

Abstract
Herein we report an enantioselective synthesis of complex cyclopentanones using aliphatic aldehydes and activated enones. With the combination of a chiral secondary amine and a chiral triazolium catalyst, high diastereoselectivity and excellent enantioselectivity can be achieved. We present evidence of a clear cooperative effect when these two catalysts are present simultaneously in the system.
Introduction
The incorporation of multiple catalytic cycles in a single procedure allows for complex compounds to be easily accessed, a concept that has been termed cascade catalysis.1–3 By eliminating the need for isolation and purification of intermediates, both time and resources are saved. This is especially important if these intermediates prove unstable upon isolation. While attractive, cascade catalysis provides unique challenges. With the possibility of multiple catalysts present simultaneously, the need for reaction selectivity is important. A solution to this problem is to use catalysts of orthogonal reactivity. Our group has previously reported that secondary amine and N-heterocyclic carbene (NHC) catalysts initiate cascade reactions of enals and β-dicarbonyls to form α-hydroxycyclopentanones (Fig 1, a).4 This reaction proceeds via iminium activation of the enal to induce a conjugate addition by the dicarbonyl followed by an intramolecular benzoin. Importantly the two catalysts work cooperatively, providing higher yields and enantioselectivities compared to a two-step process. Herein, we report the secondary amine/NHC catalyzed cascade reaction of aliphatic aldehydes and activated Michael acceptors to form complex cyclopentanones with a complementary substitution pattern.
Fig 1.

Secondary amine/NHC cascade reactions6
Whereas our previous work employed iminium catalysis, we sought to explore the use of enamine catalysis in combination with NHC’s in a cascade reaction. Ma has reported a highly diastereo- and enantioselective Michael addition of aliphatic aldehydes into activated enones with the use of a secondary amine catalyst (3).5 We speculated that the aldehyde intermediate formed could undergo an intramolecular benzoin reaction when exposed to an N-heterocyclic carbene catalyst (Fig 1, b). This would provide a cyclopentanone product in what may be considered a formal [3+2] cycloaddition. This approach provides complimentary and inaccessible substitution patterns from our previous work.
Results and Discussion
Reaction Development
The reaction conditions developed by the Ma group were repeated using the Jorgensen-Hayashi catalyst 37 in methanol at room temperature. After observing the consumption of starting material, achiral triazolium salt 58 and sodium acetate were added to catalyze the benzoin cyclization. These conditions provide no desired product (Table 1, entry 1). When methanol is replaced with chloroform, the desired product is formed in 29% yield and 96% ee but with a 3:1:<1:<1 dr (Table 1, entry 2).9 Employing a one-step protocol with all reagents present from the outset results in an increase in yield to 35% (entry 3). Increasing the temperature from 23 °C to 60 °C improves the yield to 89% while maintaining high enantioselectivity (96% ee) and a 5:1:<1:<1 diastereomeric ratio. The diastereoselectivity is further improved to 290:15:6:1 when the chiral aminoindanol based triazolium 610,11 is used (entry 5). The use of the antipode of this catalyst, 6′, results in lower diastereoselectivity (4:1:<1:<1). This is likely a result of a match/mismatch relationship.
Table 1.
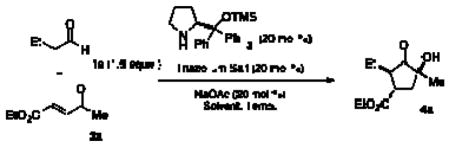 | ||||||
|---|---|---|---|---|---|---|
| Solvent | Triazolium salt | Temp. (°C) | Yield (%) | ee (%)b | drb | |
| 1c | MeOH |
 5 |
23 | 0 | -- | -- |
| 2c | CHCl3 | 5 | 23 | 29 | 96 | 3:1:<1:<1 |
| 3d | CHCl3 | 5 | 23 | 35 | 96 | 2:1:<1:<1 |
| 4d | CHCl3 | 5 | 60 | 89 | 96 | 5:1:<1:<1 |
| 5d | CHCl3 |
 6 |
60 | 87 | 95 | 19:1:<1:<1 |
| 6d | CHCl3 |
 6′ |
60 | 59 | 93 | 4:1:<1:<1 |
See supporting information for general procedure.
Enantioselectivity and diastereoselectivity were determined by GC.
20 mol % Triazolium salt was added after full consumption of 2a.
20 mol % Triazolium salt was added at the beginning of the reaction.
Synthetic Scope
With suitable conditions established, a variety of substrates were screened to explore the scope of this new cascade. Aliphatic aldehydes provide the desired products in good yield and high enantio- and diastereoselectivity (Chart 1, 4b–c). Isovaleraldehyde competitively forms the Stetter product12 in a 1:1 ratio with cyclopentanone 4d under standard conditions. This side product can be avoided when the introduction of the triazolium is delayed until after complete formation of the intermediate. Larger aldehydes are also viable but routinely require prolonged reaction times (Chart 1, 4f–g).
Chart 1.
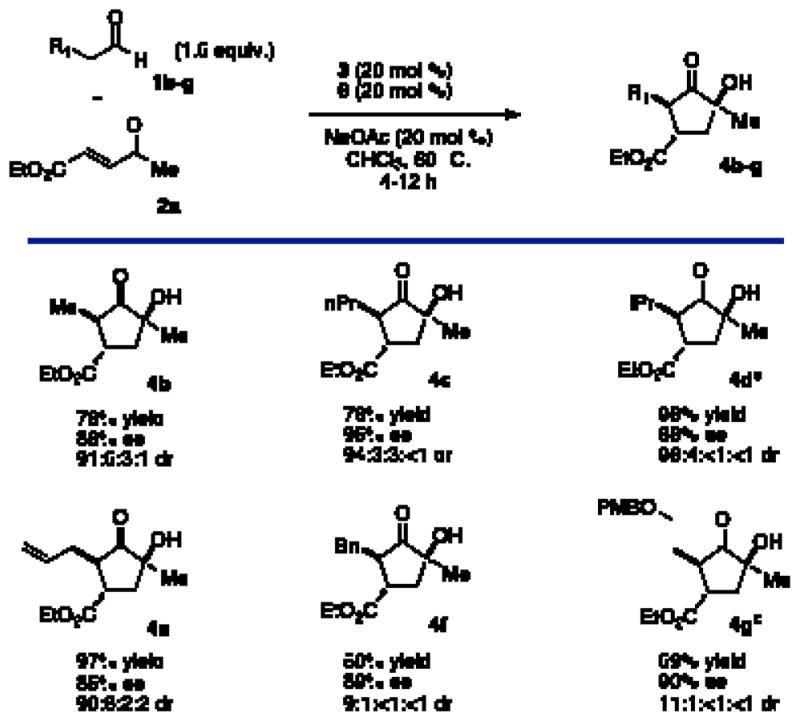
Aldehyde Scopea
aSee Table 1. bCatalyst 6 was added after consumption of starting material. cDiastereomeric ratio determined by 1H NMR
Variation in the enone component was then explored (Chart 2). Esters and tertiary amides are viable under these conditions (Chart 2, 4h–k).13 Substitution at the ketone position is also tolerated. N-Alkyl ketones provide products in excellent yields while maintaining high stereoselectivity. The isopropyl ketone failed to cyclize under standard conditions. By substituting the bulky triazolium 6 with the smaller achiral catalyst 5, the intramolecular benzoin was accomplished albeit with a curiously low enantioselectivity (4o). Phenyl ketone can also be employed, with diminished diastereoselectivity (4n). Diketones may also be used to form products (4p–q) with good selectivity despite diminished yield.
Chart 2.
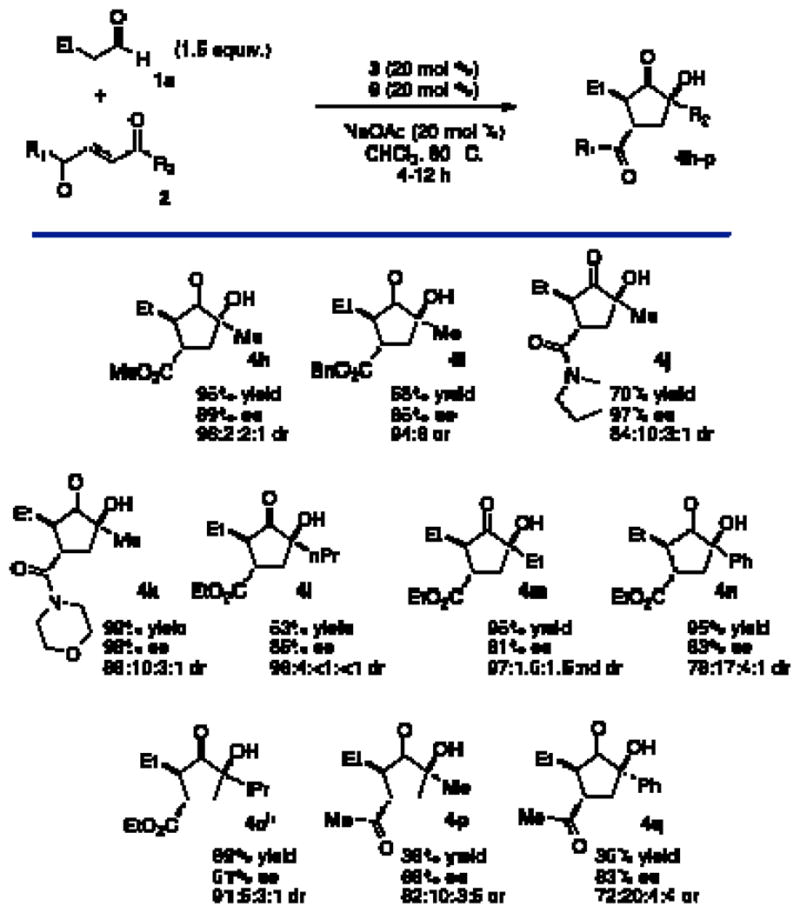
Keto-ester Scopea
aSee Table 1. bCatalyst 5 was used in place of 6.
With high complexity already built into the cyclopentanone products, functionalization should allow rapid access to even more elaborate products. The addition of sodium triacetoxyborohydride at the end of the reaction permits a diastereoselective reduction of the ketone to 1,2-diol 7. This action effectively permits the formation of a fourth stereocenter in one pot (Eqn 1).
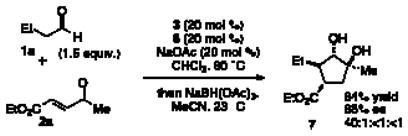 |
(1) |
Mechanistic Insights
We then explored if there is an advantage between this onestep protocol versus a two-step reaction. Aldehyde 8 was prepared by exposing butyraldehyde and enone 2a to catalyst 3, catalytic acetic acid, and chloroform at room temperature (Scheme 1). After purification, the aldehyde intermediate was isolated in moderate yield with a 2:1 diastereomeric ratio.
Scheme 1.
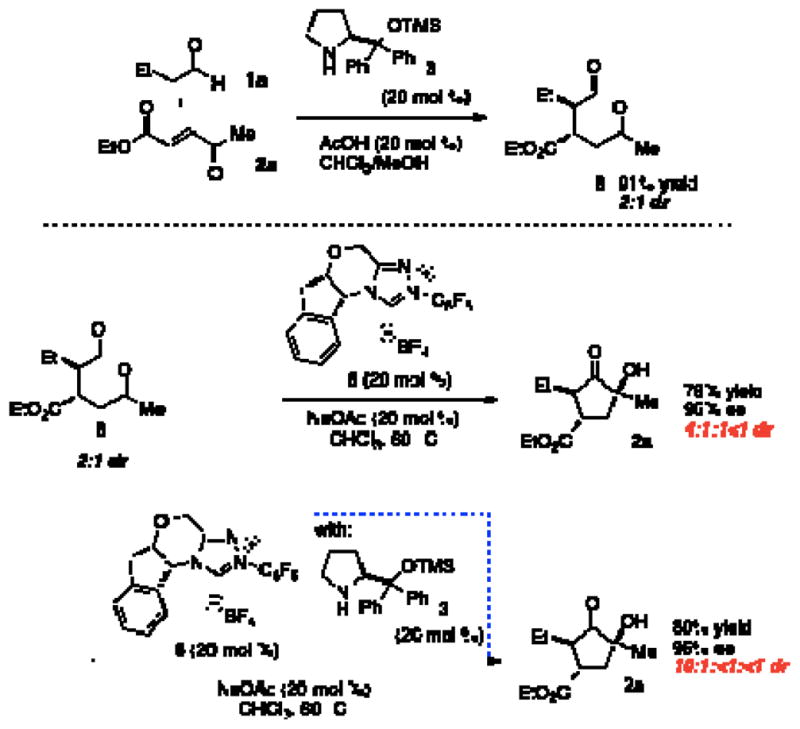
Two-Pot Reactions
Exposure of this intermediate to benzoin conditions provides the cyclized product in comparable yield and enantioselectivity, but with a low diastereomeric ratio (4:1:1:<1). This diastereoselectivity can be improved when the benzoin cyclization is performed with the addition of chiral amine catalyst 3(10:1:<1:<1 dr).
In our previous work, it was discovered that the amine catalyst is responsible for a retro-Michael reaction, which eroded enantioselectivity in the two-pot reaction. Crossover experiments indicate that this pathway is non-operative in this system.14 Instead, we propose that the secondary amine catalyst is capable of epimerizing the α-position of the intermediate aldehyde to form an equilibrium between 8 and 8′. The chiral triazolium 6 prefers cyclization with only one of these diastereomers, and this adduct continues on to the final product. The amine catalyst thus aids in converting the less reactive diastereomerinto its epimer.15 This hypothesis explains how the amine catalyst can convert aldehyde 8 with low diastereoselectivity to a diastereomerically enriched product.
To support this mechanism, the benzoin cyclization was monitored over time by NMR spectroscopy. When intermediate aldehyde 8 is exposed to catalyst 6, we see complete consumption of one diastereomer in preference over the other (Fig. 3). When amine catalyst 3 is included in this reaction, consumption of both diastereomers occurs over the course of the reaction, consistent with the hypothesis that amine catalyst 3 serves to interconvert the two diastereomers of 8.16,17
Figure 3.

NMR experiments
Conclusion
In summary, a one-pot stereoeselective Michael-Benzoin cascade reaction has been developed for the synthesis of complex cyclopentanones. The presence of both the secondary amine and triazolium catalysts is essential for excellent results, indicating a cooperative relationship between the catalysts. This provides a unique and useful method to form complicated cyclopentanes from simple starting materials.
Supplementary Material
Fig 2.

Proposed Mechanism
Acknowledgments
We thank NIGMS for generous support of this research (GM72586). K. E. O. thanks NIH (GM80442-S1 and GM096749) for funding. T.R. thanks Amgen and Roche for unrestricted support. We thank Donald Gauthier and Greg Hughes (Merck) for a generous gift of aminoindanol and Kevin M. Oberg and Derek M. Dalton (CSU) for solving the crystal structure of 9.
Footnotes
Electronic Supplementary Information (ESI) available: Experimental procedures, spectras, and crystal structues are provided. See DOI: 10.1039/b000000x/
References
- 1.For recent reviews: Grondal C, Jeanty M, Enders D. Nat Chem. 2010;2:167. doi: 10.1038/nchem.539.Zhou J. Chem Asian J. 2010;5:422. doi: 10.1002/asia.200900458.Xinhong Y, Wang W. Org Biomol Chem. 2008;6:2037. doi: 10.1039/b800245m.Enders D, Grondal C, Huttl MRM. Angew Chem Int Ed. 2007;46:1570. doi: 10.1002/anie.200603129.Walji AM, MacMillan DWC. Synlett. 2007:1477.Wasilke JC, Obrey SJ, Baker RT, Bazan GC. Chem Rev. 2005;105:1001. doi: 10.1021/cr020018n.
- 2.For some recent examples of multicatalytic systems: Quintard A, Alexakis A, Mazet C. Angew Chem Int Ed. 2011;50:2354. doi: 10.1002/anie.201007001.Trost BM, Luan X. J Am Chem Soc. 2011;133:1706. doi: 10.1021/ja110501v.Raup DEA, Cardinal-David B, Holte D, Scheidt KA. Nat Chem. 2010;2:766. doi: 10.1038/nchem.727.Yu C, Zhang Y, Zhang S, He J, Wang W. Tetrahedron Lett. 2010;51:1742.Hong BC, Dange NS, Hsu CS, Liao JH. Org Lett. 2010;12:4812. doi: 10.1021/ol101969t.Huang Y, Walji AM, Larsen CH, MacMillan DWC. J Am Chem Soc. 2009;127:15051. doi: 10.1021/ja055545d.Simmons B, Walji AM, MacMillan DWC. Angew Chem Int Ed. 2009;48:4349. doi: 10.1002/anie.200900220.Jiang H, Elsner P, Jensen KL, Falcicchio A, Marcos V, Jorgensen KA. Angew Chem Int Ed. 2009;48:6849. doi: 10.1002/anie.200901446.Belot S, Vogt KA, Besnard C, Krause N, Alexakis A. Angew Chem Int Ed. 2009;48:8923. doi: 10.1002/anie.200903905.Wang Y, Ha RA, Zhou YL, Yang S, Xu PF, Dixon DJ. Angew Chem Int Ed. 2009;48:9834. doi: 10.1002/anie.200905014.Cernak TA, Lambert TH. J Am Chem Soc. 2009;131:3124. doi: 10.1021/ja809897f.Kelly BD, Allen JM, Tundel RE, Lambert TH. Org Lett. 2009;11:1381. doi: 10.1021/ol900198r.Nicewicz DA, MacMillan DWC. Science. 2008;322:77. doi: 10.1126/science.1161976.Chi Y, Scroggins ST, Frechet JMJ. J Am Chem Soc. 2008;130:6322. doi: 10.1021/ja8013456.Lu LQ, Cao YJ, Liu XP, An J, Yao C-J, Ming Z-H, Xiao WJ. J Am Chem Soc. 2008;130:6946. doi: 10.1021/ja800746q.Zhou J, List B. J Am Chem Soc. 2007;129:7498. doi: 10.1021/ja072134j.Wang W, Li H, Wang J, Zu L. J Am Chem Soc. 2006;128:10351. doi: 10.1021/ja065187u.
- 3.For select recent examples of cascade reactions with a single catalyst: Liu L, Zhang X, Wang R, Wang W. Org Lett. 2010;12:4948. doi: 10.1021/ol102096s.Jui NT, Lee ECY, MacMillan DWC. J Am Chem Soc. 2010;132:10015. doi: 10.1021/ja104313x.McGarraugh PG, Brenner SE. Org Lett. 2009;11:5654. doi: 10.1021/ol9024293.Zu L, Zhang S, Xie H, Wang W. Org Lett. 2009;11:1627. doi: 10.1021/ol9003433.Bencivenni G, Wu LY, Mazzanti A, Giannichi B, Pesciaoli F, Song MP, Bartoli G, Melciorre P. Angew Chem Int Ed. 2009;48:7200. doi: 10.1002/anie.200903192.Wang J, Li H, Xi H, Sheng X, Wang W. Angew Chem Int Ed. 2007;46:9050. doi: 10.1002/anie.200703163.Xie H, Zu L, Li H, Wang J, Wang W. J Am Chem Soc. 2007;129:10886. doi: 10.1021/ja073262a.Zhou J, List B. J Am Chem Soc. 2007;129:7498. doi: 10.1021/ja072134j.Enders D, Huttl MRM, Grondal C, Raabe G. Nature. 2006;441:861. doi: 10.1038/nature04820.Huang Y, Walji AM, Larsen CH, MacMillan DWC. J Am Chem Soc. 2005;127:15051. doi: 10.1021/ja055545d.
- 4.Lathrop SP, Rovis T. J Am Chem Soc. 2009;131:13628. doi: 10.1021/ja905342e.For other contributions to cascade catalysis from our laboratories, see: Vora HU, Rovis T. J Am Chem Soc. 2007;129:13796. doi: 10.1021/ja0764052.Filloux CM, Lathrop SP, Rovis T. Proc Nat Acad Sci USA. 2010;107:20666. doi: 10.1073/pnas.1002830107.
- 5.Wang J, Ma A, Ma D. Org Lett. 2008;10:5425. doi: 10.1021/ol802354u. [DOI] [PubMed] [Google Scholar]
- 6.The “IM” and “EN” convention to denote iminium and enamine catalysis was first introduced by MacMillan and coworkers and subsequently adopted by others. See: Huang Y, Walji AM, Larsen CH, MacMillan DWC. J Am Chem Soc. 2005;127:15051. doi: 10.1021/ja055545d.(b) also see ref. 1a.
- 7.(a) Marigo M, Wabnitz TC, Fielenbach D, Jorgensen KA. Angew Chem Int Ed. 2005;44:794. doi: 10.1002/anie.200462101. [DOI] [PubMed] [Google Scholar]; (b) Hayashi Y, Gotoh H, Hayashi T, Shoji M. Angew Chem Int Ed. 2005;44:4212. doi: 10.1002/anie.200500599. [DOI] [PubMed] [Google Scholar]
- 8.Kerr MS, Read de Alaniz J, Rovis T. J Org Chem. 2005;70:5725. doi: 10.1021/jo050645n. [DOI] [PMC free article] [PubMed] [Google Scholar]
-
9.Absolute configuration determined by Single Crystal X-ray diffraction of the chiral salt(see SI):
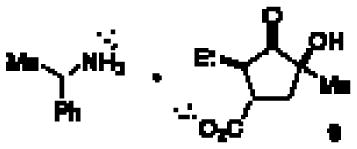
- 10.(a) Vora HU, Lathrop SP, Reynolds NT, Kerr MS, Read de Alaniz J, Rovis T. Org Synth. 2010;87:350. [Google Scholar]; (b) Kerr MS, Read de Alaniz J, Rovis T. J Am Chem Soc. 2002;124:10298. doi: 10.1021/ja027411v. [DOI] [PubMed] [Google Scholar]
- 11.(a) Hachisu Y, Bode JW, Suzuki K. J Am Chem Soc. 2003;125:8432. doi: 10.1021/ja035308f. [DOI] [PubMed] [Google Scholar]; (b) Takikawa H, Hachisu Y, Bode JW, Suzuki K. Angew Chem Int Ed. 2006;45:3492. doi: 10.1002/anie.200600268. [DOI] [PubMed] [Google Scholar]; (c) Enders D, Niemeier O, Balensiefer T. Angew Chem Int Ed. 2006;45:1463. doi: 10.1002/anie.200503885. [DOI] [PubMed] [Google Scholar]
-
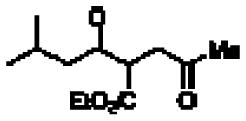
12.Stetter Product formed:
- 13.Secondary amides and thioesters do not provide desired products.
- 14.See Supporting Information for crossover experiment results.
- 15.The second step of this reaction could then be considered analogous to a dynamic kinetic resolution, which has been invoked in several other organocatalyzed transformations: Lee A, Michrowski A, Sulzer-Mosse S, List B. Angew Chem Int Ed. 2011;50:1707. doi: 10.1002/anie.201006319.Hoffman S, Nicoletti M, List B. J Am Chem Soc. 2006;128:13074. doi: 10.1021/ja065404r.Ward DE, Jheengut V, Akinnusi OT. Org Lett. 2005;7:1181. doi: 10.1021/ol050195l.
- 16.See Supporting Information for spectra from NMR experiments.
-
17.In addition, when intermediate 8 is exposed to catalyst 3 in deuterated methanol at elevated temperature, complete deuteration is observed at the α-position.
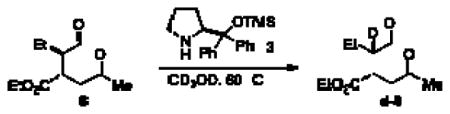
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.