

Abstract
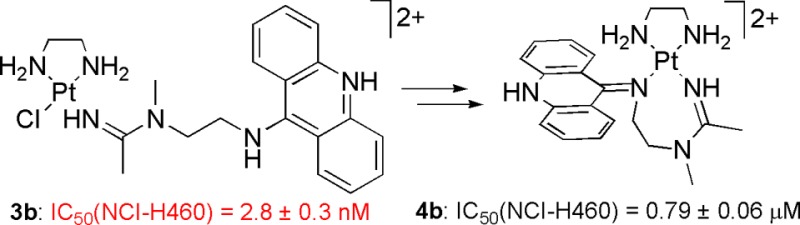
The formation of unusual seven-membered, sterically overloaded chelates [Pt(en)(L/L′)](NO3)2 (4a/4b) from the corresponding potent hybrid antitumor agents [PtCl(en)(LH/L′H)](NO3)2 (3a/3b) is described, where en is ethane-1,2-diamine and L(H) and L′(H) are (protonated) N-(2-(acridin-9-ylamino)ethyl)-N-methylpropionimidamide and N-(2-(acridin-9-ylamino)ethyl)-N-methylacetimidamide, respectively. Compounds 3a and 3b inhibit H460 lung cancer cell proliferation with IC50 values of 12 ± 2 and 2.8 ± 0.3 nM, respectively. The new derivative 3b proves to be not only the most cytotoxic platinum–acridine hybrid of this kind but also one of the most potent platinum-based anticancer agents described to date. The chelates 4a and 4b do not undergo ligand substitution reactions with nucleobase nitrogen and cysteine sulfur and do not intercalate into DNA. Despite their inertness, the two chelates appear to maintain micromolar activity in H460 cells. The results are discussed in the context of potential DNA-mediated and DNA-independent cell kill mechanisms and the potential use of the chelates as prodrugs.
Keywords: Platinum−acridine, DNA interactions, nonsmall cell lung cancer, reactivity, chelate
The design of novel chemotherapies and targeted agents for the treatment of chemoresistant cancers presents a pressing need in clinical oncology.1,2 We have recently communicated the discovery of a potent platinum-based compound as the result of extensive structure–activity relationship studies on platinum–acridine hybrid agents designed as potential treatments for cisplatin-resistant nonsmall-cell lung cancer (NSCLC).3 The prototypical complexes, [PtCl(am2)(LH)](NO3)2 [compounds 3a and 3a*; LH = N-(2-(acridin-9-ylamino)ethyl)-N-methylpropionimidamide, protonated form, am2 = en, ethane-1,2-diamine, am = NH3; Chart 1], have demonstrated high cytotoxic potency in NCI-H460 NSCLC cells at nanomolar concentrations, and 3a* was able to slow H460 tumor growth in a mouse xenograft model.3 Unfortunately, compound 3a* proved to be quite toxic to the test animals, most likely due to renal injury.3,4 The nephrotoxicity of platinum-based drugs is known to be caused by glutathione (GSH)-S-platinum species, which are formed either by direct ligand substitution or by enzymatic conjugation during glutathione transferase (GST)-mediated phase II chemoprotection.5,6 GSH-S conjugate formation is an undesired major drug detoxification mechanism in cancer cells often associated with tumor resistance to platinum drugs.7 Thus, to control this unwanted reactivity and the dose-limiting systemic toxicity of our platinum–acridine compounds, structural modifications are being made to the prototype with the objective of slowing the reaction of platinum with reactive sulfur nucleophiles. One possibility explored is the replacement of the chloro leaving group with bulky carboxylato ligands. Here, we report an unexpected reactivity feature as a result of the attempted ligand substitution in compound 3a, which led to the formation of an unusual seven-membered N,N-chelate. In the course of this study, we synthesized a new analogue by changing the ethyl group of the amidine residue in 3a into a methyl group (compound 3b, Scheme 1). The new derivative shows an enhanced cytotoxic effect in H460 cells as compared to the parent compound and proves to be one of the most potent platinum-containing anticancer agents reported to date.
Chart 1. Structures of the Platinum–Acridine Prototypes.

Scheme 1. Synthesis and Reactivity of Platinum–Acridines.

Reagents and conditions: (a) One equiv of AgNO3/DMF/room temperature/40 min, then N1-(acridin-9-yl)-N2-methylethane-1,2-diamine/–10 °C to room temperature/16 h. (b) One equiv of HNO3(aq)/MeOH. (c) One equiv of AgO2CR′/DMF/room temperature/24 h (R′ = CH3, C6H5). (d) One equiv of AgNO3/DMF/room temperature/48 h, then EtOH/reflux/48 h. R = Et for 1a–4a and R = Me for 1b–4b.
Compound 3a was synthesized from the corresponding platinum–nitrile complex 1a and N1-(acridin-9-yl)-N2-methylethane-1,2-diamine according to a previously reported method involving addition of the dangling secondary amine across the metal-activated CN triple bond, followed by protonation of intermediate 2a to produce the physiologically relevant acridinium form (pKa ≈ 9–10) (Scheme 1).3 To test if the chloro ligand can be substituted with carboxylate, compound 3a was reacted with 1 equiv of silver acetate or silver benzoate in DMF solution. While abstraction of chloride could be achieved using this procedure, as evidenced by the formation of silver chloride, the formation of the desired carboxylato derivatives (Scheme 1) of compound 3a was not observed. Instead, the reaction leads to an unusual seven-membered chelate, [Pt(en)(L)](NO3)2, compound 4a, in which both the amidine imino nitrogen and the exocyclic nitrogen (N9) of the acridine ligand are attached to the metal. This result indicates that the acridinium ligand (LH) in 3a has been deprotonated to the free base form (L), most likely by carboxylate, which serves as a weak auxiliary base in this reaction rather than a ligand. The resulting acridine free base can exist in two tautomeric forms (see compound 2, Scheme 1).8 The imino tautomer produces a strong intramolecular nucleophile (N9), which reacts with platinum to form a metal chelate. Compound 4a can also be synthesized directly from the monocationic complex 2 in the absence of carboxylate by abstracting the chloro ligand with silver nitrate.
Single crystals of compound 4a were obtained, which were suitable for X-ray crystallography (Figure 1). The compound forms discrete 2+ charged complex cations and crystallizes along with two nitrate counterions as a racemic mixture of two enantiomers in the centrosymmetric point group P21/c. While platinum shows a classical square-planar [PtN4] coordination, the most striking feature of complex 4a is the unique geometry of the chelate formed by the acridine–amidine carrier ligand. Platinum is coordinated by the amidine-NH group and the exocyclic acridine nitrogen. The acridine moiety exists in its imino tautomeric form, which causes the chromophore to significantly deviate from planarity: The angle between planes defined by the outermost C6 rings (C7–C12 and C13–C18) is 18.7(5)°. The rigid fold of the seven-membered chelate positions the ethylene linker (C4 and C5) on one side of the square coordination plane and one edge of the acridine ring on the opposite side. The latter produces an anagostic interaction9 between the acridine and the metal from an axial position of the complex (Pt1···H8 2.433 Å; Figure S1 in the Supporting Information).
Figure 1.
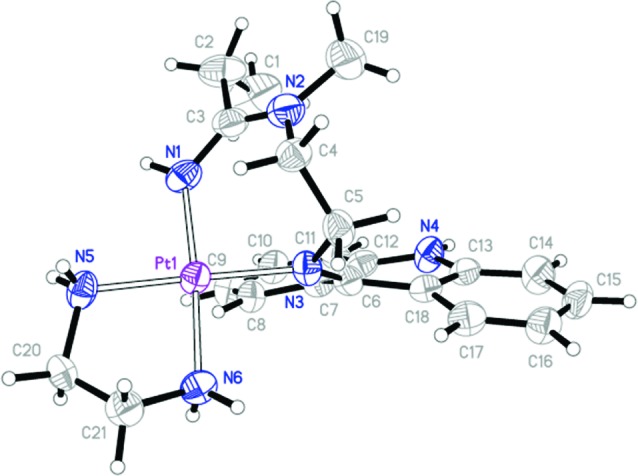
Thermal ellipsoid presentation (50% probability level) of complex 4a in the solid state with nonhydrogen atoms labeled. Nitrate counterions have been omitted for clarity.
On the basis of signal multiplicities and chemical shift anomalies observed in 1H NMR spectra of compound 4a (Supporting Information), the rigid scaffold observed in the solid state persists in solution. No inversion of the seven-membered chelate occurs on the NMR time scale (1H NMR spectra in the temperature range 25–80 °C, not shown) due to restricted dynamics in the sterically overcrowded coordination sphere. Closer examination of the molecular geometry of 4a in the solid state indicates that the bidentate binding mode positions the terminal C1 of the amidine ethyl group directly above one acridine phenyl ring (Figure S8 in the Supporting Information). To test if this steric clash contributes to the rigidity of the scaffold, we synthesized derivatives 3b and 4b in which the ethyl group was changed into a methyl group by introducing an acetimidamide donor group (R = Me) instead a propionimidamide group (R = Et) (Scheme 1). A comparison of the 1H NMR signatures observed for derivatives 4a and 4b (Supporting Information) confirms that truncation of the ethyl group has no effect on the folding geometry of the chelate.
To assess the effect of chelate formation on the biological activity of the hybrid agents, compounds 4a and 4b were studied along with the precursor complexes 3a and 3b in H460 cells using cell proliferation assays. The drug–response curves for the four derivatives are shown in Figure 2. In this study, our prototypical complex 3a inhibited cancer cell proliferation with an IC50 of 12 ± 2 nM, while the IC50 of the corresponding chelate 4a was 6.2 ± 0.8 μM. Likewise, the new hybrid agent 3b was highly cytotoxic with an IC50 of 2.8 ± 0.3 nM, and the chelate derived from it, complex 4b, showed greatly reduced activity with an IC50 of 0.79 ± 0.06 μM. These results demonstrate that the parent hybrid agents, 3a and 3b, are approximately 500 times and 300 times more potent than their respective chelates. They also show that simple removal of a methyl group on the amidine donor group in 3a to produce 3b results in a significant cytotoxic enhancement by approximately 4-fold. Complex 3b proves to be the most active hybrid agent of this kind identified thus far and shows a more than 500-fold higher activity in this cell line than cisplatin, for which an IC50 of 1.51 ± 0.16 μM was determined.10
Figure 2.
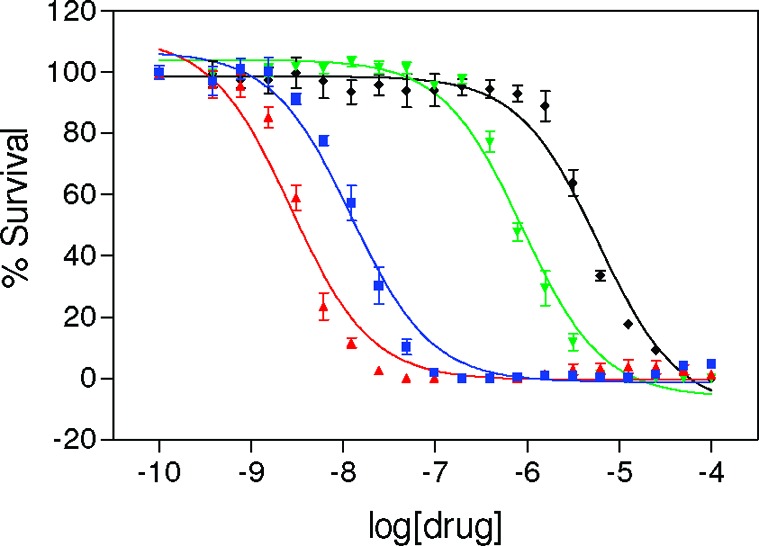
Drug–response curves for cell proliferation assays performed in NCI-H460 cancer cells with compounds 3a (blue), 3b (red), 4a (black), and 4b (green). Error bars indicate ± standard deviations for a set of three independent experiments performed in triplicate.
An interesting observation in this study is that both chelates seem to maintain activity in the micromolar concentration range. In fact, compound 4b proves to be slightly more active than cisplatin, which is surprising because the steric bulk created by the chelate and the absence of a suitable leaving group on platinum should render this complex relatively unreactive with DNA. To shed light on the reactivity and target binding properties of compounds 4a and 4b, we studied their interactions with biologically relevant nucleophiles. Both complexes were incubated at a concentration of 1 mM with excess 2′-deoxyguanosine and N-acetylcysteine at platinum-to-nucleophile ratios of 1:10 in water (or D2O) or in 10 mM phosphate buffer (pH 7.2) at 37 °C for 72 h to mimic the reactions of the chelates with DNA and GSH, respectively. Incubations were monitored by 1H NMR spectroscopy and electrospray mass spectrometry (data not shown). Using these methods, it was confirmed that complexes 4a and 4b are completely unreactive with nucleobase nitrogen and amino acid sulfur. Likewise, incubations of both complexes in dilute hydrochloric acid (pH 3) showed no sign of chelate opening under acidic conditions to produce complexes 3a and 3b. On the basis of these findings, it seems unlikely that the new chelates would act as caged precursors (prodrugs) of the open chain conjugate forms. Conversely, complexes 3a and 3b showed no sign of spontaneous conversion to their respective chelates at neutral pH. This suggests that chelate formation does not occur under physiological conditions and plays no role in the mechanism of action of the hybrid agents.
The mechanism of action of compounds 3a and 3b and related derivatives involves intercalation of the acridinium moiety into double-stranded DNA and formation of monoadducts with nucleophilic guanine and adenine nitrogen in both grooves of the biopolymer.11 While this coordinative/intercalative hybrid binding mode leads to nanomolar cytotoxicity in cancer cells, the platinum-free acridines and some intercalating complexes containing unreactive platinum moieties have also shown activity in vitro, although at significantly higher concentrations.12−14 Thus, reversible intercalation of compounds 4a and 4b alone might be responsible for the micromolar cytotoxic effect observed in NCI-H460 cells. To test if the substitution-inert chelates bind to double-stranded DNA by intercalation, we incubated 4a and 4b and their hybrid precursors 3a and 3b, with calf thymus DNA and monitored UV–visible spectral changes in the acridine region (short-axis π–π* transition within the acridine chromophore11) (Figure 3). The UV–visible features of 3a and 3b show a λmax at 416 and 414 nm, respectively, and the vibronic coupling (Δλ = 22 nm) characteristic of protonated acridine moieties.11 In the presence of double-stranded DNA, this feature in both agents experiences a pronounced bathochromic shift of 3–4 nm, which is consistent with intercalation of the chromophores into the DNA base stack (Figure 3a,c).11 By contrast, the platinum-modified iminoacridine moieties in compounds 4a and 4b show features at 410 nm, which lack the vibronic fine structure observed in the respective acridinium forms. Incubation of the two complexes with calf thymus DNA does not result in a red shift of the absorbance maximum (Figure 3b,d). Thus, intercalation of the acridine moieties in 4a and 4b as well as chelate opening and reprotonation of the chromophores in the presence of DNA can be firmly excluded. Considering the molecular structure of complex 4a, it appears that both the steric bulk and the deviation from planarity of the acridine moiety are prohibitive of intercalative binding.
Figure 3.

UV–visible spectra recorded for 50 μM solutions of complexes 3a (a), 4a (b), 3b (c), and 4b (d) alone (black traces) and after incubation with 200 μM (base pairs) calf thymus DNA (red traces) in 10 mM phosphate buffer (pH 7.2).
Our model studies with N-acetylcysteine have demonstrated that the chelated acridine ligand in compounds 4a and 4b renders the platinum center completely unreactive with cysteine sulfur, which would be a desired prodrug feature. However, we also found that the complexes are unable to form adducts or intercalate with DNA, which makes it very unlikely that the cytotoxicity caused by these agents is mediated by reversible or irreversible interactions with cellular DNA. Because of the dramatic relative differences in cytotoxicity levels observed between the hybrid agents and their corresponding chelates, it is impossible to speculate if a non-DNA binding mechanism might contribute to the cell death produced by the latter species. In fact, complex 4b for instance, itself may be a biologically inactive species. Preparations of this complex containing undetected trace impurities of compound 3b of as little as 0.2–0.3% or intracellular formation of the potent hybrid agent by an unknown mechanism by the same small percentage would explain the observed cytotoxicity levels. It should be pointed out, however, that the high-performance liquid chromatography profile recorded for the crystalline sample of 4b tested in H460 cells did not show any signs of impurities (see the Supporting Information). Likewise, chelate opening at low pH, for instance in the acidic environment of the lysosomes, seems to be an unlikely mechanism. On the other hand, it needs to be pointed out that if a cell kill mechanism existed for complexes 4a and 4b that is not mediated by DNA damage, it would be far less effective than the formation of DNA adducts by 3a and 3b. These results bear some relevance to a recent study on platinum–pyrophosphato complexes, which are relatively unreactive as compared to their precursor cisplatin derivatives and, for the most part, showed reduced activity in the high micromolar range in cancer cells.15,16
Previous biochemical studies suggested that the ability of the hybrid agents to produce high DNA adducts levels by favoring rapid platination of nucleobase nitrogen is a prerequisite for potent cytotoxicity.3,17 Here, we have generated a new derivative, 3b, by reducing the steric bulk in the platinum coordination sphere using a subtle structural modification, which resulted in a pronounced increase in cytotoxicity. This observation is in accord with previously observed structure–(re)activity relationships in this class of compounds and corroborates that DNA is the therapeutic target of these agents. Finally, the unprecedented high potency of compound 3b determined in this study confirms the unique anticancer potential of the hybrid platinum–intercalator technology. It underscores the notion that DNA damage mechanisms radically different from the common cisplatin type cross-links are needed to achieve low nanomolar cytotoxicity levels, as platinum-based agents typically produce IC50 values in the micromolar range in NCI-H460.18,19
In conclusion, efficient irreversible modification of DNA appears to be an important prerequisite for producing useful cancer cell kill. On the basis of our observations in the platinum–acridine system, the question should not be whether to target DNA but what type of DNA damage might be the most effective and how new designs can take advantage of cancer cell abnormalities to combat chemoresistant disease. In the light of the current findings, claims of non-DNA-mediated cell kill mechanisms as a new strategy for future platinum anticancer drug development should be viewed with caution.
Supporting Information Available
Experimental procedures, details of product characterization, and crystallographic details for compound 4b. This material is available free of charge via the Internet at http://pubs.acs.org.
This work was funded by the U.S. National Institutes of Health (Grant CA101880).
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Kelland L. The resurgence of platinum-based cancer chemotherapy. Nat. Rev. Cancer 2007, 7, 573–584. [DOI] [PubMed] [Google Scholar]
- Chabner B. A.; Roberts T. G. Timeline—Chemotherapy and the war on cancer. Nat. Rev. Cancer 2005, 5, 65–72. [DOI] [PubMed] [Google Scholar]
- Ma Z.; Choudhury J. R.; Wright M. W.; Day C. S.; Saluta G.; Kucera G. L.; Bierbach U. A non-cross-linking platinum-acridine agent with potent activity in non-small-cell lung cancer. J. Med. Chem. 2008, 51, 7574–7580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Z.; Rao L.; Bierbach U. Replacement of a thiourea-S with an amidine-NH donor group in a platinum-acridine antitumor compound reduces the metal’s reactivity with cysteine sulfur. J. Med. Chem. 2009, 52, 3424–3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao X.; Panichpisal K.; Kurtzman N.; Nugent K. Cisplatin nephrotoxicity: A review. Am. J. Med. Sci. 2007, 334, 115–124. [DOI] [PubMed] [Google Scholar]
- Hanigan M. H.; Devarajan P. Cisplatin nephrotoxicity: molecular mechanisms. Cancer Ther. 2003, 1, 47–61. [PMC free article] [PubMed] [Google Scholar]
- Klein A. V.; Hambley T. W. Platinum drug distribution in cancer cells and tumors. Chem. Rev. 2009, 109, 4911–4920. [DOI] [PubMed] [Google Scholar]
- Ebead Y.; Roshal A. D.; Wroblewska A.; Doroshenko A. O.; Blazejowski J. Tautomerism of acridin-9-amines substituted at the exocyclic nitrogen atom: Spectroscopic investigations and theoretical studies. Spectrochim. Acta, Part A 2007, 66, 1016–1023. [DOI] [PubMed] [Google Scholar]
- Brookhart M.; Green M. L.; Parkin G. Agostic interactions in transition metal compounds. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 6908–6914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao L.; West T. K.; Saluta G.; Kucera G. L.; Bierbach U. Probing platinum-adenine-N3 adduct formation with DNA minor-groove binding agents. Chem. Res. Toxicol. 2010, 23, 1148–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baruah H.; Wright M. W.; Bierbach U. Solution structural study of a DNA duplex containing the guanine-N7 adduct formed by a cytotoxic platinum-acridine hybrid agent. Biochemistry 2005, 44, 6059–6070. [DOI] [PubMed] [Google Scholar]
- Martins E. T.; Baruah H.; Kramarczyk J.; Saluta G.; Day C. S.; Kucera G. L.; Bierbach U. Design, synthesis, and biological activity of a novel non-cisplatin-type platinum-acridine pharmacophore. J. Med. Chem. 2001, 44, 4492–4496. [DOI] [PubMed] [Google Scholar]
- Ackley M. C.; Barry C. G.; Mounce A. M.; Farmer M. C.; Springer B. E.; Day C. S.; Wright M. W.; Berners-Price S. J.; Hess S. M.; Bierbach U. Structure-activity relationships in platinum-acridinylthiourea conjugates: effect of the thiourea nonleaving group on drug stability, nucleobase affinity, and in vitro cytotoxicity. J. Biol. Inorg. Chem. 2004, 9, 453–461. [DOI] [PubMed] [Google Scholar]
- Guddneppanavar R.; Choudhury J. R.; Kheradi A. R.; Steen B. D.; Saluta G.; Kucera G. L.; Day C. S.; Bierbach U. Effect of the diamine nonleaving group in platinum-acridinylthiourea conjugates on DNA damage and cytotoxicity. J. Med. Chem. 2007, 50, 2259–2263. [DOI] [PubMed] [Google Scholar]
- Bose R. N.; Maurmann L.; Mishur R. J.; Yasui L.; Gupta S.; Grayburn W. S.; Hofstetter H.; Salley T. Non-DNA-binding platinum anticancer agents: Cytotoxic activities of platinum-phosphato complexes towards human ovarian cancer cells. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 18314–18319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishur R. J.; Zheng C.; Gilbert T. M.; Bose R. N. Synthesis, X-ray crystallographic, and NMR characterizations of platinum(II) and platinum(IV) pyrophosphato complexes. Inorg. Chem. 2008, 47, 7972–7982. [DOI] [PubMed] [Google Scholar]
- Choudhury J. R.; Rao L.; Bierbach U. Rates of intercalator-driven platination of DNA determined by a restriction enzyme cleavage inhibition assay. J. Biol. Inorg. Chem. 2011, 16, 373–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skander M.; Retailleau P.; Bourrie B.; Schio L.; Mailliet P.; Marinetti A. N-Heterocyclic carbene-amine Pt(II) complexes, a new chemical space for the development of platinum-based anticancer drugs. J. Med. Chem. 2010, 53, 2146–2154. [DOI] [PubMed] [Google Scholar]
- Zastre J.; Anantha M.; Ramsay E.; Bally M. Irinotecan-cisplatin interactions assessed in cell-based screening assays: Cytotoxicity, drug accumulation and DNA adduct formation in an NSCLC cell line. Cancer Chemother. Pharmacol. 2007, 60, 91–102. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.