

Abstract
Suppression of lipolysis by acipimox is known to improve insulin-stimulated glucose disposal, and this is an important phenomenon. The mechanism has been assumed to be an enhancement of glucose storage as glycogen, but no direct measurement has tested this concept or its possible relationship to the reported impairment in insulin-stimulated muscle ATP production. Isoglycaemic–hyperinsulinaemic clamps with [13C]glucose infusion were performed on Type 2 diabetic subjects and matched controls with measurement of glycogen synthesis by 13C MRS (magnetic resonance spectroscopy) of muscle. 31P saturation transfer MRS was used to quantify muscle ATP turnover rates. Glucose disposal rates were restored to near normal in diabetic subjects after acipimox (6.2±0.8 compared with 4.8±0.6 mg·kgffm−1·min−1; P<0.01; control 6.6±0.5 mg·kgffm−1·min−1; where ffm, is fat-free mass). The increment in muscle glycogen concentration was 2-fold higher in controls compared with the diabetic group, and acipimox administration to the diabetic group did not increase this (2.0±0.8 compared with 1.9±1.1 mmol/l; P<0.05; control, 4.0±0.8 mmol/l). ATP turnover rates did not increase during insulin stimulation in any group, but a modest decrease in the diabetes group was prevented by lowering plasma NEFAs (non-esterified fatty acids; 8.4±0.7 compared with 7.1±0.5 μmol·g−1·min−1; P<0.05; controls 8.6±0.8 μmol·g−1·min−1). Suppression of lipolysis increases whole-body glucose uptake with no increase in the rate of glucose storage as glycogen but with increase in whole-body glucose oxidation rate. ATP turnover rate in muscle exhibits no relationship to the acute metabolic effect of insulin.
Keywords: glucose disposal, lipolysis, magnetic resonance spectroscopy, muscle glycogen, non-esterified fatty acid, Type 2 diabetes
Abbreviations: APE, atom percentage excess; BMI, body mass index; CV, coefficient of variation; ffm, fat-free mass; MR, magnetic resonance; MRS, magnetic resonance spectroscopy; NEFA, non-esterified fatty acid
INTRODUCTION
Suppression of lipolysis by acipimox or nicotinic acid has long been known to improve meal tolerance and insulin sensitivity [1,2]. Sustained suppression of plasma NEFAs (non-esterified fatty acids) is associated with a decrease in intramuscular long-chain fatty acyl-CoAs, and it is believed that this enhances glucose metabolism via substrate competition [3]. The mechanism for this interaction is well described, involving control of cellular glucose uptake in muscle [4]. Both glucose oxidation and non-oxidative metabolism are involved, and as one of the original descriptions reported increase in the active form of glycogen synthase, it has been assumed that increased rates of glucose storage as glycogen underlie the phenomenon [2]. However, there is no direct in vivo evidence that glycogen synthesis rate is enhanced by NEFA suppression. This mechanism is important in fully understanding the insulin resistance of Type 2 diabetes and evaluating potential interventions.
Recently, demonstration of subnormal insulin stimulation of ATP turnover rates in Type 2 diabetes has been postulated as a basic defect which limits glucose metabolism [5–8]. It is not known whether such a defect underlies change in glucose uptake or glycogen synthesis itself. By employing 31P MRS (magnetic resonance spectroscopy) together with 13C spectroscopy to track glycogen metabolism, it is possible to examine this fundamental problem. In the present study, we tested the hypothesis that acute suppression of lipolysis would improve the rate of glucose metabolism specifically by increasing muscle glycogen synthesis. The effect of decreasing fatty acid availability upon muscle mitochondrial ATP turnover rates was also examined.
MATERIALS AND METHODS
Subjects
Ten subjects with well-controlled Type 2 diabetes (three females and seven males) and eight physical activity-, age- and BMI (body mass index)-matched normal glucose-tolerant subjects (two females and six males) were studied (Table 1). All subjects were recruited by means of advertisement and underwent a complete medical history, clinical examination and laboratory tests to exclude hepatic and renal diseases. Patients with diabetes on insulin treatment or any oral hypoglycaemic medications apart from metformin were excluded. The individuals in the control group had neither a family history of diabetes nor were taking any medication known to affect glucose tolerance or insulin sensitivity. All subjects with diabetes and no control subjects were taking statins. Normal glucose metabolism was confirmed by a standard oral 75-g glucose tolerance test. None of the subjects performed moderate or intense exercise on a regular basis. Physical activity was assessed over 3 days using the Body Monitoring System and SenseWear Armband (BodyMedia®), which provides a measure of total daily energy expenditure and number of steps taken per day [9]. The research was carried out in accordance with the Declaration of Helsinki (2000), and the study protocol was approved by the Newcastle upon Tyne Ethics Committee No. 2. Informed consent was obtained from all subjects.
Table 1. Characteristics of subjects.
Values are means±S.E.M. HbA1c, glycated haemoglobin; NS, not significant. 1 cal≈4.184 J.
| Characteristic | Type 2 diabetic group | Control group | P value |
|---|---|---|---|
| Gender (male/female) (n) | 7/3 | 6/2 | |
| Age (years) | 57±2 | 53±3 | NS |
| BMI (kg/m2) | 28.7±1.2 | 28.1±1.1 | NS |
| Fat mass (kg) | 26.1±2.0 | 26.4±1.7 | NS |
| Fat-free mass (kg) | 54.7±3.8 | 58.5±3.6 | NS |
| Fasting glucose (mmol/l) | 7.7±0.3 | 5.1±0.1 | 0.001 |
| Fasting insulin (pmol/l) | 93±14 | 49±6 | 0.026 |
| Mean HbA1c (%) | 6.6±0.2 | 5.4±0.1 | 0.001 |
| Fasting triacylglycerol (mmol/l) | 1.6±0.2 | 1.4±0.2 | NS |
| Average daily energy expenditure (cal) | 2455±198 | 2248±76 | NS |
| Average daily steps taken (n) | 6160±385 | 5701±288 | NS |
Table 2. Plasma enrichment of [13C]glucose (APE).
Values are means±S.E.M.
| Plasma enrichment of [13C]glucose (APE) | ||||||
|---|---|---|---|---|---|---|
| Group | Time (min)… | 0 | 15 | 45 | 90 | 150 |
| Control | 0 | 3.79±0.37 | 7.50±0.72 | 12.31±0.66 | 15.15±0.70 | |
| Diabetes/placebo | 0 | 2.99±0.44 | 6.34±0.50 | 9.65±0.60 | 12.16±0.57 | |
| Diabetes/acipimox | 0 | 3.81±0.40 | 7.99±0.45 | 11.33±0.60 | 14.18±0.63 | |
Experimental protocol
All subjects refrained from planned physical exertion during the 3 days preceding the studies and fasted overnight for 12 h before the experiments. Metformin was withdrawn 3 days before each experiment. Type 2 diabetic subjects were each studied on two days, 4–8 weeks apart. Doses of acipimox (250 mg) were given at baseline and 3 h later, or on the placebo test day, identical placebo tablets were given in a double blind fashion. The body weight and lifestyle of the subjects remained unchanged during the period of the study. Control subjects were studied on 1 day only with administration of placebo tablets.
Isoglycaemic–hyperinsulinaemic clamps
For all experiments, subjects travelled to the MR (magnetic resonance) Centre by taxi and were transported within the centre by wheelchair. At 08:30 h (−270 min), one cannula was inserted into an antecubital vein for administration of glucose and insulin. A second cannula was inserted into the contralateral wrist vein for blood sampling. Use of a hand-warming device ensured arterialization of venous blood. Isoglycaemia was maintained in order to ensure that the true basal condition of each participant could be observed, and the entire study was carried out with the subject lying in the MR scanner. Isoglycaemic–hyperinsulinaemia was maintained for 150 min using the clamp technique [10]. Insulin (Actrapid; NovoNordisk) was administered as a primed continuous infusion (40 m-units·m−2·min−1). To inhibit pancreatic hormone secretion, somatostatin was infused at 0.06 μg·kg−1 of body weight·min−1 (Somatostatin-UCB; UCB-Pharma) from 5 min before the start of the insulin infusion and continued for the duration of the clamp. In order to increase sensitivity of measurement of the glycogen synthesis rate by 13C MRS, the variable glucose infusion contained 20% [1-13C]glucose (Cambridge Isotope Laboratories). Whole-body insulin sensitivity was determined from calculated whole-body glucose disposal during the last 30 min of the hyperinsulinaemic glucose clamp [10]. Whole-body glucose disposal was calculated from glucose infusion rate [11]. To assess rate of oxidation of infused glucose, breath samples for 13CO2 measurement were obtained just before the start of the clamp (0 min), at 15 min, 45 min, 90 min and at the end of the clamp (150 min). As this measure would be affected by differences in plasma glucose enrichment, the index of whole-body glucose oxidation was calculated as the ratio of breath to plasma 13C APE (atom percentage excess) [(breath APE/plasma APE)×100].
MRS examinations
MR data were acquired using a 3T Achieva scanner (Philips) with an in-built body coil used for imaging. A 14-cm diameter surface coil was used for phosphorus spectroscopy, and a 6-cm diameter 13C coil with an integral quad 1H decoupling coil (PulseTeq) was used for 13C spectroscopy. Subjects remained supine inside the MR spectrometer with each coil positioned beneath the widest part of the left calf during each investigation. The coil position was marked on the leg with indelible ink. Scout images were acquired to ensure identical coil positioning on repeat scans. To prevent movement during each study, the coil was secured in place using webbing straps around the calf. Analysis of all spectra was performed with jMRUI (version 3.0) [12] using the AMARES fitting algorithm [13].
31P MRS was carried out as described previously [14] and was acquired at baseline from −240 to −210 min and from −60 to −30 min and twice further during the isoglycaemic–hyperglycaemic clamp from 15 to 45 min and from 90 to 120 min. For 13C MRS, the distance between the 13C/1H decoupling coil surface and the muscle was recorded from scout images. The 13C pulse power was calibrated to a nominal value of 80° in the tissue of interest by observing the power-dependent variation in signal from a fiducial marker located in the coil housing, containing a sample exhibiting a 13C signal with short T1 (213 mM [2-13C]acetone and 25 mM GdCl3 in water). Spectra showing the glycogen 1-13C resonance were acquired using a non-localized 1H-decoupled 13C pulse-acquire sequence [TR (relaxation time)=200 ms, spectral width=8 kHz, 3000 averages, WALTZ decoupling, nominal tip angle=80°] over a 15-min acquisition time. Calibration of 13C spectra was performed by comparison of in vivo glycogen 1-13C signal amplitudes with that of a standard glycogen solution (100 mM glycogen, 70 mM KCl and 0.05% sodium azide). Quantification was performed by comparison of signal amplitude with those from a leg-shaped phantom acquired at a range of separations between coil and phantom, representative of the separation between coil and muscle due to skin and subcutaneous fat. The same coils, pulse sequences and tip angles as employed for in vivo spectra were used for acquisition of these calibration data. 13C MRS spectra were acquired from subjects at baseline from −205 to −185 min and from −25 to −5 min and twice further during the isoglycaemic–hyperinsulinaemic clamp from 50 to 70 min and from 130 to 150 min. The concentration of muscle glycogen at baseline, [Glyc]muscle, was calculated using the equation:
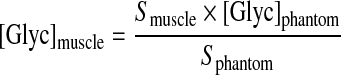 |
where Sphantom and Smuscle are the signal intensities arising from glycogen in the phantom and muscle, respectively, and [Glyc]phantom is the concentration of glycogen in the phantom (100 mmol/l). The increments in muscle glycogen concentration at 70 and 150 min of the clamp, [ΔGlyc70] and [ΔGlyc150], respectively, were calculated from the equation reported previously [15]:
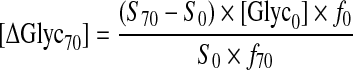 |
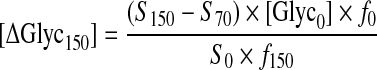 |
where S0, S70 and S150 represent the signal intensity of [13C]glycogen at 0, 70 and 150 min, [Glyc0] is the basal glycogen concentration in mmol/l. f0 corresponds to the natural abundance enrichment of [13C]glycogen at baseline (1.1%), and f70 and f150 correspond to the mean percentage 13C enrichment of plasma glucose at 70 and 150 min, respectively. Each increment was then added to the previous concentration and the slope calculated by linear regression analysis to yield the rate of glycogen synthesis [16].
Analytical techniques
Plasma glucose concentration was measured by the glucose oxidase method (YSI glucose analyser). The 13C enrichment in plasma glucose was determined by gas chromatography–MS of the pentaacetate derivatives of plasma glucose after deproteinization and deionization as previously described [17]. Plasma insulin levels were determined using ELISA kits (Dako). The plasma NEFA concentration was measured on a Roche Cobas centrifugal analyser using a Wako kit (Wako Chemicals).
Breath 13C enrichments
Breath samples for 13C enrichments were collected at five time points during the study. The subjects were asked to exhale fully through a short straw into a glass tube (Labco Exetainer). The tube was immediately stoppered. 13C enrichment of breath samples was determined by continuous flow isotope ratio MS (ABCA system; PDZ Europa). The CV (coefficient of variation) for the analysis was 0.07% and CV for the collection was 0.3%. All results of the 13C enrichment of expired air measurements are expressed as APE.
Statistical analysis
Results are presented as means±S.E.M., unless otherwise stated. Statistical analyses were performed using SPSS 15.0 software. Statistical comparisons between diabetic and control groups were performed using a Student's t test, whereas the within-group differences were determined using paired Student's t test where appropriate. Changes of sequential data within experiments were evaluated by repeated-measures ANOVA with post hoc Tukey correction where appropriate. Statistical significance was accepted at P<0.05.
RESULTS
Plasma glucose, insulin and NEFA concentrations
The experimental conditions necessary to test the hypothesis were achieved. During the 3-h baseline observation, plasma glucose concentration remained steady in the control subjects (5.1±0.1 to 5.0±0.1 mmol/l) and gradually fell as expected in the diabetic subjects (7.7±0.3 to 6.5±0.3 mmol/l; P=0.002). As per protocol, plasma glucose concentration was clamped at 5.0±0.1 mmol/l in the control group, 6.6±0.2 mmol/l in the diabetes/placebo group and 6.4±0.2 mmol/l in the diabetes/acipimox group respectively (P<0.001 diabetes compared with control; Figure 1A). Clamp plasma insulin concentrations were stable at 573±18 pmol/l (Figure 1B). Plasma NEFA concentrations rapidly suppressed below 0.2 mmol/l in the diabetes/acipimox study (P=0.005) and were stable and similar in diabetes/placebo and control groups until commencement of insulin infusion (Figure 1C).
Figure 1. Time course of plasma glucose (A), insulin (B) and NEFA (C) concentrations during the isoglycaemic–hyperinsulinaemic clamps in the control, diabetes/placebo and diabetes/acipimox groups.

Values are means±S.E.M. (○) control group; (Δ) diabetes/placebo group; (▲) diabetes/acipimox group. *P<0.05 for diabetes compared with control.
Glucose disposal rates and muscle glycogen concentrations
During the clamp, the glucose disposal rate was lower in the diabetes/placebo group compared with the control group (4.8±0.6 compared with 6.6±0.5 mg·kgffm−1·min−1, P=0.04; Figure 2A; where ffm, is fat-free mass). Prior suppression of plasma NEFAs by acipimox brought about a 23% increase in the glucose disposal rate in the Type 2 diabetic group (6.2±0.8 compared with 4.8±0.6 mg·kgffm−1·min−1, acipimox compared with placebo respectively, P=0.005; Figure 2A), so that it approached that of the control group (6.6±0.5 mg·kgffm−1·min−1; P=0.72). The fasting muscle glycogen concentration was similar in all three groups (diabetes/placebo, 67.5±4.5 mmol/l; diabetes/acipimox, 69.0±2.1 mmol/l; and control, 71.1±2.6 mmol/l) and remained unchanged during the basal period. The increment in muscle glycogen concentration after 70 min of insulin stimulation was small and not significantly different between the three groups. By the end of the clamp, insulin-stimulated muscle glycogen concentration increased by +4.0±0.8 mmol/l in the control group and +1.9±1.0 mmol/l in the diabetes/placebo group (P<0.05; Figure 2B). Prior administration of acipimox did not change the increment in muscle glycogen (+2.0±0.8 mmol/l; Figure 2B). The mean rate of muscle glycogen synthesis from 70 to 150 min was 40±7 μmol·l−1·min−1 in the control subjects, 19±9 μmol·l−1·min−1 in the diabetes/placebo group and 22±7 μmol·l−1·min−1 in the diabetes/acipimox group (P<0.05 for control compared with both diabetes groups).
Figure 2. Glucose infusion rates (A), incremental changes in muscle glycogen concentration (B) and the ratio of breath 13CO2 to plasma [13C]glucose APE (C) in the control, diabetes/placebo and diabetes/acipimox groups.

(A) Glucose infusion rates during the final 30 min in the isoglycaemic–hyperinsulinaemic clamps in the control, diabetes/placebo (D/Placebo) and diabetes/acipimox (D/Acipimox) groups. (B) Incremental changes in muscle glycogen concentration in the three studies. (C) Ratio of breath 13CO2 to plasma [13C]glucose APE at the end of the each study. *P<0.05 and **P<0.005.
Breath 13C enrichments
13C APE in expired breath increased during the clamp in all groups (control, 0.23±0.04 and 0.44±0.05; diabetes/placebo, 0.14±0.01 and 0.31±0.03; diabetes/acipimox, 0.22±0.02 and 0.44±0.03 at 90 and 150 min respectively). In order to compare rates of glucose oxidation corrected for plasma glucose enrichment, the ratio of breath to plasma 13C APE was examined. At the end of the clamp period, this index of glucose oxidation was 2.53±0.17 compared with 3.16±0.21 for diabetes/placebo and diabetes/acipimox respectively (P<0.005), and 2.87±0.24 for controls (Figure 2C).
Muscle ATP turnover rate
Baseline ATP turnover rate in the diabetes/placebo group (8.6±0.8 μmol·g−1 of muscle·min−1) was similar to that of the control subjects (8.6±0.7 μmol·g−1 of muscle·min−1; Figure 3). Muscle ATP turnover rate in the control group did not change during the basal period or during the insulin infusion (end of clamp: 8.6±1.3 μmol·g−1 of muscle·min−1). A gradual decline in ATP turnover rate was observed during the course of the diabetes/placebo group. This decline was not seen in the diabetes/acipimox group such that by the end of the clamp, ATP turnover rate was significantly higher than in the diabetes/placebo group (8.4±0.7 compared with 7.1±0.5 μmol·g−1 of muscle ·min−1; P=0.02).
Figure 3. ATP turnover rate in the control, diabetes/placebo and diabetes/acipimox groups during basal period and after 120 min of isoglycaemic–hyperinsulinaemia.

White bar, control group; hatched bar, diabetes/placebo group; black bar, diabetes/acipimox group. *P<0.05.
DISCUSSION
The results of the present study demonstrate that prior suppression of lipolysis effectively normalizes glucose infusion rate during hyperinsulinaemia in Type 2 diabetes, but has no effect upon the rate of muscle glycogen synthesis. In contrast, the subnormal rate of whole-body glucose oxidation in Type 2 diabetes was increased. The muscle ATP turnover rate was found to be normal in Type 2 diabetes but could be modulated to a degree by suppression of lipolysis.
In Type 2 diabetes, insulin-stimulated whole-body glucose uptake was observed to be associated with a 2-fold lower incorporation of [13C]glucose into glycogen. Acipimox brought about a 23% increase in glucose infusion rate during hyperinsulinaemia, similar to that reported by others [2,3]. Previously observed increases in glucose disposal rates during acipimox administration have been attributed to change in rate of glycogen synthesis [18,19]. Clearly, this was not the case over the 6 h of NEFA suppression in the present study, a period which has been shown to be sufficient for the effect upon glucose metabolism to be maximal [4]. The concept that the synthesis rate of glycogen would be increased under such circumstances derives from observation of an increase in the active form of muscle glycogen synthase, even though no change in muscle glycogen concentrations could be detected [2]. Subsequently, it has been demonstrated that change in activation state of glycogen synthase by suppression of lipolysis occurs in the absence of change in the proximal steps of insulin activation of glycogen synthase [GSK3β (glycogen synthase kinase 3β) and Akt Thr308] [19], and its relevance to substrate flux must be questioned.
Glucose infusion rate is a useful independent measure of overall glucose disposal, but does not take into account possible incomplete suppression of hepatic glucose production during the clamp. An estimate of endogenous glucose production can be obtained from the measured isotopic enrichments of infusate and plasma together with glucose infusion rate [20]. Hepatic glucose production was 0.01, 1.13 and 0.39 mg·kgffm−1·min−1 in control, diabetes/placebo and diabetes/acipimox groups. By adding this value to the glucose infusion rate, total glucose disposal rates can be estimated to be 6.61, 5.93 and 6.59 mg·kgffm−1·min−1 respectively. Hence acipimox brought about an 11% increase in total glucose disposal. Isoglycaemic–hyperinsulinaemic clamps were used in the present study in order to observe the physiological mechanisms operating at ambient plasma glucose concentrations in the diabetic and control subjects, and in respect of the primary comparison between acipimox and placebo studies in the diabetic subjects, clamp glucose levels were similar.
Whole-body glucose oxidation rate was increased approximately 25% by acipimox administration. This could not be quantified using indirect calorimetry in the present study as subjects remained in the 3T magnet throughout the test periods. However, the extent of acipimox-induced change in glucose oxidation has previously been shown to be approximately one-third of the simultaneous change in rate of whole-body glucose uptake [2]. It is likely that processes additional to glucose oxidation and muscle glycogen synthesis could explain the change in whole-body glucose uptake. Glycolysis with lactate release (Cori cycle activity) could account for some of the change. Approximately 15% of a glucose load is taken up by skeletal muscle then released as lactate in normal health [21] and accounts for approximately 45% of systemic lactate appearance [22]. In Type 2 diabetes, the proportion of glucose taken up by muscle then released as lactate is 2–3-fold higher than normal during hyperinsulinaemia [23,24]. This flux could be affected if early steps in glucose metabolism were rate limiting, so that the increased transport of glucose into the muscle cell by low fatty acid availability caused increased export from muscle as lactate, and an 18% increase in plasma lactate has been observed to accompany acipimox-induced suppression of lipolysis during a clamp [25]. Further work is required to examine specifically the acute changes in glycolysis and glucose oxidation in response to change in NEFA supply.
Although the change in plasma NEFAs is a useful general indicator of rate of lipolysis, suppression of lipolysis would be expected to exert greatest effect upon muscle metabolism by preventing breakdown of intramyocellular triacylglycerol (triglyceride) and decreasing intracellular availability of fatty acid and diacylglycerols. Evidence is accumulating for diacylglycerols to be major modulators of substrate level control of metabolism [26,27]. In the present study, the overweight control subjects were not restudied with acipimox, and although it would appear likely that similar effects would be produced, this awaits experimental verification.
No significant abnormality in muscle ATP turnover rate in overnight fasting Type 2 diabetic subjects in comparison with exercise-, weight-, age- and sex-matched non-diabetic subjects was observed in the present study, and this contrasts with reports of decreased basal mitochondrial function in Type 2 diabetes [28,29]. Matching for daily physical activity may be of importance, as the greatest deficits in ATP turnover rates have been reported in a study which explicitly compared extremes of insulin sensitivity with no matching for habitual physical activity [8]. Both basal ATP turnover rates and the independently assessed phosphocreatine recovery times have been reported to be normal in Type 2 diabetes when compared with exercise-matched controls [30,31]. However, the major reason for the apparent discrepancy is clear from the work of Schrauwen-Hinderling and co-workers [28,29], who demonstrated that in vivo basal mitochondrial function was related to fasting blood glucose, with normal values for diabetic subjects at levels similar to those reported in the present study. More recent work from the same group demonstrated that change in metabolic state alters measured mitochondrial function [32]. In the Type 2 diabetic subjects given placebo, a fall in muscle ATP turnover rate was observed during the fasting fall in plasma glucose concentration. It is possible that the downward trend occurred with increasing duration of fasting and consequent increased mobilization of fatty acids from IMCL (intramyocellular lipid) as acipimox administration prevented the fall in ATP turnover rate and would have inhibited intracellular lipolysis. This phenomenon requires further study.
The present study was designed in the expectation of inducing an increase in ATP turnover rate with insulin in the non-diabetic control subjects, as it was believed that increase in ATP turnover rates would precede or accompany the acute changes in cellular metabolism. We observed no effect of insulin upon ATP turnover rates. However, the previously reported insulin effect on mitochondrial function related to measurements averaged over 150 to 350 min of insulin stimulation [7,33–35]. The method of quantifying ATP turnover used in the present study was optimized so that measurements could be carried out frequently over 30-min periods for the first time. When it became clear that there was no stimulation of ATP turnover rates in the matched control subjects of the present study, a separate investigation of time course of stimulation of muscle ATP turnover rates was conducted and has been published [14]. We observed no change in ATP turnover rate during the first 45 min of insulin infusion when the change in glucose metabolism was maximal, yet in young insulin-sensitive subjects, there was a slow steady increase evident after 2 h. This substudy concluded that the acute metabolic effect of insulin does not depend upon measureable increases in ATP turnover rate. Processes separate from the insulin effect on glucose metabolism, such as the insulin effect on mitochondrial fusion and proliferation [36,37] or mitochondrial protein synthesis [38], may affect ATP turnover rate on a timescale of several hours of insulin stimulation. These processes are temporally dissociated from and not relevant to the early metabolic effects of insulin and led to the belief that insulin acutely changes ATP turnover. However, suppression of plasma NEFAs in the Type 2 diabetic subjects during the 5.5-h protocol did modestly affect the rate of ATP turnover, and the converse finding of elevation of plasma NEFAs and suppression of ATP turnover has been reported previously [34]. Other recent work has demonstrated decreased glucose disposal during short-term elevation of plasma NEFAs without change in ATP turnover rate in healthy subjects [35], and the time course of change in fatty acid supply requires further study. Overall, change in metabolic state rather than diabetes can affect measured ATP turnover rates in muscle.
As all subjects with Type 2 diabetes were taking statin therapy, reflecting current clinical practice, the possible effect of this on glucose metabolism must be considered. Statins do not affect glucose tolerance nor modulate 72-h glucose profiles, but may bring about a very small beneficial effect on insulin sensitivity, as shown in a randomized crossover study designed for this purpose [39]. No impact of statins would be expected on the primary comparison of acipimox or placebo exposure within the diabetic group.
The results of the present study demonstrate that lowering fatty acid availability increased whole-body glucose uptake without increasing the rate of glucose storage as glycogen, but with increase in whole-body glucose oxidation rates. A change in fasting ATP turnover rate in muscle was observed after suppression of lipolysis in Type 2 diabetes, but this change was associated with no effect on muscle glycogen synthesis.
AUTHOR CONTRIBUTION
Ee Lim researched the data, contributed to the discussion and wrote the paper. Kieren Hollingsworth researched the data, contributed to the discussion and reviewed/edited the paper prior to submission. Fiona Smith researched the data and reviewed/edited the manuscript prior to submission. Peter Thelwall contributed to the discussion and reviewed/edited the manuscript prior to submission. Roy Taylor researched the data, contributed to the discussion and reviewed/edited the manuscript prior to submission.
ACKNOWLEDGEMENTS
We are most grateful to the volunteers for their commitment during the study. We thank Mrs Jean Gerrard and Ms Mei Jun Chen, senior research nurses, for their assistance during the clamps, Ms Louis Morris and Mrs Carol Smith, senior radiographers, and Mrs Heather Cook and Mrs Annette Lane for laboratory assistance.
FUNDING
The work was supported by the Wellcome Trust [grant number 073561] and the Newcastle upon Tyne MRC Biomedical Research Centre.
References
- 1.Fulcher G. R., Walker M., Catalano C., Agius L., Alberti K. G. M. M. Metabolic effects of suppressions of non-esterified fatty acid levels with acipimox in obese NIDDM subjects. Diabetes. 1992;41:1400–1408. doi: 10.2337/diab.41.11.1400. [DOI] [PubMed] [Google Scholar]
- 2.Vaag A., Skott P., Damsbo P., Gall M. A., Richter E. A., Beck-Nielsen H. Effect of the antilipolytic nicotinic acid analogue acipimox on whole-body and skeletal muscle glucose metabolism in patients with non-insulin-dependent diabetes mellitus. J. Clin. Invest. 1991;88:1282–1290. doi: 10.1172/JCI115432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bajaj M., Suraamornkul S., Romanelli A., Cline G. W., Mandarino L. J., Shulman G. I., DeFronzo R. A. Effect of a sustained reduction in plasma free fatty acid concentration on intramuscular long-chain fatty acyl-CoAs and insulin action in type 2 diabetic patients. Diabetes. 2005;54:3148–3153. doi: 10.2337/diabetes.54.11.3148. [DOI] [PubMed] [Google Scholar]
- 4.Roden M., Price T. B., Perseghin G., Petersen K. F., Rothman D. L., Cline G. W., Shulman G. I. Mechanism of free fatty acid-induced insulin resistance in humans. J. Clin. Invest. 1996;97:2859–2865. doi: 10.1172/JCI118742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kelley D. E., He J., Menshikova E. V., Ritov V. B. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes. 2002;51:2944–2950. doi: 10.2337/diabetes.51.10.2944. [DOI] [PubMed] [Google Scholar]
- 6.Ritov V. B., Menshikova E. V., He J., Ferrell R. E., Goodpaster B. H., Kelley D. E. Deficiency of subsarcolemmal mitochondria in obesity and type 2 diabetes. Diabetes. 2005;54:8–14. doi: 10.2337/diabetes.54.1.8. [DOI] [PubMed] [Google Scholar]
- 7.Szendroedi J., Schmid A. I., Chmelik M., Toth C., Brehm A., Krssak M., Nowotny P., Wolzt M., Waldhausl W., Roden M. Muscle mitochondrial ATP synthesis and glucose transport/phosphorylation in type 2 diabetes. PLoS Med. 2007;4:e154. doi: 10.1371/journal.pmed.0040154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Petersen K. F., Dufour S., Befroy D., Garcia R., Shulman G. I. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N. Engl. J. Med. 2004;350:664–671. doi: 10.1056/NEJMoa031314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mignault D., St-Onge M., Karelis A. D., Allison D. B., Rabasa-Lhoret R. Evaluation of the Portable HealthWear Armband: a device to measure total daily energy expenditure in free-living type 2 diabetic individuals. Diabetes Care. 2005;28:225–227. doi: 10.2337/diacare.28.1.225-a. [DOI] [PubMed] [Google Scholar]
- 10.DeFronzo R. A., Tobin J. D., Andres R. Glucose clamp technique: a method for quantifying insulin secretion and resistance. Am. J. Physiol. 1979;237:E214–E223. doi: 10.1152/ajpendo.1979.237.3.E214. [DOI] [PubMed] [Google Scholar]
- 11.Rizza R. A., Mandarino L. J., Gerich J. E. Dose–response characteristics for effects of insulin on production and utilization of glucose in man. Am. J. Physiol. 1981;240:E630–E639. doi: 10.1152/ajpendo.1981.240.6.E630. [DOI] [PubMed] [Google Scholar]
- 12.Naressi A., Couturier C., Castang I., de Beer R., Graveron-Demilly D. Java-based graphical user interface for MRUI, a software package for quantitation of in vivo/medical magnetic resonance spectroscopy signals. Comput. Biol. Med. 2001;31:269–286. doi: 10.1016/s0010-4825(01)00006-3. [DOI] [PubMed] [Google Scholar]
- 13.Vanhamme L., Van Huffel S., Van Hecke P., van Ormondt D. Time-domain quantification of series of biomedical magnetic resonance spectroscopy signals. J. Magn. Reson. 1999;140:120–130. doi: 10.1006/jmre.1999.1835. [DOI] [PubMed] [Google Scholar]
- 14.Lim E. L., Hollingsworth K. G., Thelwall P. E., Taylor R. Measuring the acute effect of insulin infusion on ATP turnover rate in human skeletal muscle using phosphorus-31 magnetic resonance saturation transfer spectroscopy. NMR Biomed. 2010;23:952–957. doi: 10.1002/nbm.1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jue T., Rothman D. L., Shulman G. I., Tavitian B. A., DeFronzo R. A., Shulman R. G. Direct observation of glycogen synthesis in human muscle with 13C NMR. Proc. Natl. Acad. Sci. U.S.A. 1989;86:4489–4491. doi: 10.1073/pnas.86.12.4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shulman G. I., Rothman D. L., Jue T., Stein P., DeFronzo R. A., Shulman R. G. Quantitation of muscle glycogen synthesis in normal subjects and subjects with non-insulin-dependent diabetes by 13C nuclear magnetic resonance spectroscopy. N. Engl. J. Med. 1990;322:223–228. doi: 10.1056/NEJM199001253220403. [DOI] [PubMed] [Google Scholar]
- 17.Wolfe R. R. Tracers in metabolic research: radioisotope and stable isotope/mass spectrometry methods. Lab. Res. Methods Biol. Med. 1984;9:1–287. [PubMed] [Google Scholar]
- 18.Vaag A., Skott P., Damsbo P., Gall M. A., Richter E. A., Beck-Nielsen H. Effect of the antilipolytic nicotinic acid analogue acipimox on whole-body and skeletal muscle glucose metabolism in patients with non-insulin-dependent diabetes mellitus. J. Clin. Invest. 1991;88:1282–1290. doi: 10.1172/JCI115432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lindegaard B., Frosig C., Petersen A. M., Plomgaard P., Ditlevsen S., Mittendorfer B., Van Hall G., Wojtaszewski J. F., Pedersen B. K. Inhibition of lipolysis stimulates peripheral glucose uptake but has no effect on endogenous glucose production in HIV lipodystrophy. Diabetes. 2007;56:2070–2077. doi: 10.2337/db07-0144. [DOI] [PubMed] [Google Scholar]
- 20.Ravikumar B., Gerrard J., Dalla Man C., Firbank M. J., Lane A., English P. T., Cobelli C., Taylor R. Pioglitazone decreases fasting and postprandial endogenous glucose production in proportion to decrease in hepatic triglyceride content. Diabetes. 2008;57:2288–2295. doi: 10.2337/db07-1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kelley D., Mitrakou A., Schwenk F., Benn J., Sonnenberg G., Arcangell M., Aoki T., Sorensen J., Berger M., Sonksen P., Gerich J. Skeletal muscle glycolysis, oxidation, and storage of an oral glucose load. J. Clin. Invest. 1988;81:1563–1571. doi: 10.1172/JCI113489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Consoli A., Nurjahan N., Gerich J. E., Mandarino L. J. Skeletal muscle is a major site of lactate uptake and release during hyperinsulinemia. Metab. Clin. Exp. 1992;41:176–179. doi: 10.1016/0026-0495(92)90148-4. [DOI] [PubMed] [Google Scholar]
- 23.Mitrakou A., Kelley D., Veneman T., Jenssen T., Pangburn T., Reilly J., Gerich J. Contribution of abnormal muscle and liver glucose metabolism to postprandial hyperglycemia in NIDDM. Diabetes. 1990;39:1381–1390. doi: 10.2337/diab.39.11.1381. [DOI] [PubMed] [Google Scholar]
- 24.Capaldo B., Napoli R., Di Bonito P., Albano G., Sacca L. Glucose and gluconeogenic substrate exchange by the forearm skeletal muscle in hyperglycemic and insulin-treated type II diabetic patients. J. Clin. Endocrinol. Metab. 1990;71:1220–1223. doi: 10.1210/jcem-71-5-1220. [DOI] [PubMed] [Google Scholar]
- 25.Vaag A., Henriksen J. E., Beck-Nielsen H. Defective insulin activation of glycogen synthase in skeletal muscle in first degree relatives to patients with type 2 (non-insulin dependent) diabetes mellitus. Diabetologia. 1991;34(Suppl. 2):A70. [Google Scholar]
- 26.Chibalin A. V., Leng Y., Vieira E., Krook A., Bjornholm M., Long Y. C., Kotova O., Zhong Z., Sakane F., Steiler T., et al. Downregulation of diacylglycerol kinase δ contributes to hyperglycemia-induced insulin resistance. Cell. 2008;132:375–386. doi: 10.1016/j.cell.2007.12.035. [DOI] [PubMed] [Google Scholar]
- 27.Erion D. M., Shulman G. I. Diacylglycerol-mediated insulin resistance. Nat. Med. 2010;16:400–402. doi: 10.1038/nm0410-400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schrauwen-Hinderling V. B., Kooi M. E., Hesselink M. K., Jeneson J. A., Backes W. H., van Echteld C. J., van Engelshoven J. M., Mensink M., Schrauwen P. Impaired in vivo mitochondrial function but similar intramyocellular lipid content in patients with type 2 diabetes mellitus and BMI-matched control subjects. Diabetologia. 2007;50:113–120. doi: 10.1007/s00125-006-0475-1. [DOI] [PubMed] [Google Scholar]
- 29.Phielix E., Schrauwen-Hinderling V. B., Mensink M., Lenaers E., Meex R., Hoeks J., Kooi M. E., Moonen-Kornips E., Sels J. P., Hesselink M. K., Schrauwen P. Lower intrinsic ADP-stimulated mitochondrial respiration underlies in vivo mitochondrial dysfunction in muscle of male type 2 diabetic patients. Diabetes. 2008;57:2943–2949. doi: 10.2337/db08-0391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Trenell M. I., Hollingsworth K. G., Lim E. L., Taylor R. Increased daily walking improves lipid oxidation without changes in mitochondrial function in type 2 diabetes. Diabetes Care. 2008;31:1644–1649. doi: 10.2337/dc08-0303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.De Feyter H. M., van den Broek N. M., Praet S. F., Nicolay K., van Loon L. J., Prompers J. J. Early or advanced stage type 2 diabetes is not accompanied by in vivo skeletal muscle mitochondrial dysfunction. Eur. J. Endocrinol. 2008;158:643–653. doi: 10.1530/EJE-07-0756. [DOI] [PubMed] [Google Scholar]
- 32.Hoeks J., van Herpen N. A., Mensink M., Moonen-Kornips E., van Beurden D., Hesselink M. K., Schrauwen P. Prolonged fasting identifies skeletal muscle mitochondrial dysfunction as consequence rather than cause of human insulin resistance. Diabetes. 2010;59:2117–2125. doi: 10.2337/db10-0519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Petersen K. F., Dufour S., Shulman G. I. Decreased insulin-stimulated ATP synthesis and phosphate transport in muscle of insulin-resistant offspring of type 2 diabetic parents. PLoS Med. 2005;2:e233. doi: 10.1371/journal.pmed.0020233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brehm A., Krssak M., Schmid A. I., Nowotny P., Waldhausl W., Roden M. Increased lipid availability impairs insulin-stimulated ATP synthesis in human skeletal muscle. Diabetes. 2006;55:136–140. [PubMed] [Google Scholar]
- 35.Brehm A., Krssak M., Schmid A. I., Nowotny P., Waldhausl W., Roden M. Acute elevation of plasma lipids does not affect ATP synthesis in human skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2010;299:E33–E38. doi: 10.1152/ajpendo.00756.2009. [DOI] [PubMed] [Google Scholar]
- 36.Molina A. J., Wikstrom J. D., Stiles L., Las G., Mohamed H., Elorza A., Walzer G., Twig G., Katz S., Corkey B. E., Shirihai O. S. Mitochondrial networking protects β-cells from nutrient-induced apoptosis. Diabetes. 2009;58:2303–2315. doi: 10.2337/db07-1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang J. Y., Yeh H. Y., Lin K., Wang P. H. Insulin stimulates Akt translocation to mitochondria: implications on dysregulation of mitochondrial oxidative phosphorylation in diabetic myocardium. J. Mol. Cell. Cardiol. 2009;46:919–926. doi: 10.1016/j.yjmcc.2009.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stump C. S., Short K. R., Bigelow M. L., Schimke J. M., Nair K. S. Effect of insulin on human skeletal muscle mitochondrial ATP production, protein synthesis, and mRNA transcripts. Proc. Natl. Acad. Sci. U.S.A. 2003;100:7996–8001. doi: 10.1073/pnas.1332551100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huptas S., Geiss H. C., Otto C., Parhofer K. G. Effect of atorvastatin (10 mg/day) on glucose metabolism in patients with the metabolic syndrome. Am. J. Cardiol. 2006;98:66–69. doi: 10.1016/j.amjcard.2006.01.055. [DOI] [PubMed] [Google Scholar]