

Abstract
Joubert syndrome (JS) is a rare autosomal recessive disorder with key finding of cerebellar vermis hypoplasia with a complex brainstem malformation that comprises the molar tooth sign on axial magnetic resonance images. This syndrome is difficult to diagnose clinically because of its variable phenotype. The exact diagnosis is often not made for several years after birth. This report shows that with the availability of magnetic resonance imaging (MRI), especially in developing countries like India, it is quite feasible to make an early diagnosis which may positively affect the subsequent management and outcome. We present a case of JS in a 7-month-old girl who presented to the pediatric outpatient clinic with developmental delay and abnormal eye movements. MRI showed molar tooth configuration of superior cerebellar peduncles, the fourth ventricle shaped like a bat wing and hypoplasia of the vermis which resulted in median approach of the two cerebellar hemispheres.
Keywords: Joubert, magnetic resonance imaging, molar tooth sign
Introduction
Joubert syndrome (JS) is characterized by episodes of abnormal respiratory pattern, oculomotor findings, hypotonia, ataxia, developmental retardation with evidence of neuropathologic abnormalities of cerebellum and brainstem.[1] This clinical entity is underreported with a prevalence of less than 1 in 100,000. Only about 200 cases have been reported worldwide.[2] It is a syndrome with a variable phenotype making it difficult to establish the exact clinical diagnostic boundaries of the syndrome. Even though the clinical features of the disorder are present in the newborn period, the correct diagnosis is often not made for several months or years after birth.[3] The average age at diagnosis is 33 months.[4] The importance of early detection of the syndrome is stressed so that suitable measures can be started as early as possible. A comprehensive review of literature is also presented.
Case Report
A 7-month-old girl presented to the pediatric outpatient clinic with developmental delay and abnormal eye movements. Abnormal eye movements were noted shortly after birth and involved episodic deviation to lateral extremes of gaze, usually alternating and lasting for a few seconds. The movements were not accompanied by any change in color and activity and were present throughout the day. In between these movements, the child was unable to fixate and follow objects visually. Parents also noticed that the child was not able to keep up with developmental milestones. She had social smile at 3 months and head control at 5 months of age and was unable to sit even with support.
There was no history of seizure, abnormal breathing pattern, feeding or swallowing difficulty. She was born at term to non-consanguineous parents and suffered no significant perinatal asphyxia. She was the only child of her parents.
On examination, she appeared awake, alert, only inconsistently focusing visually. Intermittent movements of eyes to extremes of gaze were noted throughout the examination. She interacted with her parents and had social smile. She had no neurocutaneous markers. Ocular examination was normal. She showed mild facial dysmorphism in the form of forehead prominence, deep-set eyes, bilateral epicanthic folds and low frontal hairline. There was no organomegaly. Heart and lungs were normal on auscultation. Neurological examination revealed normal cranial nerves and fundus. Motor examination revealed hypotonia with normal tendon reflexes. Head circumference was normal for age.
The axial T1-weighted and T2-weighted [Figure 1] Magnetic resonance (MR) images showed abnormally oriented and thickened superior cerebellar peduncles that resulted in a molar tooth configuration. The more caudal T2- and T1-weighted axial MR images [Figure 2] showed the fourth ventricle shaped like a bat wing. Furthermore, T2-weighted axial MR images showed hypoplasia of the vermis which resulted in median approach of the two cerebellar hemispheres but without evidence of a posterior fossa cyst [Figure 3]. Based on clinical and magnetic resonance imaging (MRI) findings, diagnosis of JS was made and parents were counseled. Follow-up at 9 months of age revealed that she is able to sit without support and has no truncal ataxia or titubation. Hypotonia has diminished.
Figure 1.
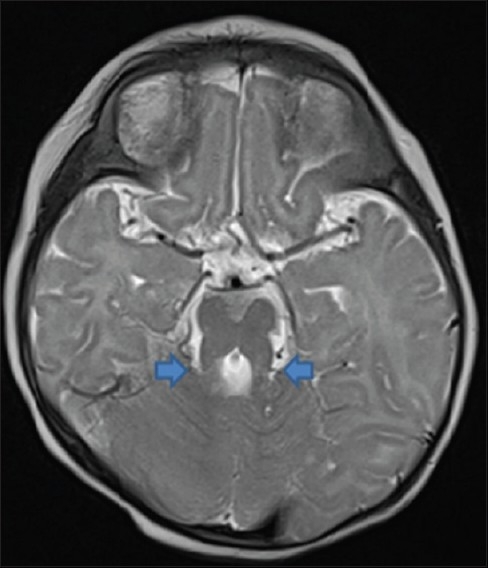
T2W axial image showing molar tooth sign (arrows)
Figure 2.
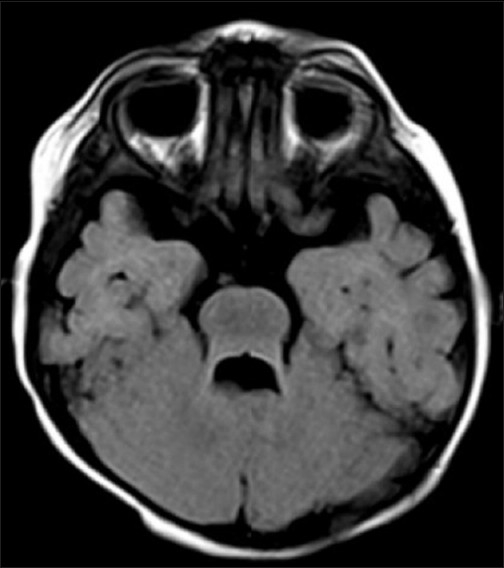
T1W axial image showing bat wing appearance of the fourth ventricle
Figure 3.
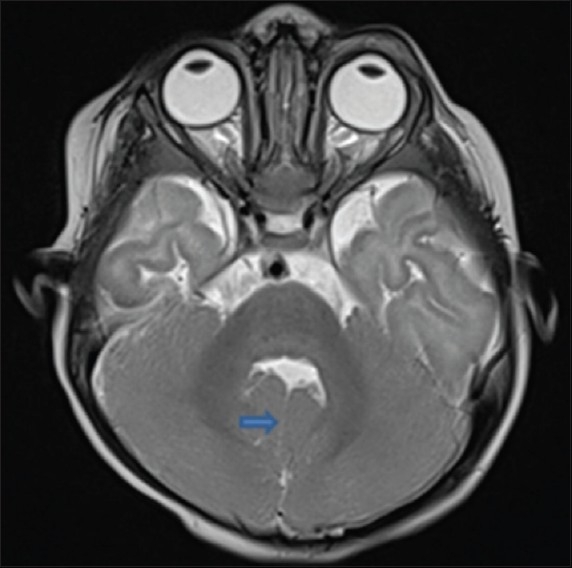
T2W axial image showing thin median cleft (arrows) separating cerebellar hemispheres
Discussion
JS is a rare autosomal recessive disorder characterized by clinical and characteristic neuroradiological findings. Key neuroimaging features of JS include deep interpeduncular fossa, narrow isthmus (the ponto-mesencephalic junction), lack of decussation of superior cerebellar peduncles, dilated, distorted, and rostrally deviated fourth ventricle giving “Bat Wing” appearance, thick vertical superior cerebellar peduncles, rostral deviation of fastigium of fourth ventricle, wide foramen of Magendie and dysplastic vermis. The brain stem, predominantly the medulla and upper cervical spinal cord, tends to be small. “Molar tooth sign” encompasses deeper than normal posterior interpeduncular fossa, prominent or thickened superior cerebellar peduncles, and vermian hypoplasia or dysplasia.[5,6]
Although the diagnostic criteria for JS have not been established, the clinical features frequently mentioned as essential for the diagnosis of classic JS comprise:[1,5,7,8]
Hypotonia in infancy.
Developmental delay/mental retardation.
-
One or both of the following (not absolutely required but helpful for the diagnosis):
- Irregular breathing pattern in infancy (intermittent tachypnea and/or apnea).
- Abnormal eye movements.
Our patient had all the clinical symptoms with the exception of breathing abnormalities which may have been overlooked.
Associated supratentorial anomalies are uncommon, but cerebral cortical dysplasia and gray matter heterotopias have been reported.[9] Moderate lateral ventricular enlargement due to atrophy has been described in 6–20% of cases.[6] Many authors have reported the prevalence of some of these associated findings, which include polydactyly (8%), ocular coloboma (4%), and hamartomas of the tongue (2%), dysmorphic facies, microcephaly, tongue protrusion, multicystic kidney disease, congenital heart disease, unsegmented midbrain tectum, retinal dystrophy and agenesis of the corpus callosum.[10–13]
This syndrome is classified into two groups on the basis of presence or absence of retinal dystrophy. Patients with retinal dystrophy have a higher prevalence of multicystic renal disease and these patients also appear to have decreased survival rates compared with those of patients without retinal dystrophy.[5] There was no evidence of retinal disease on ophthalmological examination in our patient.
Besides JS, cerebellar vermian anomalies have been reported with other disorders, such as Dandy-Walker syndrome and rhombencephalosynapsis. In Dandy-Walker malformation, the inferior part of the vermis is hypoplastic. However, the fourth ventricle is enlarged and communicates with a cyst in the posterior fossa. In addition, the ponto-mesencephalic junction, interpeduncular fossa and superior cerebellar peduncle are normal. In rhombencephalosynapsis, the cerebellar hemispheres are fused and, unlike in JS, a midline cerebellar cleft is not present.[14]
Molar tooth sign is not specific for JS. Other clinical features define the subtypes of JS termed as Joubert syndrome and related disorders (JSRD). JSRD are categorized into six phenotypic subgroups: Pure JS, JS with ocular defect, JS with renal defect, JS with oculorenal defects, JS with hepatic defect, and JS with orofaciodigital defects. Although the molar tooth sign and other important clinical features of the JS may be seen in these syndromes, they usually have supplementary prominent features. These are syndromes such as the COACH, Varadi-Papp, Dekaban-Arima, Senior-Loken, Joubert with polymicrogyria, and Malta syndromes. Patients with COACH syndrome have bilateral coloboma, hepatic fibrosis and renal calcification, and in the Varadi-Papp syndrome there is mesaxial polydactyly, Y-shaped metacarpal, cleft lip or cleft palate, lingual hamartomas and vermian hypoplasia. The Dekaban-Arima syndrome is allied with Leber's congenital amaurosis and cystic dysplastic kidneys, whereas the Senior-Loken syndrome is related with Leber's congenital amaurosis, retinitis pigmentosa and juvenile nephronophthisis. In the Malta syndrome, these patients have the molar tooth sign, occipital encephalocele, hydrocephalus, cortical renal cysts with or without coloboma, and Leber's congenital amaurosis. Few patients can have features of JS and polymicrogyria.[15,16]
Developmental outcome in JS is variable. Steinlin et al.[7] suggested that outcomes in JS can be divided into three courses: first, children who die young; second, patients who survive but are severely developmentally delayed and have a variety of visual and motor handicaps; and third, patients whose developmental quotients fall within the mildly delayed range (70–80).
With the exception of rare X-linked recessive cases, JSRD follow autosomal recessive inheritance and are genetically heterogeneous with one locus pointing to chromosome 9q. In addition, consanguinity has been documented in a few cases. Ten causative genes have been recognized so far, every single one encoding for proteins of the primary cilium or the centrosome, making JSRD part of an expanding group of diseases called “ciliopathies”.[5,12,16]
Pathological studies[17] in these patients have shown that the cerebellar vermis is hypoplastic and the dentate nucleus is fragmented. The ponto-mesencephalic junction is dysplastic, with abnormal decussation of the superior cerebellar peduncle and elongation of rostral fourth ventricle. There is a decrease in neurons of the basis pontis and reticular formation. In the medulla, the inferior olivary nucleus, tractus solitarius, the nucleus and spinal tracts of trigeminal nerves show evidence of hypoplasia. The posterior median sulcus and pyramidal decussation are not present. Besides, there is neuronal enlargement in the nucleus gracilis and cuneatus.
Once a diagnosis of JS is made in one neonate or an infant, the diagnosis of this syndrome can be made by looking for the specific imaging findings at ultrasound during a subsequent pregnancy.[9] Renal and retinal dysfunction can be progressive. In patients with retinal anomalies, the renal function should be monitored regularly and ultrasonography should be done to detect cystic renal disease.[5] Finally, the diagnosis is important for future procedures that require anesthesia. These patients are sensitive to respiratory depressant effects of anesthetic agents like opiates and nitrous oxide.[18] Hence, the use of these anesthetic agents should be avoided in these patients.
Footnotes
Source of Support: Nil.
Conflict of Interest: None declared.
References
- 1.Joubert M, Eisenring JJ, Robb JP, Andermann F. Familial agenesis of the cerebellar vermis. A syndrome of episodic hyperpnea, abnormal eye movements, ataxia and retardation. Neurology. 1969;19:813–25. doi: 10.1212/wnl.19.9.813. [DOI] [PubMed] [Google Scholar]
- 2.Choh SA, Choh NA, Bhat SA, Jehangir M. MRI findings in Joubert syndrome. Indian J Pediatr. 2009;76:231–5. doi: 10.1007/s12098-008-0232-1. [DOI] [PubMed] [Google Scholar]
- 3.Akcakus M, Gunes T, Kumandas S, Kurtoglu S, Coskun A. Joubert syndrome: Report of a neonatal case. Paediatr Child Health. 2003;8:499–502. doi: 10.1093/pch/8.8.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maria BL, Quisling RG, Rosainz LC, Yachnis AT, Gitten J, Dede D, et al. Molar tooth sign in Joubert syndrome: Clinical, radiologic, and pathologic significance. J Child Neurol. 1999;14:368–76. doi: 10.1177/088307389901400605. [DOI] [PubMed] [Google Scholar]
- 5.Saraiva JM, Baraitser M. Joubert syndrome: A review. Am J Med Genet. 1992;43:726–31. doi: 10.1002/ajmg.1320430415. [DOI] [PubMed] [Google Scholar]
- 6.Kendall B, Kingsley D, Lambert SR, Taylor D, Finn P. Joubert syndrome: A clinico-radiological study. Neuroradiology. 1990;31:502–6. doi: 10.1007/BF00340131. [DOI] [PubMed] [Google Scholar]
- 7.Steinlin M, Schmid M, Landau K, Boltshauser E. Follow-up in children with Joubert syndrome. Neuropediatrics. 1997;28:204–11. doi: 10.1055/s-2007-973701. [DOI] [PubMed] [Google Scholar]
- 8.Parisi MA, Glass IA. In: GeneReviews at GeneTests-GeneClinics: Medical Genetics Information Resource. Seattle: Copyright, University of Washington; 1997-2006. [last cited on 2010 Nov 1]. Joubert syndrome. Available from: http://www.geneclinics.org. or http:// www.genetests.org. 2006. [Google Scholar]
- 9.Barkovich AJ. Pediatric neuroimaging. 2nd ed. New York, NY: Raven; 1995. pp. 249–57. [Google Scholar]
- 10.Egger J, Bellman MH, Ross EM, Baraitser M. Joubert-Boltshauser syndrome with polydactyly in siblings. J Neurol Neurosurg Psychiatry. 1982;45:737–9. doi: 10.1136/jnnp.45.8.737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aslan H, Ulker V, Gulcan EM, Numanoglu C, Gul A, Agar M, et al. Prenatal diagnosis of Joubert syndrome: A case report. Prenat Diagn. 2002;22:13–6. doi: 10.1002/pd.220. [DOI] [PubMed] [Google Scholar]
- 12.Chance PF, Cavalier L, Satran D, Pellegrino JE, Koenig M, Dobyns WB. Clinical nosologic and genetic aspects of Joubert and related syndromes. J Child Neurol. 1999;14:660–6. doi: 10.1177/088307389901401007. [DOI] [PubMed] [Google Scholar]
- 13.Maria BL, Hoang KB, Tusa RJ, Mancuso AA, Hamed LM, Quisling RG, et al. “Joubert syndrome” revisited: Key ocular motor signs with magnetic resonance imaging correlation. J Child Neurol. 1997;12:423–30. doi: 10.1177/088307389701200703. [DOI] [PubMed] [Google Scholar]
- 14.Van Beek EJ, Majoie CB. Case 25: Joubert syndrome. Radiology. 2000;216:379–82. doi: 10.1148/radiology.216.2.r00au34379. [DOI] [PubMed] [Google Scholar]
- 15.Gleeson JG, Keeler LC, Parisi MA, Marsh SE, Chance PF, Glass IA, et al. Molar tooth sign of the midbrain-hindbrain junction: Occurrence in multiple distinct syndromes. Am J Med Genet A. 2004;125A:125–34. doi: 10.1002/ajmg.a.20437. [DOI] [PubMed] [Google Scholar]
- 16.Brancati F, Dallapiccola B, Valente EM. Joubert Syndrome and related disorders. Orphanet J Rare Dis. 2010;5:20. doi: 10.1186/1750-1172-5-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yachnis AT, Rorke LB. Neuropathology of Joubert syndrome. J Child Neurol. 1999;14:655–9. doi: 10.1177/088307389901401006. [DOI] [PubMed] [Google Scholar]
- 18.Habre W, Sims C, D’Souza M. Anaesthetic management of children with Joubert syndrome. Paediatr Anaesth. 1997;7:251–3. doi: 10.1046/j.1460-9592.1997.d01-65.x. [DOI] [PubMed] [Google Scholar]