

Abstract
The genomes of multicellular organisms are under constant assault from a host of environmental agents. The efficient elimination of cells harboring damage is essential to avoid the accumulation of deleterious changes that may promote tumorigenesis. Consequently, a complex and elaborate series of damage responses have evolved to either ensure that correct repair of the DNA has been carried out, or alternatively, to initiate programmes that result in the ablation of the damaged cell. Apoptosis is recognized as both a fast an efficient way of disposing of damaged or unwanted cells before they accumulate changes that may result in the acquisition of neoplastic autonomy. The mitochondrial apoptotic pathway relies upon two effector proteins of the Bcl2 family, Bax and Bak, that when activated form pores in the outer mitochondrial membrane that release cytochrome c and other apoptogenic factors. We have recently shown that the initiation of Bak activation is controlled by dephosphorylation. In particular, we found that a specific tyrosine dephosphorylation was required for Bak activation to proceed and that tyrosine phosphatases may serve to integrate apoptotic signals that culminate in Bak dephosphorylation. Here, we discuss these findings and present additional data underlining the importance of dephosphorylation in the Bak activation process and how the modulation of Bak phosphorylation status may be modified to enhance cell killing.
Key words: apoptosis, mitochondria, phosphatase, BH3, p53, Bak, signaling, phosphorylation
Introduction
The inactivation of cell death pathways during tumor progression is a hallmark of cancer cells.1 Understanding the processes and identifying underlying mechanisms by which otherwise healthy cells become tumorigenic and fail to respond to endogenous or therapeutic signals to kill them is vital. As loss of function of apoptotic pathways often contributes towards drug resistance and this poses a major barrier to effective treatment. The ‘decision’ of a cell whether to commit to enter apoptosis is based on the receipt of incoming signals that may emanate from cellular damage and micro-environmental cues. The mitochondrial apoptotic pathway is controlled by members of the Bcl-2 family,2 which regulate the commitment to apoptosis by their ability to govern mitochondrial outer membrane (MOM) permeabilization that releases apoptotic factors including cytochrome c, AIF, Omi and Smac. Pro-survival Bcl-2 family members such as Bcl-2, Mcl-1 and BCL-XL contain up to four homology domains (BH1-BH4), while pro-apoptotic Bid, Bim and Puma contain only the BH3 death domain that is required for the activation of the effector proteins Bax and Bak.3,4 Current proposed models of Bax and Bak activation remain somewhat controversial and are incompletely understood, but involve a series of conformational changes followed by multimerization and pore formation. In the indirect activation model, BH3-only proteins bind to and neutralize prosurvival Bcl-2 proteins relieving their effect and enabling Bak or Bax to be activated. Alternatively, direct activation has been proposed, where ‘activator’ class BH3 proteins bind directly to Bax/Bak whereas ‘sensitizer’ BH3 proteins bind to and neutralize anti-apoptotic Bcl-2 proteins. In addition, in human cell lines cytosolic p53 can also bind to Bak at a site distinct from that bound by BH3-only proteins that also results in the induction of the N-terminal change of Bak conformation and the initiation of apoptosis.5–11 The association of Bak with BH3-only proteins causing conformational change is necessarily transitory, as the BH3 binding surface is also required to mediate Bak-Bak interactions during the formation of dimers,12 that then go on to multimerize mediated by interactions at different sites.13,14
Whereas inactive Bax is a cytosolic protein that becomes inserted into the MOM upon activation, Bak is an integral mitochondrial membrane protein implying different mechanisms of activation. Both Bak and Bax are known to undergo a series of conformational changes early during the activation process that ultimately result in formation of homo-oligomeric complexes that permeabilize the MOM,14–17 suggesting that in healthy cells inactive Bak is restrained in an inactive state. One mechanism proposed for keeping Bak in check is its association with a minor isoform of the voltage dependent anion channel 2 (VDAC2),18,19 where recent evidence suggests that in healthy cells Bak forms large protein complexes in which the association with VDAC2 is mediated by the C-terminal Bak transmembrane anchor.20
Bak Dephosphorylation and Activation
Our interest in Bak and its role in apoptosis was kindled by our previous work that showed the E6 protein of human papillomavirus (HPV) was able to promote Bak proteolysis after UV damage,21 preserving mitochondrial integrity and function that presumably aids survival of virally infected cells.22,23 In our systems HPV E6 appeared to target Bak for proteolytic degradation after DNA damage, so we hypothesized multiple levels of Bak regulation over and above that provided by BH3 and p53 proteins. A key break-through was our finding that in healthy cells Bak is heavily phosphorylated and that upon DNA damage, undergoes Ser/Thr/Tyr dephosphorylation at multiple sites. We found that for Bak to undergo conformational change and subsequent multimerization Tyr dephosphorylation was required as an early step in the Bak activation process. A combinatorial approach using mutational analysis, the production of specific anti-sera and mass spectrometry, identified Tyr108 as being the critical residue involved in mediating Bak N-terminal conformational change.24 Mutation of Tyr108 to a non-phosphorylatable residue enhanced greatly not only the degree of Bak activation, but also cytochrome c release from isolated mitochodria, activation of caspase 9 and cell killing. Conversely, mitochondria isolated from cells expressing the Y108F mutant, that failed to undergo conformational change, are refractory to cytochrome c release by a Bid BH3 peptide and do not activate caspase 9 (Fig. 1). Importantly, these results imply that upon DNA damage only a proportion of the total Bak protein pool becomes activated and that the degree of Bak activation and cell killing can be actively controlled by modulation of Bak phosphorylation status. Our results showed that Tyr108 dephosphorylation is an initial, essential and obligatory rate-limiting step in the Bak activation process, generating what we have termed an ‘activation competent’ Bak state that can then go on to multimerize24—in effect in healthy cells the default state at the molecular level promotes cell survival and Bak must therefore be ‘licensed to kill.’
Figure 1.
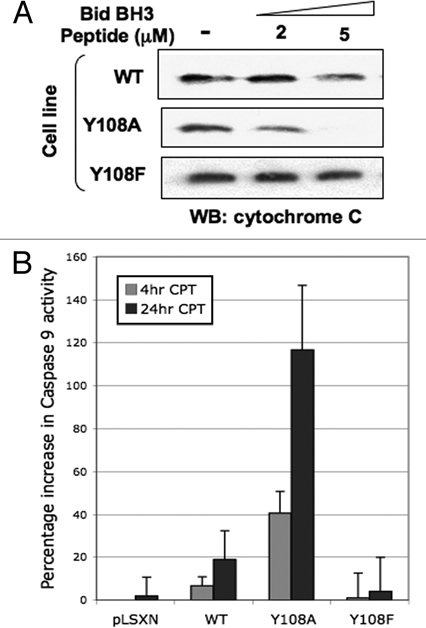
Bak Y108 status plays an important role in determining cytochrome c release from miochondria. (A) Mitochondrial preparations from HCT116 cells expressing either wild type (WT) or Bak mutated at Y108,24 by retroviral transduction (using a pLXSN vector) were incubated with a BH3 peptide derived from Bid than is known to cause cytochrome c release. Mitochondria were then harvested and the remaining cytochrome c levels determined by western blot. Mitochondria isolated from cells expressing the Bak Y108A mutant—that undergoes enhanced conformational change following DNA damage—released more cytochrome c than did WT. Conversely, the Y108F mutant is unable to undergo N-terminal conformational change and is refractory to cytochrome c release by the Bid peptide. (B) HCT116 cells expressing the Bak Y108A mutant showed enhanced activation of caspase 9 at both early and later time points following camptothecin (CPT, 6 µM) treatment than cells expressing WT Bak.
Tyrosine Phosphatases are Required to Integrate Diverse Apoptotic Signals for Bak Activation
To extend these studies, we performed a siRNA screen that identified three protein tyrosine phosphatases (PTP) that were involved in Bak activation—PTPN2, PTPN5 and PTPN23—each having been reported to be responsive to different cues. PTPN5 was the most effective in our assay systems, where we found PTPN5 gene silencing resulted in reduced cell death as measured by Annexin V positivity, while conversely, overexpression of PTPN5 resulted in enhanced Bak activation with a concomitant activation of caspase 3 compared to a PTPN5 mutant that had little effect on Bak activation.24 PTPN5 has been reported to be inactivated by phosphorylation by ERK1/2 and since the HCT116 cells used contain activating K-RAS mutations that result in constitutively activated MAPK pathway, we reasoned and then demonstrated that DNA damaging agents that result in a transient decrease in ERK1/2 signalling led to Bak underphosphorylation. Inhibition of ERK1/2 signalling with U1026 resulted in Bak underphosphorylation and in combination with DNA damage led to enhanced cell killing. These results provided a direct link between mitogenic growth signalling and the inhibition of mitochondrial apoptosis and are an important first insight into the crucial regulatory systems controlling the initiation of mitochondrial apoptosis.24 The RNAi screen also revealed possible roles for PTPN2 and PTPN23 in Bak activation in our test system, phosphatases that may be more responsive to different stimuli. Whether other phosphatases may be equally or even more important for Bak activation in different genetic backgrounds or in primary cells remains to be determined. Thus, in our new model of Bak activation we propose multiple positive apoptotic signals are required to be integrated before apoptosis can proceed without inhibition.24 These findings have important implications for interventional approaches as modulating Bak phosphorylation and hence activation potential in any given cell type, or in a pathway-dependent manner, depending on the genetic background of the cell, may unleash the pro-apoptotic activity of the protein to enhance cell killing. The conversion of Bak into the ‘activation competent’ form by dephosphorylation of Tyr108 we found did not directly per se lead to Bak activation, but instead may lower the threshold of Bak activation sufficiently to provide greater efficacy for current chemotherapeutic agents or enhance the effects of BH3 mimetic compounds that have shown great potential as clinically important agents.25 Our findings also highlight the fact that the mechanism of restraining Bak in an inactive state is markedly different from that of Bax, where recruitment of activator BH3 proteins is required to drive initiation of Bax activation leading to sequential conformational changes and subsequent membrane insertion.26,27 Indeed, while Bak can in one sense be thought to have bypassed the early steps associated with Bax activation that lead to its membrane insertion, the rapid activation response afforded by a dephosphorylation cascade mechanism is more likely to explain why the killing kinetics associated with Bak are faster than those of Bax. In addition, the inhibitory phosphorylation at Tyr108 may also serve to guard against Bak activation and unwanted cell killing in the absence of death stimuli that may otherwise occur through its inappropriate or aberrant activation by, for example, BH3 only proteins.
As both BH3-only and p53 proteins28–33 have been implicated in causing Bak N-terminal conformational change during activation, we were interested to determine whether translocation of these proteins to the mitochondria was blocked, or alternatively, whether they relocalized to the mitochondria following DNA damage induced by CPT under conditions where Bak dephosphoylation was inhibited. In HT1080 cells both p53 and tBid were found in mitochondrial cell fractions following damage induced by camptothecin (CPT), but were absent when the cells were pre-treated with sodium stibogluconate (SS) that we showed inhibited Bak dephosphorylation. A similar situation was found in A549 cells, where SS treatment blocked the relocalization of p53 and Bim to mitochondrial cell fractions (Fig. 2). This suggests that the mitochondrial relocalization of either BH3 proteins or p53 to the mitochondria is dependent on phosphatase activity. This raises the question as to whether Tyr108 phosphorylation has a role in mediating the association of Bak with Bcl-2 survival partners, Mcl-1 and Bcl-XL, whose functional activity in binding to Bak is still under debate, but whose association with Bak may account for the failure of the BH3 and p53 proteins to relocalize to mitochondria. We therefore assessed Mcl-1 protein levels following CPT treatment in cells pre-treated with SS or phenylarsine oxide (PAO) in HT1080 cells. As expected, CPT treatment led to the rapid destruction of Mcl-1, but we found no difference in Mcl-1 degradation induced by CPT treatment in the cells that had been exposed to phosphatase inhibition by SS. However the situation appeared more complex, as co-immunopreciptiation experiments revealed that SS pre-treatment maintained the Bcl-XL association with Bak following CPT treatment (Fig. 3). These initial findings suggest that the Bak phosphorylation status may alter Bak association with other cellular factors that may in turn regulate subsequent steps in Bak activation. Whether the phosphorylation status of Bak promotes its retention or release from other large protein complexes, such as those proposed to occur through VDAC2, remain to be determined.
Figure 2.
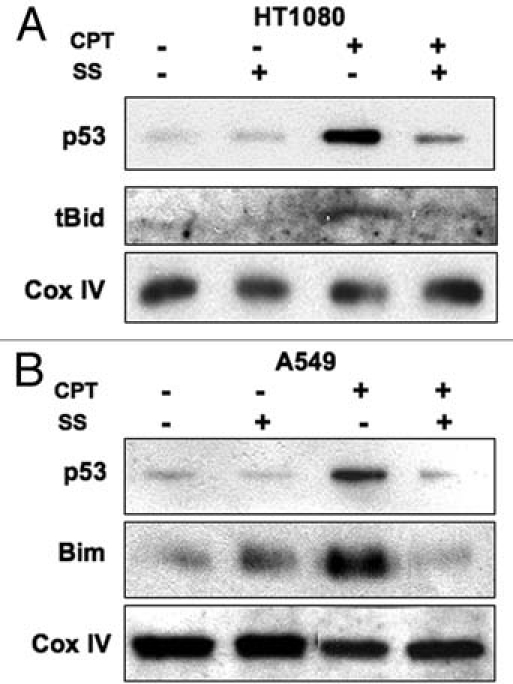
Recruitment of BH3-only or p53 proteins to mitochondria following DNA damage is blocked by phosphatase inhibition. Cell fractions enriched in mitochondria were isolated from either HT1080 (A) or A549 (B) cells following a 4 hr incubation with CPT and probed for levels of the BH3-only proteins tBid or Bim. Pre-treatment of the cells with the protein tyrosine phosphatase (PTP) inhibitor sodium stibogluconate (SS, 100 µM) blocked the recruitment of tBid, Bim or p53 to mitochondria after DNA damage. Cox IV acted as a loading control.
Figure 3.
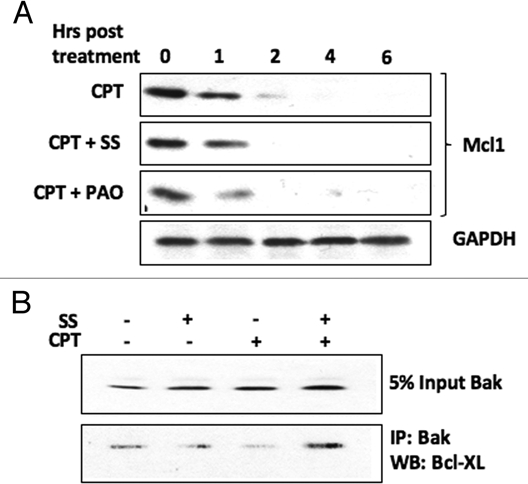
PTP inhibition differentially affects the association of Bak with Bcl2 family survival factors following DNA damage. HT1080 cells were pre-treated with PTP inhibitors SS or phenylarsine oxide (PAO, 5 µM) then incubated with CPT. (A) Mcl-1 protein levels, as determined by western blotting of cell lysates, decreased in cells treated with CPT irrespective of whether or not they has been pre-treated with PTP inhibitors. (B) Bak was immunoprecipitated from HT1080 cells and the level of bound Bcl-XL determined by western blot. Although the levels of Bcl-XL appear to be quite low in these cells, CPT treatment decreased the association of Bcl-XL with Bak, while pre-treatment of the cells with SS allowed maintainance of the Bcl-XL-Bak association following incubation with CPT. GAPDH acted as a loading control.
In addition to a role for Tyr108, we detected additional phosphorylation sites but their impact on Bak function is currently unknown and from the observed pI of Bak we infer more sites whose locations have not yet been mapped fully. As Bak only underwent only partial dephosphorylation following drug or UV treatment, we can speculate that these other sites may impinge upon other stages in the Bak activation process in apoptosis, or perhaps other Bak functions, possibly involving mitochondrial dynamics that were unaffected by loss of phosphorylation at Tyr108. Close to the phosphorylation site at Y108, we also detected an additional tyrosine phosphorylation site at residue 110, although we have not as yet been able to ascribe a role in Bak activation involving this site. As this residue is not conserved in murine Bak, any function must therefore be limited to the human protein and may serve either a more specialized or subtle function. At present the structural information on Bak is derived from protein produced in systems that have not permitted Bak phosphorylations to be detected,34,35 and hence their possible impact on Bak structure to be investigated. Further work will be necessary to enable a better understanding of the structure of inactive Bak to be elucidated, as this has the potential to reveal further important insights of the role of these modifications that will give a clearer picture of the series of conformational changes involved in Bak activation. Such efforts will also require the development of additional tools to monitor Bak phosphorylation status.
Conclusions and Future Directions
Our RNAi screen for phosphatases required for Bak activation also revealed that silencing of specific genes caused an increase in Bak activation.24 We suspect that this effect may be the result of an alteration in the activity of upstream kinase pathways that are responsible for Bak phosphorylation, hence decreasing their activity may lead to Bak underphosphorylation that in turn confers a greater capacity for activation. Thus, as Bak functions at a critical point that commits a cell to apoptosis, Tyr108 phosphorylation may serve as an integration point for multiple signal transduction systems, where a perturbation of the dynamic equilibrium of Tyr108 phosphorylation can either enhance or suppress cell killing by apoptotic inducers or survival signals (Fig. 4). At the molecular level, our findings suggest that the default mode setting for a cell is survival as multiple positive signals, including the dephosphorylation event discussed here, are required for Bak activation to proceed an initiate apoptosis. The additional involvement of posttranslational events that may be critical for Bak activation opens up new avenues of investigation and highlights that there is still much that we do not understand about the pathways that control the many steps involved. While in vitro reconstitution experiments have been useful for dissecting Bak function, such as proteinprotein interactions with BH3 proteins or the use of artificial membranes for example, we must also bear in mind that such systems may not permit Bak posttranslational modifications to occur and the spectrum of phosphorylation sites will be difficult to replicate or monitor in these in vitro assays. Factoring in how signalling pathways result in Bak modification certainly complicates our efforts to better understand Bak function in the many systems currently available, but nevertheless it presents new challenges and opportunities for manipulating Bak activity in ways previously not envisioned. As a first step to this end, the identification of Bak Tyr108 kinases now becomes an imperative as they are very likely to be suppressors of Bak activation and may be overexpressed in specific types of cancer or other diseases. Such Bak-inhibitory kinases represent excellent targets for interventional approaches, as they may be amenable to small molecule inhibition and relief from their suppressor activity may render cells more sensitive to chemotherapeutic agents. In general Bak is seldom mutated in cancer cells hence the re-activation of phosphatase signalling pathways, or conversely, suppression/inactivation of kinase pathways by novel small molecules or agents may offer new opportunities to reengage apoptotic pathways to enhance cell killing.
Figure 4.

PTPs regulate an early step in Bak activation. In healthy cells Bak is phosphorylated at Y108 an maintained in an inactive state by an as yet unidentified protein tyrosine kinase(s) (PTK). Apoptotic stimuli result in the activation of PTPs that dephosphorylate Bak at Y108, an obligatory step that then permits the recruitment of BH3-only proteins that bind to Bak causing a conformational change and exposure of Bak-BH3 helix, that inserts into the hydrophobic groove on another Bak molecule for dimerization. Bak dimers then assemble into higher order complexes leading to pore formation in the mitochondrial outer membrane and release of apoptotic factors such as cytochrome c.
Materials and Methods
All materials and experimental procedures are as described in reference 24 and accompanying Supplemental material, except for the following:
Caspase 9 assay.
Caspase 9 activity were measured by using Caspase-Glo® 9 assay kit (Promega) according to manufacturer's instructions. Briefly, cells were seeded 5,000 per well in 96 well plate and treated with CPT. The reagent and plates were equilibrated to room temperature and equal volume of reagent was added to media. Plates were rocked for 45 min to 2 hrs. Luminescence was measured with a BMG Labtech Luminator with Fluostar software. Values from wells containing only media were used as blanks.
Immunobloting and immunoprecipitation.
Immunoblotting was performed following standard procedures. Briefly, cell lysates were prepared in RIPA buffer. Antibodies used were as follows: rabbit anti Bak (Y164, abcam), anti Bcl-XL (Cell Signalling), anti p53 (DO1, CRUK), anti Bim (Cell Signalling), anti Cox IV (abcam), anti Mcl1 (Y37, abcam). For immunoprecipitation studies, total cell lysates were prepared in IP buffer (20 mM Tris-HCl pH 7.4, 135 mM NaCl, 1.5 mM MgCl2, 1 mM EGTA, 10% glycerol, Roche protease inhibitor cocktail) containing 1% CHAPS and incubated on ice for 30 min. The supernatants containing protein extracts were collected by centrifugation at 12,000 rpm for 10 min. The antibody was then added to the extracts and incubated overnight at 4°C with rotation.30 µl of a 50% slurry of either protein-A sepharose or protein G—sepharose were added to the samples and incubated at 4°C for 90 min. The beads were then washed in IP buffer for 3 times and dissolved in 25 µl of 2× SDS buffer.
Acknowledgements
We are grateful to Cancer Research UK for the funding that enabled us to undertake the research cited in this article (grant C5314). In addition, F.I. was the recipient of a Clinical Training Fellowship from the Medical Research Council, UK.
References
- 1.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 2.Adams JM, Cory S. The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene. 2007;26:1324–1337. doi: 10.1038/sj.onc.1210220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281:1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- 4.Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, et al. Proapoptotic BAX and BAK: A requisite gateway to mitochondrial dysfunction and death. Science. 2001;292:727–730. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chipuk JE, Fisher JC, Dillon CP, Kriwacki RW, Kuwana T, Green DR. Mechanism of apoptosis induction by inhibition of the anti-apoptotic BCL-2 proteins. Proc Natl Acad Sci USA. 2008;105:20327–20332. doi: 10.1073/pnas.0808036105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chipuk JE, Green DR. How do BCL-2 proteins induce mitochondrial outer membrane permeabilization? Trends Cell Biol. 2008;18:157–164. doi: 10.1016/j.tcb.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dewson G, Kluck RM. Mechanisms by which Bak and Bax permeabilise mitochondria during apoptosis. J Cell Sci. 2009;122:2801–2808. doi: 10.1242/jcs.038166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Uren RT, Dewson G, Chen L, Coyne SC, Huang DC, Adams JM, et al. Mitochondrial permeabilization relies on BH3 ligands engaging multiple prosurvival Bcl-2 relatives, not Bak. J Cell Biol. 2007;177:277–287. doi: 10.1083/jcb.200606065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van Delft MF, Huang DC. How the Bcl-2 family of proteins interact to regulate apoptosis. Cell Res. 2006;16:203–213. doi: 10.1038/sj.cr.7310028. [DOI] [PubMed] [Google Scholar]
- 10.Willis SN, Fletcher JI, Kaufmann T, van Delft MF, Chen L, Czabotar PE, et al. Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science. 2007;315:856–859. doi: 10.1126/science.1133289. [DOI] [PubMed] [Google Scholar]
- 11.Youle RJ, Strasser A. The BCL-2 protein family: Opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- 12.Dewson G, Kratina T, Sim HW, Puthalakath H, Adams JM, Colman PM, et al. To trigger apoptosis, Bak exposes its BH3 domain and homodimerizes via BH3: Groove interactions. Mol Cell. 2008;30:369–380. doi: 10.1016/j.molcel.2008.04.005. [DOI] [PubMed] [Google Scholar]
- 13.Dewson G, Kratina T, Czabotar P, Day CL, Adams JM, Kluck RM. Bak activation for apoptosis involves oligomerization of dimers via their alpha6 helices. Mol Cell. 2009;36:696–703. doi: 10.1016/j.molcel.2009.11.008. [DOI] [PubMed] [Google Scholar]
- 14.Oh KJ, Singh P, Lee K, Foss K, Lee S, Park M, et al. Conformational changes in BAK, a pore-forming proapoptotic Bcl-2 family member, upon membrane insertion and direct evidence for the existence of BH3:BH3 contact interface in BAK homooligomers. J Biol Chem. 2010;285:28924–28937. doi: 10.1074/jbc.M110.135293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Desagher S, Osen-Sand A, Nichols A, Eskes R, Montessuit S, Lauper S, et al. Bid-induced conformational change of Bax is responsible for mitochondrial cytochrome c release during apoptosis. J Cell Biol. 1999;144:891–901. doi: 10.1083/jcb.144.5.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Griffiths GJ, Corfe BM, Savory P, Leech S, Esposti MD, Hickman JA, et al. Cellular damage signals promote sequential changes at the N-terminus and BH-1 domain of the pro-apoptotic protein Bak. Oncogene. 2001;20:7668–7676. doi: 10.1038/sj.onc.1204995. [DOI] [PubMed] [Google Scholar]
- 17.Griffiths GJ, Dubrez L, Morgan CP, Jones NA, Whitehouse J, Corfe BM, et al. Cell damage-induced conformational changes of the pro-apoptotic protein Bak in vivo precede the onset of apoptosis. J Cell Biol. 1999;144:903–914. doi: 10.1083/jcb.144.5.903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheng EH, Sheiko TV, Fisher JK, Craigen WJ, Korsmeyer SJ. VDAC2 inhibits BAK activation and mitochondrial apoptosis. Science. 2003;301:513–517. doi: 10.1126/science.1083995. [DOI] [PubMed] [Google Scholar]
- 19.Roy SS, Ehrlich AM, Craigen WJ, Hajnoczky G. VDAC2 is required for truncated BID-induced mitochondrial apoptosis by recruiting BAK to the mitochondria. EMBO Rep. 2009;10:1341–1347. doi: 10.1038/embor.2009.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lazarou M, Stojanovski D, Frazier AE, Kotevski A, Dewson G, Craigen WJ, et al. Inhibition of Bak activation by VDAC2 is dependent on the Bak transmembrane anchor. J Biol Chem. 2010;285:36876–36883. doi: 10.1074/jbc.M110.159301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jackson S, Harwood C, Thomas M, Banks L, Storey A. Role of Bak in UV-induced apoptosis in skin cancer and abrogation by HPV E6 proteins. Genes Dev. 2000;14:3065–3073. doi: 10.1101/gad.182100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Akgül B, Cooke JC, Storey A. HPV-associated skin disease. J Pathol. 2006;208:165–175. doi: 10.1002/path.1893. [DOI] [PubMed] [Google Scholar]
- 23.Leverrier S, Bergamaschi D, Ghali L, Ola A, Warnes G, Akgul B, et al. Role of HPV E6 proteins in preventing UVB-induced release of pro-apoptotic factors from the mitochondria. Apoptosis. 2007;12:549–560. doi: 10.1007/s10495-006-0004-1. [DOI] [PubMed] [Google Scholar]
- 24.Fox JL, Ismail F, Azad A, Ternette N, Leverrier S, Edelmann MJ, et al. Tyrosine dephosphorylation is required for Bak activation in apoptosis. EMBO J. 2010;29:3853–3868. doi: 10.1038/emboj.2010.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Labi V, Grespi F, Baumgartner F, Villunger A. Targeting the Bcl-2-regulated apoptosis pathway by BH3 mimetics: A breakthrough in anticancer therapy? Cell Death Differ. 2008;15:977–987. doi: 10.1038/cdd.2008.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gavathiotis E, Reyna DE, Davis ML, Bird GH, Walensky LD. BH3-triggered structural reorganization drives the activation of proapoptotic BAX. Mol Cell. 2010;40:481–492. doi: 10.1016/j.molcel.2010.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim H, Tu HC, Ren D, Takeuchi O, Jeffers JR, Zambetti GP, et al. Stepwise activation of BAX and BAK by tBID, BIM and PUMA initiates mitochondrial apoptosis. Mol Cell. 2009;36:487–499. doi: 10.1016/j.molcel.2009.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marchenko ND, Wolff S, Erster S, Becker K, Moll UM. Monoubiquitylation promotes mitochondrial p53 translocation. EMBO J. 2007;26:923–934. doi: 10.1038/sj.emboj.7601560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marchenko ND, Zaika A, Moll UM. Death signalinduced localization of p53 protein to mitochondria. A potential role in apoptotic signaling. J Biol Chem. 2000;275:16202–16212. doi: 10.1074/jbc.275.21.16202. [DOI] [PubMed] [Google Scholar]
- 30.Mihara M, Erster S, Zaika A, Petrenko O, Chittenden T, Pancoska P, et al. p53 has a direct apoptogenic role at the mitochondria. Mol Cell. 2003;11:577–590. doi: 10.1016/s1097-2765(03)00050-9. [DOI] [PubMed] [Google Scholar]
- 31.Mihara M, Moll UM. Detection of mitochondrial localization of p53. Methods Mol Biol. 2003;234:203–209. doi: 10.1385/1-59259-408-5:203. [DOI] [PubMed] [Google Scholar]
- 32.Moll UM, Wolff S, Speidel D, Deppert W. Transcription-independent pro-apoptotic functions of p53. Curr Opin Cell Biol. 2005;17:631–636. doi: 10.1016/j.ceb.2005.09.007. [DOI] [PubMed] [Google Scholar]
- 33.Pietsch EC, Perchiniak E, Canutescu AA, Wang G, Dunbrack RL, Murphy ME. Oligomerization of BAK by p53 utilizes conserved residues of the p53 DNA binding domain. J Biol Chem. 2008;283:21294–21304. doi: 10.1074/jbc.M710539200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moldoveanu T, Liu Q, Tocilj A, Watson M, Shore G, Gehring K. The X-ray structure of a BAK homodimer reveals an inhibitory zinc binding site. Mol Cell. 2006;24:677–688. doi: 10.1016/j.molcel.2006.10.014. [DOI] [PubMed] [Google Scholar]
- 35.Wang H, Takemoto C, Akasaka R, Uchikubo-Kamo T, Kishishita S, Murayama K, et al. Novel dimerization mode of the human Bcl-2 family protein Bak, a mitochondrial apoptosis regulator. J Struct Biol. 2009;166:32–37. doi: 10.1016/j.jsb.2008.12.003. [DOI] [PubMed] [Google Scholar]