

Abstract
Apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC) is an important component of the inflammasome, functioning as an adaptor protein that facilitates the recruitment and activation of procaspases that in turn promote the maturation of interleukin-1β (IL-1β) and IL-18. Despite initial focus on the inflammatory properties of ASC there is emerging evidence that highlights the importance of ASC in facilitating adaptive immune responses. However, the cellular and molecular basis for the involvement of ASC in adaptive immunity remains largely unexplored. We have previously demonstrated that activated ASC-deficient T cells have dampened proliferative responses. We have therefore explored the underlying cellular mechanism(s) by which ASC regulates T-cell proliferation. We show that under activating conditions (anti-CD3/CD28 stimulation) in bulk T-cell cultures the presence of ASC−/− CD4+ T cells is sufficient to suppress the proliferative responses of neighbouring T cells. Furthermore, ASC−/− CD4+ T cells upon activation exhibit a suppressive cytokine profile, with elevated production of IL-10 and reduced secretion of T helper type 1 cytokines, interferon-γ and IL-2. This increase in IL-10 secretion within the activated ASC−/− CD4+ T-cell compartment was not associated with a proportional increase in conventional Foxp3+ regulatory T (Treg) cells. Interestingly, when equal numbers of fluorescence-activated cell sorted ASC+/+ and ASC−/− Treg cells (CD4+ CD44intermediate/high CD25+) were activated in vitro, the ASC−/− fraction produced significantly more IL-10 than their wild-type counterparts, suggesting that ASC−/− Treg cells have greater suppressive capacity. Collectively, these results imply that the ASC may influence the development and functioning of Treg cells.
Keywords: apoptosis-associated speck-like protein containing a caspase recruitment domain, interleukin-10, regulatory T cells, T cells
Introduction
Apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC) is an integral component of the inflammasome, a cytosolic multiprotein platform that facilitates the activation of pro-inflammatory caspases, which in turn promote the maturation and subsequent secretion of interleukin-1β (IL-1β) and IL-18.1,2 ASC is a simple adaptor protein with two linked protein–protein interaction domains of the death domain superfamily: an N-terminal pyrin/PAAD death domain and a C-terminal caspase recruitment domain, which interact with the different NOD-like receptors, the sensory elements of the inflammasome and pro-caspase-1, respectively.3–5 These two domains enable ASC to function as an essential link between the sensor protein and effector molecules during inflammasome assembly. The Nacht domain, leucine-rich repeat and PYD-containing protein-3 (NALP3) inflammasome, comprising the regulatory subunit NALP3, the adaptor ASC and the effector subunit caspase-1, represents the best characterized inflammasome. So far, three other inflammasome prototypes have been described: the NALP1 inflammasome, the IL-1β converting enzyme protease-activating factor (IPAF) inflammasome and recently the absent in melanoma 2 (AIM2) inflammasome.6
To date, much attention has focused around the inflammatory properties of ASC; however, recent evidence has highlighted the importance of ASC in adaptive immune responses. Studies using ASC−/− mice have revealed its significance in adaptive immunity in several physiological and pathological situations. It seems that ASC is essential to mount protective T-cell and B-cell immunity against influenza virus infection.7 Expression of ASC on dendritic cells (DCs) has also been described as being critical in T-cell priming and the subsequent induction of both antigen-specific cellular and humoral immunity and on the onset of collagen-induced arthritis.8 Furthermore, ASC has also been strongly linked to modulating joint inflammation in antigen-induced arthritis by affecting the induction of antigen-specific cellular immunity.9 Finally, ASC has been shown to contribute to disease progression in experimental autoimmune encephalomyelitis.10 However, the cellular and molecular basis behind the importance of ASC in adaptive immunity remains largely unexplored.
We have previously described how ASC−/− T cells exhibit impaired proliferative capacity in response to both antigen-specific and non-specific (anti-CD3/CD28) stimulation ex vivo.9 In this study we explored the cellular basis for the influence of ASC on T-cell proliferation and subsequent effector function.
Materials and methods
Mice
ASC−/− mice1 and NALP3−/− mice11 were backcrossed into the C57BL/6 background for at least nine generations and were compared with wild-type (WT) littermates in this study. Mice were bred under conventional, non-specific pathogen-free conditions. Mice between 8 and 12 weeks of age were used for experiments. All experiments were carried out in agreement with Institutional and Swiss regulations.
T-cell proliferation
CD3+ T cells were enriched from splenocyte suspensions by negative selection using the EasySep mouse T-cell enrichment kit (StemCell Technologies, Grenoble, France). Splenic CD4+ and CD8+ T-cell fractions were purified using magnetic antibody cell sorting CD4+ and CD8+ MicroBeads, respectively (> 95%) (Miltenyi Biotec, Bergisch Glaabach, Germany). T cells (2 × 105/200 μl per well) were cultured in 96-well plates previously coated with anti-mouse CD3 (2 μg/ml, clone 145-2C11; eBioscience, San Diego, CA) and anti-CD28 (2 μg/ml, clone 37-51; eBioscience). For co-culture experiments, different isolated T-cell fractions were plated in 96-well plates at a 1 : 1 ratio (1 × 105 T cells per fraction in 200 μl).
For IL-10 neutralizing assays, anti-mouse IL-10 (1 μg/ml, clone JES5-2A5; eBioscience) was added to cultures. For some experiments recombinant mouse IL-10 was added to T-cell cultures (1 ng/ml; eBioscience). Proliferation was assessed by pulsing cultures overnight with 0·5 μCi/well of [3H]thymidine overnight and performing scintillation counts.
Cytokine quantification
Culture supernatants were harvested daily over 4 days. Expression levels of interferon-γ, IL-2, IL-4, IL-5, IL-10, IL-17 and tumour necrosis factor-α in supernatant samples were quantified by means of a cytofluorimetry-based ELISA system according to the manufacturer's instructions (Flowcytomix; Bender Medsystem GmbH, Vienna, Austria).
Flow cytometry
Cells were suspended in FACS buffer (3% fetal calf serum, 5 mm EDTA in PBS). Cells were incubated with conjugated monoclonal antibodies in the presence of Fc blockers (clone 2.4G2). All data acquisition was performed on a FACSCalibur flow cytometer (Becton-Dickinson, San Jose, CA). The anti-mouse monoclonal antibodies used (Becton-Dickinson) were: CD4-FITC, CD44-phycoerythrin, CD62L-peridinin chlorophyll protein complex, CD25-allophycocyanin and Foxp3-phycoerythrin.
T cells were identified as CD3+ and either CD4+ CD8− for CD4 T cells or CD4− CD8+ for CD8+ T cells. CD44, CD62L and CD25 expression was used to assess T-cell activation status. For FACS, regulatory T (Treg) cells were characterized as CD4+ CD44intermediate/high CD25+ cells.12
Statistical analysis
All values were expressed as the mean ± standard error of the mean (SEM). Statistical analysis was calculated by the two-tailed unpaired t-test using graphpad prism software (GraphPad Software, La Jolla, CA). A P-value < 0·05 was considered statistically significant.
Results
To confirm that the proliferation inhibition observed among ASC−/− CD3+ T cells in response to anti-CD3/CD28 stimulation9 is specifically linked to ASC deficiency and so not a consequence of a general NALP3 inflammasome dysfunction, we initially compared the proliferative response of ASC−/− and NALP3−/− CD3+ T cells. When compared with ASC−/− CD3+ T cells, NALP3−/− CD3+ T cells did not display an impaired proliferative response to anti-CD3/CD28 stimulation (Fig. 1a), suggesting that this ASC-associated T-cell defect is NALP3 inflammasome-independent. We next investigated whether this ASC−/− T-cell phenomenon is restricted to a specific T-cell subset or if it affects T cells more globally. Therefore purified CD4+ and CD8+ T cells from ASC+/+ and ASC−/− mice were stimulated separately with plate-bound anti-CD3/CD28 and their proliferation was assessed over time. When compared with similarly stimulated WT controls, ASC−/− CD4+ (Fig. 1b) and CD8+ (Fig. 1c) T cells displayed no impairment in their proliferative response upon activation. Furthermore, no alteration in the regulation of T-cell activation markers (CD44, CD62L and CD25) was observed on ASC−/− CD4+ (Fig. 1d) and CD8+ (Fig. 1e) T cells following activation compared with WT controls.
Figure 1.
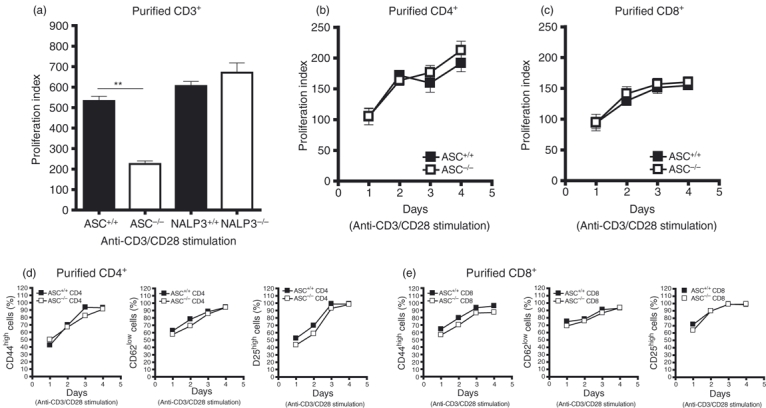
Purified ASC−/− CD4+ and CD8+ T cells when activated separately do not show any impairment in proliferation or CD44/CD62L/CD25 expression. (a) Proliferation of purified ASC−/− and NALP3−/− splenic T cells in response to stimulation with 2 μg/ml anti-CD3/CD28 at day 2 was assessed by [3H]thymidine incorporation. Graph is representative of two independent experiments. T cells were purified from splenocyte preparations pooled from two mice per group. Proliferation kinetics of purified ASC−/− splenic CD4+ (b) and CD8+ T cells (c) following stimulation with 2 μg/ml of anti-CD3/CD28 was determined by [3H]thymidine incorporation. Graphs are representative of two independent experiments. CD4+ and CD8+ T cells were purified from splenocyte preparations pooled from two mice per group. The regulation of activation markers (CD44, CD62L and CD25) on these anti-CD3/CD28 stimulated ASC+/+ and ASC−/− CD4+ (d) and CD8+ (e) T cells over the 4 days when the time–course was evaluated by flow cytometry. Graphs represent data pooled from eight different mice. The proliferation index (a–c) was calculated as the ratio of counts/min from stimulated cells over counts/min from unstimulated cells. Error bars denoted as mean values ± SEM and are representative of cultures performed in triplicate. For P-values; **P < 0·01.
We next set up CD4+ and CD8+ T-cell co-culture experiments to determine whether stimulation of purified ASC−/− CD4+ and CD8+ T cells together (1 : 1 ratio) replicates the proliferation defect observed in anti-CD3/CD28-stimulated ASC−/− bulk T-cell cultures. Indeed, when purified ASC−/− CD4+ and CD8+ T cells were stimulated for 2 days with anti-CD3/CD28 in a co-culture assay, T-cell proliferation was inhibited compared with similarly activated ASC+/+ CD4+ and CD8+ T-cell co-cultures (Fig. 2a). Working on the hypothesis that in the co-culture set-up one ASC−/− T-cell subset is able to suppress the proliferation of the other when activated, we next attempted to identify this suppressive ASC−/− T-cell subset. ASC+/+ and ASC−/− CD4+ and CD8+ T cells were purified and co-cultured with different purified T-cell fractions under activation conditions (anti-CD3/CD28 stimulation) (Fig. 2b). In this set up, significant inhibition of proliferation was observed in co-cultures that included ASC−/− CD4+ T cells. A slight, but significant reduction was also noted in some co-cultures that included ASC−/− CD8+ T cells. When the expression of CD25 (Fig. 2c), CD44 and CD62L (data not shown) were assessed in co-cultures where T-cell proliferation was impaired, no activation-induced differences were observed. Collectively, these results suggest that activated ASC−/− CD4+ T cells are able to suppress activation-induced proliferation of other neighbouring activated T cells. Furthermore, as no changes in cell surface expression of T-cell activation markers were noted following anti-CD3/CD28 stimulation we speculate that T-cell activation in the presence ASC−/− CD4+ T cells occurs normally and that inhibition of proliferative responses occurs at the phase of T-cell clonal expansion.
Figure 2.
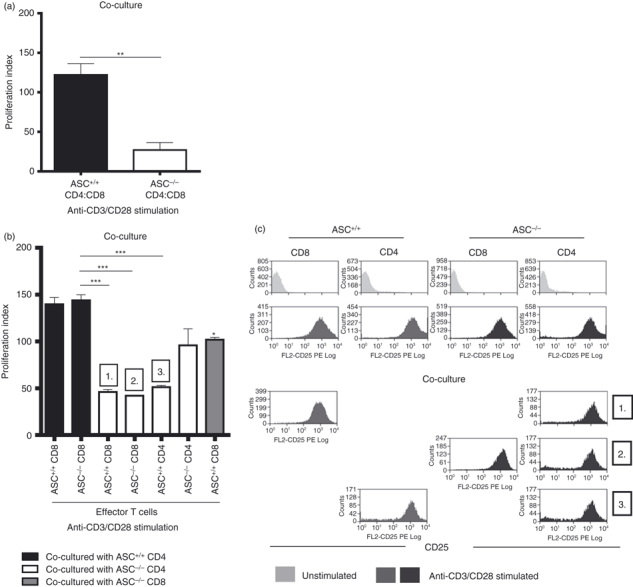
Activation of ASC−/− CD4+ T cells in a T-cell co-culture system results in reduced T-cell proliferation. (a) Proliferation of ASC+/+ and ASC−/− co-cultured T cells (purified CD8+ and CD4+ T cells only) in response to stimulation with 2 μg/ml anti-CD3/CD28 at day 2 was assessed by [3H]thymidine incorporation. Graph is representative of at least three independent experiments. T cells were purified from splenocyte preparations pooled from two mice per group. (b) Purified ASC+/+ and ASC−/− purified CD8+ and CD4+ cells were co-cultured in different combinations. At day 2, T-cell proliferation was assessed by [3H]thymidine incorporation and up-regulation of CD25 following activation was evaluated by flow cytometry (c). Graph and plots are representative of two independent experiments. T cells were purified from splenocyte preparations pooled from two mice per group. The proliferation index was calculated as the ratio of counts/min from stimulated cells over counts/min from unstimulated cells. Error bars denoted as mean values ± SEM and are representative of cultures performed in triplicate. For P-values; *P < 0·05, **P < 0·01 and ***P < 0·001.
One possible mechanism for the observed suppression of T-cell proliferation after CD3/CD28 stimulation in the presence of activated ASC−/− CD4+ T cells could be the secretion of suppressive soluble factor(s). To test this hypothesis we used WT CD4+ (Fig. 3a) and CD8+ T cells (Fig. 3b) as effector T cells. These cells were then activated (anti-CD3/CD28 stimulation) in the presence of supernatant derived from activated WT or ASC−/− CD4+ T cells. T cells stimulated in the presence of activated ASC−/− CD4+ T-cell-derived supernatant proliferated significantly less than those stimulated in the presence of supernatants derived from ASC+/+ CD4+ T cells. These results suggest that ASC−/− CD4+ T cells once activated secrete soluble factor(s) that have suppressive potential. To characterize the suppressive factor(s) involved in ASC−/− CD4+ T-cell mediated suppression, we compared the cytokine secretion profile of activated ASC+/+ and ASC−/− CD4+ T cells. Interestingly, we found that anti-CD3/CD28-activated ASC−/− CD4+ T cells produced significantly less interferon-γ over a 4-day time–course experiment when compared with their ASC+/+ counterparts (Fig. 3c). Interleukin-2 concentrations were also decreased in activated ASC−/− CD4+ T-cell cultures at day 2, which represented peak secretion of IL-2 for WT controls. Furthermore, activated ASC−/− CD4+ produced high levels of IL-10, whereas IL-10 levels in the activated ASC+/+ CD4+ T-cell cultures were undetectable. The concentrations of IL-4 and IL-5 detected in the activated CD4+ T-cell cultures were similar between the ASC+/+ and ASC−/− groups. Interleukin-6, IL-17 and tumour necrosis factor-α were undetectable in any of the culture groups. Based on these findings, we speculated that IL-10 is involved at least in part in suppressing the proliferative response of effector T cells in the context of activated ASC−/− CD4+ T-cell-mediated suppression. To test this hypothesis, we set up ASC+/+ and ASC−/− T-cell co-cultures (CD4 and CD8 T cells) in the presence of anti-CD3/CD28 and IL-10 neutralizing antibodies.13 Inclusion of IL-10 neutralizing antibodies in the ASC−/− T-cell co-cultures was able to rescue T cells from activation-induced proliferation inhibition, though this restorative effect was not complete (Fig. 3d), suggesting that other IL-10-independent mechanisms may be involved. To investigate the specific effect of IL-10 of ASC+/+ and ASC−/− T-cell cultures, purified CD4+ and CD8+ T cells were activated with anti-CD3/CD28 in the presence of exogenous recombinant IL-10. In the presence of exogenous IL-10 (1 ng/ml) activation-induced proliferation of ASC+/+ CD4+ and CD8+ and ASC−/− CD8+ T-cell cultures was significantly reduced (Fig. 3e). Inhibition of activation-induced T-cell proliferation was also achieved in the presence of 0·1 ng/ml of exogenous IL-10; however, the differences observed were not as striking as with 1 ng/ml exogenous IL-10 (data not shown). Interestingly, the addition of exogenous IL-10 appeared to have no influence on the proliferation of ASC−/− CD4+ T cells, at least at concentrations sufficient to inhibit the proliferation of the other T-cell fractions.
Figure 3.
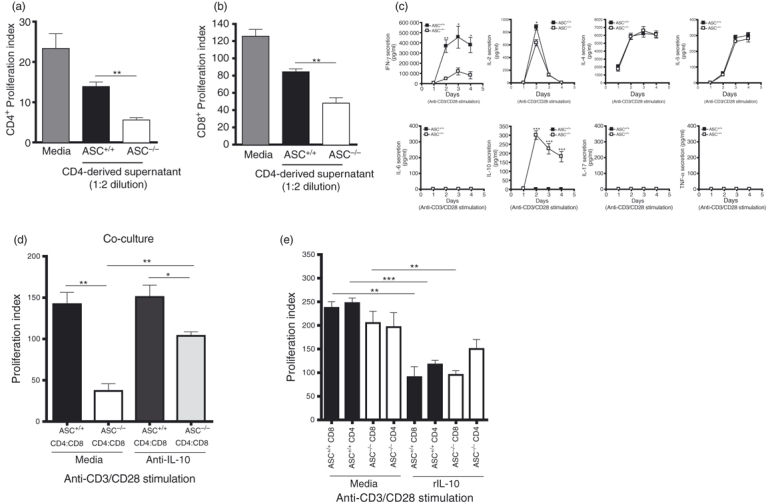
Interleukin-10 (IL-10) produced by activated ASC−/− CD4+ T cells in a T-cell co-culture system suppresses T-cell proliferation: Purified wild-type (WT) CD4+ (a) and CD8+ cells (b) were used as effector T cells and stimulated with 2 μg/ml anti-CD3/CD28 in the presence of supernatant derived from either similarly 2-day-activated ASC+/+ or ASC−/− CD4+ T cells. At day 2, T-cell proliferation was assessed by [3H]thymidine incorporation. The proliferation index was calculated as the ratio of counts/min from stimulated cells over counts/min from unstimulated cells. Graphs are representative of two independent experiments. T cells were purified from splenocyte preparations pooled from two mice per group. (c) Purified ASC+/+ and ASC−/− CD8+ and CD4+ T cells were stimulated with 2 μg/ml anti-CD3/CD28. Culture supernatant was assayed for the presence of interferon-γ (IFN-γ), IL-2, IL-4, IL-5, IL-10, IL-17 and tumour necrosis factor-α (TNF-α) over a time–course. Graphs are representative of at least three independent experiments. T cells were purified from splenocyte preparations pooled from two mice per group. (d) Proliferation of ASC+/+ and ASC−/− co-cultured T cells (purified CD8+ and CD4+ T cells only) in response to stimulation with 2 μg/ml of anti-CD3/CD28 and in the presence of IL-10 neutralizing antibodies (1 μg/ml) at day 2 was assessed by [3H]thymidine incorporation. Graph is representative of two independent experiments. T cells were purified from splenocyte preparations pooled from two mice per group. (e) Proliferation of purified ASC+/+ and ASC−/− purified CD8+ and CD4+ T cells in response to stimulation with 2 μg/ml of anti-CD3/CD28 and in the presence of exogenous recombinant IL-10 (1 ng/ml) was assessed at day 2 by [3H]thymidine incorporation. The proliferation indices (d, e) were calculated as the ratio of counts/min from stimulated cells over counts/min from unstimulated cells. Graphs are representative of two independent experiments. T cells were purified from splenocyte preparations pooled from two mice per group. Error bars denoted as mean values ± SEM and are representative of cultures performed in triplicate. For P-values; *P < 0·05, **P < 0·01 and ***P < 0·001.
The CD4+ Foxp3+ regulatory T cells are known to suppress T-cell function via IL-10 secretion14 and for this reason we considered the possibility that elevated numbers of CD4+ Foxp3+ Treg cells within the ASC−/− CD4+ compartment were responsible for mediating suppression of T-cell proliferation in our T-cell co-cultures. We first investigated Treg cell population dynamics within both purified ASC+/+ and ASC−/− CD4+ T-cell cultures following activation (Fig. 4a). Although Treg cell percentages increased following activation within both ASC+/+ and ASC−/− CD4+ fractions, no significant differences were observed between both groups. However, there was a trend towards slightly elevated percentages of Treg cells in the ASC−/− CD4+ fraction. Similarly, following arthritis induction (inflammation), Treg cell percentages increased in both ASC+/+ and ASC−/− mice when compared with steady-state levels in naive animals (Fig. 4b). Although there was also a trend towards increased levels of Treg cells in arthritic ASC−/− mice, the difference was not statistically significant. We next investigated whether ASC−/− Treg cells intrinsically have more suppressive potential. Equal numbers of FACS-sorted splenic CD4+ cells and Treg cells (CD4+ CD44intermediate/high CD25+ cells)12 from naive ASC+/+ and ASC−/− mice were cultured in the presence of anti-CD3/CD28 (Fig. 4c). FACS-sorted ASC−/− Treg cells were shown to secrete significantly greater amounts of IL-10 compared with similarly treated ASC+/+ controls. No significant differences in IL-10 production were observed between isolated ‘non-Treg’ cells from ASC+/+ and ASC−/− mice upon stimulation (data not shown).
Figure 4.
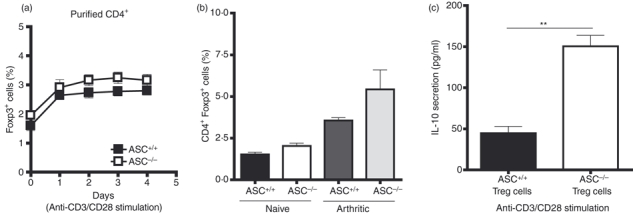
Activated ASC−/− CD4+ regulatory T (Treg) cells produced increases amounts of interleukin-10 (IL-10): (a) purified ASC+/+ and ASC−/− splenic CD4+ were cultured with 2 μg/ml anti-CD3/CD28. The population kinetics of Foxp3+ cells within both culture conditions was monitored by flow cytometry. Graph is representative of two independent experiments. T cells were purified from splenocyte preparations pooled from two mice per group. (b) The frequency of lymph node CD4+ Foxp3+ cells was compared between naive and arthritic ASC+/+ and ASC−/− using flow cytometry (n = 6). (c) FACS-sorted Treg cells (CD4+ CD44intermediate/high CD25+ cells) was activated in the presence of 3 μg/ml of anti-CD3/CD28 and culture supernatants were assayed for IL-10 levels at day 2 following activation. Graph is representative of two independent experiments. T cells were purified from splenocyte preparations pooled from two mice per group. Error bars denoted as mean values ± SEM and are representative of cultures performed in triplicate. For P-values; **P < 0·01.
Discussion
Although an inflammatory role for the ASC adaptor is widely acknowledged, its significance in the adaptive immune response is not well understood. We have previously reported an important role of ASC in regulating activation-induced T-cell proliferation.9 In this study we further demonstrate that in the context of ASC deficiency, activation of a CD4+ regulatory T-cell population(s) results in the production of high levels of IL-10, which contributes toward the suppression of activation-induced proliferative responses of neighbouring T cells. Although the frequency of ASC−/− CD4+ Foxp3+ Treg cells remained unchanged relative to WT controls under both steady-state and inflammatory conditions, our data indicate that ASC−/− Treg cells (defined as CD4+ CD44intermediate/high CD25+) have a more suppressive phenotype. We would speculate that an ASC-deficient in vivo environment skews T-cell development towards unique population(s) of suppressive T cells, though the basis of this enhanced CD4+ suppressive activity in ASC−/− mice remains unexplored.
The impact of ASC on T-cell function has recently been highlighted in different murine models of autoimmune disease. ASC has been implicated in the pathogenesis of collagen-induced arthritis, with ASC−/− mice protected against collagen-induced arthritis whereas NALP3−/− and Capase-1−/− mice were susceptible.8 The authors demonstrated reduced antigen-induced CD4+ T-cell activation and subsequent proliferation in the presence of ASC−/− DCs. Direct ligation of CD3/CD28 induced normal proliferative responses from ASC−/− CD4+ T cells, suggesting that perhaps the ASC adaptor protein is more critical on DCs than on T cells in the context of T-cell activation. We also noted no reduction in anti-CD3/CD28-specific proliferation when purified CD4+ and CD8+ T cells were stimulated separately. This defective ability to prime T-cell responses by ASC−/− DCs reported by the authors was not associated with any alterations in cell surface expression of MHCII and CD86, suggesting that perhaps the defective T-cell priming by DCs in the presence of ASC deficiency represents a downstream impairment in antigen processing, intracellular trafficking or peptide loading on MHC molecules and not a defect in initial antigen uptake and DC maturation. However, our in vitro results would indicate that ASC−/− DCs are not necessary for impaired activation-induced ASC−/− T-cell proliferation at least in the context of anti-CD3/CD28 stimulation, because we were able to observe reduced proliferative responses from co-cultured ASC−/− CD4+ and CD8+ T cells following anti-CD3/CD28 ligation. It is possible that the authors failed to identify an intrinsic T-cell modulation in ASC−/− mice, because none of their experiments were aimed at investigating this ASC−/− T-cell phenotype. When considering these results collectively, we could further speculate that along with a functional impairment in the ability of ASC−/− DCs to prime effector T cells, in ASC−/− mice there exists a differentiation bias among the CD4+ T-cell compartment that results in the development of suppressive CD4+ T-cell subset(s). The physiological significance and contribution of these potential mechanisms in autoimmunity remains to be investigated.
Experimental autoimmune encephalomyelitis (T-cell-dependent model) is another disease model in which reduced antigen-specific T-cell responses are seen in ASC−/− mice.10 From assessing the presence of adoptively transferred ASC−/− CD4+ T cells in peripheral sites (blood, lymph node and spleen of lethally irradiated WT recipients) the authors conclude that ASC deficiency confers a survival disadvantage on CD4+ T cells. They also demonstrated that fewer antigen-specific T cells are present in the draining lymph nodes and central nervous system of diseased ASC−/− mice. However, the authors have not convincingly ruled out a T-cell trafficking defect among ASC−/− T cells. A more systematic look at the frequency of adoptively transferred ASC−/− CD4+ T cells in the periphery of lethally irradiated WT recipients would need to be undertaken to confirm that these cells are not sequestered anywhere in the periphery. If survival and subsequently cell death did apply then one would expect that adoptively transferred ASC−/− CD4+ T-cell numbers would be reduced in all peripheral organs. We have previously demonstrated no increase in apoptotic markers in vitro and in vivo at the level of antigen-primed bulk ASC−/− splenocytes.9 However, we would have to specifically assess apoptosis levels within similarly treated T-cell populations to exclude the possibility that ASC−/− T cells have a survival defect.
Kinetic experiments revealed that IL-10 is secreted by purified ASC−/− CD4 T cells following activation. Furthermore, this endogenous IL-10 production by ASC−/− CD4+ T cells accounts in part for the low proliferative capacity of effector T cells in response to CD3/CD28 stimulation when co-cultured with ASC−/− CD4+ T cells, as proliferation of these T cells was augmented in the presence of IL-10 neutralizing antibodies. This finding is consistent with our observation that exogenous IL-10 prevents anti-CD3/CD28-specific T-cell proliferation and the observations of previous studies that indicate that IL-10 prevents or inhibits T-cell proliferation.14,15 Interestingly, exogenous IL-10, at least at the concentrations used for this study, did not have a significant effect on the proliferative response of ASC−/− CD4+ T cells, considering they themselves produce large amounts of IL-10. As IL-10 has been reported to promote the induction of Foxp3+ regulatory T cells,16 it may act as a growth factor for ASC−/− CD4+ T cells. However, in such a situation, one would expect elevated proliferation and an increase in Foxp3+ numbers within the ASC−/− CD4+ T-cell fraction following activation and subsequent IL-10 secretion, which we do not observe. The fact that no increase in actual Foxp3+ regulatory cells was observed in our study does not exclude the possibility that the expansion of a different regulatory CD4+ population is supported by IL-10 in a feedback loop like IL-2 for conventional T cells. Alternatively, it would be conceivable that the ASC−/− CD4+ T-cell population is less responsive to their own suppressive cytokine(s) to effectively function as regulatory T cells and that a higher IL-10 threshold is required to inhibit their proliferation and function, which may also act as a physiological mechanism to damp their influence. Therefore, perhaps under our co-culture conditions, IL-10 concentrations never reach high enough levels to actually be suppressive to ‘non-regulatory’ ASC−/− CD4+ T cells within the culture (note that IL-10 levels start to decline after day 2 of culture).
The observation that enhanced IL-10 production within the activated ASC−/− CD4+ T-cell fraction is mirrored by a significant decrease in IL-2 secretion (in earlier time-points) may be characteristic of a Treg cell phenotype.17–19 However, with no significant increase in Foxp3+ Treg cell levels within the ASC−/− CD4+ T-cell population both from in vitro and ex vitro analysis, it is probable that Foxp3− Treg cells are responsible for IL-10 production and subsequent inhibition of effector T-cell proliferation. Indeed there is evidence that Foxp3− Treg cells are able to produce IL-10 and inhibit naive T-cell proliferation in a similar manner to Foxp3+ CD4+ CD25+ Treg cells.17
Altogether, we demonstrate that ASC influences the development and functionality of CD4+ Treg cells. These findings reveal a novel relationship between ASC and Treg cells. A better comprehension of the basis of this association may explain how ASC might regulate T-cell and adaptive immune responses in general. Although we demonstrated that NALP3 does not influence T-cell development and functionality, non-NALP3 inflammasomes have been described and we cannot exclude their involvement in the generation of T-cell responses.
Acknowledgments
Technical assistance provided by Nathaliane Bagnoud in genotyping transgenic mice is greatly appreciated. This work has been supported by a grant from the Fonds National Suisse de la Recherche Scientifique (310030-130085/1) and by the Jean and Linette Warnery Foundation.
Disclosures
None of the authors of this paper have conflicts of interest to disclose.
References
- 1.Mariathasan S, Newton K, Monack DM, et al. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature. 2004;430:213–8. doi: 10.1038/nature02664. [DOI] [PubMed] [Google Scholar]
- 2.Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10:417–26. doi: 10.1016/s1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- 3.Martinon F. Detection of immune danger signals by NALP3. J Leukoc Biol. 2008;83:507–11. doi: 10.1189/jlb.0607362. [DOI] [PubMed] [Google Scholar]
- 4.Srinivasula SM, Poyet JL, Razmara M, Datta P, Zhang Z, Alnemri ES. The PYRIN-CARD protein ASC is an activating adaptor for caspase-1. J Biol Chem. 2002;277:21119–22. doi: 10.1074/jbc.C200179200. [DOI] [PubMed] [Google Scholar]
- 5.Stehlik C, Lee SH, Dorfleutner A, Stassinopoulos A, Sagara J, Reed JC. Apoptosis-associated speck-like protein containing a caspase recruitment domain is a regulator of procaspase-1 activation. J Immunol. 2003;171:6154–63. doi: 10.4049/jimmunol.171.11.6154. [DOI] [PubMed] [Google Scholar]
- 6.Petrilli V, Papin S, Tschopp J. The inflammasome. Curr Biol. 2005;15:R581. doi: 10.1016/j.cub.2005.07.049. [DOI] [PubMed] [Google Scholar]
- 7.Ichinohe T, Lee HK, Ogura Y, Flavell R, Iwasaki A. Inflammasome recognition of influenza virus is essential for adaptive immune responses. J Exp Med. 2009;206:79–87. doi: 10.1084/jem.20081667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ippagunta SK, Brand DD, Luo J, et al. Inflammasome-independent role of apoptosis-associated speck-like protein containing a CARD (ASC) in T cell priming is critical for collagen-induced arthritis. J Biol Chem. 2010;285:12454–62. doi: 10.1074/jbc.M109.093252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kolly L, Karababa M, Joosten LA, et al. Inflammatory role of ASC in antigen-induced arthritis is independent of caspase-1, NALP-3, and IPAF. J Immunol. 2009;183:4003–12. doi: 10.4049/jimmunol.0802173. [DOI] [PubMed] [Google Scholar]
- 10.Shaw PJ, Lukens JR, Burns S, Chi H, McGargill MA, Kanneganti TD. Cutting edge: critical role for PYCARD/ASC in the development of experimental autoimmune encephalomyelitis. J Immunol. 2010;184:4610–4. doi: 10.4049/jimmunol.1000217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237–41. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- 12.Guarda G, Dostert C, Staehli F, Cabalzar K, Castillo R, Tardivel A, Schneider P, Tschopp J. T cells dampen innate immune responses through inhibition of NLRP1 and NLRP3 inflammasomes. Nature. 2009;460:269–73. doi: 10.1038/nature08100. [DOI] [PubMed] [Google Scholar]
- 13.Tsuboi Y, Abe H, Nakagawa R, et al. Galectin-9 protects mice from the Shwartzman reaction by attracting prostaglandin E2-producing polymorphonuclear leukocytes. Clin Immunol. 2007;124:221–33. doi: 10.1016/j.clim.2007.04.015. [DOI] [PubMed] [Google Scholar]
- 14.Groux H, O'Garra A, Bigler M, Rouleau M, Antonenko S, de Vries JE, Roncarolo MG. A CD4+ T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature. 1997;389:737–42. doi: 10.1038/39614. [DOI] [PubMed] [Google Scholar]
- 15.de Waal Malefyt R, Haanen J, Spits H, et al. Interleukin 10 (IL-10) and viral IL-10 strongly reduce antigen-specific human T cell proliferation by diminishing the antigen-presenting capacity of monocytes via downregulation of class II major histocompatibility complex expression. J Exp Med. 1991;174:915–24. doi: 10.1084/jem.174.4.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heo YJ, Joo YB, Oh HJ, et al. IL-10 suppresses Th17 cells and promotes regulatory T cells in the CD4+ T cell population of rheumatoid arthritis patients. Immunol Lett. 2010;127:150–6. doi: 10.1016/j.imlet.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 17.Vieira PL, Christensen JR, Minaee S, et al. IL-10-secreting regulatory T cells do not express Foxp3 but have comparable regulatory function to naturally occurring CD4+ CD25+ regulatory T cells. J Immunol. 2004;172:5986–93. doi: 10.4049/jimmunol.172.10.5986. [DOI] [PubMed] [Google Scholar]
- 18.Barrat FJ, Cua DJ, Boonstra A, et al. In vitro generation of interleukin 10-producing regulatory CD4+ T cells is induced by immunosuppressive drugs and inhibited by T helper type 1 (Th1)- and Th2-inducing cytokines. J Exp Med. 2002;195:603–16. doi: 10.1084/jem.20011629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sundstedt A, O'Neill EJ, Nicolson KS, Wraith DC. Role for IL-10 in suppression mediated by peptide-induced regulatory T cells in vivo. J Immunol. 2003;170:1240–8. doi: 10.4049/jimmunol.170.3.1240. [DOI] [PubMed] [Google Scholar]