

Abstract
Bacterial biofilms are complex microbial communities that are common in nature and are being recognized increasingly as an important determinant of bacterial virulence. However, the structural determinants of bacterial aggregation and eventual biofilm formation have been poorly defined. In Gram-negative bacteria, a major subgroup of extracellular proteins called self-associating autotransporters (SAATs) can mediate cell–cell adhesion and facilitate biofilm formation. In this study, we used the Haemophilus influenzae Hap autotransporter as a prototype SAAT to understand how bacteria associate with each other. The crystal structure of the H. influenzae HapS passenger domain (harbouring the SAAT domain) was determined to 2.2 Å by X-ray crystallography, revealing an unprecedented intercellular oligomerization mechanism for cell–cell interaction. The C-terminal SAAT domain folds into a triangular-prism-like structure that can mediate Hap–Hap dimerization and higher degrees of multimerization through its F1–F2 edge and F2 face. The intercellular multimerization can give rise to massive buried surfaces that are required for overcoming the repulsive force between cells, leading to bacterial cell–cell interaction and formation of complex microcolonies.
Keywords: bacterial aggregation and biofilm formation, Haemophilus influenzae Hap adhesin, oligomerization, self-associating autotransporter, structural biology
Introduction
Intercellular interaction is important for the development, function, and survival of all cell types. In bacteria, cells can adhere to each other to form biofilms, complex communities that enhance survival in harsh environments, including infected hosts (Hall-Stoodley et al, 2004). Bacteria living in biofilms exhibit up to a thousand-fold increase in resistance to detergents and antibiotics (Stewart and Costerton, 2001). In addition, biofilms have been shown to be a key factor in the pathogenesis of a wide range of bacterial infections, including the upper and lower respiratory tract infections, urinary tract infections, endocarditis, and infections of permanently implanted prostheses and devices (Donlan and Costerton, 2002; Hall-Stoodley et al, 2004; Moons et al, 2009).
The factors that mediate biofilm formation remain poorly understood. In enteropathogenic Escherichia coli, enterohaemorrhagic E. coli, Haemophilus influenzae, and a variety of other pathogenic bacteria, a group of virulence factors known as self-associating autotransporters (SAATs) promote bacterial aggregation and microcolony formation, important steps in the process of biofilm formation (Klemm et al, 2006). Similar to other autotransporter proteins, SAATs contain three basic domains, including an N-terminal signal peptide (SP), an internal passenger domain, and a C-terminal outer membrane β-barrel domain (Henderson et al, 1998; Dautin and Bernstein, 2007). In all autotransporters characterized to date, the region responsible for effector function as protease, adhesin, toxin, or invasin is found in the passenger domain (Henderson and Nataro, 2001). In the H. influenzae Hap SAAT, the SP corresponds to residues 1–25, the passenger domain (referred to as HapS) corresponds to residues 26–1036, and the β-barrel domain (referred to as Hapβ) corresponds to residues 1037–1394; Figure 1A). Hap has been demonstrated to mediate bacterial adherence to respiratory epithelial cells and extracellular matrix proteins, bacterial entry into epithelial cells, and bacterial aggregation (Figure 1B; Rao et al, 1999). The C-terminal 511 amino acids of HapS (residues 525–1036) are responsible for interactions with fibronectin, laminin, and collagen IV, and the C-terminal 311 amino acids of HapS (residues 725–1036) are responsible for interactions with epithelial cells (Fink et al, 2002). The C-terminal 311 amino acids of HapS are also capable of mediating Hap–Hap interaction, triggering bacterial aggregation and microcolony formation (Hendrixson and St Geme, 1998; Fink et al, 2003), (Figure 1B). Hap also contains an N-terminal serine protease domain (residues 26–290, with the canonical catalytic triad, including His98, Asp140, and Ser243) (Fink et al, 2002). The serine protease domain mediates intermolecular autoproteolysis at L1036-N1037, L1046-T1047, F1077-A1078, and F1067-S1068 (termed the primary, secondary, tertiary, and quaternary cleavage sites, respectively), resulting in release of the HapS passenger domain from the bacterial surface and modulating bacterial adherence and aggregation (Hendrixson and St Geme, 1998; Fink and St Geme, 2003). Autoproteolytic activity is inhibited by secretory leukocyte protease inhibitor (SLPI) (Koizumi et al, 2008), a serine protease inhibitor that is present in varying amounts in the upper and lower respiratory tract and that results in enhanced Hap adhesive activity (Figure 1B) (Hendrixson and St Geme, 1998).
Figure 1.

Haemophilus influenzae Hap adhesin. (A) Domain arrangement of Hap adhesin. Similar to other autotransporters, Hap contains three general domains: a signal peptide (SP), a passenger domain (HapS), and an outer membrane translocator domain (Hapβ). (B) Hap-mediated Haemophilus influenzae pathogenesis. Red oval for bacterium and blue stick for Hap adhesin. The SAAT domain in the passenger domain mediates adherence to epithelial cells, invasion of epithelial cells, and bacterial aggregation. The ECM-binding domain mediates adherence to fibronectin, laminin, and collagen IV. A host protein known as secretory leukocyte protease inhibitor (SLPI) inhibits Hap intermolecular autoproteolysis through its N-terminal serine protease domain and results in accumulation of HapS on the bacterial surface, resulting in enhanced bacterial adhesive activity and microcolony formation (Hendrixson and St Geme, 1998).
In order to understand the Hap adhesin as a prototype SAAT that mediates bacterial aggregation and microcolony formation, the crystal structure of the HapS passenger domain was determined to 2.2 Å by X-ray crystallography. The structure reveals a ‘Dane Axe’-like architecture with the serine protease domain and the SAAT domain lying in the same plane. Interestingly, crystal packing and structural simulation of the HapS/SLPI complex suggest an unprecedented intercellular oligomerization mechanism for bacterial aggregation. The SAAT domains in this remarkable multimer are located in a trans configuration that is flanked by the F1–F2 edge and the F2 face, which act as nucleation sites for recruitment of more Hap molecules. Through intercellular multimerization, Hap seems to be able to generate enough mechanical force in the form of buried surfaces to mediate stable cell–cell interactions. On the basis of our structural data, we propose a polymerization/depolymerization model that might allow pathogenic bacteria to modulate interactions with the biofilm community during various stages of colonization and spread. Sequence alignment and structural similarity between the HapS structure and other SAAT domains suggest that our results may have broad implications for other SAATs. Overall, the HapS structure described in this report provides important insights into the mechanism of bacterial cell–cell interaction and eventual biofilm formation.
Results
Overall architecture of HapS
To determine the structure of H. influenzae HapS, the protein was expressed and purified from H. influenzae strain DB117/pJS106. Secreted HapS was harvested from the culture medium and subjected to further purification using sepharose caion-exchange, phenyl, and gel-filtration chromatography. Initially HapS crystals tended to grow into small-stacked plates. Increasing the protein concentration to >10 mg/ml allowed the growth of a singular protein crystal that diffracted to 2.2 Å. The structure of HapS was solved by molecular replacement, using the published E. coli Hbp (Otto et al, 2005) and H. influenzae IgA1 protease coordinates (Johnson et al, 2009) as initial search templates (Supplementary Figure 1A). The unit cell contains one HapS polypeptide chain per asymmetric unit. Residues 1–25 represent the SP and hence are not present in the structure of HapS. Residues 265–272, 851–873, and 977–1036 yielded no electron density, either because they are disordered (265–272, 851–873) or were cleaved during the process of Hap secretion or purification (977–1036), making them unavailable for model building.
The crystal structure of HapS reveals a ‘Dane Axe’-like architecture (Figure 2 and Supplementary Figure 1). The overall structure can be partitioned into two parts, an N-terminal serine protease domain and a C-terminal β-spine with a height of ∼120 Å. On the basis of three-dimensional arrangement of the β-strands, the β-spine can be further divided into two parts, a coiled β-helix with 54 strands (residues 294–885) and a C-terminal domain with an Ig-like fold (this domain is also known as the autochaperone domain, a conserved region necessary for folding of the passenger domain in many monomeric autotransporters) (Oliver et al, 2003; Otto et al, 2005). The interaction between the coiled β-helix and the autochaperone domain is mediated by β67, β68, β69, and β71 through β-strand augmentation (Figure 2). The β67–β69 pair contributes three hydrogen bonds and the β68–β71 pair contributes four hydrogen bonds to this junction. As a result of these interactions, the autochaperone domain appears to be a short extension of the β-helix by adding two extra strands on each face.
Figure 2.

Crystal structure of H. influenzae HapS. Ribbon diagram of HapS is coloured using the rainbow colour scheme implemented in Pymol (DeLano, 2002), with the N terminus in blue and the C terminus in red. Serine protease domain, SD1-4 subdomains, the ECM-binding domain, the SAAT domain, and the autochaperone domain are bracketed and labelled. Residues 977–1036 linking HapS to the membrane-embedded Hapβ domain (green box) are represented with a dashed line. The outer membrane (OM) is depicted schematically with a bilayer.
Other factors influencing the HapS architecture are the subdomains protruding from the β-spine. From N terminus to C terminus, they are subdomain 1 (residues 271–364 between strands β18 and β19, termed SD1), subdomain 2 (residues 540–561 between β35 and β36, termed SD2), subdomain 3 (residues 616–649 between β40 and β41, termed SD3), and subdomain 4 (residues 683–725 between β46 and β49, termed SD4). The biological functions of these subdomains are not yet clear. The current structural data suggest that these subdomains appear to have structural roles in coordinating the relative orientation of the serine protease domain and the β-spine, shaping the overall structures into different architectures (Figure 2 and Supplementary Figure 2). The SD1 domain is a typical hairpin-like loop, acting as a ‘molecular Velcro’ and clipping the serine protease domain into the same plane that contains the β-spine. The SD3 and SD4 domains are conserved in the Hbp and IgA1 protease structures (Otto et al, 2005; Johnson et al, 2009) and form intensive hydrogen bonds with surface residues from the serine protease domain (Supplementary Figure 3). From a structural/architectural point of view, these two subdomains together with the β-spine appear to act as a pedestal, on which the serine protease domain can rest. Interestingly, this mode of interaction appears to be conserved in Hap, Hbp, and IgA1 protease autotransporters (Supplementary Figure 2C and D). In comparison to the relative positions of subdomains SD1, SD3, and SD4, subdomain SD2 appears to be a bit isolated, forming a loose hairpin-like structure protruding away from the β-spine, perpendicular to a bended plane containing most of the ‘Dane Axe’-like HapS molecule. Interestingly, in the Hbp and IgA1 protease structures, the equivalent subdomain is much bigger, containing over 70 residues. As a result, the overall architectures of Hbp and IgA1 protease are more branched, with a ‘Y’-shape morphology (Supplementary Figure 2A and B).
Serine protease domain
As the catalytic mechanism of serine proteases is well characterized and HapS shares the same fold and the same catalytic triad as canonical serine proteases, such as trypsin, chymotrypsin, and Hbp (for example, the root mean square deviations (RMSDs) in Cα positions between Hap and Hbp/IgA1 serine protease domains are 1.1 Å and 1.3 Å, respectively), we will focus our description of the HapS structure on HapS substrate binding. The molecular surface of the HapS serine protease domain together with structural superimposition of HapS, Hbp, and IgA1 protease reveals a ‘V’-shaped substrate-binding groove meandering half the circumference of the globular domain (Figure 3A). The catalytic triad (i.e., H98, D140 and S243) is located in the central turning point of the binding groove, facing away from the β-spine (Supplementary Figure 1C). In addition, as the Hap serine protease domain and human neutrophil elastase are both inhibited by SLPI and share the same fold with a RMSD deviation in Cα positions of 2.5 Å, the published SLPI/elastate structure (PDB code: 2Z7F; Koizumi et al, 2008) was used to predict the SLPI/HapS, structure and residues in the HapS active site (Figure 3B and C). The substrate-binding pocket surrounding the P1 position of the substrate/inhibitor (termed the S1-subsite) is composed of the side chains of S243 and L263 and the main chains of G238, S239, K240, G241, D242, and R264, with a leucine side chain at the bottom (Figure 3C). As observed in other serine proteases (Evnin et al, 1990; Johnson et al, 2009), the bottom residue in the S1-subsite is crucial to the overall depth of the binding pocket and hence is important for substrate recognition. For example, in trypsin, a much deeper S1-subsite with an aspartic acid residue at the bottom of the pocket is thought to be essential for interaction with arginine and lysine on the substrate peptide (Evnin et al, 1990). Interestingly, the perfect match between the leucine side chain and the shallow S1-subsite appears to explain why HapS preferentially cleaves peptides with a leucine residue in the P1 position. HapS can also tolerate a much larger side chain such as a phenylalanine in the S1-subsite (Hendrixson et al, 1997; Kenjale et al, 2009). On the basis of the HapS structure, this observation may be due to the presence of a disordered/mobile loop connecting strands β13 and β14, loop265−275 (Figure 3A). Although the electron density for loop265−275 is not available for model building, the covalent linkages to R264 and F276 suggest that this loop is likely to participate in the formation of the S1-subsite. Indeed, mutations of E265 and N274 to Ala, Trp, and Arg in this loop disrupt autoproteolysis, leading to accumulation of Hap precursor in the outer membrane (Kenjale et al, 2009). On the basis of the mobile nature of loop265−275 and the position in the active site of loop265−275, it is plausible to envision that this loop undergoes an induced-fit mechanism to accommodate a Leu or Phe side chain in the S1-subsite upon the binding of substrates with different signatures in the P1 position. Consistent with this possibility, connecting loops are important for substrate selectivity in other members of the serine protease family (Perona and Craik, 1995, 1997).
Figure 3.

Serine protease domain. (A) Molecular surface of the HapS serine protease domain reveals a V-shaped binding groove (indicated by dashed line). The active site loop265−275 (coloured in grey) is modelled based on the equivalent loop in the Hbp structure through structural superimposition. (B) Simulated complex of SLPI (cyan) bound to the HapS serine protease domain (red). The upper part of the helical spine and the protruding subdomains that mediate the orientation of the HapS serine protease domain are shown in cartoon diagram and coloured in grey and green, respectively. (C) Active site. Left panel: the serine protease domain is shown in electrostatic surface, and SLPI is shown in ribbon diagram (cyan). Right panel: close-up view of the active site. Simulated SLPI and surrounding residue from HapS are shown in stick and coloured in green and grey, respectively.
SAAT domain
The uncleaved Hap precursor embedded in the outer membrane can mediate cell–cell interaction in a self-associating manner (Hendrixson and St Geme, 1998). In earlier work, examination of a series of Hap deletion mutants allowed identification of a SAAT domain corresponding to residues 725–1036 (Fink et al, 2003). The SAAT domain is a complete β-structure, consisting of 48 strands and no helices (Figure 4). These strands fold into two independent subdomains, namely, an N-terminal 7-turn right-handed β-helix, with ∼19 residues in each turn, and a C-terminal Ig-like autochaperone domain. The transition between the β-helix and the autochaperone domain is mediated by β-augmentation. The exterior of the SAAT domain is decorated with hydrophilic edges with stacked Asn/Asp ladders (Figure 4). This is a common feature also observed in other autotransporters (Otto et al, 2005; Johnson et al, 2009). Compared with other autotransporter structures, the C-terminal region of HapS corresponding to the SAAT domain is much straighter and more regular, giving rise to a triangular prism morphology with ∼9 strands in each vertical face. These flat faces are designated F1, F2, and F3, moving clockwise from the bottom of the HapS molecule (Figure 4). The inner core of the SAAT domain is packed with relatively conserved hydrophobic/aromatic residues along the axis of the β-helix, including the sequence motif (I/L)XLXXXXX(A/F)X(V/L), in which X represents a random amino acid. As shown in Supplementary Figure 4, HapS appears to adopt a strictly conserved inner core to assemble/extend the β-helix region of the SAAT domain. The orientations of the side chains of the Ile, Leu, Ala, and Val residues in the β-helix turns of residues 732–742 and residues 772–782 are nearly identical. The same applies to the Val, Leu, Phe, and Leu residues in the β-helix turns of residues 752–762 and 791–801. Interestingly, this repetitive structural feature is not seen in the inner core of the Hbp and IgA1 autotransporters, which lack SAAT activity, provoking the thought that this feature may have an important structural role shaping the prism-like morphology that appears to be critical for the SAAT activity (see discussion below).
Figure 4.

Self-association of HapS dimer in trans configuration. The close-up views of the packing interfaces between SAAT domains are show in the right panel. The F1/F2/F3 faces and the F1–F2 edge in the prism-like SAAT domain are labelled.
Consistent with the role of HapS in cell–cell adhesion, the crystal packing reveals a remarkable oligomerization of (HapS–HapS)n related by a crystallographic two-fold screw axis, perpendicular to the axis of the β-helix and in parallel with the bacterial membrane (Figure 4). The interaction between HapS molecules is mediated primarily by the SAAT domains. Two HapS molecules pack against each other in a trans configuration (Figure 4), and the primary interaction site lies between the F1–F2 edge (from molecule A) and the F2 face (from molecule B), explaining our earlier observation that perturbations of this region disrupt the self-associating activity (Fink et al, 2003). The intermolecular distance between the F1–F2 edge and the F2 face is 4–6 Å (top-right panel in Figure 4), and the only hydrogen bond between HapS molecules involves Gln940 in the SAAT domain and Asn577 in the N-terminal β-spine above the SAAT domain (bottom-right panel in Figure 4). The fact that deletion of residues 26–725 has no effect on Hap-mediated bacterial settling (Fink et al, 2003) suggests that this hydrogen bond is not essential to HapS–HapS interactions. Accordingly, the region flanking the primary interaction site is termed the secondary interaction site.
As suggested by crystal lattice formation and previous characterization of HapS–HapS interaction in native gels (Hendrixson and St Geme, 1998), HapS–HapS multimerization is not restricted to a dimeric assembly. A HapS dimer can act as a nucleus to recruit more HapS molecules horizontally (Figure 5), giving rise to a formidable mega-Dalton complex with a massive buried intermolecular surface. On the basis of the calculations using the program AREAIMOL (CCP4, 1994), the buried surface of a HapS dimer in trans configuration is estimated to be 1173 Å2. As oligomerization proceeds to a tetramer, the buried surface increases nearly seven-fold to a staggering 7053 Å2 (Figure 5A). Interestingly, the mulitmerization of HapS molecules appears to bring a significant portion of N-terminal Hap into effect, contributing 1779 Å2 of buried surface (between two HapS molecules in cis configuration) into the overall packing (Figure 5A). HapS–HapS multimers appear to have the capacity to ‘grow’ upon demand via the F1–F2 edge and the F2 face in the SAAT domains (Figure 5B). The two HapS molecules at the edges of the tetrameric complex become the new growing points for the assembly of a HapS hexamer or an even higher order HapS multimer required for overcoming the repulsive force between two adjacent cells.
Figure 5.

The growing self-association surfaces in the HapS multimer. (A) For the purpose of illustration, only four HapS molecules are shown in surface (coloured in yellow and by electrostatic surface potential) and cartoon (black and magenta) representations, respectively. (B) Slab view of the packing interface of these HapS–HapS multimer at the cross-section of D776-N777. The F1/F2/F3 faces are labelled. The F2 face and F1–F2 edge at the growing ends of the multimer are highlighted in red.
Functional characterization of self-associating activity
To elucidate how the overall architecture of HapS influences its ability as a cell linker and to define the minimum structure required for SAAT activity, we used the HapS structure and we generated a series of truncated mutants (Figure 6B).
Figure 6.

Structure-based functional characterization of H. influenzae HapS self-associating activity. (A) Scanning electron micrographs of Hap-mediated bacterial aggregation. Scanning electron microscopy demonstrated Hap-mediated bacterial aggregation and formation of complex microcolonies, as assessed in a tube settling assay (D) after incubation for 1 h, 2 h. (B) Location of the deletions in the HapS mutants in this study. HapS–HapS dimer in trans configuration is coloured in grey. Progressive deletions starting from residue 751 are coloured by red, yellow, blue, green, and magenta, respectively. Asp776 located in the heart of the primary interaction site is labelled and shown in stick representation (cyan). (C) Outer membrane proteins corresponding to the Hap-deletion constructs detected by western blot analysis with a polyclonal antiserum directed against HapS. DB117 represents the H. influenzae host strain with vector alone. (D) Tube setting assay of H. influenzae strain DB117 harbouring empty vector (DB117), DB117/pLS88-HapS243A, and DB117 expressing HapS243A-deletion constructs (HapΔ751–758, HapΔ751–770, HapΔ751–789, HapΔ751–808, and HapΔ751–827). Bacterial suspensions were incubated standing at room temperature, and bacterial aggregation was quantified by measuring the absorbance of the suspension at 600 nm.
The mutagenesis data are shown in Figure 6C and D. All of the truncates were derived from a common HapS243A mutant and contain a deletion beginning at residue 751. In HapΔ751–758, a single β-strand is deleted, resulting in a change in the relative orientation of the N- and C-terminal regions of the HapS molecule. HapΔ751–770, HapΔ751–789, HapΔ751–808, and HapΔ751–827 have progressively larger deletions of β-helix turns, shortening the length of the SAAT domain with little perturbation of the overall architecture. These mutant proteins were stably expressed and were successfully localized to the H. influenzae outer membrane (Figure 6C), suggesting that the deletions have little effect on the overall conformation or secretion of the protein. To assess self-associating activity of these proteins and their ability to mediate bacterial aggregation, we performed tube settling assays, using H. influenzae strain DB11/pHapS243A as a control. As shown in Figure 6D, elimination of one turn in the β-helix in HapΔ751–770 had no effect on self-associating activity. In contrast, larger deletions that approached the putative autochaperone domain (HapΔ751–789, HapΔ751–808, and HapΔ751–827) were associated with a significant loss in self-associating activity, approaching the effect of a change in the relative orientation of the N-terminal and C-terminal regions of HapS (HapΔ751–758). To further assess the effect of mutation within the SAAT domain on aggregation, we examined latex beads coated with either purified HapS or HapSΔ751–827. Visualization by light microscopy revealed that beads coated with HapS formed clusters, while beads coated with HapSΔ751–827 remained isolated, similar to beads coated with BSA or incubated in buffer alone (Figure 7). Taken together, these data highlight the importance of the SAAT region in bacterial aggregation.
Figure 7.

Visualization of HapS-coated beads. Latex beads were coated with either BSA (A), HapS (C), or HapSΔ751–827 (D) and were viewed by light microscopy. Beads coated with HapS formed aggregates, while beads coated with HapSΔ751–827 remained isolated, similar to controls (BSA-coated beads and beads incubated in PBS alone (B)).
Discussion
β-Helix is a versatile fold
This work demonstrates that the structure of the H. influenzae HapS molecule is very similar to the structures of E. coli Hbp and H. influenzae IgA1 protease (Otto et al, 2005; Johnson et al, 2009) (Supplementary Figure 2). The serine protease domain, the β-helix, and the autochaperone domain are conserved and arranged in the same order in these three structures. More strikingly, the subdomains appear to leave and return to the central β-spine at the same positions (Supplementary Figure 2). The subtle changes in the Hbp/IgA1 protease architectures seem to bring unwanted structural clashes into the observed oligomerization, accounting for the lack of self-associating activity. Superimposition of HapS and Hbp suggests that the bulky subdomain 2 and the decorating structure in the C-terminal β-helix in Hbp would create structural clashes if Hbps were assembled into a HapS-like multimer (Figure 8A–C). Indeed the structure of an Hbp mutant illustrates this structural clash (Nishimura et al, 2010). As shown in Supplementary Figure 5, deletion of subdomain 2 in Hbp enables assembly into a SAAT-like packed structure. In IgA1 protease, the bulky subdomain 2, the relatively short β-helix, and variations in the C-terminal domain provide explanations for the lack of self-associating activity (Figure 8D–F). In comparison, the HapS architecture has a variety of features that potentiate intermolecular interaction, resulting in multimerization and interbacterial association: (1) the hairpin-like subdomain 1 restricts intramolecular movement and leads to steady engagement of two serine protease domains when they are brought into proximity via oligomerization of the SAAT domains; (2) the subdomain 2 is relatively small, allowing space for intermolecular interaction; (3) the C-terminal domain has a triangular prism-like morphology that facilitates high-order intercellular [HapS–HapS]n multimerization; (4) the insertion in the C-terminal region (residues 851–873, disordered in the present structure) is located away from the packing interface and does not interfere with multimerization. All of these structural characteristics appear to act in a concerted manner, shaping a proteolytic autotransporter into a powerful cellular crosslinker. Interestingly, according to the Conserved Domain Database (http://www.ncbi.nlm.nih.gov/Structure/cdd/cddsrv.cgi?uid=29329; Marchler-Bauer et al, 2009), HapS and the SPATE-like proteases are classified as type-1 monomeric autotransporters (Henderson and Nataro, 2001), and the SAATs such as Ag43, TibA, and AIDA-I are classified as type-2 autotransporters (Supplementary Figure 6A). Given the structure of HapS, it is intriguing to consider the evolutionary relationship between Hap and other autotransporters with SAAT activity (Supplementary Figure 6A). The SAAT domain in the AIDA-I adhesin (Sherlock et al, 2004; Girard et al, 2010) is predicted to be very similar to the SAAT domain in HapS presented in this manuscript, with an RMSD of 1.8 Å in Cα positions (Supplementary Figure 6B). Furthermore, when compared to the SAAT domain in the HapS–HapS multimer, the SAAT domain in AIDA-I has no structural clashes, suggesting that other SAAT-type autotransporters might adopt a similar intercellular oligomerization mechanism to mediate bacterial aggregation. Considering all of these observations, we conclude that (1) the β-helix is a very versatile fold, and (2) the HapS structure may represent a missing evolutionary link between type-1 and type-2 monomeric autotransporters.
Figure 8.

Structural comparison between H. influenzae HapS, H. influenzae IgA1 protease and E. coli Hbp in the context of HapS–HapS multimer. (A, B, C) Superimposition between HapS (grey) and Hbp (blue). (D, E, F) Superimposition between HapS and IgA1 protease (green). The structural variations between HapS and Hbp/IgA1 protease are boxed. Subdomain 2 (SD2) in Hbp and IgA1 are labelled.
Crystal packing reveals an unprecedented Hap–Hap oligomerization mechanism for bacterial aggregation
Several lines of evidence indicate that the HapS–HapS multimer observed by crystallography is a genuine reflection of Hap–Hap oligomerization between two adjacent cells (Figure 9 and Supplementary Figure 7). First, the crystallographically related trans configuration involving the HapS C terminus in (HapS–HapS)n multimers and the intermolecular distance of 112 Å are both consistent with cell-linkers observed elsewhere (Freigang et al, 2000; Aricescu et al, 2007; Himanen et al, 2010). Second, the measured distance of the overhang in the trans configuration is consistent with the predicted length of the linker loop attached to the cell membrane (Supplementary Figure 7). Assuming that the average distance of Cα–Cα in a stretched conformation is about 2.3 Å, we can estimate that the theoretical length of residues 977–1036, the bridge between the C-terminal end of HapS and the cell anchor (Hapβ), is ∼135 Å, much longer than the overhang in the trans configuration, giving ample space for Hap–Hap interactions. Third, intermolecular distance between the Hap dimer in cis configuration derived from self-association is ∼41 Å. This is in perfect agreement with the intermolecular distance (∼34 Å) between the published Hbpβ (a Hapβ homologue) dimer structure (Supplementary Figure 8). Fourth, the fact that deletion of residues 25–725 in the N-terminal half of HapS has little effect on Hap–Hap interactions (Fink et al, 2003) is consistent with the structural observation that the SAAT domains residing in the C-terminal region of Hap molecules are in immediate contact with each other in the trans configuration (Figure 4 and Supplementary Figure 7). Fifth, all the functional studies (Fink et al, 2003) show that deletion of β-strands in the HapS SAAT domain disrupts critical interacting faces and dramatically reduces Hap-mediated bacterial aggregation and microcolony formation. Of note, site-directed mutagenesis of the Asn/Asp ladders to Ala in the primary interface had no significant effect on Hap SAAT activity (data not shown). This observation appears to be in agreement with the structural observation presented in this manuscript. No direct hydrogen bonding is identified in the primary interface and the closest distance between Asp/Asn ladders in the primary interface is 4–6 Å, suggesting that interaction of Hap molecules in HapS–HapS multimers is not restricted to specific residues and instead is likely mediated by van der Waal forces derived from self-complementary interacting surfaces. Interestingly, the Hbp and IgA1 structures also have Asp/Asn-like hydrophilic edges. However, due to the lack of self-complementary surfaces, Hbp and IgA1 protease cannot interact with each other. Consistently, the Hap–Hap-like assembly observed in Hbp mutant is much more loosely packed than the interaction in Hap multimer. The intermolecular distances are >10 Å (in the trans configuration) and >20 Å (in the cis configuration) (Supplementary Figure 5), suggesting that the self-complementary surface is unique in HapS structure. Sixth, Hap-mediated bacterial aggregation is enhanced by SLPI, which inhibits Hap serine protease activity and autoproteolysis, and results in accumulation of the Hap SAAT domain on the bacterial surface (Hendrixson and St Geme, 1998). If the Hap multimer revealed by crystal packing is biologically relevant, the simulated SLPI molecules should fit nicely into the HapS–HapS multimer, causing no structural clashes. The simulated SLPI molecules are located in the diagonal positions of the HapS–HapS multimer related by a two-fold screw axis, with SLPI L72 occupying the Hap S1-subsite and no structural clashes in [HapS/SLPI–HapS/SLPI]n (Figure 9G and Supplementary Figure 7). Seventh, the homology models of the SAAT domains of the Ag43 (Hasman et al, 1999), TibA (Sherlock et al, 2005), and AIDA-I type-2 autotransporters (Sherlock et al, 2004) superimpose well with the observed HapS–HapS multimer (Supplementary Figure 6). The structurally conserved SAAT domain and the trans oligomerization of the SAAT domain in HapS appear to explain why SAATs are able to participate in heterologous interactions with other SAAT partners (Sherlock et al, 2004). Therefore, all existing experimental data are in good agreement with our structural observation to support the hypothesis that the HapS–HapS multimers revealed by crystal packing are biologically relevant. Furthermore, it has been shown that the membrane-anchoring domain in autotransporter proteins is mobile in lipid bilayers (Jose and Meyer, 2007). Hence, it is reasonable to envisage that the mobile HapS domain forms multiple arrays of oligomers at the cross-section of cell junctions upon bacterial aggregation. These ‘lines’ of HapS multimer can act like ‘stitches’ to seal cells together, leading to biofilm formation.
Figure 9.

Crystal packing of H. influenzae HapS structures reveals an unprecedented intercellular oligomerization (A–F) for complex microcolony formation and eventual biofilm formation catalysed by the presence of SLPI (G). For the purpose of illustration, only two SLPI molecules are shown (boxed, cyan) (G). The C terminus of Hap molecules in A are labelled to indicate the location of the bacterium.
The repetitive nature of the HapS crystal lattice and the two-fold crystallographic axis observed in (HapS–HapS)n multimers implies a possible polymerization/depolymerization model for Hap–Hap interaction and interbacterial association. As illustrated in Figure 9, the recruitment of a HapS molecule into an existing multimer might proceed in an intermittent manner between two adjacent cells. The formation of a stable HapS–HapS dimer acting as a nucleus is critical for further oligomerization. However, no hydrogen bond is formed in the primary interaction site in the HapS–HapS dimer, suggesting that (HapS–HapS)2 might not be stable in solution. Indeed, HapS seems to be a monomer at low protein concentration, as assessed by gel filtration analysis (data not shown), and a multimer with molecular mass >640 kDa at high concentration, as assessed in Blue native gels (Hendrixson and St Geme, 1998). This information implies that formation of a HapS–HapS dimer and a mega-Dalton multimer might be a thermodynamic process constantly associated with a polymerization/depolymerization mechanism, reminiscent of growth of an actin filament. We further tested this hypothesis using dynamic light scattering (DLS) assay. If the formation of Hap multimers is driven mainly by apolar interaction, as suggested in Figure 9, the polymerization should be temperature dependent and much weaker at 4 °C than at higher temperatures (Schellman, 1997). The DLS results presented in Supplementary Table 1 clearly show that this is the case. As the temperature rises, the hydrodynamic radius (Rd) increases from 986 to 1902 nm, suggesting that the protein has undergone a temperature-dependent polymerization. In the context of H. influenzae pathogenicity, cleaved and released HapS is a monomer and is highly soluble, potentially an advantage in migrating through the extracellular matrix to cleave host proteins. In contrast, membrane-associated HapS can undergo multimerization, and the resulting [HapS–HapS]n multimers may be critical to generate sufficient mechanical force to pull two cells together. Depolymerization of [HapS–HapS]n and autoproteolytic cleavage, leading to the release of HapS, may be important to allow individual organisms to separate from a microcolony or a biofilm and spread to other sites.
Conclusion
Bacterial aggregation and biofilm formation are being recognized increasingly as important virulence properties that contribute to the pathogenesis of disease. Recent work has identified a major subgroup of autotransporter proteins called SAATs that mediate interbacterial interaction via protein multimerization. In this study, we have solved the crystal structure of the H. influenzae Hap passenger domain and have discovered a C-terminal triangular prism-like structure that appears to be conserved among SAAT-type autotransporters, that mediates formation of HapS–HapS dimers, and that enables higher-order oligomerization through its F1–F2 edge and F2 face. The unprecedented intercellular oligomerization is important to generate massive buried surfaces and formidable interactions among Hap molecules that are required for overcoming the repulsive force between bacteria, leading to bacterial aggregation and formation of complex bacterial microcolonies. Overall, our results provide important structural insights into SAATs, H. influenzae pathogenesis, and the mechanistic principles of bacterial aggregation and biofilm formation, potentially facilitating the design of novel therapeutics that target bacterial aggregation and biofilm formation.
Materials and methods
Bacterial strains, plasmids, and culture conditions used in this study
The bacterial strains used in this study are derivatives of H. influenzae strain DB117 (Setlow et al, 1968). Strains were grown in supplemented brain–heart infusions (BHIs) broth or on BHI-DB or chocolate agar and were frozen at −80 °C in BHI broth containing 20% glycerol. The plasmid pJS106 contains the wild-type hap gene from H. influenzae strain N187 and was used to overexpress and purify HapS (Fink et al, 2003). The plasmid pLS88:HapS243A encodes Hap with an alanine in place of the catalytic serine at amino acid 243, resulting in a lack of autoproteolytic activity and retention of the HapS passenger domain on the bacterial surface. Deletions in HapS243A were generated using the pLS88:HapS243A as template and the QuikChange® Site-Mutagenesis Kit (Stratagene) according to the manufacturer's instructions. Deletions were confirmed by nucleotide sequencing, and plasmids were then introduced into strain DB117 using the MII/MIV transformation method (Herriott et al, 1970).
Expression, purification, and crystallization
In order to purify HapS, H. influenzae strain DB117/pJS106 was grown on BHI-DB agar containing 5 μg/ml of tetracycline at 37 °C with 5% CO2 overnight. A single colony was picked and inoculated into 1 litre of BHIs broth. The 1 litre culture was incubated at 37 °C with shaking at 250 r.p.m. until the OD(600) reached 0.9, and was then inoculated into a fermentor containing 10-litre of BHIs broth with the aeration parameter set at 8%. The fermentor culture was incubated at 37 °C until the OD(600) reached 3.0. Bacteria were pelleted by centrifugation at 4000 g for 20 min, and the supernatant was concentrated to ∼500 ml using a transverse flow cartridge with a 50 kDa cut-off (Millipore). The concentrated supernatant was dialfiltrated into 8 litre of 20 mM sodium phosphate, pH 6.4, buffer and then applied to a sepharose cation-exchange column (GE healthcare) equilibrated with 20 mM sodium phosphate, pH 6.4, buffer. Protein was eluted with a linear sodium chloride gradient (pH 8.0, 0 to 1 M NaCl), and peak fractions containing HapS were pooled and confirmed by western blot analysis. The protein sample was mixed with ammonium sulphate with a final concentration of 1.3 M and was then loaded onto a phenyl sepharose column (GE healthcare) equilibrated with buffer containing 20 mM MES, pH 6.4, 1.3 M ammonium sulphate. Fractions were eluted with a reverse ammonium sulphate gradient (pH 6.4, 1 to 0 M ammonium sulphate). HapS containing fractions were pooled and dialysed into a buffer containing 20 mM Tris, pH 7.0, 50 mM NaCl. Purified HapS was concentrated and then loaded onto an S200 gel-filtration column (GE healthcare), which was equilibrated with 20 mM Tris, pH 8.0, 100 mM NaCl. The peak fraction was estimated to be ∼95% pure, as indicated by Coomassie blue-stained SDS–PAGE.
For crystallization, purified HapS was concentrated to ∼10 mg/ml using an Amicon Ultra 10 concentrator with a 10 kDa cut-off (Millipore). HapS crystals with dimensions of 0.1 mm × 0.1 mm × 0.03 mm were obtained at room temperature using the hanging-drop vapour diffusion method. The reservoir solution contained 100 mM sodium citrate, pH 5.6, 14% (w/v) PEG4000, and 100 mM ammonium sulphate. The hanging drop contained a 1:1 (v/v) ratio of reservoir and protein solutions. Crystals were flash-cooled to 100 K by liquid nitrogen using 20% PEG400 as cryoprotectant. Crystals of HapS diffracted to 2.2 Å and were in space group P212121 with cell dimensions a=41.4 Å b=137.2 Å c=209.6 Å, and one molecule in the asymmetric unit.
Data collection and phasing
Diffraction data for HapS native crystals were recorded on BL17U at the Shanghai Synchrotron Radiation Facility (Shanghai, China). HapS data were integrated and scaled using MOSFLM/SCALA (CCP4, 1994). The statistics of the data collection are reported in Table I.
Table 1. Data collection and structure refinement statistics of H. influenzae HapS.
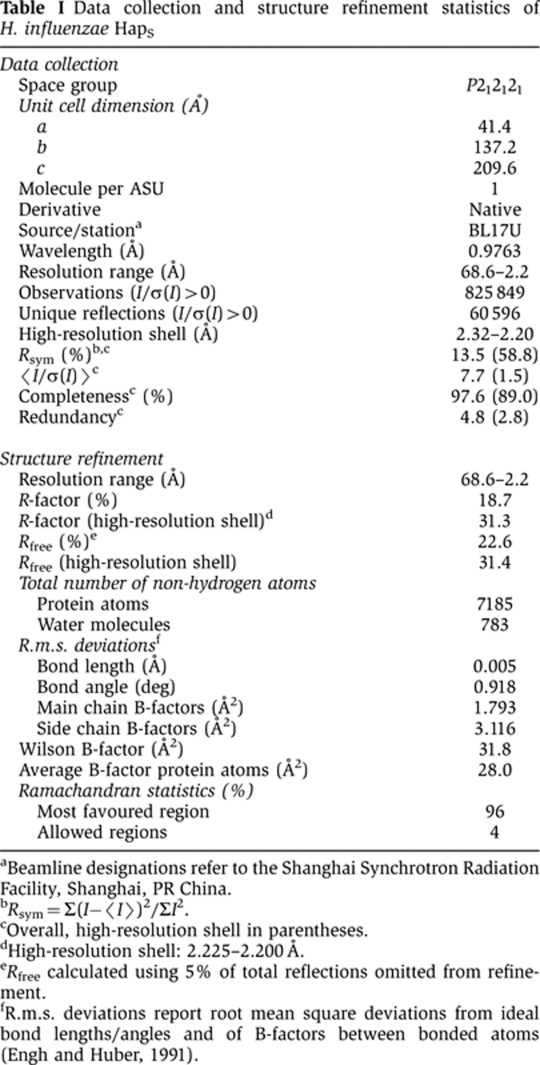
aBeamline designations refer to the Shanghai Synchrotron Radiation Facility, Shanghai, PR China.
bRsym=Σ(I−〈I〉)2/ΣI2.
cOverall, high-resolution shell in parentheses.
dHigh-resolution shell: 2.225–2.200 Å.
eRfree calculated using 5% of total reflections omitted from refinement.
fR.m.s. deviations report root mean square deviations from ideal bond lengths/angles and of B-factors between bonded atoms (Engh and Huber, 1991).
The initial set of phases was obtained by molecular replacement using H. influenzae IgA1 protease (pdb code: 3H09) and E. coli Hbp (pdb code: 2WXR) as search models. To prepare the search models, HapS sequence was first aligned with H. influenzae IgA1 protease and E. coli Hbp sequences using ClustalW (http://www.ebi.ac.uk/Tools/clustalw2/), respectively. The resulting sequence alignment was supplied to program CHAINSAW (CCP4, 1994) to remove the non-conserved residues, i.e., residues that differ in HapS and IgA1 protease/Hbp were changed to alanine. The pruned models were then used as search templates in PHASER (CCP4, 1994). The solutions with a Z-score above five and little crystal clashes were selected for further refinement. Refmac5 (CCP4, 1994) and PHENIX.REFINE (Adams et al, 2010), together with intermittent manual building in COOT (CCP4, 1994) were used to correct and improve the initial models produced by PHASER. The electron density map of HapS residues 265–272, 851–873, and 977–1036 are not available for model building.
Structure refinement
The structure of HapS was refined by conjugate-gradient minimization (REFMAC5) (CCP4, 1994), with intermittent manual rebuilding, refining individual B-factors applying a TLS correction (1 TLS group, 20 parameters) (Winn et al, 2001). The final model of HapS contains 920 residues and 783 water molecules. Ramachandran statistics of HapS calculated by PROCHECK (Laskowski et al, 1993) indicate that 96.0% of the atoms are in the most favoured region, and 4.0% are in the allowed regions. The detailed structure refinement statistics are reported in Table I. Coordinate of the HapS structure has been deposited into the Protein Database Bank (entry code 3SYJ).
Modelling of SLPI–HapS complex and AIDA-I structures
The docking was carried out based on the following two observations: (1) SLPI is known to be the common inhibitor of HapS and human neutrophil elastase proteolytic activity; (2) the HapS serine protease domain shares the same fold and a nearly identical active site with human neutrophil elastase. The initial model of SLPI–HapS was obtained via structural superimposition of the serine protease domains of HapS and human neutrophil elastase. The resulting complex was then subjected to energy minimization using SYBYL (Tripos, St Louis, MO) to remove structural clashes derived from the manual docking. The (SLPI–HapS)n multimer was generated using the same methodology as described above. The AIDA-I homology model was generated using the standard protocol implemented in SwissModel (http://swissmodel.expasy.org).
Analysis of bacterial outer membrane fractions
Outer membrane proteins were isolated on the basis of sarkosyl insolubility using the method of Carlone et al (1986), and were resolved by SDS–PAGE as described earlier (Fink et al, 2003).
Bacterial aggregation assays
Bacterial aggregation was assessed using the tube settling assay as described earlier (Fink et al, 2003). In brief, bacteria were resuspended from plates into tubes containing 2 ml of BHIs broth to an OD(600) of 0.2 and were grown at 37 °C to an OD(600) of 0.8–0.9. Subsequently, tubes were allowed to stand at room temperature for 2 h, and OD(600) was measured every 15 min. Using this assay, as bacterial aggregation develops, bacterial settling occurs, resulting in a decrease in the OD(600).
Scanning electron microscopy
In order to examine bacterial aggregates by scanning electron microscopy, samples containing bacterial aggregates were removed from the bottoms of the culture tubes in tube settling assays and were spotted onto glass coverslips. Samples on coverslips were fixed with 0.5 ml of 2.5% glutaraldehyde in 0.1 M sodium phosphate buffer for 1 h, then dehydrated in a critical-point dryer, evaporated with gold, and visualized with a Hitachi S-450 scanning electron microscope (Hendrixson and St Geme, 1998).
Latex beads assay
Purified protein was passively adsorbed onto 2 μm polystyrene latex beads (Sigma). A 1% solution of beads in 25 mM MES, pH 6.5, was incubated with 5 μg protein for 2 days at room temperature with gentle agitation. The coupling reaction was stopped by washing the beads three times with the same buffer and resuspending in 100 μl BHI broth. After a 6-h incubation at room temperature, beads were viewed by light microscopy using a Nikon Labophot.
Dynamic light scattering
Purified HapS was concentrated to 3 mg/ml before it was subjected to analysis by dynamic light scattering (DLS, Wyat Technology). The instrument was calibrated and washed using the manufacturer’s protocol and was then set to 4 °C. As the temperature stabilized, 50 μl purified HapS was precooled to 4 °C and was then loaded into the machine. Over a period of ∼17 min, 200 measurements were recorded at this temperature. The same protein sample and the same measuring protocol were used for the DLS assays at 10, 16, 22, and 37 °C. The values of hydrodynamic radius (Rd) and polydispersity at different temperatures were obtained by software DYNAMICS (Wyat Technology).
Supplementary Material
Acknowledgments
This work was supported by ‘The Program for Professor of Special Appointment (Eastern Scholar) at Shanghai Institute of Higher Learning’ (to GM), a research Grant 31070645 from the National Natural Science Foundation of China (to GM), and NIH Grant R01 AI49322 (to JWS). We thank the personnel of beamline BL17U (Shanghai, China) for help during data collection. We also thank Drs Manwu Zha and Jianping Ding for their generous sharing of equipment in Shanghai Institutes of Biological Sciences.
Author contributions: GM and JWS conceived and designed the experiments; GM, NS, and RK performed the experiments; GM, NS, RK, GW, and JWS analysed the data; and GM and JWS wrote the paper.
Footnotes
The authors declare that they have no conflict of interest.
References
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66(Part 2): 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aricescu AR, Siebold C, Choudhuri K, Chang VT, Lu W, Davis SJ, van der Merwe PA, Jones EY (2007) Structure of a tyrosine phosphatase adhesive interaction reveals a spacer-clamp mechanism. Science 317: 1217–1220 [DOI] [PubMed] [Google Scholar]
- Carlone GM, Thomas ML, Rumschlag HS, Sottnek FO (1986) Rapid microprocedure for isolating detergent-insoluble outer membrane proteins from Haemophilus species. J Clin Microbiol 24: 330–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- CCP4 (1994) The CCP4 suite: programs for protein crystallography. Acta Cryst D 50: 760–763 [DOI] [PubMed] [Google Scholar]
- Dautin N, Bernstein HD (2007) Protein secretion in Gram-negative bacteria via the autotransporter pathway. Annu Rev Microbiol 61: 89–112 [DOI] [PubMed] [Google Scholar]
- DeLano WL (2002) The PyMOL Molecular Graphics System. Palo Alto, CA, USA: DeLano Scientific [Google Scholar]
- Donlan RM, Costerton JW (2002) Biofilms: survival mechanisms of clinically relevant microorganisms. Clin Microbiol Rev 15: 167–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engh RA, Huber R (1991) Accurate bond and angle parameters for X-ray protein structure refinement. Acta Crystallogr A 47: 392–400 [Google Scholar]
- Evnin LB, Vasquez JR, Craik CS (1990) Substrate specificity of trypsin investigated by using a genetic selection. Proc Natl Acad Sci USA 87 (17): 6659–6663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink DL, Buscher AZ, Green B, Fernsten P, St Geme JW III (2003) The Haemophilus influenzae Hap autotransporter mediates microcolony formation and adherence to epithelial cells and extracellular matrix via binding regions in the C-terminal end of the passenger domain. Cell Microbiol 5: 175–186 [DOI] [PubMed] [Google Scholar]
- Fink DL, Green BA, St Geme JW III (2002) The Haemophilus influenzae Hap autotransporter binds to fibronectin, laminin, and collagen IV. Infect Immun 70: 4902–4907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink DL, St Geme JW III (2003) Chromosomal expression of the Haemophilus influenzae Hap autotransporter allows fine-tuned regulation of adhesive potential via inhibition of intermolecular autoproteolysis. J Bacteriol 185: 1608–1615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freigang J, Proba K, Leder L, Diederichs K, Sonderegger P, Welte W (2000) The crystal structure of the ligand binding module of axonin-1/TAG-1 suggests a zipper mechanism for neural cell adhesion. Cell 101: 425–433 [DOI] [PubMed] [Google Scholar]
- Girard V, Cote JP, Charbonneau ME, Campos M, Berthiaume F, Hancock MA, Siddiqui N, Mourez M (2010) Conformation change in a self-recognizing autotransporter modulates bacterial cell-cell interaction. J Biol Chem 285 (14): 10616–10626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall-Stoodley L, Costerton JW, Stoodley P (2004) Bacterial biofilms: from the natural environment to infectious diseases. Nat Rev Microbiol 2: 95–108 [DOI] [PubMed] [Google Scholar]
- Hasman H, Chakraborty T, Klemm P (1999) Antigen-43-mediated autoaggregation of Escherichia coli is blocked by fimbriation. J Bacteriol 181 (16): 4834–4841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson IR, Nataro JP (2001) Virulence functions of autotransporter proteins. Infect Immun 69: 1231–1243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson IR, Navarro-Garcia F, Nataro JP (1998) The great escape: structure and function of the autotransporter proteins. Trends Microbiol 6: 370–378 [DOI] [PubMed] [Google Scholar]
- Hendrixson DR, de la Morena ML, Stathopoulos C, St Geme JW III (1997) Structural determinants of processing and secretion of the Haemophilus influenzae hap protein. Mol Microbiol 26: 505–518 [DOI] [PubMed] [Google Scholar]
- Hendrixson DR, St Geme JW III (1998) The Haemophilus influenzae Hap serine protease promotes adherence and microcolony formation, potentiated by a soluble host protein. Mol Cell 2: 841–850 [DOI] [PubMed] [Google Scholar]
- Herriott RM, Meyer EM, Vogt M (1970) Defined nongrowth media for stage II development of competence in Haemophilus influenzae. J Bacteriol 101: 517–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Himanen JP, Yermekbayeva L, Janes PW, Walker JR, Xu K, Atapattu L, Rajashankar KR, Mensinga A, Lackmann M, Nikolov DB, Dhe-Paganon S (2010) Architecture of Eph receptor clusters. Proc Natl Acad Sci USA 107 (24): 10860–10865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson TA, Qiu J, Plaut AG, Holyoak T (2009) Active-site gating regulates substrate selectivity in a chymotrypsin-like serine protease the structure of haemophilus influenzae immunoglobulin A1 protease. J Mol Biol 389: 559–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jose J, Meyer TF (2007) The autodisplay story, from discovery to biotechnical and biomedical applications. Microbiol Mol Biol Rev 71: 600–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenjale R, Meng G, Fink DL, Juehne T, Ohashi T, Erickson HP, Waksman G, St Geme JW III (2009) Structural determinants of autoproteolysis of the Haemophilus influenzae Hap autotransporter. Infect Immun 77 (11): 4704–4713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemm P, Vejborg RM, Sherlock O (2006) Self-associating autotransporters, SAATs: functional and structural similarities. Int J Med Microbiol 296: 187–195 [DOI] [PubMed] [Google Scholar]
- Koizumi M, Fujino A, Fukushima K, Kamimura T, Takimoto-Kamimura M (2008) Complex of human neutrophil elastase with 1/2SLPI. J Synchrotron Radiat 15(Part 3): 308–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski RA, MacArthur MW, Moss DS, Thornton JM (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Cryst 26: 283–291 [Google Scholar]
- Marchler-Bauer A, Anderson JB, Chitsaz F, Derbyshire MK, DeWeese-Scott C, Fong JH, Geer LY, Geer RC, Gonzales NR, Gwadz M, He S, Hurwitz DI, Jackson JD, Ke Z, Lanczycki CJ, Liebert CA, Liu C, Lu F, Lu S, Marchler GH et al. (2009) CDD: specific functional annotation with the Conserved Domain Database. Nucleic Acids Res 37 (Database issue): D205–D210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moons P, Michiels CW, Aertsen A (2009) Bacterial interactions in biofilms. Crit Rev Microbiol 35: 157–168 [DOI] [PubMed] [Google Scholar]
- Nishimura K, Yoon YH, Kurihara A, Unzai S, Luirink J, Park SY, Tame JR (2010) Role of domains within the autotransporter Hbp/Tsh. Acta Crystallogr D Biol Crystallogr 66(Pt 12): 1295–1300 [DOI] [PubMed] [Google Scholar]
- Oliver DC, Huang G, Nodel E, Pleasance S, Fernandez RC (2003) A conserved region within the Bordetella pertussis autotransporter BrkA is necessary for folding of its passenger domain. Mol Microbiol 47: 1367–1383 [DOI] [PubMed] [Google Scholar]
- Otto BR, Sijbrandi R, Luirink J, Oudega B, Heddle JG, Mizutani K, Park SY, Tame JR (2005) Crystal structure of hemoglobin protease, a heme binding autotransporter protein from pathogenic Escherichia coli. J Biol Chem 280 (17): 17339–17345 [DOI] [PubMed] [Google Scholar]
- Perona JJ, Craik CS (1995) Structural basis of substrate specificity in the serine proteases. Protein Sci 4: 337–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perona JJ, Craik CS (1997) Evolutionary divergence of substrate specificity within the chymotrypsin-like serine protease fold. J Biol Chem 272 (48): 29987–29990 [DOI] [PubMed] [Google Scholar]
- Rao VK, Krasan GP, Hendrixson DR, Dawid S, St Geme JW III (1999) Molecular determinants of the pathogenesis of disease due to non-typable Haemophilus influenzae. FEMS Microbiol Rev 23: 99–129 [DOI] [PubMed] [Google Scholar]
- Schellman JA (1997) Temperature, stability, and the hydrophobic interaction. Biophys J 73: 2960–2964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Setlow JK, Brown DC, Boling ME, Mattingly A, Gordon MP (1968) Repair of deoxyribonucleic acid in Haemophilus influenzae. I. X-ray sensitivity of ultraviolet-sensitive mutants and their behavior as hosts to ultraviolet-irradiated bacteriophage and transforming deoxyribonucleic acid. J Bacteriol 95: 546–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherlock O, Schembri MA, Reisner A, Klemm P (2004) Novel roles for the AIDA adhesin from diarrheagenic Escherichia coli: cell aggregation and biofilm formation. J Bacteriol 186 (23): 8058–8065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherlock O, Vejborg RM, Klemm P (2005) The TibA adhesin/invasin from enterotoxigenic Escherichia coli is self recognizing and induces bacterial aggregation and biofilm formation. Infect Immun 73: 1954–1963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart PS, Costerton JW (2001) Antibiotic resistance of bacteria in biofilms. Lancet 358: 135–138 [DOI] [PubMed] [Google Scholar]
- Winn MD, Isupov MN, Murshudov GN (2001) Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Cryst D 57: 122–133 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.