

Abstract
An overview of issues associated with the design and development of low molecular weight inhibitors of protein-protein interactions is presented. Areas discussed include information on the nature of protein-protein interfaces, methods to characterize those interfaces and methods by which that information is applied towards ligand identification and design. Specific examples of the strategy for the identification of inhibitors of protein-protein interactions involving the proteins p56lck kinase, ERK2 and the calcium-binding protein S100B are presented. Physical characterization of the inhibitors identified in those studies shows them to have drug-like and lead-like properties, indicating their potential to be developed into therapeutic agents.
Introduction: Principles of Protein-Protein Interactions
Protein-protein interactions (PPIs) are critical for the maintenance of normal cellular functions such as signal transduction, immune response, gene regulation, structural organization, proliferation, and apoptosis.[1-4] As our understanding of the molecular mechanisms driving complex biological processes grows, the determination of how proteins function together to influence cellular processes presents a unique task. Towards meeting this challenge, the field of chemical biology offers potential, allowing for the inhibition of specific protein-protein interactions via chemical means. Hence, the elucidation and modulation of cellular pathways through the design of low molecular weight compounds to specifically inhibit protein-protein interactions will lead to a better understanding of biological systems.[5-8] In addition, such efforts offer attractive opportunities for novel therapeutic interventions.
The prediction of protein interactions and the de novo discovery of small molecules capable of modulating such complexes have been fraught with challenges, yielding very few inhibitors.[9-14] This suggests that the combination of biochemical data with three-dimensional structural information will be required to design low-molecular weight compounds to investigate networks of protein function via inhibition of protein-protein interactions.[15, 16] As a consequence, it is essential to understand the structural features of protein interfaces that dictate their interaction with other proteins. This review will summarize progress in the delineation of protein-protein molecular interactions with challenges and opportunities in developing low molecular weight antagonists of such interactions. While a number of recent successes in this field have been reported[17-19], emphasis will be placed on successes in our own laboratories in targeting highly specific protein-protein interactions with low molecular weight inhibitors.
The characteristics of protein-protein interfaces make their targeting by small molecules a difficult undertaking. Contact surfaces involved in these macromolecular interactions are large (∼1,500–3,000 Å2)[20, 21] and often relatively flat compared with those involved in protein–ligand interactions[22] the with surface areas of ∼300–1,000 Å).[17] Additionally, in determining the structure-function characteristics of proteins, an understanding of the physicochemical nature of their intermolecular interactions is required. Determinants of the strength of these associations can include the composition of the interface, the surface area that is buried by the interacting molecules, fraction of nonpolar molecules; hydrogen bonding and salt bridges across the interface; buried water molecules; residue conservation; the shape of the binding interface; and the types of secondary structures, all of which can vary significantly among proteins.[3, 23-27] These properties have lead to protein-protein interfaces being considered “undruggable” (i.e., not able to bind to a drug and inhibit the targeted protein-protein interaction).[28] This was due to the simple assumption that a small molecule could not effectively compete with the large interface associated with protein-protein interactions. However, a low molecular weight compound does not have to “block” the entire protein-protein interface, but rather perturb the interaction to a large enough extent to elicit a biological response. For example, from thermodynamic arguments based on the vant Hoff equation, disrupting a protein-protein interaction by only 2.7 kcal/mol, an the amount of energy associated with a typical hydrogen bond, will shift the equilibrium towards the unbound state by a factor of 100. If such a change in equilibrium can effectively alter the biological process associated with that interaction, a successful outcome will be achieved. Furthermore, inhibition of protein-protein interactions involved in signaling may be facilitated by such complexes being heterodimers with relatively low binding affinities (e.g. 10-6 M), versus homodimers which may have affinities in the range of 10-9-10-12 M.[12]
An essential step in blocking protein-protein interactions is the ability to identify regions of the protein surfaces involved in the interfaces when experimental data on the complexes is lacking. Such interfaces may be identified based on the presence of complimentary residues; i.e. the pattern of polar vs. nonpolar residues. Jones and Thornton have analyzed the hydrophobicity, accessible surface area, shape, and residue propensities between interior, surface, and interface constituents in oligomeric proteins.[29, 30] Results of their analyses revealed that large hydrophobic and uncharged polar residues were more prevalent in the interfaces of heterocomplexes in comparison with the rest of the protein surface. Interestingly, charged residues were observed more frequently on the exposed, outer surface and only a small fraction of the interface residues may contribute to the binding energy.[31] Such patterns may acts as the basis of specific protein-protein interactions.
A variety of information is needed for identifying critical residues at protein-protein interfaces. Residue conservation in the central region of interfaces has been observed to be greater in comparison with residues in the periphery of the interface as well as the outer rim of the interface.[32] Recent mutagenesis work by Wells and colleagues have illustrated that the stability of protein-protein association is largely dependent on amino acids at the interface, as evidenced by alanine scanning.[33] These studies led to the characterization of “hot spots” to describe residues whose substitution by alanine leads to a significant decrease in the binding free energy (ΔΔG > 2 kcal/ mol) and are purported to contribute to the molecular recognition of structurally diverse binding partners.[34] These hotspots are usually located at the center of the contact interface and represent less than half of the contact surface.[35] Further investigation by various groups has broadened the scope of knowledge in this area by correlating structural effects of hot spot substitutions with the energetics of binding.[36-38] The identification of hot spot binding regions holds promise for enhancing the specificity of small molecule inhibitors by proximally targeting these areas, which often include well-defined grooves, to disrupt protein associations. However, while advances have been made in identifying residues of importance for protein-protein interactions, it appears that strict rules are not present such that each system must be investigated individually.
Identification and characterization of low molecular weight surface inhibitors that disrupt protein-protein interactions
Currently, several avenues are being pursued for targeting protein-protein interactions. Historically, large peptidomimetic and natural products represented modulators of protein-protein interactions.[39] In recent years, progress in the identification of small molecules that perturb protein associations and possess “disease-modifying” or “druggable” properties consistent with those of therapeutic agents have been made using a variety of experimental approaches. These include NMR[40], high throughput screening (HTS) and fragment assembly.[41] NMR based methods are often used in the early stages of the lead identification process as well in the development stages to determine the affinities and kinetics of drug binding, distinguishing modes of drug binding, and understanding the specificity of target-ligand interactions.[42] A field of particular utility incorporating NMR methodology, fragment-based screening, utilizes very low molecular weight compounds (50–250 Da) to independently analyze the binding requirements within multiple regions of a protein-protein interface. Ligand-induced changes in protein chemical shifts as measured by 15N-1H HSQC, a two-dimensional (2D) NMR method, is the basis for this “SAR by NMR” strategy, in which weakly binding ligands are first detected, and then chemically linked together to construct high affinity leads.[43]
The first successful drug design strategy incorporating SAR by NMR utilizing fragment leads was described in Shuker et al.[42] 2D isotope edited NMR spectroscopy was used to identify two fragment leads binding at proximal sites on a protein surface. 3D structural information of the target bound ligands facilitated the linking of these fragments to produce a high affinity ligand. This strategy is commonly referred to as the linked-fragment method. In this approach, the binding energy of the linked compound should equal the sum of the binding affinity of the individual pieces, excluding potential entropic contributions to energy gain. Essentially, linking two leads with KD values of 100 μM each (ΔG = −5.5 kcal/mol) should result in a 10 nM lead (ΔG̣ = −11 kcal/mol).[44]
High throughput screening [45-47] using combinatorial chemistry libraries represents another powerful approach to drug discovery. Despite the cost, time, and resource requirements of automated biochemical screening, the use of HTS to discover active compounds is still an integral component in ligand discovery and has led to a number of pharmaceutical drug candidates[45, 48-56] Once a protein target has been identified, a HTS of a relatively large (∼106) compound collection typically can serve as the entry point to lead identification and chemical optimization. This approach often fails to deliver quality small molecule therapeutics as the identified compounds often lack the appropriate physicochemical properties required for clinical progression as a viable drug candidate.[57-59] Biochemical HTS methods have produced larger, more hydrophobic and less soluble leads, though often of high binding affinity, when compared to time-tested “active” compounds specifically designed[60] for a protein (eg. based on the structure of the protein's substrate). [61] Thus, many discovery programs, formerly driven by the pursuit of potency at the expense of solubility, stability, and oral absorption are prioritizing chemically diverse “lead-like” compounds (e.g., lower mass and hydrophobicity) that possess “drug-like” properties after successive rounds of chemical optimization [62].
Choosing the appropriate lead compounds for drug discovery has traditionally been inspired by Lipinski's rule of five (Ro5) establishing the upper (ninety percentile) limits for active compounds as follows: 500 Da for molecular weight (MW); 5 for CLogP, the calculated logarithm of the octanol/water partition coefficient, 5 for the number of hydrogen bond donors (HDO); and 10 for the sum of hydrogen bond acceptors (HAC) accounted for by nitrogen and oxygen.[63] The pharmaceutical industry enforces compliance with Ro5 criteria with the premise that if any two of the above conditions are violated, the molecule is unlikely to be an orally active drug. However, as the aim of HTS is to identify chemical leads amenable to medicinal chemistry optimization, more stringent criteria for Ro5 compliance have been proposed to filter out compounds that are not “leadlike.”[64] These more restrictive criteria include MW ≤ 460, -4 ≤̣ ClogP ≤ 4.2, HDO ≤̣ 5, HAC ≤̣ 9, aqueous solubility (LogSw ≥̣ -5), flexibility (number of rotatable bonds, RTB ≤ 10), and complexity (number of rings, RNG ≤̣ 4). While these criteria serve as a guideline primarily in the lead identification stage, deriving the appropriate combination of potency, pharmacokinetic properties, toxicity and biological activity of lead compounds would require the monitoring of ADME (absorption, distribution, metabolism, and excretion) properties, which is not feasible. However, such analysis is appropriate as a major component of the lead optimization process.[47] More recently, computer-based methods have streamlined structure processing based on Ro5 principles to prioritize structures of interest for bioscreening and chemical optimization.[65, 66]
As an alternative to purely experimental drug discovery efforts, computer-aided drug design (CADD) offers a significant cost and time savings approach with an efficient means to target novel biological molecules for chemical intervention. CADD may be used in the lead identification process via searching through in silico databases of 1 million or more compounds, identifying a subset of candidate compounds (∼100) that have a higher probability of binding to a target protein. The selected compounds are subsequently subjected to experimental assays to identify those with the desired activity. In contrast to biochemical HTS methods, which have a hit rate of 0.01% (i.e. 1 in 10,000) or less, CADD based screening followed by experimental analysis often have hit rates of 5% or more.[67, 68] Lead compounds are then chemically optimized to increase binding affinity, selectivity, and specificity. Towards these goals, CADD offers the advantage of allowing chemical modifications to be computationally optimized prior to synthesis to predict structure activity relationships (SAR) while minimizing the number of compounds synthesized and experimentally analyzed. Computationally guided approaches integrating synthetic chemistry have revolutionized the drug development processes by capitalizing on successive rounds of ligand or target model analysis, synthesis, and biological testing.[69, 70]
CADD methods can follow either a ligand-based or structure-based drug design (SBDD) strategy to identify lead chemical compounds and improve their affinity, specificity, and ADME properties. In cases where a structure of the target protein-ligand complex is not available, a ligand-based CADD approach can be applied to aid in the chemical optimization of small molecule inhibitors.[71] Quantitative structure activity relationship (QSAR)[72, 73] analyses and pharmacophore modeling are ligand-based approaches that are frequently utilized to correlate structural modifications with biological activity.[74, 75] [76-78] Accordingly, biological evaluation of compounds by the use of in vitro cellular and biochemical assays are crucial in progressing compounds into clinical development and refining SAR.[79-82] Although ligand-based approaches are of great utility in the optimization of novel small molecules, this process is driven primarily by the experimental data provided from the biological analysis of a training set of compounds. Such data is often difficult to obtain, especially in the beginning of a drug discovery process, making the application of ligand based approaches difficult. In addition, in the total absence of any lead compound, ligand based methods are of no utility.
Alternatively, SBDD typically has greater utility for the identification and optimization of lead compounds that have potential to be inhibitors of PPIs. When a 3D structure of either one of the targeted proteins or the protein-protein complex is available[46, 83] CADD may be used to select compounds from 3D chemical databases that have an increased probability for binding to the target molecule [46, 84-86] or create novel compounds de novo using in silico methods [74, 87] This aspect of the design cycle has proven to be one of the most powerful implementation of computational approaches in the development of PPIs and is used throughout the examples presented in the remainder of this review. Notably, SBDD has the advantage that a large training set of compounds for which biological activity data is available is not required for chemical optimization. Therefore, structure-based CADD may be applied to expedite the selection of lead compounds at an early stage of a drug discovery as well as facilitate the optimization of the lead compounds to improve their biological efficacy.[62, 70] However, it should be emphasized that for effective application of SBDD for lead optimization it is highly recommended that the structure of the ligand-protein complex be available from NMR or X-ray crystallography to assure that the modeling is being initiated from the correct starting point.
Examples of low-molecular weight inhibitors of protein-protein interactions
In the remainder of this overview, a few examples are presented to illustrate how the above concepts are being successfully reduced to practice to yield novel small molecule compounds capable of modulating PPIs, and highlight the value of CADD in combination with experimental methods in the drug-design process. Systems that will be discussed include small molecule ligands targeting the S100B-p53 interaction,[88, 89] ERK-protein substrate interactions[13, 90], and the p56Lck SH2 domain–phosphopeptide interaction.[72, 91] In these studies, CADD methods have successfully identified new inhibitors targeting these biologically interesting and pharmaceutically relevant proteins.
ERK2
ERK1/2, c-Jun-N-terminal kinase (JNK) and p38 kinase are three known mitogen activated (MAP) protein kinases[92, 93] whose perturbed activation has been implicated in cancer and inflammation [94]. The extracellular signal-regulated kinases (ERK1/2) are signaling mediators involved in cell proliferation and represent an interesting system as it is involved in a variety of PPIs. Upon activation, ERK proteins undergo a conformational change [95] leading to phosphorylation of associated substrate proteins.[95] Thus, inhibiting the ability of ERK to phosphorylate its substrates may be of utility with respect to the development of cancer therapeutics. However, ERK proteins have been implicated to interact with and regulate through phosphorylation over 50 substrate proteins, which include other kinases, transcription factors, and structural proteins [96] like RSK-1 and ELK-1, as well as others involved in other diverging signaling pathways. Given the wide range of substrates and the importance of ERK signaling in normal cell functions, inhibitors that target ERK protein's phosphorylation site would therefore lead to a lack of specificity in regards to substrates involved in normal or pathological conditions. To overcome this potential problem, our laboratory has devised a drug design strategy that targets the docking regions of ERK2 involved in the binding of the different substrate proteins.[13, 97] Motivation for this drug design strategy comes from a variety of biological studies indicating that different regions of the docking domain are important for determining the binding specificity of different substrate proteins to ERK2.[95, 96, 98-100]. CADD virtual screening (VS) was applied to identify low molecular weight molecules that could target specific ERK docking domains and thereby selectively disrupt substrate interactions. [92]. VS initially involved database screening of 800,000 commercially available low molecular weight compounds against a specific site on the docking domain. Screening involved a hierarchical approach that included two rounds of docking, followed by chemical similarity analysis and final compound selection based on Lipinski's Ro5. A total of 80 compounds were then obtained and tested for biological activity. Several compounds inhibited the phosphorylation of two ERK substrates, RSK-1 and ELK-1 with direct binding of the inhibitors to the protein confirmed by fluorescence spectroscopy. Examples of the ERK inhibitors are shown in Table 1. Active compounds also displayed dose-dependent inhibition of cellular proliferation in several cancer cell lines. This approach is being extended to identify and optimize low molecular weight molecules that specifically disrupt ERK and other MAP kinase interactions with substrates involved in cancer and inflammatory syndromes without affecting substrates involved in normal cellular functions.
Table 1. Physicochemical properties of low molecular weight PPI inhibitor compounds.
| Structure | Compound name | MW | ClogP | HDO | HAC |
|---|---|---|---|---|---|
| SBi1 | 340 | 2.31 | 4 | 2 | |
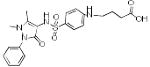 |
SBi2 | 445 | 0.25 | 3 | 4 |
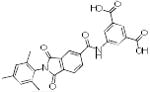 |
SBi3 | 472 | 3.74 | 3 | 5 |
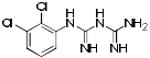 |
SBi4 | 246 | 2.60 | 5 | 0 |
 |
SBi5 | 316 | 3.30 | 5 | 1 |
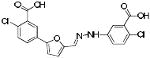 |
SBi6 | 419 | 4.89 | 3 | 3 |
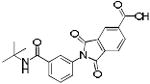 |
SBi7 | 366 | 2.62 | 2 | 4 |
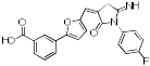 |
p56 Lck-73 | 390 | 4.07 | 2 | 3 |
| p56 Lck-99 | 388 | 3.22 | 3 | 3 | |
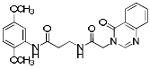 |
p56 Lck-139 | 410 | 1.05 | 2 | 5 |
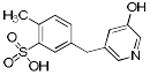 |
p56 Lck-160 | 279 | 0.71 | 2 | 2 |
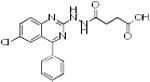 |
p56 Lck-195 | 371 | 3.37 | 3 | 2 |
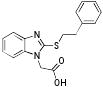 |
p56 Lck-201 | 312 | 4.21 | 1 | 2 |
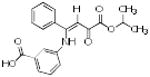 |
p56 Lck-88 | 353 | 3.29 | 2 | 4 |
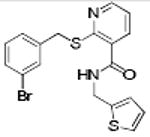 |
p56 Lck-146 | 419 | 4.60 | 1 | 3 |
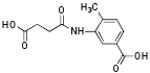 |
p56 Lck-162 | 251 | 0.83 | 3 | 3 |
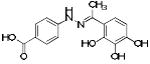 |
p56 Lck-196 | 302 | 1.67 | 5 | 1 |
| ERK2-17 | 354 | 3.07 | 2 | 3 | |
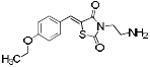 |
ERK2-76 | 292 | 2.35 | 1 | 4 |
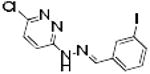 |
ERK2-86 | 359 | 3.97 | 1 | 0 |
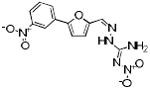 |
ERK2-89 | 318 | -1.20 | 2 | 5 |
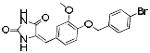 |
ERK2-95 | 403 | 3.29 | 2 | 4 |
MW=Molecular weight, ClogP= calculated log octanol/water partition coefficient, HDO=hydrogen bond donors, HAC=hydrogen bond acceptors
p56Lck Kinase
Our laboratory has also been involved in the successful identification of low molecular weight compounds to inhibit the PPI between the SH2 domain of the p56 L cell tyrosine kinase (Lck) protein and immunoreceptor tyrosine-based activation motifs (ITAMs), [101, 102] peptides involved in T-cell mediated immune response. [103, 104] Lck is a member of the Src family tyrosine kinases and plays an essential role in T-cell response thru the phosphorylation of immunoreceptor tyrosine-based activation motifs (ITAM) on CD3 chains. This action involves the binding of Lck to the phosphotyrosine (pY) residues on the ITAMs via its SH2 domain. Failure of the Lck SH2 domain to bind to ITAMs will ultimately block downstream T cell activation signaling cascades.[105, 106] Therefore, small molecules that perturb Lck SH2 domain-dependent protein-protein interactions may have immunotherapeutic activity and potentially serve as effective treatments for T-cell leukemias, lymphomas, and autoimmune diseases.[102]
To exploit this therapeutic potential, our laboratory in collaboration with Hayashi and colleagues has successfully identified non-peptide specific drug-like molecules targeting the Lck SH2 domain using CADD methodologies.[91, 101] Inspection of the X-ray structures of the Lck SH2 domain complexed with the ITAM pY-E-E-I peptide revealed two cavities on the SH2 domain into which the pY and Ile residues bind; the later site being referred to as the pY+3 binding site. These results along with other experimental mutation data[107] suggest that the pY+3 binding pocket confers specificity of the p56LcK-ITAM interaction and, therefore, represents a novel druggable molecular target amenable to chemical intervention versus efforts to target the pY site, which is present a large number of SH2 domains. Using CADD VS methodologies, a chemical database of 2 million compounds was docked into the pY+3 binding site.[91] In total, 194 compounds were obtained for biological evaluation. Thirteen compounds were active in biological assays as demonstrated by inhibition of ITAM-Lck SH2 binding, mixed lymphocyte reaction, and fluorescence titration assays. Additional lead validation studies were conducted to identify which of the active compounds were candidates for chemical optimization based on chemical similarity.[72] This approach is based on the assumption that a compound that is a member of a structurally similar family of biologically active compounds is viable for lead optimization studies. In addition, these studies yield first generation SAR data that is of utility to rationally direct the lead optimization process. Examples of active compounds that are inhibitors of Lck are shown in Table 1.
S100B-p53 complex
S100B is a calcium binding protein that inhibits tumor suppression by binding to the C terminus of the tumor suppressor protein p53.[108] [109-113] Binding of S100B to p53 leads to degradation of the tumor suppressor protein, facilitating cell proliferation. High endogenous protein levels of S100B has been recently shown to not only be a prognostic marker, but also correlated with cancer progression in malignant melanoma.[114-116]. Confirmation of the S100B-p53 interaction was demonstrated by crystal and NMR experimental structures.[88] In these analyses, it was shown that when S100B binds to calcium, a conformational change is observed, exposing a well-defined ‘hot-spot’ on the protein, comprised of hydrophobic residues that interact with p53. The availability of 3D structures makes S100B an ideal target for the development of PPI inhibitors via a SBDD approach. S100B is particularly attractive for inhibiting complex formation versus targeting p53 itself, as inhibitors of p53 may also inhibit its tumor suppressor function.
In silico chemical database searching was performed to identify low molecular weight chemical compounds with a high probability for binding S100B and inhibiting its interactions with p53. An initial CADD screening of 640,000 compounds was carried out, from which the 20,000 top compounds were selected based on their interaction energies with the protein. Following an additional round of docking, 60 potential inhibitors were selected based on their desirable physicochemical properties and were subjected to fluorescent binding/competition assays and NMR perturbation experiments. Of the 60 tested compounds, seven were determined to bind directly in the p53 binding pocket of S100B, with KD values between 1 and 120 μM.[117] (Table 1) Five compounds halted growth of melanoma in micromolar concentrations and have been selected for further chemical derivation to optimize anticancer activity and produce the next series of small molecule PPI inhibitors.[117] These low molecular weight compounds may have therapeutic utility in the medical management of malignant melanoma.
Drug-like properties of PPI inhibitors
From the three studies presented above, a collection of compounds that inhibit PPIs have been identified (Table 1). The question remains, do these compounds have drug-like and lead-like characteristics as defined by Lipinksi and Oprea,[51, 61, 62, 65] respectively? To answer this question, a collection of physical properties of the compounds identified in the studies discussed above are presented in Table 1. As may be seen, all of the compounds fulfill Lipinksi's Ro5, though several approach upper limits. For example SBi6 has a MW over 400 and the ClogP approaches 5, though it does formally fulfill the rules. With respect to Oprea's rules for lead-like compounds, most of the compounds do fulfill those criteria, though exceptions are present. SBi3 has a MW of over 460 while p56 Lck-146 and SBi6 have ClogP over 4.6. In general, there are advantages to following these standard rules; however, there are always exceptions. Accordingly when doing screening studies combined with similarity clustering, if clusters are identified whose members do not fulfill the rules, our approach is to still test at least one member. With respect to MW, the field of fragment based drug design is based on the premise that it is generally more effective to optimize lower molecular weight compounds,[40, 41] a consideration that should be taken into account in studies where extensive optimization efforts are anticipated.
In summary, signaling checkpoints involved in aberrant cell growth and viability that are mediated by PPIs will provide druggable targets for cancer therapy and other disease states. Modulating these processes with small molecules presents a challenge in the field of chemical biology, requiring an integrated approach to understand the structural and biochemical characteristics of PPIs and applying that knowledge to identify inhibitors. As shown in this review, the use of CADD as a means to rationally discover drug candidates that target PPIs in combination with the appropriate biological assays is certainly feasible. The current successes reported here combined with the promise of even more exciting results as new biologically important PPIs are identified further indicates that the development of small molecules to inhibit protein-protein interactions will have a significant impact on the drug design process while increasing the pipeline of targeted therapeutic agents.
Acknowledgments
Support from the NIH (CA107331 to DJW, CA120215 to PSW and ADM), the Samuel Waxman Cancer Foundation to ADM and the University of Maryland Computer-Aided Drug Design Center are acknowledged.
References
- 1.Schulte U. CNS Neurol Disord Drug Targets. 2008;7:172–86. doi: 10.2174/187152708784083795. [DOI] [PubMed] [Google Scholar]
- 2.Medico E. Tumori. 2008;94:172–8. doi: 10.1177/030089160809400207. [DOI] [PubMed] [Google Scholar]
- 3.Keskin O, Gursoy A, Ma B, Nussinov R. Chem Rev. 2008;108:1225–44. doi: 10.1021/cr040409x. [DOI] [PubMed] [Google Scholar]
- 4.Gomez SM, Choi K, Wu Y. Curr Protoc Bioinformatics. 2008;Chapter 8(Unit 8 2) doi: 10.1002/0471250953.bi0802s22. [DOI] [PubMed] [Google Scholar]
- 5.Laudet B, Moucadel V, Prudent R, Filhol O, Wong YS, Royer D, Cochet C. Mol Cell Biochem. 2008 doi: 10.1007/s11010-008-9821-6. [DOI] [PubMed] [Google Scholar]
- 6.Chen X, Zhong S, Zhu X, Dziegielewska B, Ellenberger T, Wilson GM, MacKerell AD, Jr, Tomkinson AE. Cancer Res. 2008;68:3169–77. doi: 10.1158/0008-5472.CAN-07-6636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Walsh CT. Nat Chem Biol. 2005;1:122–4. doi: 10.1038/nchembio0805-122. [DOI] [PubMed] [Google Scholar]
- 8.Lampson MA, Kapoor TM. Nat Chem Biol. 2006;2:19–27. doi: 10.1038/nchembio757. [DOI] [PubMed] [Google Scholar]
- 9.Domling A. Curr Opin Chem Biol. 2008;12:281–91. doi: 10.1016/j.cbpa.2008.04.603. [DOI] [PubMed] [Google Scholar]
- 10.Hardcastle IR, Ahmed SU, Atkins H, Farnie G, Golding BT, Griffin RJ, Guyenne S, Hutton C, Kallblad P, Kemp SJ, Kitching MS, Newell DR, Norbedo S, Northen JS, Reid RJ, Saravanan K, Willems HM, Lunec J. J Med Chem. 2006;49:6209–21. doi: 10.1021/jm0601194. [DOI] [PubMed] [Google Scholar]
- 11.Gadek TR, Nicholas JB. Biochem Pharmacol. 2003;65:1–8. doi: 10.1016/s0006-2952(02)01479-x. [DOI] [PubMed] [Google Scholar]
- 12.Cochran AG. Chem Biol. 2000;7:R85–94. doi: 10.1016/s1074-5521(00)00106-x. [DOI] [PubMed] [Google Scholar]
- 13.Hancock CN, Macias AT, Mackerell AD, Jr, Shapiro P. Med Chem. 2006;2:213–22. doi: 10.2174/157340606776056151. [DOI] [PubMed] [Google Scholar]
- 14.Fletcher S, Hamilton AD. Curr Top Med Chem. 2007;7:922–7. doi: 10.2174/156802607780906735. [DOI] [PubMed] [Google Scholar]
- 15.Micallef J, Gajadhar A, Wiley J, DeSouza LV, Michael Siu KW, Guha A. Neurosurgery. 2008;62:539–55. doi: 10.1227/01.neu.0000317302.85837.61. discussion 539-55. [DOI] [PubMed] [Google Scholar]
- 16.Schueler-Furman O, Wang C, Bradley P, Misura K, Baker D. Science. 2005;310:638–42. doi: 10.1126/science.1112160. [DOI] [PubMed] [Google Scholar]
- 17.Wells JA, McClendon CL. Nature. 2007;450:1001–9. doi: 10.1038/nature06526. [DOI] [PubMed] [Google Scholar]
- 18.Betzi S, Restouin A, Opi S, Arold ST, Parrot I, Guerlesquin F, Morelli X, Collette Y. Proc Natl Acad Sci U S A. 2007;104:19256–61. doi: 10.1073/pnas.0707130104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rajapakse HA. Curr Top Med Chem. 2007;7:966–71. doi: 10.2174/156802607780906816. [DOI] [PubMed] [Google Scholar]
- 20.Lo Conte L, Chothia C, Janin J. J Mol Biol. 1999;285:2177–98. doi: 10.1006/jmbi.1998.2439. [DOI] [PubMed] [Google Scholar]
- 21.Cheng AC, Coleman RG, Smyth KT, Cao Q, Soulard P, Caffrey DR, Salzberg AC, Huang ES. Nat Biotechnol. 2007;25:71–5. doi: 10.1038/nbt1273. [DOI] [PubMed] [Google Scholar]
- 22.Jones S, Thornton JM. Proc Natl Acad Sci U S A. 1996;93:13–20. doi: 10.1073/pnas.93.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nooren IM, Thornton JM. J Mol Biol. 2003;325:991–1018. doi: 10.1016/s0022-2836(02)01281-0. [DOI] [PubMed] [Google Scholar]
- 24.Reichmann D, Rahat O, Cohen M, Neuvirth H, Schreiber G. Curr Opin Struct Biol. 2007;17:67–76. doi: 10.1016/j.sbi.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 25.Reichmann D, Cohen M, Abramovich R, Dym O, Lim D, Strynadka NC, Schreiber G. J Mol Biol. 2007;365:663–79. doi: 10.1016/j.jmb.2006.09.076. [DOI] [PubMed] [Google Scholar]
- 26.Neuvirth H, Raz R, Schreiber G. J Mol Biol. 2004;338:181–99. doi: 10.1016/j.jmb.2004.02.040. [DOI] [PubMed] [Google Scholar]
- 27.Glaser F, Rosenberg Y, Kessel A, Pupko T, Ben-Tal N. Proteins. 2005;58:610–7. doi: 10.1002/prot.20305. [DOI] [PubMed] [Google Scholar]
- 28.Hopkins AL, Groom CR. Nat Rev Drug Discov. 2002;1:727–30. doi: 10.1038/nrd892. [DOI] [PubMed] [Google Scholar]
- 29.Jones S, Thornton JM. J Mol Biol. 1997;272:133–43. doi: 10.1006/jmbi.1997.1233. [DOI] [PubMed] [Google Scholar]
- 30.Jones S, Thornton JM. J Mol Biol. 1997;272:121–32. doi: 10.1006/jmbi.1997.1234. [DOI] [PubMed] [Google Scholar]
- 31.Bordner AJ, Abagyan R. Proteins. 2005;60:353–66. doi: 10.1002/prot.20433. [DOI] [PubMed] [Google Scholar]
- 32.Nimrod G, Glaser F, Steinberg D, Ben-Tal N, Pupko T. Bioinformatics. 2005;21 1:i328–37. doi: 10.1093/bioinformatics/bti1023. [DOI] [PubMed] [Google Scholar]
- 33.Cunningham BC, Wells JA. Science. 1989;244:1081–5. doi: 10.1126/science.2471267. [DOI] [PubMed] [Google Scholar]
- 34.Clackson T, Ultsch MH, Wells JA, de Vos AM. J Mol Biol. 1998;277:1111–28. doi: 10.1006/jmbi.1998.1669. [DOI] [PubMed] [Google Scholar]
- 35.Moreira IS, Fernandes PA, Ramos MJ. Proteins. 2007;68:803–12. doi: 10.1002/prot.21396. [DOI] [PubMed] [Google Scholar]
- 36.Guerois R, Nielsen JE, Serrano L. J Mol Biol. 2002;320:369–87. doi: 10.1016/S0022-2836(02)00442-4. [DOI] [PubMed] [Google Scholar]
- 37.Cho S, Swaminathan CP, Yang J, Kerzic MC, Guan R, Kieke MC, Kranz DM, Mariuzza RA, Sundberg EJ. Structure. 2005;13:1775–87. doi: 10.1016/j.str.2005.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kortemme T, Baker D. Proc Natl Acad Sci U S A. 2002;99:14116–21. doi: 10.1073/pnas.202485799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pagliaro L, Felding J, Audouze K, Nielsen SJ, Terry RB, Krog-Jensen C, Butcher S. Curr Opin Chem Biol. 2004;8:442–9. doi: 10.1016/j.cbpa.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 40.Hajduk PJ. Mol Interv. 2006;6:266–72. doi: 10.1124/mi.6.5.8. [DOI] [PubMed] [Google Scholar]
- 41.Braisted AC, Oslob JD, Delano WL, Hyde J, McDowell RS, Waal N, Yu C, Arkin MR, Raimundo BC. J Am Chem Soc. 2003;125:3714–5. doi: 10.1021/ja034247i. [DOI] [PubMed] [Google Scholar]
- 42.Shuker SB, Hajduk PJ, Meadows RP, Fesik SW. Science. 1996;274:1531–4. doi: 10.1126/science.274.5292.1531. [DOI] [PubMed] [Google Scholar]
- 43.Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, Joseph MK, Kitada S, Korsmeyer SJ, Kunzer AR, Letai A, Li C, Mitten MJ, Nettesheim DG, Ng S, Nimmer PM, O'Connor JM, Oleksijew A, Petros AM, Reed JC, Shen W, Tahir SK, Thompson CB, Tomaselli KJ, Wang B, Wendt MD, Zhang H, Fesik SW, Rosenberg SH. Nature. 2005;435:677–81. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 44.Erlanson DA, McDowell RS, O'Brien T. J Med Chem. 2004;47:3463–82. doi: 10.1021/jm040031v. [DOI] [PubMed] [Google Scholar]
- 45.Webb TR. Curr Opin Drug Discov Devel. 2005;8:303–8. [PubMed] [Google Scholar]
- 46.Martin YC, Kofron JL, Traphagen LM. J Med Chem. 2002;45:4350–8. doi: 10.1021/jm020155c. [DOI] [PubMed] [Google Scholar]
- 47.Jenkins KM, Angeles R, Quintos MT, Xu R, Kassel DB, Rourick RA. J Pharm Biomed Anal. 2004;34:989–1004. doi: 10.1016/j.jpba.2003.08.001. [DOI] [PubMed] [Google Scholar]
- 48.Keseru GM, Makara GM. Drug Discov Today. 2006;11:741–8. doi: 10.1016/j.drudis.2006.06.016. [DOI] [PubMed] [Google Scholar]
- 49.Fattori D, Squarcia A, Bartoli S. Drugs R D. 2008;9:217–27. doi: 10.2165/00126839-200809040-00002. [DOI] [PubMed] [Google Scholar]
- 50.Diller DJ. Curr Opin Drug Discov Devel. 2008;11:346–55. [PubMed] [Google Scholar]
- 51.Oprea TI. Curr Opin Chem Biol. 2002;6:384–9. doi: 10.1016/s1367-5931(02)00329-0. [DOI] [PubMed] [Google Scholar]
- 52.Martin YC. J Comb Chem. 2001;3:231–50. doi: 10.1021/cc000073e. [DOI] [PubMed] [Google Scholar]
- 53.Wood ER, Truesdale AT, McDonald OB, Yuan D, Hassell A, Dickerson SH, Ellis B, Pennisi C, Horne E, Lackey K, Alligood KJ, Rusnak DW, Gilmer TM, Shewchuk L. Cancer Res. 2004;64:6652–9. doi: 10.1158/0008-5472.CAN-04-1168. [DOI] [PubMed] [Google Scholar]
- 54.Stamos J, Sliwkowski MX, Eigenbrot C. J Biol Chem. 2002;277:46265–72. doi: 10.1074/jbc.M207135200. [DOI] [PubMed] [Google Scholar]
- 55.Goldberg M, Mahon K, Anderson D. Adv Drug Deliv Rev. 2008;60:971–8. doi: 10.1016/j.addr.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Usui T, Ban HS, Kawada J, Hirokawa T, Nakamura H. Bioorg Med Chem Lett. 2008;18:285–8. doi: 10.1016/j.bmcl.2007.10.084. [DOI] [PubMed] [Google Scholar]
- 57.Teague SJ, Davis AM, Leeson PD, Oprea T. Angew Chem Int Ed Engl. 1999;38:3743–3748. doi: 10.1002/(SICI)1521-3773(19991216)38:24<3743::AID-ANIE3743>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 58.Hann MM, Leach AR, Harper G. J Chem Inf Comput Sci. 2001;41:856–64. doi: 10.1021/ci000403i. [DOI] [PubMed] [Google Scholar]
- 59.Lipinski CA. J Pharmacol Toxicol Methods. 2000;44:235–49. doi: 10.1016/s1056-8719(00)00107-6. [DOI] [PubMed] [Google Scholar]
- 60.Inglese J, Shamu CE, Guy RK. Nat Chem Biol. 2007;3:438–41. doi: 10.1038/nchembio0807-438. [DOI] [PubMed] [Google Scholar]
- 61.Oprea TI, Allu TK, Fara DC, Rad RF, Ostopovici L, Bologa CG. J Comput Aided Mol Des. 2007;21:113–9. doi: 10.1007/s10822-007-9105-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Oprea TI, Davis AM, Teague SJ, Leeson PD. J Chem Inf Comput Sci. 2001;41:1308–15. doi: 10.1021/ci010366a. [DOI] [PubMed] [Google Scholar]
- 63.Lipinski CA, L F, Dominy BW, Feeney PJ. Adv Drug Deliv Rev. 1997;23 doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 64.Hann MM, Oprea TI. Curr Opin Chem Biol. 2004;8:255–63. doi: 10.1016/j.cbpa.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 65.Oprea TI. Mol Divers. 2002;5:199–208. doi: 10.1023/a:1021368007777. [DOI] [PubMed] [Google Scholar]
- 66.Oprea TI, Matter H. Curr Opin Chem Biol. 2004;8:349–58. doi: 10.1016/j.cbpa.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 67.Chen IJ, Neamati N, MacKerell AD., Jr Curr Drug Targets Infect Disord. 2002;2:217–34. doi: 10.2174/1568005023342380. [DOI] [PubMed] [Google Scholar]
- 68.McInnes C. Curr Opin Chem Biol. 2007;11:494–502. doi: 10.1016/j.cbpa.2007.08.033. [DOI] [PubMed] [Google Scholar]
- 69.Gundla R, Kazemi R, Sanam R, Muttineni R, Sarma JA, Dayam R, Neamati N. J Med Chem. 2008;51:3367–77. doi: 10.1021/jm7013875. [DOI] [PubMed] [Google Scholar]
- 70.Barril X, Hubbard RE, Morley SD. Mini Rev Med Chem. 2004;4:779–91. doi: 10.2174/1389557043403675. [DOI] [PubMed] [Google Scholar]
- 71.Stahura FL, Bajorath J. Curr Pharm Des. 2005;11:1189–202. doi: 10.2174/1381612053507549. [DOI] [PubMed] [Google Scholar]
- 72.Macias AT, Mia MY, Xia G, Hayashi J, MacKerell AD., Jr J Chem Inf Model. 2005;45:1759–66. doi: 10.1021/ci050225z. [DOI] [PubMed] [Google Scholar]
- 73.Hall LH, Hall LM. SAR QSAR Environ Res. 2005;16:13–41. doi: 10.1080/10629360412331319853. [DOI] [PubMed] [Google Scholar]
- 74.Feher M, Gao Y, Baber JC, Shirley WA, Saunders J. Bioorg Med Chem. 2008;16:422–7. doi: 10.1016/j.bmc.2007.09.026. [DOI] [PubMed] [Google Scholar]
- 75.Dean PM, Lloyd DG, Todorov NP. Curr Opin Drug Discov Devel. 2004;7:347–53. [PubMed] [Google Scholar]
- 76.Bernard D, Coop A, MacKerell AD., Jr J Am Chem Soc. 2003;125:3101–7. doi: 10.1021/ja027644m. [DOI] [PubMed] [Google Scholar]
- 77.Bernard D, Coop A, MacKerell AD., Jr J Med Chem. 2005;48:7773–80. doi: 10.1021/jm050785p. [DOI] [PubMed] [Google Scholar]
- 78.Bernard DC, A, Mackerell AD. Drug Design Rev. 2005;2:277–291. [Google Scholar]
- 79.Ekins S, Mestres J, Testa B. Br J Pharmacol. 2007;152:21–37. doi: 10.1038/sj.bjp.0707306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Smet C, Duckert JF, Wieruszeski JM, Landrieu I, Buee L, Lippens G, Deprez B. J Med Chem. 2005;48:4815–23. doi: 10.1021/jm0500119. [DOI] [PubMed] [Google Scholar]
- 81.Tang G, Yang CY, Nikolovska-Coleska Z, Guo J, Qiu S, Wang R, Gao W, Wang G, Stuckey J, Krajewski K, Jiang S, Roller PP, Wang S. J Med Chem. 2007;50:1723–6. doi: 10.1021/jm061400l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ding K, Lu Y, Nikolovska-Coleska Z, Wang G, Qiu S, Shangary S, Gao W, Qin D, Stuckey J, Krajewski K, Roller PP, Wang S. J Med Chem. 2006;49:3432–5. doi: 10.1021/jm051122a. [DOI] [PubMed] [Google Scholar]
- 83.Martin YC. Methods Enzymol. 1991;203:587–613. doi: 10.1016/0076-6879(91)03031-b. [DOI] [PubMed] [Google Scholar]
- 84.Gohlke H, Gundisch D, Schwarz S, Seitz G, Tilotta MC, Wegge T. J Med Chem. 2002;45:1064–72. doi: 10.1021/jm010936y. [DOI] [PubMed] [Google Scholar]
- 85.Taylor RD, Jewsbury PJ, Essex JW. J Comput Aided Mol Des. 2002;16:151–66. doi: 10.1023/a:1020155510718. [DOI] [PubMed] [Google Scholar]
- 86.Kick EK, Roe DC, Skillman AG, Liu G, Ewing TJ, Sun Y, Kuntz ID, Ellman JA. Chem Biol. 1997;4:297–307. doi: 10.1016/s1074-5521(97)90073-9. [DOI] [PubMed] [Google Scholar]
- 87.Schneider G, Fechner U. Nat Rev Drug Discov. 2005;4:649–63. doi: 10.1038/nrd1799. [DOI] [PubMed] [Google Scholar]
- 88.Markowitz J, Chen I, Gitti R, Baldisseri DM, Pan Y, Udan R, Carrier F, MacKerell AD, Jr, Weber DJ. J Med Chem. 2004;47:5085–93. doi: 10.1021/jm0497038. [DOI] [PubMed] [Google Scholar]
- 89.Markowitz J, Mackerell AD, Jr, Carrier F, Charpentier TH, Weber DJ. Curr Top Med Chem. 2005;5:1093–108. doi: 10.2174/156802605774370865. [DOI] [PubMed] [Google Scholar]
- 90.Hancock CN, Dangi S, Shapiro P. J Biol Chem. 2005;280:11590–8. doi: 10.1074/jbc.M408273200. [DOI] [PubMed] [Google Scholar]
- 91.Huang N, Nagarsekar A, Xia G, Hayashi J, MacKerell AD., Jr J Med Chem. 2004;47:3502–11. doi: 10.1021/jm030470e. [DOI] [PubMed] [Google Scholar]
- 92.Zhang F, Strand A, Robbins D, Cobb MH, Goldsmith EJ. Nature. 1994;367:704–11. doi: 10.1038/367704a0. [DOI] [PubMed] [Google Scholar]
- 93.Reuter CW, Morgan MA, Bergmann L. Blood. 2000;96:1655–69. [PubMed] [Google Scholar]
- 94.Shapiro P. Crit Rev Clin Lab Sci. 2002;39:285–330. doi: 10.1080/10408360290795538. [DOI] [PubMed] [Google Scholar]
- 95.Zhang J, Zhang F, Ebert D, Cobb MH, Goldsmith EJ. Structure. 1995;3:299–307. doi: 10.1016/s0969-2126(01)00160-5. [DOI] [PubMed] [Google Scholar]
- 96.Lewis TS, Shapiro PS, Ahn NG. Adv Cancer Res. 1998;74:49–139. doi: 10.1016/s0065-230x(08)60765-4. [DOI] [PubMed] [Google Scholar]
- 97.Hancock CN, Macias A, Lee EK, Yu SY, Mackerell AD, Jr, Shapiro P. J Med Chem. 2005;48:4586–95. doi: 10.1021/jm0501174. [DOI] [PubMed] [Google Scholar]
- 98.Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K, Cobb MH. Endocr Rev. 2001;22:153–83. doi: 10.1210/edrv.22.2.0428. [DOI] [PubMed] [Google Scholar]
- 99.Nichols A, Camps M, Gillieron C, Chabert C, Brunet A, Wilsbacher J, Cobb M, Pouyssegur J, Shaw JP, Arkinstall S. J Biol Chem. 2000;275:24613–21. doi: 10.1074/jbc.M001515200. [DOI] [PubMed] [Google Scholar]
- 100.Tanoue T, Adachi M, Moriguchi T, Nishida E. Nat Cell Biol. 2000;2:110–6. doi: 10.1038/35000065. [DOI] [PubMed] [Google Scholar]
- 101.Tong L, Warren TC, King J, Betageri R, Rose J, Jakes S. J Mol Biol. 1996;256:601–10. doi: 10.1006/jmbi.1996.0112. [DOI] [PubMed] [Google Scholar]
- 102.Broadbridge RJ, Sharma RP. Curr Drug Targets. 2000;1:365–86. doi: 10.2174/1389450003349074. [DOI] [PubMed] [Google Scholar]
- 103.Lawrence DS, Niu J. Pharmacol Ther. 1998;77:81–114. doi: 10.1016/s0163-7258(97)00052-1. [DOI] [PubMed] [Google Scholar]
- 104.Neel BG, Tonks NK. Curr Opin Cell Biol. 1997;9:193–204. doi: 10.1016/s0955-0674(97)80063-4. [DOI] [PubMed] [Google Scholar]
- 105.Straus DB, Weiss A. Cell. 1992;70:585–93. doi: 10.1016/0092-8674(92)90428-f. [DOI] [PubMed] [Google Scholar]
- 106.Straus DB, Chan AC, Patai B, Weiss A. J Biol Chem. 1996;271:9976–81. doi: 10.1074/jbc.271.17.9976. [DOI] [PubMed] [Google Scholar]
- 107.Songyang Z, Cantley LC. Trends Biochem Sci. 1995;20:470–5. doi: 10.1016/s0968-0004(00)89103-3. [DOI] [PubMed] [Google Scholar]
- 108.Rustandi RR, Baldisseri DM, Weber DJ. Nat Struct Biol. 2000;7:570–4. doi: 10.1038/76797. [DOI] [PubMed] [Google Scholar]
- 109.Levine AJ, Momand J, Finlay CA. Nature. 1991;351:453–6. doi: 10.1038/351453a0. [DOI] [PubMed] [Google Scholar]
- 110.Levine AJ. Cell. 1997;88:323–31. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- 111.Lin J, Blake M, Tang C, Zimmer D, Rustandi RR, Weber DJ, Carrier F. J Biol Chem. 2001;276:35037–41. doi: 10.1074/jbc.M104379200. [DOI] [PubMed] [Google Scholar]
- 112.Grigorian M, Andresen S, Tulchinsky E, Kriajevska M, Carlberg C, Kruse C, Cohn M, Ambartsumian N, Christensen A, Selivanova G, Lukanidin E. J Biol Chem. 2001;276:22699–708. doi: 10.1074/jbc.M010231200. [DOI] [PubMed] [Google Scholar]
- 113.Lin J, Yang Q, Yan Z, Markowitz J, Wilder PT, Carrier F, Weber DJ. J Biol Chem. 2004;279:34071–7. doi: 10.1074/jbc.M405419200. [DOI] [PubMed] [Google Scholar]
- 114.Maelandsmo GM, Florenes VA, Mellingsaeter T, Hovig E, Kerbel RS, Fodstad O. Int J Cancer. 1997;74:464–9. doi: 10.1002/(sici)1097-0215(19970822)74:4<464::aid-ijc19>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 115.Boni R, Meuli C, Dummer R. Br J Dermatol. 1997;137:833–4. doi: 10.1111/j.1365-2133.1997.tb01136.x. [DOI] [PubMed] [Google Scholar]
- 116.Hansson LO, von Schoultz E, Djureen E, Hansson J, Nilsson B, Ringborg U. Anticancer Res. 1997;17:3071–3. [PubMed] [Google Scholar]
- 117.Markowitz J, MacKerell AD, Jr, Weber DJ. Mini Rev Med Chem. 2007;7:609–16. doi: 10.2174/138955707780859422. [DOI] [PubMed] [Google Scholar]