

Abstract
Pseudoxanthoma elasticum (PXE) is a heritable multisystem disorder manifesting with ectopic calcification of peripheral connective tissues, caused by mutations in the ABCC6 gene. Alterations in vitamin K metabolism have been suggested to contribute to the pathomechanisms of the mineralization process. In this study we administered vitamin K or its glutathione conjugate (K3-GSH) into Abcc6−/− mice that recapitulate features of PXE. Oral administration of vitamin K2, in dosages that vastly exceed the amounts in control diet or the recommended amounts for humans, did not alter the ectopic mineralization in Abcc6−/− mice. Similarly, intravenous administration of K3-GSH did not alter the degree of mineralization. Testing of vitamin K2, K3 and K3-GSH in an in vitro calcification system provided no evidence of mineralization inhibition. Collectively, our data suggest that vitamin K deficiency in the peripheral tissues is not a simple explanation for development of mineral deposits in PXE.
Key words: pseudoxanthoma elasticum, ectopic mineralization, vitamin K, Abcc6 knock-out mouse, Gla proteins
Introduction
Pseudoxanthoma elasticum (PXE), a multi-system heritable disorder, is characterized by ectopic mineralization of connective tissues leading to clinical manifestations in the skin, the eyes and cardiovascular system. PXE is caused by mutations in the ABCC6 gene, which is expressed primarily in the liver and the kidneys, and at very low level, if at all, in tissues clinically affected in PXE.1 This and several experimental observations in Abcc6−/− knock-out mice, which recapitulate the genetic, histopathologic and ultrastructural features of PXE, have suggested that PXE is a metabolic disorder, the primary site for the molecular pathology being the liver.2–4 Specifically, it has been postulated that ABCC6, a putative efflux pump located at the baso-lateral surface of hepatocytes, is physiologically transporting molecules from the hepatocytes to the circulation that either directly or indirectly serve as anti-mineralization factors, and in the absence of functional ABCC6, deposition of calcium-phosphate complexes ensues.
Recent evidence has suggested that vitamin K or its derivatives may play a role in the pathophysiology of PXE. First, patients with mutations in the GGCX gene, encoding γ-glutamyl carboxylase, can develop phenotypic manifestations not unlike those encountered in PXE.5,6 γ-Glutamyl carboxylase requires vitamin K in its reduced form (KH2) as a co-factor for the carboxylation reaction.7 One of the target proteins for γ-glutamyl carboxylation is matrix Gla protein (MGP) which in its activated (Gla) form is a potent anti-mineralization factor.8 Thus, in the absence of γ-glutamyl carboxylation either due to GGCX mutations or deficiency in KH2, MGP remains in inactive (Glu) form potentially allowing the mineralization process to take place. Secondly, recent studies have reported that serum levels of vitamin K in patients with PXE are reduced, suggesting that administration of vitamin K or its derivatives could potentially counteract the pathologic mineralization of peripheral connective tissues in PXE.9
Development of Abcc6−/− mice has provided a useful model system to study the pathomechanisms of PXE.10,11 One of the characteristic features of this mouse model is extensive mineralization of connective tissues in several organs, in most cases associated with elastic fiber structures. An early site of mineralization in these mice is the connective tissue capsule surrounding the vibrissae, which demonstrates histopathological evidence of mineralization first at 5–6 weeks of age of the Abcc6−/− mice, and the amount of calcium-phosphate complexes progressively increases with advancing age.12,13 Assessment of the mineral content can be performed by histopathologic staining for calcium/phosphate (von Kossa and Alizarin Red stains) coupled with computerized morphometric analysis, or by direct chemical assay of calcium and/or phosphate in biopsies taken from the muzzle skin containing the vibrissae. Thus, the mineralization of the mouse vibrissae serves as a quantifiable biomarker of the progression of PXE in this model, and provides a system to test various treatment modalities that might interfere with this process.
Based on these and similar observations, Borst et al.14 posed in this Journal a question whether the absence of ABCC6 in patients with PXE prevents the liver from providing sufficient vitamin K to the peripheral tissues. Considering the fact that ABCC6 can serve as a transporter of small molecular weight compounds conjugated with glutathione,15 the hypothesis put forward by Borst et al. could be interpreted to suggest that following the uptake of vitamin K to the liver, critical components are not transported further to the circulation and then to the peripheral tissues in the absence of ABCC6 activity. Since the dietary vitamin K1 can be converted to vitamin K2 either directly or through vitamin K3 intermediate, and vitamin K3 can be conjugated to form a glutathione conjugate (K3-GSH)7,16 (Fig. 1A), in this study we have utilized the Abcc6−/− mouse model to study the effect of administration of vitamin K2 and K3-GSH for their efficacy to interfere with the mineralization process in PXE.
Figure 1.

Schematic representation of conversion between different forms of vitamin K (A, left) and synthesis of vitamin K3-GSH conjugate (A, right). Purity analysis of synthetic K3-GSH was done with high-performance liquid chromatography demonstrating a single peak (B, left). The absorption spectrum of the conjugate by spectrophotometer indicated the highest absorbance at 258 nm (B, right).
Results
In the first set of experiments the Abcc6−/− mice, which initially demonstrate mineralization of the connective tissue capsule surrounding the vibrissae around 5–6 weeks of age, were placed on a diet supplemented with vitamin K2, 1.5 mg per gram of food. This concentration is ∼440-fold higher than the vitamin K concentration in the control diet (3.04 µg per gram).17 Also, the calculated intake of vitamin K, ∼0.225 gram per gram of mouse body weight (based on average consumption of 3 g food per day and total body weight of ∼20 g), is ∼375-fold higher than the recommended dose for human vitamin K2 intake (based on 45 mg/day; reviewed in ref. 18). The control mice were kept on standard baseline diet. The mice were placed on these diets at 4 weeks of age, i.e., well before the initial mineralization of vibrissae, and the muzzle skin was biopsied and histopathological sections were stained with hematoxylin eosin and Alizarin Red at 12 weeks. At 12 week point, the serum concentrations of vitamin K were found to be significantly elevated in comparison to the levels before vitamin K2 administration both in wild type (WT) and knockout (KO) mice. Specifically, the serum vitamin K concentration in mice kept on control diet was essentially undetectable, while the values in WT and KO mice fed with vitamin K-containing diet were 24.4 ± 6.6 and 44.4 ± 18.3 ng/ml, respectively (n = 5–6 mice per group; p < 0.03). Significant levels of vitamin K were also detected in the liver of the same mice fed with vitamin K supplemented diet (265.0 ± 150.2 and 605.6 ± 485.3 ng/g tissue in WT and KO mice, respectively; p = 0.13). Thus, the ratio of vitamin K in the liver vs. serum in WT and KO mice was not statistically different (10.8 ± 5.1 vs. 13.6 ± 13.2; p = 0.38).
Histopathologic examination demonstrated that the WT mice maintained on control diet showed no evidence of mineralization of the vibrissae (Fig. 2A). In contrast, the KO mice maintained on control diet demonstrated significant degree of mineralization. However, supplementation of the diet with vitamin K2 (KO mice + K2) showed the same degree of mineralization by histopathologic staining as noted in the KO mouse group (Fig. 2A). The histopathologically detectable mineralization was quantitated by computerized morphometric analysis, which confirmed significant increase in mineralization in KO mice, both male and female, in comparison to the WT mice, both kept on control diet (Fig. 2B). However, computerized morphometric analysis of the degree of mineralization in mice fed on diet supplemented with vitamin K2 (KO mice + K2) showed no difference from KO mice maintained on control diet. These conclusions were verified by independent measurements of calcium and phosphate contents in skin biopsies taken from the corresponding mice (Fig. 2C).
Figure 2.

Mineralization of vibrissae in Abcc6−/− mice fed either control diet or diet supplemented with vitamin K2. The mice were placed on the diets at 4 weeks of age, and the muzzle skin was biopsied and histopathologic sections were stained with hematoxylin-eosin (upper row) and Alizarin Red (lower row) at 12 weeks (A). For comparison, wild-type (WT) Abcc6+/+ mice on control diet at 12 weeks of age were included and had no evidence of mineralization (A, left parts). The Abcc6−/− mice on control diet (A, KO-middle parts) or on vitamin K2 supplemented diet (A, KO + K2-right parts) showed similar degree of mineralization (arrows). Quantitation of connective tissue mineralization in the vibrissae was accomplished either by computerized morphometric analysis (B) or by chemical assay for calcium and phosphate (C) in the muzzle skin biopsies. The bars represent mean ± SE, n = 5–9 in each group. The statistical significance between different groups is indicated by an asterisk (p < 0.05) or two asterisks (p < 0.01).
To examine the hypothesis that the critical vitamin K derivative absent from the peripheral tissues would be K3-GSH, a putative transport substrate for Abcc6 in the liver, we performed experiments in which K3-GSH was delivered into circulation of the mice through intravenous retro-orbital injections. Since the vitamin K3-GSH conjugate was not commercially available, we chemically synthesized this molecule, and its purity was confirmed by high-performance liquid chromatography with the spectrophotometric peak absorbance at 258 nm, attesting to the purity of the compound (Fig. 1B). This compound was injected to the Abcc6−/− mice twice a week commencing at 4 weeks of age for a total of 8 weeks. The degree of mineralization of vibrissae was then determined by histopathologic staining coupled with computerized morphometric analysis and by chemical assay of calcium and phosphate in muzzle skin as above. The degree of mineralization of vibrissae in mice administered K3-GSH (n = 12) was not different from that measured in KO mice administered saline alone (n = 11), as determined by either assay method (Table 1). Furthermore, examination of male and female control and experimental populations separately, did not reveal any significant difference as a result of K3-GSH administration. To examine the degree of mineralization in the Abcc6−/− mice injected with K3-GSH, other tissues besides the vibrissae were also examined (Table 2). The results indicated that significant mineralization was detected both in experimental (K3-GSH) and in control KO mice injected with saline. The differences in these two groups of mice with respect to degree of mineralization were not statistically significant.
Table 1.
Quantitation of connective tissue mineralization in vibrissae of the Abcc6−/− mice injected with vitamin K3-GSH conjugate
| % Vibrissae Mineralized1 | % Area Mineralization1 | Ca x P (nmol2/mg tissue)2 | |
| Control-all (n = 11) | 25.2 ± 10.3 | 1.55 ± 0.415 | 56.1 ± 3.2 |
| Experimental-all (n = 12) | 29.8 ± 5.7 | 1.73 ± 0.29 | 38.1 ± 1.4 |
| Control-male (n = 5) | 22.6 ± 8.5 | 1.21 ± 0.70 | 40.1 ± 6.6 |
| Experimental-male (n = 7) | 27.0 ± 6.3 | 1.30 ± 0.33 | 33.4 ± 1.4 |
| Control-female (n = 6) | 27.8 ± 12.1 | 1.89 ± 0.57 | 69.5 ± 5.9 |
| Experimental-female (n = 5) | 32.7 ± 5.2 | 2.16 ± 0.5 | 44.7 ± 4.8 |
Skin sections containing vibrissae were stained with hematoxylin and eosin stain and examined by computerized morphometric analyses.
Mineralization was determined by chemical assay of calcium and phosphate in muzzle skin.
1,2 Values presented as mean ± SE. Note: The data from gender mixed groups are in bold; There was no statistically significant difference (p > 0.05).
Table 2.
Soft tissue mineralization of Abcc6−/− mice injected with vitamin K3-GSH conjugate1
| Mineralization in soft tissues2 | ||||
| Kidneys | Eyes | Heart | Spleen | |
| Control | 9/11 | 4/11 | 6/11 | 4/11 |
| Experimental | 6/12 | 1/12 | 2/12 | 3/10 |
Abcc6−/− animals were injected with either vitamin K3-GSH or NaCl (for details see Materials and Methods) twice a week for 8 weeks beginning at the age of 4 weeks.
Number of mice with tissue mineralization/total number of mice examined in each group, as determined by histopathologic examination of hematoxylin-eosin-stained tissue sections.
Finally, the potential effects of vitamin K and its glutathione conjugate (K3-GSH) on the mineralization process were tested in an in vitro system in which human aortic smooth muscle cells are grown in culture in the presence of 1.8 mM calcium, and once the cells reach ∼80% confluence, inorganic phosphate (+Pi) is added eliciting precipitation of calcium/phosphate complexes on the cell layer (Fig. 3A). This in vitro system has been utilized to test the efficacy of a number of compounds potentially interfering with the mineralization process, including anti-mineralization factors fetuin-A and magnesium.4,19 In this system, addition of vitamin K2 in three different concentrations (10, 50 or 100 µg/mL) together with Pi to the culture medium did not interfere with the precipitation of calcium phosphate, as determined by direct chemical assay of calcium associated with the cell layer (Fig. 3B). Similar results were obtained when vitamin K3 or its glutathione conjugate (K3-GSH) were tested in the same in vitro system (results not shown).
Figure 3.
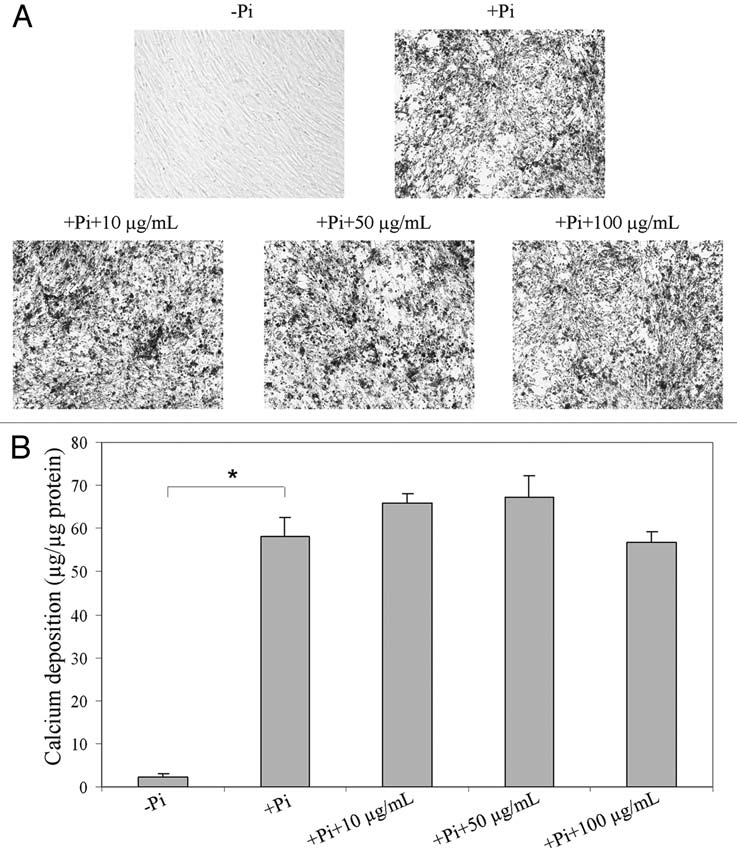
Demonstration that vitamin K2 has no influence on calcium deposition in an in vitro mineralization system. Human aortic smooth muscle cells were cultured on 96-well tissue culture plates in a medium containing 10% FBS. After the cultures reached ∼80% of confluence, 2 mM inorganic phosphate (+Pi) was added and the degree of mineralization was assessed either by (A) phase contrast light microscopy or (B) chemical assay of calcium deposition in the cell layer. Distinct calcium deposits were noted in the presence of Pi in 2 weeks but not in absence of Pi (A, top parts). Addition of vitamin K2 in concentrations indicated into the Pi-containing culture media did not prevent the calcium deposition even with a dose up to 100 µg/ml (A, bottom parts). The values are mean ± SE; n = 3; the statistical significance between different groups is indicated by an asterisk (p < 0.01).
Discussion
PXE is a systemic mineralization disorder caused by mutations in the ABCC6 gene. In fact, well over 300 distinct mutations have been disclosed representing ∼800 mutant alleles.1 The spectrum of genetic lesions in ABCC6 include nonsense mutations, small insertions or deletions leading to premature termination codon of translation, and a number of missense mutations primarily residing within the nucleotide binding folds, critical for the function of the ABCC6 protein as a transmembrane transporter. As a result of absent ABCC6 transporter activity, reflecting loss-of-function mutations in both alleles in homozygous or compound heterozygous individuals, late-onset, yet progressive, mineralization of peripheral connective tissues ensues. While identification of specific mutations in the ABCC6 gene has assisted in diagnosis and allowed presymptomatic identification of individuals at risk for PXE,1,20 relatively little progress has been made towards treatment of this complex, often devastating disease. In fact, while vascular endothelial growth factor antagonists have shown promise for treatment of the ophthalmologic complications due to neovascularization in the retina,21,22 no global, pathophysiology related approaches are currently available for treatment of PXE.
Significant progress has been recently made in counteracting the mineralization processes using Abcc6−/− mouse model as a preclinical platform. Such studies have shown, for example, that transgenic overexpression of anti-mineralization factors, such as fetuin-A, can reduce the degree of mineralization in these mice.4 Furthermore, manipulation of the mouse diet by increasing the magnesium content by five-fold has been shown to completely prevent the mineralization in this mouse model of PXE.19 Based on the latter observations, clinical trials are currently being initiated to test the efficacy of magnesium supplementation of diet for the treatment of patients with PXE. Finally, preliminary studies testing oral phosphate binders have suggested that some of them may be effective in counteracting the progress of cutaneous involvement in these patients.23
The role of vitamin K in the pathomechanism of PXE was initially suggested by the observation that patients with vitamin K-dependent coagulation factor deficiency developed cutaneous signs reminiscent of those in PXE.24 These patients were subsequently shown to harbor mutations in the GGCX gene, which encodes γ-glutamyl carboxylase, an enzyme requiring reduced vitamin K as a cofactor.5,6 One of the target proteins for γ-glutamyl carboxylation by this enzyme is MGP, which in its carboxylated form can serve as a powerful anti-mineralization factor.8 It was postulated, therefore, that a vitamin K derivative, such as vitamin K3-glutathione conjugate, could be transported from the intracellular milieu of hepatocytes to the circulation under physiological conditions.14 Such form of vitamin K would then facilitate the activation of MGP in peripheral tissues while in the absence of functional ABCC6 transporter, the peripheral tissues would become depleted of reduced vitamin K, thus preventing the activation of MGP and resulting in ectopic mineralization. We postulated, therefore, that administration of vitamin K to the Abcc6−/− mice could counteract the ectopic mineralization.
The Abcc6−/− mice recapitulate features of PXE, including late-onset, slowly progressing mineralization of peripheral connective tissue.11 First, feeding of these mice with vitamin K2 in concentrations that well exceeded the vitamin K concentrations in the regular mouse diet and the recommended daily intake for humans was initiated at 4 weeks of age, before the first signs of mineralization in peripheral tissues is observed, and was continued for an additional 8 weeks. No difference in the degree of mineralization between the KO mice kept on regular mouse diet and those on a diet supplemented with vitamin K was noted by two independent measurements of mineralization, viz., histopathology coupled with computerized morphometric analysis and direct chemical assay of calcium and phosphate in the skin of these mice. In subsequent studies, we tested the specific hypothesis that vitamin K3-GSH conjugate would be a substrate for transport by ABCC6. Specifically, we administered this compound directly into circulation with the notion that if there is a block in K3-GSH transport from the liver to circulation in the absence of active Abcc6, this approach by-passing the liver would correct the mineralization. Again, no difference in the mineralization between the mice administered K3-GSH and those KO mice injected with saline alone could be noted. Finally, testing of the efficacy of vitamin K or its glutathione conjugate in inhibiting mineralization in an in vitro cell-based assay system did not show any effect in concentrations up to 100 µg/ml. Collectively, these observations suggest that vitamin K deficiency in the peripheral tissues is not a simple explanation for development of mineral deposits in PXE. While assay of vitamin K in serum clearly revealed high concentrations of vitamin K upon oral feeding, it is conceivable that a specific metabolite of vitamin K, possibly synthesized in the liver, is the critical factor required for activation of MGP. Such metabolite is apparently not vitamin K3-GSH, since administration of large quantities of this conjugate directly into circulation of the Abcc6−/− mice did not result in modulation of the extent of mineralization. Alternatively, the transported molecule(s) may have a direct effect on the function of reduced vitamin K, thereby inhibiting the carboxylation reaction and activation of MGP. Identification and testing of other vitamin K metabolites as well as other small-molecular-weight compounds would require systematic metabolomics approach to decipher the key molecule(s) that are being transported by Abcc6 to the circulation in mice. Alternatively, observed changes in vitamin K in patients with PXE, such as reduced serum concentrations, may not reflect the pathogenic role of vitamin K or its derivatives, but rather serve as a marker of the ongoing mineralization process, similar to that noted in vascular mineralization processes in mice with fully functional Abcc6 transporter.25,26
Materials and Methods
Synthesis of vitamin K3-GSH conjugate.
Vitamin K3-GSH conjugate was synthesized as described previously in reference 27 and 28. Vitamin K3 (0.1 M in ethanol, total volume 50 ml; Sigma-Aldrich) was mixed with GSH (0.025 M in water, 10 ml; Sigma-Aldrich) at 4°C overnight in the dark. The yellow-orange crystals were formed the next day and then collected and washed twice with chloroform to dissolve excess vitamin K3. The precipitate was filtered and dried. Purity of the synthetic product was analyzed by high-performance liquid chromatography (5 µm C18 100A AstroSil®). The absorption spectrum of vitamin K3-GSH conjugate was examined with a standard spectrophotometer.
Mice and vitamin K administration.
The PXE mouse model was developed by targeted ablation of the Abcc6 gene, as described previously in reference 11. Abcc6−/− mice at 4 weeks of age were placed on a standard rodent diet (Laboratory Rodent Diet 5010; PMI Nutrition, Brentwood, MO) or the same diet with vitamin K2 supplementation (1.5 mg vitamin K2/g food; Sigma-Aldrich, Saint Louis, MO) for 8 weeks. To monitor proper food intake and subsequent weight gain, food was checked daily and mice were weighed weekly. The mice, housed in the Animal Facility of Thomas Jefferson University, had free access to water and were maintained in a temperature- and humidity-controlled environment under 12-hours light/dark cycle. The animal studies were approved by the Institutional Animal Care and Use Committee of Thomas Jefferson University. The delivery of vitamin K was monitored by assay of its concentrations in serum and tissues of mice at 12 weeks of age with an HPLC system.29
Vitamin K3-GSH conjugate was delivered into Abcc6−/− mice beginning at 4 weeks of age for a total of 8 weeks through intravenous retro-orbital injection. One hundred µl of vitamin K3-GSH containing solution (1.6 µM in 0.9% NaCl/0.05% DMSO) was injected twice a week switching between the two eyes, and the control Abcc6−/− mice were injected with 100 µl 0.9% NaCl/0.05% DMSO with the same frequency and for the same time period.
Cell cultures and induction of mineralization in vitro.
Human aortic smooth muscle cells (Cascade Biologics, Portland, OR) were cultured in DMEM. At ∼80% confluence, the cells were placed in calcification-inducing medium (DMEM supplemented with 2 mM Pi) together with 0, 10, 50 or 100 µg/ml of vitamin K2, K3 or K3-GSH and maintained for an additional 2 weeks.30 The medium was changed every 2 days. All experiments were performed in triplicate. At day 14 of cell culture after adding vitamin K, the media were removed and the cell layers were decalcified with 0.6 N HCl for 24 hours at room temperature. The calcium content of the HCl supernatants was determined colorimetrically by the o-cresolphthalein complex-one method (Calcium (CPC) Liquicolor; Stanbio Laboratory, Boerne, TX). After decalcification, the cells were rinsed three times with phosphate-buffered saline and solubilized with 0.1 N NaOH/0.1% SDS at room temperature. The protein content was measured with Protein Assay Kit (Bio-Rad, Hercules, CA), and the calcium content of the cell layer was normalized to the protein content.
Chemical quantification of calcium and phosphate deposition.
To quantify the mineral deposition in mouse vibrissae, the muzzle skin, which contains the vibrissae, was harvested and decalcified with 0.15 N HCl for 48 hours at room temperature. The calcium content was measured as above in the in vitro study, and the phosphate content was determined with Malachite Green Phosphate Assay kit (BioAssay Systems, Hayward, CA). The values for calcium and phosphate were normalized to tissue weight.
Quantification of tissue mineralization by computerized morphometric analysis.
For histopathologic analysis of mineralization of vibrissae, muzzle skin was fixed in 10% phosphate-buffered formalin, embedded in paraffin, sectioned (5 µm), and stained with H&E or Alizarin Red using standard techniques. Organ specimens from eye, heart, spleen and kidney were processed and stained using the same techniques. Computerized morphometric quantification was used to examine H&E-stained sections of muzzle skin.19,31 The sections were examined with a Nikon model Te2000 microscope furnished with an Auto Quant Imaging system (Watervliet, New York, NY). The number of vibrissae with and without perceptible mineralization was established in all sections, and the extent of mineralization was determined as the percentage of area of mineralization per total area of vibrissae.
Statistical analysis.
The statistical differences between the means in groups of cultured cells or mice were calculated by the Student's two-tailed t-test or Kruskal-Wallis ranking test.
Acknowledgements
The authors thank Carol Kelly for assistance. Sarah Cannon, Sophia Siu and Kathleen Zendell, Medical Students at Jefferson Medical College, contributed to this project. These studies were supported by NIH/NIAMS Grants R01 AR28450 and R01 AR55225 (J.U.). Dr. Jiang is the recipient of the NIAMS Clinical Career Development Award K08 AR57099, and Dr. Li is recipient of Dermatology Foundation Research Career Development Award.
Abbreviations
- PXE
pseudoxanthoma elasticum
- MGP
matrix Gla protein
- K3-GSH
vitamin K3 glutathione conjugate
- WT
wild type
- KO
knockout
References
- 1.Uitto J, Li Q, Jiang Q. Pseudoxanthoma elasticum—molecular genetics and putative pathomechanisms. J Invest Dermatol. 2010;130:661–670. doi: 10.1038/jid.2009.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jiang Q, Endoh M, Dibra F, Wang F, Uitto J. Pseudoxanthoma elasticum is a metabolic disease. J Invest Dermatol. 2009;129:348–353. doi: 10.1038/jid.2008.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jiang Q, Oldenburg R, Otsuru S, Grand-Pierre A, Horwitz E, Uitto J. Parabiotic heterogenetic pairing of Abcc6−/− and wild-type mice halts progression of ectopic mineralization in a murine model of pseudoxanthoma elasticum. Am J Pathol. 2010;176:1855–1862. doi: 10.2353/ajpath.2010.090983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jiang Q, Dibra F, Lee MD, Oldenburg R, Uitto J. Overexpression of fetuin-A counteracts ectopic mineralization in a mouse model of pseudoxanthoma elasticum (Abcc6−/−) J Invest Dermatol. 2010;130:1288–1296. doi: 10.1038/jid.2009.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vanakker OM, Martin L, Gheduzzi D, Leroy BP, Loeys BL, Guerci VI, et al. Pseudoxanthoma elasticum-like phenotype with cutis laxa and multiple coagulation factor deficiency represents a separate genetic entity. J Invest Dermatol. 2007;127:581–587. doi: 10.1038/sj.jid.5700610. [DOI] [PubMed] [Google Scholar]
- 6.Li Q, Schurgers LJ, Smith AC, Tsokos M, Uitto J, Cowen EW. Co-existent pseudoxanthoma elasticum and vitamin K-dependent coagulation factor deficiency: compound heterozygosity for mutations in the GGCX gene. Am J Pathol. 2009;174:534–540. doi: 10.2353/ajpath.2009.080865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berkner KL. Vitamin K-dependent carboxylation. Vitam Horm. 2008;78:131–156. doi: 10.1016/S0083-6729(07)00007-6. [DOI] [PubMed] [Google Scholar]
- 8.Luo G, Ducy P, McKee MD, Pinero GJ, Loyer E, Behringer RR, et al. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature. 1997;386:78–81. doi: 10.1038/386078a0. [DOI] [PubMed] [Google Scholar]
- 9.Vanakker OM, Martin L, Schurgers LJ, Quaglino D, Costrop L, Vermeer C, et al. Low serum vitamin K in PXE results in defective carboxylation of mineralization inhibitors similar to the GGCX mutations in the PXE-like syndrome. Lab Invest. 2010;90:895–905. doi: 10.1038/labinvest.2010.68. [DOI] [PubMed] [Google Scholar]
- 10.Gorgels TG, Hu X, Scheffer GL, van der Wal AC, Toonstra J, de Jong PT, et al. Disruption of Abcc6 in the mouse: novel insight in the pathogenesis of pseudoxanthoma elasticum. Hum Mol Genet. 2005;14:1763–1773. doi: 10.1093/hmg/ddi183. [DOI] [PubMed] [Google Scholar]
- 11.Klement JF, Matsuzaki Y, Jiang QJY, Terlizzi J, Choi HY, Fujimoto N, et al. Targeted ablation of the ABCC6 gene results in ectopic mineralization of connective tissues. Mol Cell Biol. 2005;25:8299–8310. doi: 10.1128/MCB.25.18.8299-8310.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jiang Q, Li Q, Uitto J. Aberrant mineralization of connective tissue in a mouse model of pseudoxanthoma elasticum: Systemic and local regulatory factors. J Invest Dermatol. 2007;127:1392–1402. doi: 10.1038/sj.jid.5700729. [DOI] [PubMed] [Google Scholar]
- 13.Li Q, Jiang Q, Pfendner E, Váradi A, Uitto J. Pseudoxanthoma elasticum: clinical phenotypes, molecular genetics and putative pathomechanisms. Exp Derm. 2009;18:1–11. doi: 10.1111/j.1600-0625.2008.00795.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Borst P, van de Wetering K, Schlingemann R. Does the absence of ABCC6 (multidrug resistance protein 6) in patients with pseudoxanthoma elasticum prevent the liver from providing sufficient vitamin K to the periphery? Cell Cycle. 2008;7:1575–1579. doi: 10.4161/cc.7.11.6005. [DOI] [PubMed] [Google Scholar]
- 15.Illiás A, Urbán Z, Seidl TL, Le Saux O, Sinkó E, Boyd CD, et al. Loss of ATP-dependent transport activity in pseudoxanthoma elasticum-associated mutants of human ABCC6 (MRP6) J Biol Chem. 2002;277:16860–16867. doi: 10.1074/jbc.M110918200. [DOI] [PubMed] [Google Scholar]
- 16.Thijssen HHW, Vervoort LMT, Schurgers LJ, Shearer MJ. Menadione is a metabolite of oral vitamin K. Brit J Nutr. 2006;95:260–266. doi: 10.1079/bjn20051630. [DOI] [PubMed] [Google Scholar]
- 17.Rodent Diets: Overview. PMI Nutrition International 1996–2008. [cited 2010 December 21] Available at: www.labdiet.com/rodent_diet.html.
- 18.Iwamoto J, Sato Y, Takeda T, Matsumoto H. High-dose vitamin K supplementation reduces fracture incidence in postmenopausal women: a review of the literature. Nutr Res. 2009;29:221–228. doi: 10.1016/j.nutres.2009.03.012. [DOI] [PubMed] [Google Scholar]
- 19.LaRusso J, Li Q, Uitto J. Elevated dietary magnesium prevents connective tissue mineralization in a mouse model of pseudoxanthoma elasticum (Abcc6−/−) J Invest Dermatol. 2009;129:1388–1394. doi: 10.1038/jid.2008.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li Q, Török L, Kocsis L, Uitto J. Mutation analysis (ABCC6) in a family with pseudoxanthoma elasticum—presymptomatic testing with prognostic implications. Brit J Dermatol. 2010;163:641–643. doi: 10.1111/j.1365-2133.2010.09856.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Verbraak FD. Antivascular endothelial growth factor treatment in pseudoxanthoma elasticum patients. Dev Ophthalmol. 2010;46:96–106. doi: 10.1159/000320012. [DOI] [PubMed] [Google Scholar]
- 22.Farjo KM, Ma JX. The potential of nanomedicine therapies to treat neovascular disease in the retina. J Angiogenes Res. 2010;2:21–35. doi: 10.1186/2040-2384-2-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sherer DW, Singer G, Uribarri J, Phelps RG, Sapadin AN, Freund KB, et al. Oral phosphate binders in the treatment of pseudoxanthoma elasticum. J Am Acad Dermatol. 2005;53:610–615. doi: 10.1016/j.jaad.2004.11.066. [DOI] [PubMed] [Google Scholar]
- 24.Neldner KH. Pseudoxanthoma elasticum. Clin Dermatol. 1988;6:1–159. doi: 10.1016/0738-081x(88)90003-x. [DOI] [PubMed] [Google Scholar]
- 25.Wallin R, Schurgers L, Wajih N. Effects of the blood coagulation vitamin K as an inhibitor of arterial calcification. Thrombosis Research. 2008;122:411–417. doi: 10.1016/j.thromres.2007.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fodor D, Albu A, Poanta L, Porojan M. Vitamin K and vascular calcifications. Acta Physiol Hung. 2010;97:256–266. doi: 10.1556/APhysiol.97.2010.3.2. [DOI] [PubMed] [Google Scholar]
- 27.Anderson ME, Meister A. Intracellular delivery of cysteine. Methods Enzymol. 1987;143:313–325. doi: 10.1016/0076-6879(87)43059-0. [DOI] [PubMed] [Google Scholar]
- 28.Chang M, Shi M, Forman HJ. Exogenous glutathione protects endothelial cells from menadione toxicity. Am J Physiol (Lung Cell Mol Physiol 6) 1992:637–643. doi: 10.1152/ajplung.1992.262.5.L637. [DOI] [PubMed] [Google Scholar]
- 29.Spronk HM, Soute BA, Schurgers LJ, Thijssen HH, De Mey JG, Vermeer C. Tissue-specific utilization of menaquinone-4 results in the prevention of arterial calcification in warfarin-treated rats. J Vasc Res. 2003;40:531–537. doi: 10.1159/000075344. [DOI] [PubMed] [Google Scholar]
- 30.Jono S, McKee MD, Murry CE, Shioi A, Nishizawa Y, Mori K, et al. Phosphate regulation of vascular smooth muscle cell calcification. Circ Res. 2000;87:10–17. doi: 10.1161/01.res.87.7.e10. [DOI] [PubMed] [Google Scholar]
- 31.Li Q, Uitto J. The mineralization phenotype in Abcc6−/− mice is affected by Ggcx gene deficiency and genetic background—a model for pseudoxanthoma elasticum. J Mol Med. 2010;88:173–181. doi: 10.1007/s00109-009-0522-8. [DOI] [PMC free article] [PubMed] [Google Scholar]