or indirectly: vesicle coat components such as clathrin AP complexes, vesicle fission factors, tethers, SNARE proteins, fusion factors, and small GTPases.110 Specific AP complexes are responsible for the formation of clathrin coats and the recruitment of vesicle cargo at various post-Golgi membrane compartments. AP-2 is specifically associated with internalization at the PM, whereas AP-1 directs the transport between the TGN and endosomes and AP-3 directs cargo to late endosomes and lysosomes. The breadth and specificity of these pathways can be modulated by various factors, including clathrin-associated sorting proteins (CLASPs), some of which recognize ubiquitin as a sorting signal.111 Ubiquitin can function as a endocytic signal, or it can bind various proteins of the endosomal sorting complex required for transport (ESCRT) to mediate cargo delivery to late endosomes and multivesicular bodies.112–114 Nef and Vpu redirect their targets along these pathways within the endosomal system, essentially by co-opting several of these cellular regulatory mechanisms.
Nef.
Nef localizes throughout the endosomal system including the PM but appears most concentrated in the juxtanuclear region of the cell.115 To upregulate targets such as MHC-II-Ii or DC-SIGN, Nef likely sequesters AP complexes, particularly AP-2, away from the targets, such that the proteins are displaced to the PM by default.116 To downregulate molecules from the cell surface, Nef alters their trafficking by recruiting endosomal coat proteins to the target. This activity requires the formation of ternary complexes between Nef, the target, and the endosomal coat proteins, specifically AP-1, AP-2, and β-COP.26 The recruitment of AP-2 stimulates the rate of endocytosis of CD4, whereas (at least in T cells), the recruitment of AP-1 results in the retention of MHC-I in the TGN117 (Fig. 2). Recruitment of β-COP results in targeting to late endosomes and ultimately the partial degradation of CD4 and MHC-I in lysosomes.88,90
Several motifs in Nef are responsible for these interactions with endosomal coat proteins. The highly conserved ExxxLL motif in Nef is required for the binding of Nef to AP complexes,86,87,118 whereas other motifs recruit additional factors such as β-COP and possibly ARF-1 (Fig. 1). Still others, specifically the EEEE65 sequence, appear to allow Nef to bind AP complexes when in ternary complex with its target.80 Two distinct models have been proposed by different research groups to explain the downregulation of MHC-I by Nef. One incorporates the interactions above; in this model Nef recruits AP-1 (and β-COP) to MHC-I, trapping MHC-I in the TGN and directing it to lysosomes for degradation.80,90,117,119 Another model is more complex and invokes signaling interactions via unidentified Src-family kinase(s) as noted above, with ultimate modulation of protein sorting via PI-3-kinase.83 Like CD4, the effects of Nef on CD8 and CD28 seem primarily to involve enhanced endocytosis and likely depend on AP-2 as a cellular cofactor, although recent RNA-interference experiments support a role for AP-1.120 CD1d appears to be downregulated by both enhanced endocytosis and retention in the TGN.121 CD80 and 86 appear to be redistributed to the TGN by Nef by an uncharacterized pathway that includes actin polymerization and is Rab11-dependent.41,122,123 Notably, the modulation of CD1d, CD80, and CD86 by Nef has not been as extensively studied as has the modulation of CD4 and MHC-I, and recent data using chimeric molecules suggests that the cytoplasmic domains of CD1d, CD80, and CD86 are not well recognized by Nef.120 SIV Nef proteins are now known to down-regulate non-human primate BST-2; this occurs via the formation of a ternary complex between AP-2, Nef, and BST-2.11–13,124 Interestingly, although Nef is not known to induce ubiquitination of its targets, it is itself ubiquitinated, and proteins of the ESCRT system are involved in the downregulation and targeting of CD4 by Nef to lysosomes.125,126
Overall, these data support the overarching notion that Nef co-opts endosomal coat proteins and trafficking pathways to misdirect cellular proteins away from the plasma membrane. An intriguing aspect of Nef's mechanism of action is that the ternary interactions formed between Nef, its target (MHC-I or CD4), and AP complexes (AP-1 in the case of MHC-I and AP-2 in the case of CD4) appear to be cooperative in nature.80,127 For example, when Nef is fused to the cytoplasmic domain of MHC-I, binding of the fusion-protein to AP-1 (via the µ1 subunit) is more avid than the binding of either Nef or the cytoplasmic domain of MHC-I alone to AP-1.80,81 Similarly, Nef binds via its ExxxLL motif to the α/σ2 hemicomplex (two of the four subunits of the AP-2 complex) more avidly in the presence of the cytoplasmic domain of CD4. Moreover, these cellular targets of Nef have sequences that are “would be” AP-binding motifs within their cytoplasmic domains, and these are required for modulation by Nef. For example, the cytoplasmic domain of MHC-I contains a tyrosine required for Nef-responsiveness but whose sequence context lacks the hydrophobic residue typical of Yxxφ AP-binding motifs. Similarly, the cytoplasmic domain of CD4 contains a required dileucine whose sequence context lacks the acidic residue(s) typical of ExxxLφ AP-binding motifs. Nef appears to facilitate the interaction of these “would be” binding sequences in its targets to the AP complexes; thus, Nef can be regarded conceptually as a virally encoded CLASP, which alters the breadth and specificity of clathrin-mediated endosomal trafficking to the advantage of the virus.80
Vpu.
Despite its ability to downregulate target proteins from the cell surface, Vpu proteins of HIV-1 subtype B, which are commonly used in many laboratories, display predominantly intracellular rather than PM localization; that is, Vpu resides in the ER, TGN and endosomes.105,128–130 Residence of Vpu in the ER is consistent with its ability to degrade CD4 by an ERAD-related mechanism. Vpu could potentially interfere with BST-2 trafficking at any of the above compartments, but localization to the TGN has been associated with Vpu-mediated enhancement of virion release.105
Ion channel formation. Vpu forms ion channels via oligomerization of its TMD.131 This activity is separable from CD4 downregulation, which maps to the cytoplasmic domain (CD) of Vpu, but overlaps with anti-BST-2 activity, insofar as both require an intact TMD. Ion channel formation could in principle affect the endosomal transport of BST-2, but recent data indicate that certain amino acid substitutions associated with loss of channel activity do not impair the downregulation of BST-2 or the enhancement of virion release.104,132 While this leaves little room for the role of ion channel formation during the downregulation of BST-2, channel activity could still play a role in the modulation of other targets such as CD1d.
Vpu serine residues 52 and 56 of the DSGxxS motif; β-TrCP; and the ubiquitination and degradation of BST-2. Much effort has been invested in assessing the role of protein degradation in the Vpu-dependent downregulation of BST-2, as well as the roles of Vpu serine residues 52 and 56 and β-TrCP in these processes. Nevertheless, controversy concerning these subjects persists.
Degradation. At first reported as the principle effect underlying Vpu-mediated downregulation of BST-2 and the counteraction of restricted virion release,132 the degradation of BST-2 is instead emerging as a potential secondary mechanism: several reports describe downregulation of BST-2 from the cell surface without significant depletion of intracellular levels.57,60–62 Degradation can be observed when Vpu and BST-2 are expressed at high levels in cells by transient transfection,58,133 and this mechanism appears to be substantial in at least one of the primary HIV-1 target cells—macrophages, which express high endogenous levels of BST-2.60,134 The Vpu-induced degradation of BST-2 in HeLa cells and T lymphocytes is not as pronounced and varies between reports.57,59–61,133 The variability of these results might be caused by differences in the BST-2 species measured: immature (high-mannose) and mature (fully glycosylated) forms of BST-2 are affected differently by Vpu in a manner that relates partly to whether BST-2 is endogenously expressed or is expressed by transient transfection.133 Specifically, immature forms, which predominate when BST-2 is expressed by transient transfection, are directed by Vpu to an ERAD-related, proteasomal degradation pathway. In contrast, the transport of mature forms, which predominate when BST-2 is endogenously and constitutively expressed, appears blocked in post-ER membranes by Vpu, and these forms might be degraded in lysosomes by default. These differences also complicate the analysis of the DSGxxS motif and the role of ubiquitination in Vpu activity with respect to BST-2. Potential acceptor sites for ubiquitin in the BST-2 CD have been studied toward the goal of enlightening the debate regarding BST-2 degradation. Mutation of lysine residues (classic ubiquitin acceptors) in the BST-2 CD (KK18,21RR) uncouples the degradation of BST-2 from cell surface downregulation and from the counteraction of restricted virion release; that is, degradation is prevented but Vpu nevertheless reduces the levels of BST-2 at the cell surface and enhances virion release.62,135 On the other hand, mutation of serines and threonines (of an STS sequence), which are alternative ubiquitin acceptors in the BST-2 CD, renders BST-2 much less efficiently downregulated from the cell surface by Vpu and impairs the ability of Vpu to enhance virion release, although these effects do not correlate with the total cellular levels of BST-2.63 As noted below, these data suggest that ubiquitin mediated trafficking within the endosomal system might be modulated by Vpu and that this need not lead to protein degradation. In contrast to BST-2, the downregulation of CD4 by Vpu appears exclusively linked to ER retention, extraction from the ER, ubiquitination of the CD of CD4, and the degradation of CD4 by the proteasome.51
Lysosomal versus proteosomal degradation. Whether Vpu-induced degradation of BST-2, when it happens, occurs in lysosomes or proteosomes should provide a clue to deciphering the specific trafficking defect(s) caused by Vpu. The use of proteosomal and lysosomal inhibitors in attempts to rescue BST-2 from Vpu-induced degradation or downregulation from the cell surface yielded a variety of conclusions.57–59,132,135 Although prolonged exposure of cells to proteasome inhibitors blocks the Vpu-induced degradation of BST-2 and inhibits the downregulation of BST-2 from the cell surface,132 these results arguably reflect ubiquitin depletion and inhibition of ubiquitin-mediated trafficking rather than implicating proteasomal degradation as Vpu's primary mechanism of action.57 In support of a trafficking defect, Vpu mis-localizes BST-2 from the plasma membrane and various endosomes to a cathepsin-D-positive compartment, when expressed in simian cells, suggesting a lysosomal degradation mechanism.136 However, in HeLa cells, Vpu does not seem to mis-direct BST-2 to lysosomes but rather redistributes it to perinuclear endosomes including the TGN.59,61,135,137 Nevertheless, consistent with lysosomal degradation or at least directional transport down the endosomal pH gradient, bafilomycin A, an inhibitor of lysosomal acidification, reduces Vpu-mediated downregulation of BST-2 from the cell surface,57 and conconamycin A rescues Vpu-induced degradation of BST-2.59 Moreover, several lines of investigation support a post-ER mechanism of Vpu action with respect to BST-2.104,133 First, when Vpu is restricted to the ER by appending a specific retention signal to its C-terminus, it is unable to downregulate BST-2 but retains substantial activity with respect to CD4.104 Second, downregulation of BST-2 from the surface of HeLa cells occurs much faster (a 6-fold reduction within 6 h) than it would if it were based exclusively on the degradation (or retention) of BST-2 in the ER: when the egress of BST-2 from the ER is blocked using brefeldin A (BFA), cell surface levels decrease by only 2-fold after 8 h.104 Third, in the case of BST-2 expressed endogenously in T cells, treatment with BFA does not enable Vpu to accelerate the degradation of ER-associated immature forms.133 Thus, a post-ER mechanism involving altered trafficking, lysosomal degradation, or both seems to be involved in the Vpu-dependent counteraction of BST-2, at least when BST-2 is constitutively expressed. Nevertheless, as noted above, ER-associated degradation can occur when BST-2 is expressed exogenously and abundantly by transient transfection of cells such as HEK-293T, as determined by pulse-chase analysis of immature BST-2 species. This effect is presumably caused by fast accumulation of de novo synthesized BST-2, which triggers ER degradation of immature BST-2 forms.133 Whether this scenario might take place under physiological conditions, such as in macrophages where the expression of BST-2 is high, or in T cells in which the expression of BST-2 is induced by interferon during the innate immune response, remains to be determined.
Vpu serines 52 and 56 and β-TrCP. The preceding arguments apply to the intriguing role of the interaction between Vpu and the E3 ubiquitin ligase complex substrate adaptor β-TrCP during the downregulation of BST-2. Mutating the key serines in Vpu required for this interaction (residues 52 and 56; “Vpu2/6” mutant) partially impairs the downregulation of BST-2 from the cell surface and the enhancement of virion release, at least when BST-2 is expressed at relatively high levels, such as in HeLa cells, macrophages, and HEK 293T cells following transient transfection.54,57–59,101,134,136 This partial effect contrasts strikingly with the case of CD4 downregulation, for which Vpu2/6 is completely inactive. Interestingly, while CD1d and NTB-A are down-regulated from the cell surface, their intracellular levels are not decreased in the presence of Vpu,64,65 and Vpu2/6 displays a wild-type-like phenotype with respect to the downregulation of NTB-A. Although one might speculate that the extent of the Vpu2/6 phenotype with respect to BST-2 simply reflects the extent of degradation, as discussed above this degradation is not necessarily ER-associated. Therefore, the Vpu-mediated downregulation of BST-2 might rely instead on β-TrCP-based ubiquitination followed by lysosomal degradation, as in the case of the ligand-induced internalization and degradation of IFNAR1, the type 1 interferon receptor.138 Moreover, the Vpu-β-TrCP interaction might be required not only for degradation but also for the trafficking effects that presumably underlie the robust downregulation of cell surface BST-2 that can be observed despite minimal depletion of total cellular protein.57 These observations imply that Vpu-β-TrCP complex formation and substrate ubiquitination are involved, at least in the case of BST-2, in the regulation of endosomal trafficking. In contrast, β-TrCP was recently proposed as dispensable for the downregulation of BST-2 based on a failure of RNA interference targeting β-TrCP to block this Vpu activity under conditions that were sufficient to impair the down-regulation of CD4.54 However, these data should be considered cautiously, because a β-TrCP mutant lacking the F-box domain (ΔF-box β-TrCP) required for interaction with the E3 ligase complex inhibits Vpu activity. Since over-expression of the wild-type β-TrCP does not inhibit Vpu, the notion that ΔF-box β-TrCP acts simply as an inhibitory ligand of the Vpu CD seems less likely. Nevertheless, this report raised the formal possibility that serines 52 and 56 in the CD of Vpu might be required for interaction with a factor other than β-TrCP that is important for the downregulation of BST-2 (Fig. 2).
Specific trafficking defects induced by Vpu. To downregulate BST-2 and other proteins from the cell surface, Vpu must alter their normal trafficking. Vpu does not increase the rate of internalization of any of its known targets (BST-2, CD1d or NTB-A).57,64,65 Although potential adaptor protein-binding motifs exist in the CD of Vpu, no AP complexes or other trafficking factors have been reported to interact with Vpu so far and mutating the putative AP-binding sequences in the Vpu cytosolic domain does not markedly impair BST-2 downregulation or enhancement of virion release105,106 (and our unpublished data). Likewise, mutating the AP-binding YxY sequence in the cytosolic domain of BST-2 (reportedly required for internalization in the absence of Vpu) does not impair Vpu-mediated downregulation.61,135 Nevertheless, components of the clathrin-based internalization machinery—the µ subunit of AP-2 (but not of AP-1 or AP-3), the clathrin assembly protein AP180, and the vesicle “pinch-ase” dynamin 2—are each required for optimal downregulation of BST-2 by Vpu57,136 (and our unpublished data). Currently, the precise role of the internalization machinery in the downregulation of BST-2 by Vpu remains to be deciphered. However, one possibility is that the effects noted above are indirect; for example, the actual effect of Vpu might be a block to the recycling of BST-2 that has been internalized constitutively as discussed below.
While not affecting the internalization rate of BST-2, Vpu does slow the resupply of BST-2 to the PM. This effect appears to involve both newly synthesized BST-2 and BST-2 that has been internalized from the plasma membrane and would otherwise return there via a recycling pathway.61,139 A Vpu-mediated recycling defect was supported using a flow cytometric assay that measures newly deposited BST-2 at the PM. The inhibition of egress of BST-2 from the ER by brefeldin A had no effect on this assay, which weighed against the possibility that newly synthesized BST-2 contributed substantially to the observed Vpu-effect.139 A similar flow cytometric approach suggested that Vpu reduces the rate of recycling of CD1d, preventing the return of CD1d from an EEA-1-positive, early endosomal compartment to the PM.64
While trapping BST-2 in intracellular compartments, Vpu could consequently or independently re-route BST-2 into the MVB/lysosomal pathway (Fig. 1). Indeed, a recent study suggested that the ESCRT machinery, and specifically the formation of a ternary complex between Vpu, BST-2 and the ESCRT 0 component HRS, promotes the Vpu-mediated counteraction of BST-2.140 This model is consistent with a role for ubiquitination in the downregulation of BST-2 by Vpu. In addition to HRS, the ESCRT components Tsg101 and Vps4 are involved in this process and further link it to lysosomal degradation. Thus, the combination of retention of newly synthesized BST-2 in the TGN, defective recycling, and enhanced trafficking down the MVB/lysosomal pathway appears to underlie the Vpu-mediated downregulation of BST-2. Whether BST-2 that is directed down the MVB/lysosomal pathway includes newly synthesized protein or is exclusively derived from the PM remains to be determined.
Final Remarks
The recent discovery of new host cell factors that are modulated by Vpu (BST-2, CD1d and NTB-A) and the study of the mechanisms underlying these effects have confirmed Vpu as a modifier of intracellular trafficking, a role long ascribed to Nef. Although Vpu and Nef share certain cellular targets such as CD4, the two viral proteins have distinct mechanisms of action: Nef recruits endosomal coat proteins such as AP complexes to its targets whereas Vpu recruits ubiquitination machinery. These differences might reflect a need for the antagonism of specific cellular proteins from specific compartments, such as the Vpu-mediated disruption of the interaction between CD4 and Env in the ER. Nevertheless, the net effect of all the perturbations reviewed here involves the altered expression of plasma membrane proteins. Interestingly, the envelope protein of HIV-2, a virus that lacks Vpu, evolved in certain isolates to downregulate BST-2 from the cell surface, directing the accumulation of BST-2 in a perinuclear compartment.141 Moreover, mutations in the SIV envelope can lead to the acquisition of a BST-2 modulating activity that compensates for the absence of Nef during viral replication in rhesus macaques.142 Tyrosine-based AP-binding motifs (Yxxφ) in the membrane-proximal region of gp41 are essential for the activity of these envelope glycoproteins. Thus, Env is yet another viral protein that is capable of modulating endosomal trafficking to counteract host defenses.
The multifaceted mechanisms underlying the functions of Nef and Vpu present many challenging questions. For example, both Nef and Vpu interact with cellular co-factors even in the absence of their targets. This could in principle cause their mislocalization and/or degradation. Presumably, these viral proteins are either intensely replenished or designed to avoid such suicide inhibition. For example, Vpu itself may not be a substrate for β-TrCP-dependent ubiquitination and degradation, although this is controversial.53,143–145
In the case of Vpu, what is the contribution of degradation versus altered trafficking to the downregulation of host targets and what is the physiological meaning of this balance? What is the contribution of altered anterograde transport versus altered recycling? What is the role of clathrin-mediated endocytosis and ubiquitination in these processes? Like Vpu, Nef appears to modulate different proteins by mechanisms that are themselves distinct yet share common elements. How and why does Nef adapt its interactions to meet the needs of different targets? For example, why does the modulation of CD4 by Nef involve AP binding via a canonical ExxxLL motif, yet this motif is dispensable for the modulation of MHC-I?
Perhaps the answers to some of these questions reside within the CDs of the target proteins and of Vpu and Nef. Identifying the components of the endosomal traffcking machinery that interact with these domains will doubtlessly aid in dissecting the mechanisms of altered traffcking. So will determining the sequences within the CDs that are necessary for this modulation. This seems particularly critical to a more complete understanding of Vpu. The complexities of such dissections are exemplifed by the distinct roles in degradation and surface downregulation played by lysine, serine, and threonine residues in the cytosolic domain of BST-2, all of which are ubiquitinated by Vpu.62,63,135 However, the potentially cooperative nature of the interactions between the viral accessory protein, its cellular co-factors, and its target proteins may render the search for novel interactions challenging.
Can our understanding of how Vpu and Nef affect the trafficking of cellular transmembrane proteins be put to clinical use? Since these viral proteins contribute to the virulence of HIV-1 as a pathogen, in principle they represent novel antiretroviral drug targets. Understanding the relevant targets of Vpu and Nef can lead to drug screening assays. Moreover, the structural basis of the ternary interactions between the viral accessory protein, its cellular targets, and its cellular cofactors, might enable rational drug design. Most importantly, both Vpu and Nef are involved in immune evasion, and these functions undoubtedly contribute to the success of HIV-1 as an agent of chronic, persistent infection. We are intrigued by the possibility that the inhibition of Vpu and Nef could empower the host's innate and adaptive immune responses to better clear the infection. In the best case, such empowerment would be sufficient to enable viral clearance and the resolution of infection.
Acknowledgments
This work was supported by grants from the California HIV/AIDS Research Program to A.A.T. (F09-VMRF-206) and from the National Institutes of Health to J.C.G. (AI081668 and AI038201). We thank Stefanie Homann for critical reading of the manuscript.
References
- 1.Stremlau M, Owens CM, Perron MJ, Kiessling M, Autissier P, Sodroski J. The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in Old World monkeys. Nature. 2004;427:848–853. doi: 10.1038/nature02343. [DOI] [PubMed] [Google Scholar]
- 2.Song B. TRIM5alpha. Curr Top Microbiol Immunol. 2009;339:47–66. doi: 10.1007/978-3-642-02175-6_3. [DOI] [PubMed] [Google Scholar]
- 3.Malim MH, Emerman M. HIV-1 accessory proteins-ensuring viral survival in a hostile environment. Cell Host Microbe. 2008;3:388–398. doi: 10.1016/j.chom.2008.04.008. [DOI] [PubMed] [Google Scholar]
- 4.Neil S, Bieniasz P. Human immunodeficiency virus, restriction factors, and interferon. J Interferon Cytokine Res. 2009;29:569–580. doi: 10.1089/jir.2009.0077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Neil SJ, Zang T, Bieniasz PD. Tetherin inhibits retro-virus release and is antagonized by HIV-1 Vpu. Nature. 2008;451:425–430. doi: 10.1038/nature06553. [DOI] [PubMed] [Google Scholar]
- 6.Van Damme N, Goff D, Katsura C, Jorgenson RL, Mitchell R, Johnson MC, et al. The interferon-induced protein BST-2 restricts HIV-1 release and is downregu-lated from the cell surface by the viral Vpu protein. Cell Host Microbe. 2008;3:245–252. doi: 10.1016/j.chom.2008.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tokarev A, Skasko M, Fitzpatrick K, Guatelli J. Antiviral activity of the interferon-induced cellular protein BST-2/tetherin. AIDS Res Hum Retroviruses. 2009;25:1197–1210. doi: 10.1089/aid.2009.0253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dube M, Bego MG, Paquay C, Cohen EA. Modulation of HIV-1-host interaction: role of the Vpu accessory protein. Retrovirology. 2010;7:114. doi: 10.1186/1742-4690-7-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Evans DT, Serra-Moreno R, Singh RK, Guatelli JC. BST-2/tetherin: a new component of the innate immune response to enveloped viruses. Trends Microbiol. 2010;18:388–396. doi: 10.1016/j.tim.2010.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Andrew A, Strebel K. The interferon-inducible host factor bone marrow stromal antigen 2/tetherin restricts virion release, but is it actually a viral restriction factor? J Interferon Cytokine Res. 2011;31:137–144. doi: 10.1089/jir.2010.0108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang F, Wilson SJ, Landford WC, Virgen B, Gregory D, Johnson MC, et al. Nef proteins from simian immunodeficiency viruses are tetherin antagonists. Cell Host Microbe. 2009;6:54–67. doi: 10.1016/j.chom.2009.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jia B, Serra-Moreno R, Neidermyer W, Rahmberg A, Mackey J, Fofana IB, et al. Species-specific activity of SIV Nef and HIV-1 Vpu in overcoming restriction by tetherin/BST2. PLoS Pathog. 2009;5:e1000429. doi: 10.1371/journal.ppat.1000429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sauter D, Schindler M, Specht A, Landford WN, Munch J, Kim KA, et al. Tetherin-driven adaptation of Vpu and Nef function and the evolution of pandemic and nonpandemic HIV-1 strains. Cell Host Microbe. 2009;6:409–421. doi: 10.1016/j.chom.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schwartz O, Marechal V, Le Gall S, Lemonnier F, Heard JM. Endocytosis of major histocompatibility complex class I molecules is induced by the HIV-1 Nef protein. Nat Med. 1996;2:338–342. doi: 10.1038/nm0396-338. [DOI] [PubMed] [Google Scholar]
- 15.Collins KL, Chen BK, Kalams SA, Walker BD, Baltimore D. HIV-1 Nef protein protects infected primary cells against killing by cytotoxic T lymphocytes. Nature. 1998;391:397–401. doi: 10.1038/34929. [DOI] [PubMed] [Google Scholar]
- 16.Stumptner-Cuvelette P, Morchoisne S, Dugast M, Le Gall S, Raposo G, Schwartz O, et al. HIV-1 Nef impairs MHC class II antigen presentation and surface expression. Proc Natl Acad Sci USA. 2001;98:12144–12149. doi: 10.1073/pnas.221256498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aiken C, Konner J, Landau NR, Lenburg ME, Trono D. Nef induces CD4 endocytosis: requirement for a critical dileucine motif in the membrane-proximal CD4 cytoplasmic domain. Cell. 1994;76:853–864. doi: 10.1016/0092-8674(94)90360-3. [DOI] [PubMed] [Google Scholar]
- 18.Lama J, Mangasarian A, Trono D. Cell-surface expression of CD4 reduces HIV-1 infectivity by blocking Env incorporation in a Nef- and Vpu-inhibitable manner. Curr Biol. 1999;9:622–631. doi: 10.1016/s0960-9822(99)80284-x. [DOI] [PubMed] [Google Scholar]
- 19.Michel N, Allespach I, Venzke S, Fackler OT, Keppler OT. The Nef protein of human immunodeficiency virus establishes superinfection immunity by a dual strategy to downregulate cell-surface CCR5 and CD4. Curr Biol. 2005;15:714–723. doi: 10.1016/j.cub.2005.02.058. [DOI] [PubMed] [Google Scholar]
- 20.Deacon NJ, Tsykin A, Solomon A, Smith K, Ludford-Menting M, Hooker DJ, et al. Genomic structure of an attenuated quasi species of HIV-1 from a blood transfusion donor and recipients. Science. 1995;270:988–991. doi: 10.1126/science.270.5238.988. [DOI] [PubMed] [Google Scholar]
- 21.Kestler HW, 3rd, Ringler DJ, Mori K, Panicali DL, Sehgal PK, Daniel MD, et al. Importance of the Nef gene for maintenance of high virus loads and for development of AIDS. Cell. 1991;65:651–662. doi: 10.1016/0092-8674(91)90097-i. [DOI] [PubMed] [Google Scholar]
- 22.Laguette N, Bregnard C, Benichou S, Basmaciogullari S. Human immunodeficiency virus (HIV) type-1, HIV-2 and simian immunodeficiency virus Nef proteins. Mol Aspects Med. 2010;31:418–433. doi: 10.1016/j.mam.2010.05.003. [DOI] [PubMed] [Google Scholar]
- 23.Foster JL, Denial SJ, Temple BR, Garcia JV. Mechanisms of HIV-1 Nef function and intracellular signaling. J Neuroimmune Pharmacol. 2011;6:230–246. doi: 10.1007/s11481-011-9262-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kirchhoff F, Schindler M, Specht A, Arhel N, Munch J. Role of Nef in primate lentiviral immunopathogenesis. Cell Mol Life Sci. 2008;65:2621–2636. doi: 10.1007/s00018-008-8094-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kirchhoff F. Immune evasion and counteraction of restriction factors by HIV-1 and other primate lentivi-ruses. Cell Host Microbe. 2010;8:55–67. doi: 10.1016/j.chom.2010.06.004. [DOI] [PubMed] [Google Scholar]
- 26.Roeth JF, Collins KL. Human immunodeficiency virus type 1 Nef: adapting to intracellular trafficking pathways. Microbiol Mol Biol Rev. 2006;70:548–563. doi: 10.1128/MMBR.00042-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lama J, Ware CF. Human immunodeficiency virus type 1 Nef mediates sustained membrane expression of tumor necrosis factor and the related cytokine LIGHT on activated T cells. J Virol. 2000;74:9396–9402. doi: 10.1128/jvi.74.20.9396-9402.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sol-Foulon N, Moris A, Nobile C, Boccaccio C, Engering A, Abastado JP, et al. HIV-1 Nef-induced upregulation of DC-SIGN in dendritic cells promotes lymphocyte clustering and viral spread. Immunity. 2002;16:145–155. doi: 10.1016/s1074-7613(02)00260-1. [DOI] [PubMed] [Google Scholar]
- 29.Folks TM, Clouse KA, Justement J, Rabson A, Duh E, Kehrl JH, et al. Tumor necrosis factor alpha induces expression of human immunodeficiency virus in a chronically infected T-cell clone. Proc Natl Acad Sci USA. 1989;86:2365–2368. doi: 10.1073/pnas.86.7.2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Geijtenbeek TB, Kwon DS, Torensma R, van Vliet SJ, van Duijnhoven GC, Middel J, et al. DC-SIGN, a dendritic cell-specific HIV-1-binding protein that enhances trans-infection of T cells. Cell. 2000;100:587–597. doi: 10.1016/s0092-8674(00)80694-7. [DOI] [PubMed] [Google Scholar]
- 31.Roche PA, Teletski CL, Karp DR, Pinet V, Bakke O, Long EO. Stable surface expression of invariant chain prevents peptide presentation by HLA-DR. EMBO J. 1992;11:2841–2847. doi: 10.1002/j.1460-2075.1992.tb05351.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cohen GB, Gandhi RT, Davis DM, Mandelboim O, Chen BK, Strominger JL, et al. The selective down-regulation of class I major histocompatibility complex proteins by HIV-1 protects HIV-infected cells from NK cells. Immunity. 1999;10:661–671. doi: 10.1016/s1074-7613(00)80065-5. [DOI] [PubMed] [Google Scholar]
- 33.Venzke S, Michel N, Allespach I, Fackler OT, Keppler OT. Expression of Nef downregulates CXCR4, the major coreceptor of human immunodeficiency virus, from the surfaces of target cells and thereby enhances resistance to superinfection. J Virol. 2006;80:11141–11152. doi: 10.1128/JVI.01556-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wildum S, Schindler M, Munch J, Kirchhoff F. Contribution of Vpu, Env, and Nef to CD4 down-modulation and resistance of human immunodeficiency virus type 1-infected T cells to superinfection. J Virol. 2006;80:8047–8059. doi: 10.1128/JVI.00252-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Benson RE, Sanfridson A, Ottinger JS, Doyle C, Cullen BR. Downregulation of cell-surface CD4 expression by simian immunodeficiency virus Nef prevents viral super infection. J Exp Med. 1993;177:1561–1566. doi: 10.1084/jem.177.6.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stove V, Van de Walle I, Naessens E, Coene E, Stove C, Plum J, et al. Human immunodeficiency virus Nef induces rapid internalization of the T-cell coreceptor CD8alphabeta. J Virol. 2005;79:11422–11433. doi: 10.1128/JVI.79.17.11422-11433.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu Y, Liu H, Kim BO, Gattone VH, Li J, Nath A, et al. CD4-independent infection of astrocytes by human immunodeficiency virus type 1: requirement for the human mannose receptor. J Virol. 2004;78:4120–4133. doi: 10.1128/JVI.78.8.4120-4133.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lanzavecchia A. Mechanisms of antigen uptake for presentation. Curr Opin Immunol. 1996;8:348–354. doi: 10.1016/s0952-7915(96)80124-5. [DOI] [PubMed] [Google Scholar]
- 39.Vigerust DJ, Egan BS, Shepherd VL. HIV-1 Nef mediates post-translational down-regulation and redistribution of the mannose receptor. J Leukoc Biol. 2005;77:522–534. doi: 10.1189/jlb.0804454. [DOI] [PubMed] [Google Scholar]
- 40.Swigut T, Shohdy N, Skowronski J. Mechanism for down-regulation of CD28 by Nef. EMBO J. 2001;20:1593–1604. doi: 10.1093/emboj/20.7.1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chaudhry A, Das SR, Jameel S, George A, Bal V, Mayor S, et al. A two-pronged mechanism for HIV-1 Nef-mediated endocytosis of immune costimulatory molecules CD80 and CD86. Cell Host Microbe. 2007;1:37–49. doi: 10.1016/j.chom.2007.01.001. [DOI] [PubMed] [Google Scholar]
- 42.Erdtmann L, Janvier K, Raposo G, Craig HM, Benaroch P, Berlioz-Torrent C, et al. Two independent regions of HIV-1 Nef are required for connection with the endocytic pathway through binding to the mu 1 chain of AP1 complex. Traffic. 2000;1:871–883. doi: 10.1034/j.1600-0854.2000.011106.x. [DOI] [PubMed] [Google Scholar]
- 43.Madrid R, Janvier K, Hitchin D, Day J, Coleman S, Noviello C, et al. Nef-induced alteration of the early/recycling endosomal compartment correlates with enhancement of HIV-1 infectivity. J Biol Chem. 2005;280:5032–5044. doi: 10.1074/jbc.M401202200. [DOI] [PubMed] [Google Scholar]
- 44.Stephens EB, McCormick C, Pacyniak E, Griffin D, Pinson DM, Sun F, et al. Deletion of the vpu sequences prior to the env in a simian-human immunodeficiency virus results in enhanced Env precursor synthesis but is less pathogenic for pig-tailed macaques. Virology. 2002;293:252–261. doi: 10.1006/viro.2001.1244. [DOI] [PubMed] [Google Scholar]
- 45.Singh DK, Griffin DM, Pacyniak E, Jackson M, Werle MJ, Wisdom B, et al. The presence of the casein kinase II phosphorylation sites of Vpu enhances the CD4(+) T cell loss caused by the simian-human immunodeficiency virus SHIV(KU-lbMC33) in pig-tailed macaques. Virology. 2003;313:435–451. doi: 10.1016/s0042-6822(03)00339-8. [DOI] [PubMed] [Google Scholar]
- 46.Hout DR, Gomez ML, Pacyniak E, Gomez LM, Inbody SH, Mulcahy ER, et al. Scrambling of the amino acids within the transmembrane domain of Vpu results in a simian-human immunodeficiency virus (SHIVTM) that is less pathogenic for pig-tailed macaques. Virology. 2005;339:56–69. doi: 10.1016/j.virol.2005.04.038. [DOI] [PubMed] [Google Scholar]
- 47.Willey RL, Maldarelli F, Martin MA, Strebel K. Human immunodeficiency virus type 1 Vpu protein induces rapid degradation of CD4. J Virol. 1992;66:7193–7200. doi: 10.1128/jvi.66.12.7193-7200.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen MY, Maldarelli F, Karczewski MK, Willey RL, Strebel K. Human immunodeficiency virus type 1 Vpu protein induces degradation of CD4 in vitro: the cytoplasmic domain of CD4 contributes to Vpu sensitivity. J Virol. 1993;67:3877–3884. doi: 10.1128/jvi.67.7.3877-3884.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Meusser B, Sommer T. Vpu-mediated degradation of CD4 reconstituted in yeast reveals mechanistic differences to cellular ER-associated protein degradation. Mol Cell. 2004;14:247–258. doi: 10.1016/s1097-2765(04)00212-6. [DOI] [PubMed] [Google Scholar]
- 50.Binette J, Dube M, Mercier J, Halawani D, Latterich M, Cohen EA. Requirements for the selective degradation of CD4 receptor molecules by the human immunodeficiency virus type 1 Vpu protein in the endoplasmic reticulum. Retrovirology. 2007;4:75. doi: 10.1186/1742-4690-4-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Magadan JG, Perez-Victoria FJ, Sougrat R, Ye Y, Strebel K, Bonifacino JS. Multilayered mechanism of CD4 downregulation by HIV-1 Vpu involving distinct ER retention and ERAD targeting steps. PLoS Pathog. 2010;6:e1000869. doi: 10.1371/journal.ppat.1000869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Buonocore L, Rose JK. Blockade of human immunodeficiency virus type 1 production in CD4+ T cells by an intracellular CD4 expressed under control of the viral long terminal repeat. Proc Natl Acad Sci USA. 1993;90:2695–2699. doi: 10.1073/pnas.90.7.2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Margottin F, Bour SP, Durand H, Selig L, Benichou S, Richard V, et al. A novel human WD protein, h-beta TrCp, that interacts with HIV-1 Vpu connects CD4 to the ER degradation pathway through an F-box motif. Mol Cell. 1998;1:565–574. doi: 10.1016/s1097-2765(00)80056-8. [DOI] [PubMed] [Google Scholar]
- 54.Tervo HM, Homann S, Ambiel I, Fritz JV, Fackler OT, Keppler OT. beta-TrCP is dispensable for Vpu's ability to overcome the CD317/Tetherin-imposed restriction to HIV-1 release. Retrovirology. 2011;8:9. doi: 10.1186/1742-4690-8-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Besnard-Guerin C, Belaidouni N, Lassot I, Segeral E, Jobart A, Marchal C, et al. HIV-1 Vpu sequesters beta-transducin repeat-containing protein (betaTrCP) in the cytoplasm and provokes the accumulation of beta-catenin and other SCFbetaTrCP substrates. J Biol Chem. 2004;279:788–795. doi: 10.1074/jbc.M308068200. [DOI] [PubMed] [Google Scholar]
- 56.Bour S, Perrin C, Akari H, Strebel K. The human immunodeficiency virus type 1 Vpu protein inhibits NF-kappa B activation by interfering with beta TrCP-mediated degradation of Ikappa B. J Biol Chem. 2001;276:15920–15928. doi: 10.1074/jbc.M010533200. [DOI] [PubMed] [Google Scholar]
- 57.Mitchell RS, Katsura C, Skasko MA, Fitzpatrick K, Lau D, Ruiz A, et al. Vpu antagonizes BST-2-mediated restriction of HIV-1 release via beta-TrCP and endolysosomal trafficking. PLoS Pathog. 2009;5:e1000450. doi: 10.1371/journal.ppat.1000450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mangeat B, Gers-Huber G, Lehmann M, Zufferey M, Luban J, Piguet V. HIV-1 Vpu neutralizes the antiviral factor Tetherin/BST-2 by binding it and directing its beta-TrCP2-dependent degradation. PLoS Pathog. 2009;5:e1000574. doi: 10.1371/journal.ppat.1000574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Douglas JL, Viswanathan K, McCarroll MN, Gustin JK, Fruh K, Moses AV. Vpu directs the degradation of the human immunodeficiency virus restriction factor BST-2/Tetherin via a {beta}TrCP-dependent mechanism. J Virol. 2009;83:7931–7947. doi: 10.1128/JVI.00242-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Miyagi E, Andrew AJ, Kao S, Strebel K. Vpu enhances HIV-1 virus release in the absence of Bst-2 cell surface down-modulation and intracellular depletion. Proc Natl Acad Sci USA. 2009;106:2868–2873. doi: 10.1073/pnas.0813223106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dube M, Roy BB, Guiot-Guillain P, Binette J, Mercier J, Chiasson A, et al. Antagonism of tetherin restriction of HIV-1 release by Vpu involves binding and sequestration of the restriction factor in a perinuclear compartment. PLoS Pathog. 2010;6:e1000856. doi: 10.1371/journal.ppat.1000856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Goffinet C, Homann S, Ambiel I, Tibroni N, Rupp D, Keppler OT, et al. Antagonism of CD317 restriction of human immunodeficiency virus type 1 (HIV-1) particle release and depletion of CD317 are separable activities of HIV-1 Vpu. J Virol. 2010;84:4089–4094. doi: 10.1128/JVI.01549-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tokarev AA, Munguia J, Guatelli JC. Serine-threonine ubiquitination mediates downregulation of BST-2/tetherin and relief of restricted virion release by HIV-1 Vpu. J Virol. 2011;85:51–63. doi: 10.1128/JVI.01795-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Moll M, Andersson SK, Smed-Sorensen A, Sandberg JK. Inhibition of lipid antigen presentation in dendritic cells by HIV-1 Vpu interference with CD1d recycling from endosomal compartments. Blood. 2010;116:1876–1884. doi: 10.1182/blood-2009-09-243667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shah AH, Sowrirajan B, Davis ZB, Ward JP, Campbell EM, Planelles V, et al. Degranulation of natural killer cells following interaction with HIV-1-infected cells is hindered by downmodulation of NTB-A by Vpu. Cell Host Microbe. 2010;8:397–409. doi: 10.1016/j.chom.2010.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vincent MJ, Abdul Jabbar M. The human immunodeficiency virus type 1 Vpu protein: a potential regulator of proteolysis and protein transport in the mammalian secretory pathway. Virology. 1995;213:639–649. doi: 10.1006/viro.1995.0035. [DOI] [PubMed] [Google Scholar]
- 67.Kerkau T, Bacik I, Bennink JR, Yewdell JW, Hunig T, Schimpl A, et al. The human immunodeficiency virus type 1 (HIV-1) Vpu protein interferes with an early step in the biosynthesis of major histocompatibility complex (MHC) class I molecules. J Exp Med. 1997;185:1295–1305. doi: 10.1084/jem.185.7.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Varthakavi V, Heimann-Nichols E, Smith RM, Sun Y, Bram RJ, Ali S, et al. Identification of calcium-modulating cyclophilin ligand as a human host restriction to HIV-1 release overcome by Vpu. Nat Med. 2008;14:641–647. doi: 10.1038/nm1778. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 69.Kuhl A, Munch J, Sauter D, Bertram S, Glowacka I, Steffen I, et al. Calcium-modulating cyclophilin ligand does not restrict retrovirus release. Nat Med. 2010;16:155–156. doi: 10.1038/nm0210-155. author reply 7. [DOI] [PubMed] [Google Scholar]
- 70.Hussain A, Wesley C, Khalid M, Chaudhry A, Jameel S. Human immunodeficiency virus type 1 Vpu protein interacts with CD74 and modulates major histocompatibility complex class II presentation. J Virol. 2008;82:893–902. doi: 10.1128/JVI.01373-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Callahan MA, Handley MA, Lee YH, Talbot KJ, Harper JW, Panganiban AT. Functional interaction of human immunodeficiency virus type 1 Vpu and Gag with a novel member of the tetratricopeptide repeat protein family. J Virol. 1998;72:8461. [PMC free article] [PubMed] [Google Scholar]
- 72.Hsu K, Seharaseyon J, Dong P, Bour S, Marban E. Mutual functional destruction of HIV-1 Vpu and host TASK-1 channel. Mol Cell. 2004;14:259–267. doi: 10.1016/s1097-2765(04)00183-2. [DOI] [PubMed] [Google Scholar]
- 73.Strebel K. HIV-1 Vpu: putting a channel to the TASK. Mol Cell. 2004;14:150–152. doi: 10.1016/s1097-2765(04)00205-9. [DOI] [PubMed] [Google Scholar]
- 74.Kwak YT, Raney A, Kuo LS, Denial SJ, Temple BR, Garcia JV, et al. Self-association of the Lentivirus protein, Nef. Retrovirology. 2010;7:77. doi: 10.1186/1742-4690-7-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Poe JA, Smithgall TE. HIV-1 Nef dimerization is required for Nef-mediated receptor downregulation and viral replication. J Mol Biol. 2009;394:329–342. doi: 10.1016/j.jmb.2009.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Grzesiek S, Stahl SJ, Wingfield PT, Bax A. The CD4 determinant for downregulation by HIV-1 Nef directly binds to Nef. Mapping of the Nef binding surface by NMR. Biochemistry. 1996;35:10256–10261. doi: 10.1021/bi9611164. [DOI] [PubMed] [Google Scholar]
- 77.Piguet V, Wan L, Borel C, Mangasarian A, Demaurex N, Thomas G, et al. HIV-1 Nef protein binds to the cellular protein PACS-1 to downregulate class I major histocompatibility complexes. Nat Cell Biol. 2000;2:163–167. doi: 10.1038/35004038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Blagoveshchenskaya AD, Thomas L, Feliciangeli SF, Hung CH, Thomas G. HIV-1 Nef downregulates MHC-I by a PACS-1- and PI3K-regulated ARF6 endocytic pathway. Cell. 2002;111:853–866. doi: 10.1016/s0092-8674(02)01162-5. [DOI] [PubMed] [Google Scholar]
- 79.Atkins KM, Thomas L, Youker RT, Harriff MJ, Pissani F, You H, et al. HIV-1 Nef binds PACS-2 to assemble a multikinase cascade that triggers major histocompatibility complex class I (MHC-I) down-regulation: analysis using short interfering RNA and knock-out mice. J Biol Chem. 2008;283:11772–11784. doi: 10.1074/jbc.M707572200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Noviello CM, Benichou S, Guatelli JC. Cooperative binding of the class I major histocompatibility complex cytoplasmic domain and human immunodeficiency virus type 1 Nef to the endosomal AP-1 complex via its mu subunit. J Virol. 2008;82:1249–1258. doi: 10.1128/JVI.00660-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Singh RK, Lau D, Noviello CM, Ghosh P, Guatelli JC. An MHC-I cytoplasmic domain/HIV-1 Nef fusion protein binds directly to the mu subunit of the AP-1 endosomal coat complex. PLoS One. 2009;4:e8364. doi: 10.1371/journal.pone.0008364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Saksela K, Cheng G, Baltimore D. Proline-rich (PxxP) motifs in HIV-1 Nef bind to SH3 domains of a subset of Src kinases and are required for the enhanced growth of Nef+ viruses but not for down-regulation of CD4. EMBO J. 1995;14:484–491. doi: 10.1002/j.1460-2075.1995.tb07024.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hung CH, Thomas L, Ruby CE, Atkins KM, Morris NP, Knight ZA, et al. HIV-1 Nef assembles a Src family kinase-ZAP-70/Syk-PI3K cascade to downregulate cell-surface MHC-I. Cell Host Microbe. 2007;1:121–133. doi: 10.1016/j.chom.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 84.Hiyoshi M, Takahashi-Makise N, Yoshidomi Y, Chutiwitoonchai N, Chihara T, Okada M, et al. HIV-1 Nef perturbs the function, structure, and signaling of the Golgi through the Src kinase Hck. J Cell Physiol. 2011 doi: 10.1002/jcp.22825. In press. [DOI] [PubMed] [Google Scholar]
- 85.Casartelli N, Giolo G, Neri F, Haller C, Potesta M, Rossi P, et al. The Pro78 residue regulates the capacity of the human immunodeficiency virus type 1 Nef protein to inhibit recycling of major histocompatibility complex class I molecules in an SH3-independent manner. J Gen Virol. 2006;87:2291–2296. doi: 10.1099/vir.0.81775-0. [DOI] [PubMed] [Google Scholar]
- 86.Craig HM, Pandori MW, Guatelli JC. Interaction of HIV-1 Nef with the cellular dileucine-based sorting pathway is required for CD4 down-regulation and optimal viral infectivity. Proc Natl Acad Sci USA. 1998;95:11229–11234. doi: 10.1073/pnas.95.19.11229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Greenberg M, DeTulleo L, Rapoport I, Skowronski J, Kirchhausen T. A dileucine motif in HIV-1 Nef is essential for sorting into clathrin-coated pits and for downregulation of CD4. Curr Biol. 1998;8:1239–1242. doi: 10.1016/s0960-9822(07)00518-0. [DOI] [PubMed] [Google Scholar]
- 88.Piguet V, Gu F, Foti M, Demaurex N, Gruenberg J, Carpentier JL, et al. Nef-induced CD4 degradation: a diacidic-based motif in Nef functions as a lysosomal targeting signal through the binding of beta-COP in endosomes. Cell. 1999;97:63–73. doi: 10.1016/s0092-8674(00)80715-1. [DOI] [PubMed] [Google Scholar]
- 89.Faure J, Stalder R, Borel C, Sobo K, Piguet V, Demaurex N, et al. ARF1 regulates Nef-induced CD4 degradation. Curr Biol. 2004;14:1056–1064. doi: 10.1016/j.cub.2004.06.021. [DOI] [PubMed] [Google Scholar]
- 90.Schaefer MR, Wonderlich ER, Roeth JF, Leonard JA, Collins KL. HIV-1 Nef targets MHC-I and CD4 for degradation via a final common beta-COP-dependent pathway in T cells. PLoS Pathog. 2008;4:e1000131. doi: 10.1371/journal.ppat.1000131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Geyer M, Yu H, Mandic R, Linnemann T, Zheng YH, Fackler OT, et al. Subunit H of the V-ATPase binds to the medium chain of adaptor protein complex 2 and connects Nef to the endocytic machinery. J Biol Chem. 2002;277:28521–28529. doi: 10.1074/jbc.M200522200. [DOI] [PubMed] [Google Scholar]
- 92.Lindwasser OW, Smith WJ, Chaudhuri R, Yang P, Hurley JH, Bonifacino JS. A diacidic motif in human immunodeficiency virus type 1 Nef is a novel determinant of binding to AP-2. J Virol. 2008;82:1166–1174. doi: 10.1128/JVI.01874-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Arold ST, Baur AS. Dynamic Nef and Nef dynamics: how structure could explain the complex activities of this small HIV protein. Trends Biochem Sci. 2001;26:356–363. doi: 10.1016/s0968-0004(01)01846-1. [DOI] [PubMed] [Google Scholar]
- 94.Geyer M, Fackler OT, Peterlin BM. Structure—function relationships in HIV-1 Nef. EMBO Rep. 2001;2:580–585. doi: 10.1093/embo-reports/kve141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Park SH, Mrse AA, Nevzorov AA, Mesleh MF, Oblatt-Montal M, Montal M, et al. Three-dimensional structure of the channel-forming trans-membrane domain of virus protein “u” (Vpu) from HIV-1. J Mol Biol. 2003;333:409–424. doi: 10.1016/j.jmb.2003.08.048. [DOI] [PubMed] [Google Scholar]
- 96.Federau T, Schubert U, Flossdorf J, Henklein P, Schomburg D, Wray V. Solution structure of the cytoplasmic domain of the human immunodeficiency virus type 1 encoded virus protein U (Vpu) Int J Pept Protein Res. 1996;47:297–310. doi: 10.1111/j.1399-3011.1996.tb01359.x. [DOI] [PubMed] [Google Scholar]
- 97.Wray V, Federau T, Henklein P, Klabunde S, Kunert O, Schomburg D, et al. Solution structure of the hydrophilic region of HIV-1 encoded virus protein U (Vpu) by CD and 1H NMR spectroscopy. Int J Pept Protein Res. 1995;45:35–43. doi: 10.1111/j.1399-3011.1995.tb01565.x. [DOI] [PubMed] [Google Scholar]
- 98.Wittlich M, Koenig BW, Willbold D. Structural consequences of phosphorylation of two serine residues in the cytoplasmic domain of HIV-1 VpU. J Pept Sci. 2008;14:804–810. doi: 10.1002/psc.1004. [DOI] [PubMed] [Google Scholar]
- 99.Wittlich M, Koenig BW, Stoldt M, Schmidt H, Willbold D. NMR structural characterization of HIV-1 virus protein U cytoplasmic domain in the presence of dodecylphosphatidylcholine micelles. FEBS J. 2009;276:6560–6575. doi: 10.1111/j.1742-4658.2009.07363.x. [DOI] [PubMed] [Google Scholar]
- 100.Rong L, Zhang J, Lu J, Pan Q, Lorgeoux RP, Aloysius C, et al. The transmembrane domain of BST-2 determines its sensitivity to down-modulation by human immunodeficiency virus type 1 Vpu. J Virol. 2009;83:7536–7546. doi: 10.1128/JVI.00620-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Vigan R, Neil SJ. Determinants of tetherin antagonism in the transmembrane domain of the human immunodeficiency virus type 1 Vpu protein. J Virol. 2010;84:12958–12970. doi: 10.1128/JVI.01699-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Banning C, Votteler J, Hoffmann D, Koppensteiner H, Warmer M, Reimer R, et al. A flow cytometry-based FRET assay to identify and analyse protein-protein interactions in living cells. PLoS One. 2010;5:e9344. doi: 10.1371/journal.pone.0009344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kobayashi T, Ode H, Yoshida T, Sato K, Gee P, Yamamoto SP, et al. Identification of amino acids in the human tetherin transmembrane domain responsible for HIV-1 Vpu interaction and susceptibility. J Virol. 2011;85:932–945. doi: 10.1128/JVI.01668-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Skasko M, Tokarev A, Chen CC, Fischer WB, Pillai SK, Guatelli J. BST-2 is rapidly down-regulated from the cell surface by the HIV-1 protein Vpu: evidence for a post-ER mechanism of Vpu-action. Virology. 2011;411:65–77. doi: 10.1016/j.virol.2010.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Dube M, Roy BB, Guiot-Guillain P, Mercier J, Binette J, Leung G, et al. Suppression of Tetherin-restricting activity upon human immunodeficiency virus type 1 particle release correlates with localization of Vpu in the trans-Golgi network. J Virol. 2009;83:4574–4590. doi: 10.1128/JVI.01800-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hill MS, Ruiz A, Schmitt K, Stephens EB. Identification of amino acids within the second alpha helical domain of the human immunodeficiency virus type 1 Vpu that are critical for preventing CD4 cell surface expression. Virology. 2010;397:104–112. doi: 10.1016/j.virol.2009.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hao M, Maxfield FR. Characterization of rapid membrane internalization and recycling. J Biol Chem. 2000;275:15279–15286. doi: 10.1074/jbc.275.20.15279. [DOI] [PubMed] [Google Scholar]
- 108.Hsu VM, Prekeris R. Transport at the recycling endosome. Curr Opin Cell Biol. 2010;22:528–534. doi: 10.1016/j.ceb.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Grant BD, Donaldson JG. Pathways and mechanisms of endocytic recycling. Nat Rev Mol Cell Biol. 2009;10:597–608. doi: 10.1038/nrm2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Bonifacino JS, Glick BS. The mechanisms of vesicle budding and fusion. Cell. 2004;116:153–166. doi: 10.1016/s0092-8674(03)01079-1. [DOI] [PubMed] [Google Scholar]
- 111.Traub LM. Common principles in clathrin-mediated sorting at the Golgi and the plasma membrane. Biochim Biophys Acta. 2005;1744:415–437. doi: 10.1016/j.bbamcr.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 112.Bonifacino JS, Traub LM. Signals for sorting of trans-membrane proteins to endosomes and lysosomes. Annu Rev Biochem. 2003;72:395–447. doi: 10.1146/annurev.biochem.72.121801.161800. [DOI] [PubMed] [Google Scholar]
- 113.Hicke L, Dunn R. Regulation of membrane protein transport by ubiquitin and ubiquitin-binding proteins. Annu Rev Cell Dev Biol. 2003;19:141–172. doi: 10.1146/annurev.cellbio.19.110701.154617. [DOI] [PubMed] [Google Scholar]
- 114.Miranda M, Sorkin A. Regulation of receptors and transporters by ubiquitination: new insights into surprisingly similar mechanisms. Mol Interv. 2007;7:157–167. doi: 10.1124/mi.7.3.7. [DOI] [PubMed] [Google Scholar]
- 115.Craig HM, Reddy TR, Riggs NL, Dao PP, Guatelli JC. Interactions of HIV-1 nef with the mu subunits of adaptor protein complexes 1, 2, and 3: role of the dileucine-based sorting motif. Virology. 2000;271:9–17. doi: 10.1006/viro.2000.0277. [DOI] [PubMed] [Google Scholar]
- 116.Mitchell RS, Chaudhuri R, Lindwasser OW, Tanaka KA, Lau D, Murillo R, et al. Competition model for upregulation of the major histocompatibility complex class II-associated invariant chain by human immunodeficiency virus type 1 Nef. J Virol. 2008;82:7758–7767. doi: 10.1128/JVI.02668-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Roeth JF, Williams M, Kasper MR, Filzen TM, Collins KL. HIV-1 Nef disrupts MHC-I trafficking by recruiting AP-1 to the MHC-I cytoplasmic tail. J Cell Biol. 2004;167:903–913. doi: 10.1083/jcb.200407031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Bresnahan PA, Yonemoto W, Ferrell S, Williams-Herman D, Geleziunas R, Greene WC. A dileucine motif in HIV-1 Nef acts as an internalization signal for CD4 downregulation and binds the AP-1 clathrin adaptor. Curr Biol. 1998;8:1235–1238. doi: 10.1016/s0960-9822(07)00517-9. [DOI] [PubMed] [Google Scholar]
- 119.Wonderlich ER, Williams M, Collins KL. The tyrosine binding pocket in the adaptor protein 1 (AP-1) mu1 subunit is necessary for Nef to recruit AP-1 to the major histocompatibility complex class I cytoplasmic tail. J Biol Chem. 2008;283:3011–3022. doi: 10.1074/jbc.M707760200. [DOI] [PubMed] [Google Scholar]
- 120.Leonard JA, Filzen T, Carter CC, Schaefer M, Collins KL. HIV-1 Nef disrupts intracellular trafficking of MHC-I, CD4, CD8, and CD28 by distinct pathways that share common elements. J Virol. 2011 doi: 10.1128/JVI.00229-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Chen N, McCarthy C, Drakesmith H, Li D, Cerundolo V, McMichael AJ, et al. HIV-1 down-regulates the expression of CD1d via Nef. Eur J Immunol. 2006;36:278–286. doi: 10.1002/eji.200535487. [DOI] [PubMed] [Google Scholar]
- 122.Chaudhry A, Das SR, Hussain A, Mayor S, George A, Bal V, et al. The Nef protein of HIV-1 induces loss of cell surface costimulatory molecules CD80 and CD86 in APCs. J Immunol. 2005;175:4566–4574. doi: 10.4049/jimmunol.175.7.4566. [DOI] [PubMed] [Google Scholar]
- 123.Chaudhry A, Das SR, Jameel S, George A, Bal V, Mayor S, et al. HIV-1 Nef induces a Rab11-dependent routing of endocytosed immune costimulatory proteins CD80 and CD86 to the Golgi. Traffic. 2008;9:1925–1935. doi: 10.1111/j.1600-0854.2008.00802.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Zhang F, Landford WN, Ng M, McNatt MW, Bieniasz PD, Hatziioannou T. SIV Nef Proteins Recruit the AP-2 Complex to Antagonize Tetherin and Facilitate Virion Release. PLoS Pathog. 2011;7:e1002039. doi: 10.1371/journal.ppat.1002039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Jin YJ, Cai CY, Zhang X, Burakoff SJ. Lysine 144, a ubiquitin attachment site in HIV-1 Nef, is required for Nef-mediated CD4 down-regulation. J Immunol. 2008;180:7878–7886. doi: 10.4049/jimmunol.180.12.7878. [DOI] [PubMed] [Google Scholar]
- 126.daSilva LL, Sougrat R, Burgos PV, Janvier K, Mattera R, Bonifacino JS. Human immunodeficiency virus type 1 Nef protein targets CD4 to the multivesicular body pathway. J Virol. 2009;83:6578–6590. doi: 10.1128/JVI.00548-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Chaudhuri R, Mattera R, Lindwasser OW, Robinson MS, Bonifacino JS. A basic patch on alpha-adaptin is required for binding of human immunodeficiency virus type 1 Nef and cooperative assembly of a CD4-Nef-AP-2 complex. J Virol. 2009;83:2518–2530. doi: 10.1128/JVI.02227-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Klimkait T, Strebel K, Hoggan MD, Martin MA, Orenstein JM. The human immunodeficiency virus type 1-specific protein vpu is required for efficient virus maturation and release. J Virol. 1990;64:621–629. doi: 10.1128/jvi.64.2.621-629.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Varthakavi V, Smith RM, Martin KL, Derdowski A, Lapierre LA, Goldenring JR, et al. The pericentriolar recycling endosome plays a key role in Vpu-mediated enhancement of HIV-1 particle release. Traffic. 2006;7:298–307. doi: 10.1111/j.1600-0854.2005.00380.x. [DOI] [PubMed] [Google Scholar]
- 130.Van Damme N, Guatelli J. HIV-1 Vpu inhibits accumulation of the envelope glycoprotein within clathrin-coated, Gag-containing endosomes. Cell Microbiol. 2008;10:1040–1057. doi: 10.1111/j.1462-5822.2007.01101.x. [DOI] [PubMed] [Google Scholar]
- 131.Schubert U, Ferrer-Montiel AV, Oblatt-Montal M, Henklein P, Strebel K, Montal M. Identification of an ion channel activity of the Vpu transmembrane domain and its involvement in the regulation of virus release from HIV-1-infected cells. FEBS Lett. 1996;398:12–18. doi: 10.1016/s0014-5793(96)01146-5. [DOI] [PubMed] [Google Scholar]
- 132.Goffinet C, Allespach I, Homann S, Tervo HM, Habermann A, Rupp D, et al. HIV-1 antagonism of CD317 is species specific and involves Vpu-mediated proteasomal degradation of the restriction factor. Cell Host Microbe. 2009;5:285–297. doi: 10.1016/j.chom.2009.01.009. [DOI] [PubMed] [Google Scholar]
- 133.Andrew AJ, Miyagi E, Strebel K. Differential effects of human immunodeficiency virus type 1 Vpu on the stability of BST-2/tetherin. J Virol. 2011;85:2611–2619. doi: 10.1128/JVI.02080-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Schindler M, Rajan D, Banning C, Wimmer P, Koppensteiner H, Iwanski A, et al. Vpu serine 52 dependent counteraction of tetherin is required for HIV-1 replication in macrophages, but not in ex vivo human lymphoid tissue. Retrovirology. 2010;7:1. doi: 10.1186/1742-4690-7-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Pardieu C, Vigan R, Wilson SJ, Calvi A, Zang T, Bieniasz P, et al. The RING-CH ligase K5 antagonizes restriction of KSHV and HIV-1 particle release by mediating ubiquitin-dependent endosomal degradation of tetherin. PLoS Pathog. 2010;6:e1000843. doi: 10.1371/journal.ppat.1000843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Iwabu Y, Fujita H, Kinomoto M, Kaneko K, Ishizaka Y, Tanaka Y, et al. HIV-1 accessory protein Vpu internalizes cell-surface BST-2/tetherin through transmem-brane interactions leading to lysosomes. J Biol Chem. 2009;284:35060–35072. doi: 10.1074/jbc.M109.058305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Hauser H, Lopez LA, Yang SJ, Oldenburg JE, Exline CM, Guatelli JC, et al. HIV-1 Vpu and HIV-2 Env counteract BST-2/tetherin by sequestration in a peri-nuclear compartment. Retrovirology. 2010;7:51. doi: 10.1186/1742-4690-7-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Kumar KG, Tang W, Ravindranath AK, Clark WA, Croze E, Fuchs SY. SCF(HOS) ubiquitin ligase mediates the ligand-induced down-regulation of the interfer-on-alpha receptor. EMBO J. 2003;22:5480–5490. doi: 10.1093/emboj/cdg524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Schmidt S, Fritz JV, Bitzegeio J, Fackler OT, Keppler OT. HIV-1 Vpu blocks recycling and biosynthetic transport of the intrinsic immunity factor CD317/ tetherin to overcome the virion release restriction. MBio. 2011;2:e00036-11. doi: 10.1128/mBio.00036-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Janvier K, Pelchen-Matthews A, Renaud JB, Caillet M, Marsh M, Berlioz-Torrent C. The ESCRT-0 component HRS is required for HIV-1 Vpu-mediated BST-2/tetherin down-regulation. PLoS Pathog. 2011;7:e1001265. doi: 10.1371/journal.ppat.1001265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Le Tortorec A, Neil SJ. Antagonism to and intracellular sequestration of human tetherin by the human immunodeficiency virus type 2 envelope glycoprotein. J Virol. 2009;83:11966–11978. doi: 10.1128/JVI.01515-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Serra-Moreno R, Jia B, Breed M, Alvarez X, Evans DT. Compensatory changes in the cytoplasmic tail of gp41 confer resistance to tetherin/BST-2 in a pathogenic nefdeleted SIV. Cell Host Microbe. 2011;9:46–57. doi: 10.1016/j.chom.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Schubert U, Strebel K. Differential activities of the human immunodeficiency virus type 1-encoded Vpu protein are regulated by phosphorylation and occur in different cellular compartments. J Virol. 1994;68:2260–2271. doi: 10.1128/jvi.68.4.2260-2271.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Estrabaud E, Le Rouzic E, Lopez-Verges S, Morel M, Belaidouni N, Benarous R, et al. Regulated degradation of the HIV-1 Vpu protein through a betaTrCP-independent pathway limits the release of viral particles. PLoS Pathog. 2007;3:e104. doi: 10.1371/journal.ppat.0030104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Belaidouni N, Marchal C, Benarous R, Besnard-Guerin C. Involvement of the betaTrCP in the ubiquitination and stability of the HIV-1 Vpu protein. Biochem Biophys Res Commun. 2007;357:688–693. doi: 10.1016/j.bbrc.2007.03.195. [DOI] [PubMed] [Google Scholar]
- 146.Grzesiek S, Bax A, Clore GM, Gronenborn AM, Hu JS, Kaufman J, et al. The solution structure of HIV-1 Nef reveals an unexpected fold and permits delineation of the binding surface for the SH3 domain of Hck tyrosine protein kinase. Nat Struct Biol. 1996;3:340–345. doi: 10.1038/nsb0496-340. [DOI] [PubMed] [Google Scholar]
- 147.Geyer M, Munte CE, Schorr J, Kellner R, Kalbitzer HR. Structure of the anchor-domain of myristoylated and non-myristoylated HIV-1 Nef protein. J Mol Biol. 1999;289:123–138. doi: 10.1006/jmbi.1999.2740. [DOI] [PubMed] [Google Scholar]
- 148.Willbold D, Hoffmann S, Rosch P. Secondary structure and tertiary fold of the human immunodeficiency virus protein U (Vpu) cytoplasmic domain in solution. Eur J Biochem. 1997;245:581–588. doi: 10.1111/j.1432-1033.1997.t01-1-00581.x. [DOI] [PubMed] [Google Scholar]
- 149.Coleman SH, Hitchin D, Noviello CM, Guatelli JC. HIV-1 Nef stabilizes AP-1 on membranes without inducing ARF1-independent de novo attachment. Virology. 2006;345:148–155. doi: 10.1016/j.virol.2005.09.045. [DOI] [PubMed] [Google Scholar]
- 150.Schindler M, Wurfl S, Benaroch P, Greenough TC, Daniels R, Easterbrook P, et al. Down-modulation of mature major histocompatibility complex class II and up-regulation of invariant chain cell surface expression are well-conserved functions of human and simian immunodeficiency virus nef alleles. J Virol. 2003;77:10548–10556. doi: 10.1128/JVI.77.19.10548-10556.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Le Gall S, Erdtmann L, Benichou S, Berlioz-Torrent C, Liu L, Benarous R, et al. Nef interacts with the mu subunit of clathrin adaptor complexes and reveals a cryptic sorting signal in MHC I molecules. Immunity. 1998;8:483–495. doi: 10.1016/s1074-7613(00)80553-1. [DOI] [PubMed] [Google Scholar]
- 152.Greenberg ME, Iafrate AJ, Skowronski J. The SH3 domain-binding surface and an acidic motif in HIV-1 Nef regulate trafficking of class I MHC complexes. EMBO J. 1998;17:2777–2789. doi: 10.1093/emboj/17.10.2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Mangasarian A, Piguet V, Wang JK, Chen YL, Trono D. Nef-induced CD4 and major histocompatibility complex class I (MHC-I) down-regulation are governed by distinct determinants: N-terminal alpha helix and proline repeat of Nef selectively regulate MHC-I trafficking. J Virol. 1999;73:1964–1973. doi: 10.1128/jvi.73.3.1964-1973.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Akari H, Arold S, Fukumori T, Okazaki T, Strebel K, Adachi A. Nef-induced major histocompatibility complex class I down-regulation is functionally dissociated from its virion incorporation, enhancement of viral infectivity, and CD4 down-regulation. J Virol. 2000;74:2907–2912. doi: 10.1128/jvi.74.6.2907-2912.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Williams M, Roeth JF, Kasper MR, Filzen TM, Collins KL. Human immunodeficiency virus type 1 Nef domains required for disruption of major histocompatibility complex class I trafficking are also necessary for coprecipitation of Nef with HLA-A2. J Virol. 2005;79:632–636. doi: 10.1128/JVI.79.1.632-636.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Guy B, Kieny MP, Riviere Y, Le Peuch C, Dott K, Girard M, et al. HIV F/3′ orf encodes a phosphorylated GTP-binding protein resembling an oncogene product. Nature. 1987;330:266–269. doi: 10.1038/330266a0. [DOI] [PubMed] [Google Scholar]
- 157.Rhee SS, Marsh JW. Human immunodeficiency virus type 1 Nef-induced down-modulation of CD4 is due to rapid internalization and degradation of surface CD4. J Virol. 1994;68:5156–5163. doi: 10.1128/jvi.68.8.5156-5163.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Chaudhuri R, Lindwasser OW, Smith WJ, Hurley JH, Bonifacino JS. Downregulation of CD4 by human immunodeficiency virus type 1 Nef is dependent on clathrin and involves direct interaction of Nef with the AP2 clathrin adaptor. J Virol. 2007;81:3877–3890. doi: 10.1128/JVI.02725-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Henderson WW, Ruhl R, Lewis P, Bentley M, Nelson JA, Moses AV. Human immunodeficiency virus (HIV) type 1 Vpu induces the expression of CD40 in endothelial cells and regulates HIV-induced adhesion of B-lymphoma cells. J Virol. 2004;78:4408–4420. doi: 10.1128/JVI.78.9.4408-4420.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Willey RL, Buckler-White A, Strebel K. Sequences present in the cytoplasmic domain of CD4 are necessary and sufficient to confer sensitivity to the human immunodeficiency virus type 1 Vpu protein. J Virol. 1994;68:1207–1212. doi: 10.1128/jvi.68.2.1207-1212.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Vincent MJ, Raja NU, Jabbar MA. Human immunodeficiency virus type 1 Vpu protein induces degradation of chimeric envelope glycoproteins bearing the cytoplasmic and anchor domains of CD4: role of the cytoplasmic domain in Vpu-induced degradation in the endoplasmic reticulum. J Virol. 1993;67:5538–5549. doi: 10.1128/jvi.67.9.5538-5549.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Paul M, Jabbar MA. Phosphorylation of both phospho-acceptor sites in the HIV-1 Vpu cytoplasmic domain is essential for Vpu-mediated ER degradation of CD4. Virology. 1997;232:207–216. doi: 10.1006/viro.1997.8541. [DOI] [PubMed] [Google Scholar]