

Abstract
The synthesis, X-ray structure, solution stability, and photophysical properties of several trivalent lanthanide complexes of Yb(III) and Nd(III) using both tetradentate and octadentate ligand design strategies and incorporating the 1-methyl-3-hydroxy-pyridin-2-one (Me-3,2-HOPO) chelate group are reported. Both the Yb(III) and Nd(III) complexes have emission bands in the Near Infra-Red (NIR) region, and this luminescence is retained in aqueous solution (). Furthermore, the complexes demonstrate very high stability (pYb ~ 18.8 – 21.9) in aqueous solution, making them good candidates for further development as probes for NIR imaging. Analysis of the low temperature (77 K) photophysical measurements for a model Gd(III) complex were used to gain an insight into the electronic structure, and were found to agree well with corresponding TD-DFT calculations at the B3LYP/6-311G++(d,p) level of theory for a simplified model monovalent sodium complex.
Keywords: X-ray structure, Ytterbium, Neodymium, NIR luminescence, solution stability
Introduction
There is a growing interest in organic complexes of Ln(III) metal ions with emission in the Near Infra-Red (NIR) region (eg. Ln = Nd, Yb).1,2 While their usage as the gain medium in solid state laser technologies (eg. Nd:YAG) has been well known for some time, organic complexes of these metals have more recently attracted considerable interest, a major impetus being the development of NIR emitting probes for bioimaging.3,4 This interest can be traced to the fact that biological tissues are considerably more transparent at these low energy wavelengths, which facilitates deeper penetration of excitation and emitted radiation. Moreover, the lack of endogenous NIR emissive fluorophores allows for much greater sensitivity when compared to visible emitters, by virtually eliminating autofluorescence.5 When compared to NIR emission from organic molecules such as indocyanine green, an additional benefit of Ln(III) complexes are their significantly longer luminescence lifetimes (eg. Yb(III), τrad ~ 1.2 – 1.3 ms),6 which can be used with time-gated techniques to remove background emission and scattered excitation, further improving signal to noise ratios. This latter methodology has been successfully applied in luminescence microscopy, using complexes of Eu(III), and the ongoing development of time resolved imaging equipment with higher sensitivity in the NIR region will allow its direct application at these longer wavelengths.
As a result of their weak (Laporte forbidden) f-f absorption, the direct excitation of Ln(III) cations is not efficient. Instead, sensitization of the metal emission is more effectively achieved using the so-called `antennae' effect, wherein electronic excitation is initially absorbed by a strongly absorbing proximal chromophore, and this excitation is then transferred to the metal centre via an energy transfer process.7 Transition metal complexes, in particular with Ru(II) and Re(I), have been used with great success to sensitise emission from NIR emitting Ln(III) cation, as reviewed recently by Ward.8 Alternately, several other groups have also used organic chromophores, such as 8-hydroxyquinoline derivatives, as the light absorbing antennae.9,10 We have recently examined several tetradentate and octadentate complexes of Eu(III), featuring the organic 1-hydroxypyridin-2-one (1,2-HOPO) chelate, which also acts as an antennae chromophore.11,12 While the 1,2-HOPO isomer was found to strongly sensitize Eu(III) emission, the analogous Me-3,2-HOPO isomer (see Chart 1) does not, which has prompted our further recent13,14 investigations of the photophysical properties for this chromophore. Herein, we describe the synthesis, structure, aqueous stabilities and photophysical properties of two bis(tetradentate) and octadentate Me-3,2-HOPO based ligands, in complex with Yb(III) and Nd(III), which display strong NIR emission.
Chart1.tif.

Chart 1 Chemical structure of tetradentate 5LIO-Me-3,2-HOPO (left) and octadentate H(2,2)-Me-3,2-HOPO (right) ligands for sensitization of Yb(III) and Nd(III).
Experimental
General
All solvents for reactions were dried using standard methodologies. Thin-layer chromotography (TLC) was performed using precoated Kieselgel 60 F254 plates. Flash chromatography was performed using EM Science Silica Gel 60 (230– 400 mesh). NMR spectra were obtained using a Bruker AV-400 spectrometer operating at 400.1 (or 100.6) MHz for 1H (or 13C) respectively. 1H (or 13C) chemical shifts are reported in ppm relative to the solvent resonances, taken as δ = 7.26 ppm (δ = 77.0 ppm) and δ = 2.49 ppm (δ = 39.5 ppm) respectively for CDCl3 and (CD3)2SO while coupling constants (J) are reported in Hz. Fast-atom bombardment mass spectra (FABMS) were performed using 3-nitrobenzyl alcohol (NBA) or thioglycerol/glycerol (TG/G) as the matrix. Elemental analyses were performed by the Microanalytical Laboratory, University of California, Berkeley, CA.
Synthesis
Detailed synthesis of relevant precursors including the important 2-mercaptothiazoline activated carboxyl derivative, 1-methyl-3-benzyloxy-4-(2-thioxo-thiazolidine-3-carbonyl)-pyridin-2-one (Me-3,2-HOPO-Bn-thiaz) and the free base hexamine backbone, N,N,N',N'-tetrakis-(2-aminoethyl)-ethane-1,2-diamine (H(2,2)-amine), have been previously described.15,16 Similarly, a detailed synthesis of 5LIO-Me-3,2-HOPO (Chart 1) has been previously reported.17 Herein, the detailed synthesis of the H(2,2)-Me-3,2-HOPO ligand (Chart 1) and synthesis of the [Ln(5LIO-Me-3,2-HOPO)2] and [Ln(H(2,2)-Me-3,2-HOPO)] complexes isolated as the charge neutral complex and the dicationic chloride salts respectively with Ln = Yb(III) and Nd(III) are described. Complexes with Gd(III) were prepared in situ by mixing a standardized aqueous solution of GdCl3·6H2O with the appropriate stoichiometric amount of the 5LIOMe-3,2-HOPO or H(2,2)-Me-3,2-HOPO free ligands, and diluting with 0.1 M aqueous TRIS buffer or 1:1 EtOH/MeOH (v/v).
H(2,2)-Me-3,2-HOPO-Bn
To a solution of the H(2,2)-amine (0.152 g, 0.65 mmol) and five molar equivalents of Me-3,2-HOPO-Bn-thiaz in CH2Cl2 (50 mL) was added NEt3 (0.274 g, 2.70 mmol). The reaction mixture was stirred in the dark under N2 for 5 days, then extracted (3 × 50 mL CH2Cl2) with 0.1 M KOH. The organic extracts were combined, dried over MgSO4, and filtered. The solvent was removed in vacuo, and the resulting residue was purified by column chromatography on silica gel, initially by eluting with 3 % MeOH in CH2Cl2 then with increasing polarity to 5–7 % MeOH in CH2Cl2 to elute the crude product. This crude residue was subsequently repurified by column chromatography on silica gel eluting with 5–7 % MeOH in CH2Cl2, yielding 0.47 g (60 %) of the product as an off-white solid after removal of the solvent. 1H NMR (400.1 MHz, CDCl3): δ 2.27 (bt, 12H, CH2CH2NH and NCH2CH2N), 3.13 (q, 8H, CH2CH2NH), 3.56 (s, 12H, HOPO CH3), 5.30 (s, 8H, HOPO Bn CH2), 6.71 (d, 4H, HOPO Ar-H), 7.09 (d, 4H, HOPO Ar-H), 7.25–7.37 (m, 20H, Bn Ar-H), 7.89 (t, 4H, CH2NHCO). 13C NMR (100.6 MHz, CDCl3): δ163.3, 159.5, 146.3, 136.3, 132.1, 130.8, 128.9, 128.7, 128.6, 104.8, 74.6, 52.3, 50.7, 37.7, 37.5.
H(2,2)-Me-3,2-HOPO
The protected ligand, H(2,2)-Me-3,2-HOPO-Bn (0.47 g, 0.39 mmol), was dissolved in a mixture of concentrated HCl (10 mL) and glacial acetic acid (10 mL). The solution was stirred under N2 for two days at which point the volatiles were removed in vacuo by co-evaporation with MeOH (5 × 10 mL) to yield the hydrochloride salt of the product (0.41 g, 92 %) as an off-white solid. 1H NMR (400.1 MHz, DMSO-d6): δ 3.43 (br m, 20H, CH2CH2NH), 3.73 (s, 12H, HOPO CH3), 6.50 (d, 4H, HOPO Ar-H), 7.13 (d, 4H, HOPO Ar-H), 8.73 (br m, 4H, CH2NHCO). 13CNMR (100.6 MHz, DMSO-d6): δ166.4, 158.4, 147.9, 128.1, 117.3, 103.2, 52.0, 37.3, 34.5. Anal. Calc'd. (Found) for C38H48N10O12·9H2O·4HCl (1144.84 g.mol−1): C, 39.87 (39.96); H, 6.16 (6.00); N, 12.23 (11.95).
[Yb(5LIO-Me-3,2-HOPO)2]
To a suspension of the 5LIO-Me-3,2-HOPO ligand (22.0 mg, 54.1 μmol) in MeOH (5 mL) was added 100 μL of pyridine and this was heated gently to assist dissolution of the solid. To the resulting solution was added 0.53 equivs. of YbCl3·6H2O (11.0 mg, 28.4 μmol) in MeOH (1 mL). This mixture was refluxed for 4 hrs, then left to cool to room temperature. Addition of diethyl ether (ca. 10 mL) induced precipitation of a white solid, which was collected by vacuum filtration, washed with diethyl ether and air dried to yield the desired product (17.0 mg, 30 %). Anal. Calc'd. (Found) for [Yb(C18H20N4O7)(C18H21N4O7))]·3.5H2O (1045.86 g.mol−1): C, 41.34 (41.36); H, 4.63 (4.93); N, 10.71 (10.55). Single crystals suitable for X-ray analysis of this material were recrystallised by slow diffusion of diethyl ether into a solution of the complex in 5 % aqueous 1:1 (v/v) DMF:MeOH.
[Nd(5LIO-Me-3,2-HOPO)2]
This complex was prepared using an analogous procedure to that for the Yb(III) complex, starting with the 5LIO-Me-3,2-HOPO ligand (27.0 mg, 66.4 μmol), substituting 0.53 equivs. of NdCl3·6H2O (12.60 mg, 35.1 μmol) where appropriate. The desired complex was obtained as a very faintly pinkish/purple solid (12.0 mg, 34.0 %). Anal. Calc'd. (Found) for [Nd(C18H20N4O7)(C18H21N4O7)]·6H2O (1062.09 g.mol−1): C, 40.71 (40.88); H, 5.03 (4.53); N, 10.55 (10.40).
[Yb(H(2,2)-Me-3,2-HOPO)]
To a suspension of the H(2,2)-Me-3,2-HOPO ligand (24.1 mg, 21.1 μmol) in MeOH (10 mL) was added 100 μL pyridine. To this mixture was added a clear solution of YbCl3·6H2O (9.5 mg, 24.5 μmol) in MeOH (1 mL). The solution was heated at reflux overnight during which time a white solid precipitated. Upon cooling to room temperature, the desired complex was collected by vacuum filtration and dried overnight resulting in a faint beige colored solid (10.0 mg, 39.9 %). Anal. Calc'd. (Found) for [Yb(C38H47N10O12)]Cl2·6H2O (1187.88 g.mol−1): C, 38.42 (38.29); H, 5.01 (4.92); N, 11.79 (11.43).
[Nd(H(2,2)-Me-3,2-HOPO)]
This complex was prepared using an analogous procedure to that for the Yb(III) complex, starting with the H(2,2)-Me-3,2-HOPO ligand (26.0 mg, 22.7 μmol), substituting NdCl3·6H2O (8.7 mg, 24.3 μmol) where appropriate. In this case, the desired complex did not precipitate from solution. Instead, the dropwise addition of diethyl ether was required to induce precipitation of the solid, which was isolated by vacuum filtration and air dried to yield a faint purple colored solid (17.0 mg, 60.9 %). Anal. Calc'd. (Found) for [Nd(C38H47N10O12)]Cl2·10H2O (1231.14 g.mol−1): C, 37.07 (37.08); H, 5.49 (4.99); N, 11.38 (10.94).
Physical Methods
Crystallography
Crystals suitable for X-ray analysis were mounted on a Kapton loop using Paratone N hydrocarbon oil and measured at low temperature using a Siemens SMART CCD18 area detector with graphite monochromated Mo-Kα radiation. Cell constants and orientation matrices were obtained from least-squares refinement. The resulting data were integrated by the program SAINT19 and corrected for Lorentz and polarization effects. Data were also analyzed for agreement and possible absorption using XPREP.20 An empirical absorption correction based on the comparison of redundant and equivalent reflections was applied using SADABS.21 Equivalent reflections where appropriate were merged and no decay correction was applied. The structures were solved within the WinGX22 package by direct methods using SIR9223 and expanded using full-matrix least-squares techniques with SHELXL-97.24 Hydrogen atoms were positioned geometrically, with C–H = 0.93 Å for CH aromatic, C–H = 0.97 Å for CH2 methylene, N–H = 0.89 Å, and C–H = 0.96 Å for CH3 methyl. Hydrogen atoms were constrained to ride on their parent atoms, with Uiso(H) values set at 1.2 times Ueq(C) for all H atoms. Resulting drawings of molecules were produced with ORTEP-3.25
Solution Thermodynamics
The general procedure used to compare aqueous stabilities of the Ln(III) complexes herein was that of competition batch titration as described previously26,27 using DTPA as the known competitor. Briefly, varying volumes of standardized DTPA stock solution were added to aqueous 0.1 M TRIS (or 0.1 M HEPES) buffer (pH 7.4) containing 0.1 M KCl electrolyte and a fixed amount of the appropriate ligand and Ln(III) cation delivered by Eppendorf pipette (either 2:1 or 1:1 stoichiometry for the 5LIO-Me-3,2-HOPO and H(2,2)-Me-3,2-HOPO ligands respectively). If necessary, the pH of the solutions was readjusted to 7.4 with HCl and/or KOH and the solutions were diluted to identical volumes with additional 0.1 M TRIS (or 0.1 M HEPES) buffer (pH 7.4) containing 0.1 M KCl. The initial concentrations of DTPA relative to the Ln(III) complex ranged from ca. 1:10 to 3000:1. After standing for 24 h to ensure thermodynamic equilibrium, the concentration of the free and complexed ligand were evaluated. For 5LIO-Me-3,2-HOPO, this was accomplished by absorbance measurements, using the integral of the spectra in the wavelength range between two isobestic points at 290–370 nm, and the spectra of the free and fully complexed 5LIO-Me-3,2-HOPO as references. For H(2,2)-Me-3,2-HOPO, the integrated intensity of the observed NIR emission spectra at ca. 980 nm after excitation at 345 nm was used to determine the equilibrium concentration of the [Yb(H(2,2)-Me-3,2-HOPO)] complex directly, since the free ligand is not luminescent at this wavelength and the spectra of [Yb(H(2,2)-Me-3,2-HOPO)] in the absence of DTPA competitor was used as the reference.
Computational Studies
Ground state density functional theory (DFT) and time-dependent DFT (TD-DFT) calculations were performed at the Molecular Graphics and Computational Facility, College of Chemistry, University of California, Berkeley. In both cases, the B3LYP/6-311G++(d,p) basis set provided in Gaussian'0328 was used, with simplified input structures derived from a previously reported15 crystal structure. All calculations were done in the gas phase and geometry optimizations were performed with no symmetry restraints.
Photophysics
Typical sample concentrations for absorption and fluorescence measurements were ca. 10−5 M. UV-Visible absorption spectra were recorded on Varian Cary 300 double beam absorption spectrometer using quartz cells of 1.0 cm path length. Steady state emission spectra were acquired on a HORIBA Jobin Yvon IBH FluoroLog-3 (FL-3) spectrofluorimeter. The excitation light source was a 450 W Xe arc lamp and spectral selection was achieved by passage through a double grating excitation monochromator (1200 grooves/mm) blazed at 330 nm. Spectra were reference corrected for the excitation light source variation (lamp and grating). Emission spectra were collected at 90° to the excitation, and emission was collected using a Hamamatsu H9170-75 NIR as the detector. Spectral selection of the emission was achieved by passage through a double grating emission monochromator (600 grooves/mm) blazed at 1 μm. The emission spectral response of the Hamamatsu H9170-75 NIR detector is almost linear across the 950–1700 nm wavelength range, and since the Ln(III) emission is essentially monochromatic and quite narrow (ca. 60 nm at FWHM), the observed emission signals were not corrected for the efficiency of the grating. Quantum yields were determined using the following equation;
where A is the absorbance at the excitation wavelength (λ), I is the intensity of the excitation light at the same wavelength, η is the refractive index and D is the integrated luminescence intensity. The subscripts `x' and `r' refer to the sample and reference respectively. By using a common excitation wavelength of 345 nm in the present case, the I(λr)/I(λx) term is removed. Hence, a plot of integrated emission intensities for both sample and reference (i.e. Dr or Dx) vs. absorbance at 345 nm (i.e. Ar(λr) or Ax(λx)) yield linear plots with slopes equal to or . Values of η = 1.33 and η = 1.49 were used for solutions in water and toluene respectively. The emission of [Yb(TTA)3(H2O)2] in toluene (Φr = 0.35 %) was used as a reference29, 30 for the [Yb(5LIO-Me-3,2-HOPO)2] quantum yield determination, which was then used as a relative standard for the [Yb(H(2,2)-Me-3,2-HOPO)] complex, and the estimated error on these values is ± 25 %.
Luminescence lifetimes were determined with the same HORIBA Jobin Yvon IBH FluoroLog-3 spectrofluorimeter, adapted for time resolved measurements. An N2 laser (LN1000, Laser Photonics, Inc) was used as the light source (λex = 337.1 nm), coupled to the entrance port of the FluoroLog-3. The laser output pulse energy was ca. 1.5 mJ/pulse, with an optical pulse duration of less than 800 ps FWHM. A portion of this excitation was sampled with a quartz beam sampling plate, which was focused onto the entrance of a UV-sensitive photodiode (DET210, Thor Labs). The small amplitude analogue output from the photodiode was processed into a TAC Start signal (NIM) using a TB-01 pulse converter module from IBH. A Hamamatsu H9170-75 NIR fast rise time PMT operating at −800 V and −60°C was used as the detector, and the output signal from the PMT was processed using a TB-02 0.5 GHz pre-amplifier module from IBH, and a 100 MHz Constant Fraction Discriminator (CFD) (Model 6915, Phillips Scientific), yielding appropriate TAC Stop signals (NIM). These were acquired using a 2 ns PCI Multi Channel Scaling (MCS) card (Model P7888-1E, FAST ComTec GmbH), and processed using the DataStation software package from IBH. Data analysis was performed using the commercially available DAS 6 decay analysis software package from HORIBA Jobin Yvon IBH. Goodness of fit was assessed by minimizing the reduced chi squared function, χ2, and a visual inspection of the weighted residuals. Each trace contained at least 5,000 points and the reported lifetime values result from three independent measurements. The estimated error on these values is ± 10 %.
Results and Discussion
Synthesis and Structure
The 5LIO-Me-3,2-HOPO and H(2,2)-Me-3,2-HOPO ligands (Chart 1) were previously reported as potential actinide sequestering agents.17,31 As a result of our renewed interest, however, we have now obtained X-ray quality crystals of the former ligand. Although the collected data set was incomplete (completeness to θ of ca. 85 %) due to crystal decomposition, a tenable solution was able to be obtained from the frames collected, and two views of the resulting structure are shown in Figure 1. The two Me-3,2-HOPO moieties are slightly offset but remain parallel to each other, and the two amide functional groups are coplanar with the aromatic ring system. The aromatic C-C bonds of the ring system show no evidence of localisation, with all bond lengths within the typical expected range of 1.3 to 1.4 Å. The resonance form for the Me-3,2-HOPO chelate group, as drawn in Chart 1, was confirmed in the solid state structure, typified by a much shorter C-O bond length in the 2-oxo position at ca. 1.24 Å, compared to the longer 3-hydroxyl C-O bond length of ca. 1.35 Å. Each of the hydroxyl protons are hydrogen bonded with the adjacent keto oxygen, with a separation of ca. 2.26 Å. An additional strong hydrogen bonding interaction is apparent between the amide proton and the hydroxyl group oxygen, with an average N-H---O separation of 1.95 Å. Lastly, two solvent water molecules complete the unit cell, each of which are associated with the amide carbonyl oxygen atom via an intramolecular hydrogen bond.
Figure 1.

Top-down view (left) and side-on view (right) of the X-ray crystal structure for 5LIO-Me-3,2-HOPO. Thermal ellipsoids of non-H atoms are shown at the 50 % probability level (C = black, O = red, N = blue).
The corresponding Ln(III) complexes of both ligands with the NIR emitting metal ions (Ln = Yb, Nd) were prepared using well established methodologies, as described in the experimental section. The desired complexes with 5LIO-Me-3,2-HOPO were obtained in analytically pure form as the monoprotonated charge neutral LnL2H complexes, while the complexes with H(2,2)-Me-3,2-HOPO were isolated as chloride salts of the triply protonated dicationic LnLH3 complex. This behaviour is somewhat different to the results obtained32 with analogous 1,2-HOPO ligands, such as 5LIO-1,2-HOPO, which yielded anionic Ln(III) complexes isolated as pyridinium or alkylammonium salts, but can be rationalised in terms of the greater basicity of the Me-3,2-HOPO chelate (vide infra) and the pKa of the base used to deprotonated the phenol group (i.e. pyridine, pKa ~ 5.2).
A single crystal suitable for X-ray analysis was grown from a 5 % aqueous 1:1 (v/v) DMF:MeOH solution of the [Yb(5LIO-Me-3,2-HOPO)2] complex diffused with diethyl ether, and the resulting structure is shown in Figure 2. The compound crystallized in the asymmetric space group P21, and includes two independent complex molecules in the asymmetric unit, together with several molecules of co-crystallized solvent water and methanol. Notably, the crystal structure with Yb(III) is very similar to that reported31 for the same ligand with Ce(IV), and other Ln(III) complexes formed with the isomeric 5LIO-1,2-HOPO ligand. As expected, both ytterbium atoms are coordinated by a total of eight oxygen atoms from the two 5LIO-Me-3,2-HOPO ligands, with a sandwich-like structure. The average Yb-O bond lengths, considering either complex, were identical within experimental error at ca. 2.33 Å whereas the geometries of the two coordination polyhedra for both Yb(III) ions were slightly different. Shape analysis as discussed elsewhere33,34 indicated that Yb(1) and Yb(2) were best described as idealized bicapped trigonal prism (C2v) and square antiprism (D4d), respectively. As with our previous reports using analogous ligands,11 the least-square faces of the `top' and `bottom' ligands for both complexes are offset and rotated by about 123° relative to one another. Importantly, a consideration of the charge requirements for the isolated complex reveals it must be either protonated or anionic with an associated cation in order to maintain the required charge balance. However, other than isolated solvent H2O and CH3OH molecules, no other peaks were observed in the difference map that we could attribute to a cation such as Na+ or K+ as we have observed previously.12,14 As a result, we conclude the crystallized complex is protonated, which is in accordance with the results from elemental analysis of the bulk material. Unfortunately, despite the excellent quality of the X-ray data, the position of this proton on the complex could not be unequivocally located from the difference map.
Figure 2.

X-ray crystal structures showing views of the two independent Yb(1) (left) and Yb(2) (right) complexes found in the asymmetric unit for the [Yb(5LIO-Me-3,2-HOPO)2] complex. Co-crystallized solvent molecules, and selected H atoms have been omitted for clarity and non-H atoms are drawn at the 50 % probability level (C = black, O = red, N = blue, Yb = green). Only the major component of the disordered ligand backbone is shown for the Yb(2) complex. The position of the proton necessary for charge neutrality could not be located from the difference map (see text).
Solution Thermodynamics
The aqueous stability of the tetradentate 5LIO-Me-3,2-HOPO and octadentate H(2,2)-Me-3,2-HOPO ligands in complex with Yb(III) were assessed in terms of their pM, which by analogy to pH, is defined as the negative log of the concentration of free metal in solution (pM = −log10[M]free) at a specified set of standard conditions (typically [M]total = 1 μM, [L]total = 10 μM, and pH= 7.4). Competition batch titrations using DTPA as the competitor with a known35 pYb of 19.4 were performed. For 5LIO-Me-3,2-HOPO, the absorption spectra in Figure 3 show two isobestic points at ca. 289 and 375 nm, confirming the presence of only the ligand and fully formed [Yb(5LIO-Me-3,2-HOPO)2]− complex under the conditions of the experiment. From the analysis, a conditional stability constant of pYb = 18.8(1) was obtained, which is comparable to the reported31 value of pCe = 16.3 for the same ligand with Ce(III). Using the known31 pKa values for the 5LIO-Me-3,2-HOPO ligand of 7.14(3) and 5.91(4), we can also evaluate the formation constant for the Yb(III) complex to be logβ120 ~ 23.4(1), which is similarly comparable with the logβ120 reported31 for Ce(III) of 20.9(2). The corresponding values for DTPA demonstrate that such an increase in stability over the lanthanide series is plausible. For example, the determined35 logβ110 values of DTPA rise from 20.3 with Ce(III) to 22.6 with Yb(III) representing a similar difference in stability as observed here, with the increase due to the decreasing size and increased Lewis acidity of the metal cation.
Figure 3.

(a) Absorption spectra (left) for solutions of [Yb(5LIO-Me-3,2-HOPO)2]− (26 μM in 0.1 M KCl; 0.01 M HEPES at pH 7.4; black line) and after addition of up to ca. 500 equivalents of DTPA (red). (b) Plot of equilibrium concentration (right) determined from absorption spectra versus equilibrium concentration of log DTPA (mM−1) and corresponding fit used to evaluate equal speciation between 5LIO-Me-3,2-HOPO and DTPA (i.e. 50 % [Yb(DTPA)] at ca. 0.013 mM, which corresponds to a pYb = 18.8 (1) for the [Yb(5LIO-Me-3,2-HOPO)2 complex.
For the H(2,2)-Me-3,2-HOPO ligand, the corresponding conditional stability constant was evaluated using fluorescence techniques, and the Yb(III) emission spectra in the NIR region upon increasing addition of DTPA are shown in Figure 4. From the analysis of these spectra, a pYb of 21.9(1) was determined, which is significantly improved when compared to the corresponding value for 5LIO-Me-3,2-HOPO, as we would expect due to the improved chelate effect of the octadentate versus bis(tetradentate) ligand topology. The magnitude of the differences are also similar to those observed for the structurally analogous ligands using the identical octadentate (H(2,2)-) and tetradentate (5LIO-) ligand backbones but with the isomeric 1,2-HOPO chelating unit, where an increase of ΔpEu = 2.6 log units was seen, comparable to the ΔpYb = 3.1 log units observed herein.
Figure 4.

(a) Luminescence spectra (left) for solutions of [Yb(H(2,2)-Me-3,2-HOPO)]– (ca. 1.5 μM in 0.1 M TRIS at pH 7.4; black line) and after addition of up to ca. 500 equivalents of DTPA (red). (b) Double log plot (right) of equilibrium metal complex concentrations log[[Yb(DTPA)]/[Yb(H(2,2)-Me-3,2-HOPO)]] determined from luminescence spectra versus equilibrium concentrations of log[[DTPA]/[H(2,2)-Me-3,2-HOPO]] and corresponding fit used to evaluate equal speciation between H(2,2)-Me-3,2-HOPO and DTPA (i.e. 50 % [Yb(DTPA)] at ca. 0.013 mM, which corresponds to a pYb = 21.9 (1) for the [Yb(H(2,2)-Me-3,2-HOPO)] complex.
Electronic Structure Calculations
Density Functional Theory (DFT) calculations were performed using Gaussian'0328 in order to elucidate the ground state geometry and electronic structure of the Me-3,2-HOPO chromophore. As a simplified model, only the 6-methyl amide of Me-3,2-HOPO was used as the input, and this was first geometry optimized with no symmetry constraints to give the relaxed output geometry shown in Figure 5. The results obtained show all non-H atoms lying essentially in a single plane, and the calculated structure reproduces the expected strong H-bonding interactions between the hydroxyl proton and adjacent keto oxygen (O1-H---O2 = 1.99 Å) and, similarly, between the amide proton and the hydroxyl oxygen atom (N2-H---O1 = 1.98 Å). Using Time-Dependent techniques (TD-DFT) and employing the same basis set, the electronic structure was calculated and the results are summarized in Table 2. A consideration of this data reveals the nature of the first excited singlet state of the Me-3,2-HOPO chromophore can be described principally by a HOMO → LUMO excitation, with a smaller contribution from the HOMO-3 → LUMO+2 excitation. An inspection of the relevant Kohn–Sham molecular orbitals, also shown in Figure 5, reveals these transitions to be of 1π-π* character. Similarly, the nature of the lowest energy triplet state (T1) can be described principally by a HOMO → LUMO excitation, i.e. a 3ππ* transition.
Figure 5.

(a) Calculated output geometries obtained from static B3LYP/6-311G++ (d,p) geometry optimization and relevant molecular orbital diagrams from TD-DFT electronic structure calculations for 6-MeAmide-Me-3,2-HOPO (top) and (b) a model [Na(6-MeAmide-Me-3,2-HOPO)] complex (bottom). The metal centered character of the LUMO in the latter is an artifact of the B3LYP functional calculation (see text).
Table 2.
Summary of electronic structure calculations for Me-3,2-HOPO chromophore and a model [Na(6-MeAmide-Me-3,2-HOPO)] complex.
| Excited State | Multiplicity | Energy(eV) | Wavelength (nm) | Oscillator Strength | Orbital(s) Involved(n → n') | Transition Coefficient | |
|---|---|---|---|---|---|---|---|
| 6-MeAmide-Me-3,2-HOPO | 1 | T1 | 2.580 | 480.49 | 0.0000 | 45 → 49 | 0.1076 |
| 48 → 49 | 0.7776 | ||||||
| 48 → 51 | −0.1226 | ||||||
|
| |||||||
| 2 | T2 | 3.756 | 330.06 | 0.0000 | 45 → 49 | −0.2497 | |
| 47 → 49 | 0.1471 | ||||||
| 48 → 49 | 0.1136 | ||||||
| 48 → 51 | 0.6922 | ||||||
|
| |||||||
| 3 | T3 | 3.864 | 320.90 | 0.0000 | 47 → 49 | 0.6870 | |
| 47 → 51 | 0.1157 | ||||||
| 47 → 56 | 0.1041 | ||||||
| 47 → 60 | 0.1958 | ||||||
| 48 → 51 | −0.1724 | ||||||
|
| |||||||
| 4 | T4 | 3.942 | 314.56 | 0.0000 | 46 → 49 | 0.6999 | |
| 46 → 60 | 0.1276 | ||||||
|
| |||||||
| 5 | S1 | 4.045 | 306.53 | 0.1043 | 48 → 49 | 0.6286 | |
| 45 → 51 | 0.1374 | ||||||
|
| |||||||
| Na(6-MeAmide-Me-3,2-HOPO)] | 1 | T1 | 2.369 | 532.42 | 0.0000 | 53 → 55 | 0.7897 |
|
| |||||||
| 2 | T2 | 2.831 | 438.02 | 0.0000 | 53 → 54 | 0.7066 | |
|
| |||||||
| 3 | S1 | 2.839 | 436.77 | 0.0039 | 53 → 54 | 0.7065 | |
|
| |||||||
| 4 | T3 | 3.586 | 345.79 | 0.0000 | 50 → 55 | −0.1738 | |
| 50 → 61 | −0.1228 | ||||||
| 53 → 61 | 0.7052 | ||||||
| 53 → 63 | −0.1172 | ||||||
| 53 → 67 | −0.1337 | ||||||
|
| |||||||
| 5 | S2 | 3.844 | 322.58 | 0.1816 | 50 → 61 | 0.1299 | |
| 53 → 55 | 0.6199 | ||||||
Additional static and time dependant DFT calculations using the same methodologies were also undertaken with Gaussian'03 in order to characterize the excited state of the anionic Me-3,2-HOPO chromophore, in complex with a metal cation. We have used an identical approach to that reported11 previously for the 1,2-HOPO chromophore, wherein the ionizable hydroxyl proton of the Me-3,2-HOPO chromophore has been replaced with a monovalent Na+ cation, chelated in a bidentate mode between the oxygen atoms, as shown in Figure 5. As we have noted, while this is a somewhat crude approximation, this simplification avoids computationally expensive calculations involving a lanthanide cation, and can nonetheless be a useful tool for understanding the ligand's electronic structure. It must be stressed that the goal of this “calculation” is not to determine the electronic states of the Ln(III) cation but rather to approximate the energy for the lowest excited singlet and triplet states of the ligand. Similarly, use of the same basis set allows a direct comparison of the theoretical results obtained for the 1,2-HOPO and Me-3,2-HOPO chromophores. The resulting output geometry is shown in Figure 5, where it is evident that all non-H atoms are lying essentially in the plane of the aromatic ring, and the energy minimized geometry of the chelate group is similar to that observed in the [Yb(5LIO-Me-3,2-HOPO)2] structure by X-ray crystallography. The bidentate coordination mode of the monovalent Na+ cation is retained with Na-O bondlengths of 2.15 Å and 2.18 Å to the phenolic and keto oxygen atoms respectively, mirroring the binding mode of a Ln(III) cation, and a strong intramolecular H-bond between the amide proton and the phenolic oxygen atom is also readily apparent at 1.84 Å. The results from the corresponding TD-DFT electronic structure calculations for the model [Na(6-MeAmide-Me-3,2-HOPO)] complex are also summarized in Table 2. A consideration of this data reveals the first excited singlet state of the Me-3,2-HOPO chromophore can be described by a HOMO → LUMO excitation. However, an inspection of the relevant Kohn–Sham molecular orbitals, also shown in Figure 5, shows this transition to be of 1LMCT (ligand to metal charge transfer) character. These results are analogous to our previous reported11 work using TD-DFT analysis of a simplified Na+ complex of the 1,2-HOPO chromphore, wherein we noted these LMCT transitions are typically underestimated by the B3LYP/6-311G++ basis set due to self interaction errors with the simple exchange correlational (xc) functional used,36 and a similar argument is applicable in the present instance. Hence, the real location of this 1LMCT transition is likely at significantly higher energy, while the predicted energy of ligand centered (LC) transitions such as that of the S0 → S2 transition at 322.6 nm, which can be described principally by a HOMO → LUMO+1 excitation, is in good agreement with experimentally observed electronic spectra maxima of ca. 340 nm (vide infra). Similarly, the nature of the lowest energy triplet state (T1) can also be described by a HOMO → LUMO +1 excitation, and an inspection of the relevant molecular orbitals reveals this to be of distinctly ligand centered π → π* character (i.e. a 3ππ* transition) with a predicted energy of 523.4 nm (19,105 cm−1).
Photophysics
The absorption spectra of the 5LIO-Me-3,2-HOPO and H(2,2)-Me-3,2-HOPO ligands are essentially identical (data not shown), displaying a broad electronic envelope in the UV, which is red shifted upon deprotonation and is centered at ca. 340 nm in aqueous solution at pH 7.4. From corresponding TD-DFT analysis (vide infra), we assign this band to the lowest energy singlet transitions with essentially π-π* character.
Upon complexation with Ln(III) cations, the vibrational fine structure of this band is improved slightly, and a comparison of the spectra of both ligands with Yb(III) as examples are shown in Figure 6. We can attribute this sharpening to a loss of the homogeneous line broadening normally present due to variable hydrogen bonding interactions involving the phenolate in aqueous solution, which is replaced by a more well defined geometry for the chromophore upon Ln(III) binding. Notably, the Ln(III) complexes of Me-3,2-HOPO containing ligands also display improved vibrational structure when compared with the corresponding 1,2-HOPO complexes. This may be a reflection of the stronger aromaticity and π-π* character of this transition in the former. Previously, the 1,2-HOPO chelate has been considered as a cyclic hydroxamic acid37 rather than an aromatic systems and the lowest energy transition has been assigned to have mixed n-π* and π-π* parentage.11 Upon closer inspection of the electronic spectra, we also note a slight shift in λmax between the two differing ligands in complex with Yb(III). Hence, for [Yb(5LIO-Me-3,2-HOPO)2], the apparent λmax is located at 345.6 nm (28,940 cm−1) whereas for [Yb(H(2,2)-Me-3,2-HOPO)], this value is blue shifted to 344.7 nm (29,010 cm−1). While this difference is only slight (Δ≈ 75 cm−1), it is nonetheless reproduced in the spectra of the corresponding Nd(III) complexes which are otherwise indistinguishable from their Yb(III) counterparts, and we attribute the difference to subtle changes in the coordination geometries supported by the bis(tetradentate) versus octadentate ligand topologies.
Figure 6.

(a) The absorption spectra of the [Yb(5LIO-Me-3,2-HOPO)2] (red) and [Yb(H(2,2)-Me-3,2-HOPO)] (black) complexes in 0.1 M aqueous TRIS buffer, pH 7.4. (b) Emission spectra (λex = 345 nm) at room temperature (red, × 10) and 77 K (black) of the [Gd(5LIO-Me-3,2-HOPO)2]− complex measured in a 1:1 (v/v) EtOH:MeOH glassing solvent, and corresponding fit (blue)to a series of overlapping gaussians to evaluate the zero phonon transition.
A comparison of the room temperature and 77 K emission spectra for [Gd(5LIO-Me-3,2-HOPO)2] in 1:1 EtOH:MeOH (v/v) glassing solvent is also shown in Figure 6. Since Gd(III) lacks an appropriately positioned metal centered energy accepting level, its complexes may be used to assess the energetic position of the ligand centered lowest energy triplet state. At room temperature, the emission of the complex is quite weak, with an apparent maximum at ca. 400 nm which we attribute to residual singlet emission from the Me-3,2-HOPO chromophore. However, upon cooling to 77 K, a much more intense and vibrationally structured band appears, with peak maxima at ca. 536, 579 and 624 nm. We can confidently attribute this emission to phosphorescence from the ligand centered lowest energy triplet excited state. Concomitant with this new band, we also observe an increase in intensity and considerable sharpening of the residual singlet excited state emission band in the UV region, with new peaks at 376 and 393 nm, and a weaker shoulder at ca. 415 nm. It is worth noting that the apparent zero phonon energy of the phosphorescence, when taken as the first peak maxima of the vibrational progression (ca. 18,657 cm−1), is in good agreement with that predicted value for the Me-3,2-HOPO chromophore of 19,105 cm−1 as obtained from TD-DFT analysis of the model [Na(6-MeAmide-Me-3,2-HOPO)] complex.
The luminescence spectra in the NIR region for the Yb(III) and Nd(III) complexes with both the 5LIO-Me-3,2-HOPO and H(2,2)-Me-3,2-HOPO ligands in 0.1 M TRIS buffered aqueous solution at pH 7.4 are shown in Figure 7. The spectra of both Nd(III) complexes are remarkably similar, each displaying the characteristic 4F3/2 → 4I11/2 and 4F3/2 → 4I13/2 emission bands at 1064 and 1332 nm respectively. By contrast, the spectra of the two Yb(III) complexes show distinct differences in the characteristic 2F5/2 → 2F7/2 electronic transition at ca. 1μm. For the [Yb(5LIO-Me-3,2-HOPO)2] complex, the emission band is highly structured and relatively symmetrical, with several minor peaks evident due to crystal field splitting of the single electronic transition. For the corresponding [Yb(H(2,2)-Me-3,2-HOPO)] complex, the splitting of the 2F5/2 → 2F7/2 transition clearly differs, with the appearance of a significantly more intense shoulder at ca. 1060 nm. The lack of a corresponding peak at 1332 nm can be used as evidence to exclude the possibility of cross contamination with Nd(III) and, moreover, we have found that the intensity of this ca. 1060 nm peak is also pH dependent (see Fig. S1, Supp. Info.), implicating the associated protonation equilibria of the adjacent amine backbone as an important factor. It is intuitive that the presence or absence of a formal positive charge in close proximity to the Yb(III) cation will have a dramatic effect on the subsequent crystal field splitting.
Figure 7.
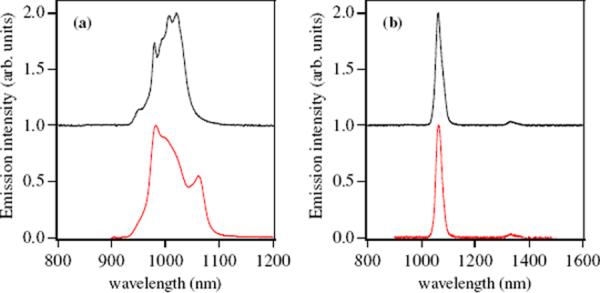
(a) Observed emission spectra (left) of [Yb(5LIO-Me-3,2-HOPO)2]− (black) and [Yb(H(2,2)-Me-3,2-HOPO)]− (red) in 0.1 M TRIS buffered aqueous solution at pH 7.4. (b) Corresponding emission spectra (right) for [Nd(5LIO-Me-3,2-HOPO)2]− (black) and [Nd(H(2,2)-Me-3,2-HOPO)]− (red) in 0.1 M TRIS buffered aqueous solution at pH 7.4. Spectra of the 5LIO-Me-3,2-HOPO complexes are horizontally offset for clarity.
To further examine this phenomenon, we have performed low temperature (77 K) measurements for these two complexes in a 1:1 (v/v) EtOH:MeOH glassing solvent, with resulting spectra that are remarkably different (Figure 8) and also behave very differently to the presence and absence of 1 % piperidine acting as a strong base (pKa ~ 11.2). For the [Yb(5LIO-Me-3,2-HOPO)2] complex, the spectra with and without added base present are indistinguishable as was the case in aqueous solution at differing pH's values. Moreover, the resulting emission spectra are significantly sharper at 77 K, and the spectrum can be successfully approximated by three overlapping Voigt functions (see Figure 8) to account for both homogeneous (Lorentzian) and inhomogeneous (Gaussian) line broadening effects. It is pertinent that we have previously11 assigned local D2d symmetry to the structurally related Eu(III) complex with 5LIO-1,2-HOPO on the basis of the observed crystal field splitting of the metal centered 5D0 → 7F0,1 transitions in solution. By contrast, further splitting of the spectra for the monoprotonated [Yb(H(2,2)-Me-3,2-HOPO)] complex suggests a lower symmetry at the metal centre. It has been shown that the splitting of Eu(III) emission bands can be useful to determine the site symmetry of this metal cation,38 and we have previously assigned local C2 symmetry to the related [Eu(H(2,2)-1,2-HOPO)] complex via detailed analysis of its' emission spectrum.12. The results with the Yb(III) cation and the H(2,2)-Me-3,2-HOPO ligand in this case are also consistent with this point group, since under C2 symmetry, the 2F7/2 ground state will be split into four doubly degenerate levels.39 Under the assumption that at 77 K there is insufficient thermalisation to populate the higher Stark levels of the emitting 2F5/2 state, we should thus anticipate four lines in the NIR emission spectrum, as are observed experimentally (Figure 8). Upon the addition of piperidine acting as a base, we note a significant change to the line shape of the observed luminescence (see Fig S2, Supp Info). Nonetheless, the shape of the observed NIR emission can still be well approximated by four overlapping Voigt functions, suggesting the proposed C2 geometry in alkaline solution is retained. The change in the splitting pattern may be correlated to structural perturbations due to the loss of H-bonding interactions previously noted40 for the H(2,2)-backbone, in going from the monoprotonated to doubly deprotonated state, which are translocated to the attached Me-3,2-HOPO chelates and results in a subtle change in the crystal field of the Yb(III) cation. Our initial results reported here hint that, by analogy to the case with Eu(III), a wealth of information concerning the first coordination sphere of the Yb(III) cation may be obtained from more detailed analysis of their NIR emission spectra. Analyses of this type will be enhanced in the NIR region since, while the magnitude of crystal field splitting for Eu(III) compared to Yb(III) remain similar (eg. ~ 100–1000 cm−1), the non-linearity of the wavelength scale with energy necessitates a vast improvement in the spectral window. The reason why this sensitivity to crystal field splitting is not evidenced with the corresponding [Nd(H(2,2)-Me-3,2-HOPO)] complex is unclear, but our investigations into this unusual effect are ongoing.
Figure 8.

(a) Observed emission spectra (left) of [Yb(5LIO-Me-3,2-HOPO)2] (black circles) in 1:1 (v/v) MeOH:EtOH at 77 K and fitted curve (blue) to a series of three overlapping Voigt functions (red) and (b) corresponding emission spectra (right) for [Yb(H(2,2)-Me-3,2-HOPO)] (black circles) in 1:1 (v/v) MeOH:EtOH at 77 K and fitted curve (blue) to a series of four overlapping Voigt functions (red).
In order to quantify the luminescence efficiency of the observed NIR emission, the overall quantum yields, Φtot, were determined for the Yb(III) complexes in 0.1 M TRIS buffered aqueous solution at pH 7.4, using [Yb(TTA)3(H2O)2] in toluene as a reference29,30 (Φref = 0.35 %) (see Fig. S3 and S4, Supp. Info). The resulting values we obtain for [Yb(5LIO-Me-3,2-HOPO)2] and [Yb(H(2,2)-Me-3,2-HOPO)] were Φtot = 0.22 % and Φtot = 0.09 % respectively. The absolute magnitude of these values remains modest, due to highly competitive non-radiative deactivation in the NIR region. Also, it is quite likely that energy transfer from the ligand to metal cation in these systems is not optimized, since the energy difference between the lowest energy ligand triplet T1 state and the metal is large, at ca. 7500 cm−1. Nonetheless, they still compare very well with some of the more emissive complexes appearing in the literature, such as the modified `fluorexon' derivatives from Werts et. al.,41 with Φtot = 0.45 % in D2O and the more recent 8-hydroxyquinoline derived podands reported by Bünzli et. al.,9 with Φtot = 0.37 % in H2O. For the Nd(III) Me-3,2-HOPO based complexes, given the inherently weaker signals involved, we made no effort to measure the quantum yields, although, qualitatively, their emission was approximately an order or magnitude weaker than the corresponding Yb(III) complexes.
Another indicator of the luminescence efficiencies are the luminescence lifetimes of the Ln(III) cations, which are summarized in Table 3. In 0.1 M TRIS buffered aqueous solution at pH 7.4, the values for Yb(III) are comparable with recently reported Yb(III) complexes, with observed lifetimes in the μs time domain.41,9 For the Nd(III) complexes, the decay times are significantly shorter with observed lifetimes in the nanosecond region, due to additional non-radiative deactivation pathways for the 4F3/2 excited state involving intervening electronic states. Interestingly, the magnitudes of the decay time are reversed in aqueous solution when comparing the Yb(III) and Nd(III) complexes, such that while [Yb(H(2,2)-Me-3,2-HOPO)] has a shorter lifetime than [Yb(5LIO-Me-3,2-HOPO)2] in aqueous solution, the situation is reversed for the Nd(III) complexes. These changes are likely a result of differing solvent accessibilities for the differing metal cations, and will be further discussed below (vide infra).
Table 3.
Summary of photophysical data for Yb(III) and Nd(III) complexes of Me-3,2-HOPO ligands.
| Complex | λmax (nm) | εmax (M−1cm−1) | τH2o (μs) | τD2O (μs) | Φ (%) | q |
|---|---|---|---|---|---|---|
| [Yb(5LIO-Me-3,2-HOPO)2] | 345.6 | 28,940 | 1.55 | 25.3 | 0.22 | 0.4 |
| [Yb(H(2,2)-Me-3,2-HOPO)] | 344.7 | 29,010 | 1.18 | 16.1 | 0.09 | 0.6 |
| [Nd(5LIO-Me-3,2-HOPO)2] | 345.7 | 28,280 | 0.082 | 0.655 | N/A | 1.0 |
| [Nd(H(2,2)-Me-3,2-HOPO)] | 344.7 | 28,850 | 0.098 | 0.471 | N/A | 0.7 |
Corresponding lifetimes measured in D2O (pD ~ 7.4) for all complexes were significantly longer, as summarized in Table 3, due to well known reduced non-radiative quenching effect of vOD versus vOH solvent vibrations. From these luminescence lifetimes in deuterated solvent, we were able to estimate the number of bound inner sphere water molecules, q, using the empirical relationships proposed by Beeby42 and Faulkner;43
determined for Yb(III) and Nd(III) respectively, where A = 1 μs for Yb(III) or 130 ns for Nd(III), B = 0.2 μs−1 and C = 0.4. The Δkobs term is evaluated as Δkobs = kobs(H2O) − kobs(D2O) with kobs = 1/τobs, using units of ns and μs for the Nd(III) and Yb(III) lifetimes respectively.
Hence, for [Yb(5LIO-Me-3,2-HOPO)2], we obtain a q value of ca. 0.4. We take this value as a slight overestimate, indicating an absence of water molecules in the first coordination sphere in accordance with the observed solid state structure, and the residual is most likely due to second sphere contributions such as vC-H and vN-H vibrations of the ligand which are not correctly accounted for by the B term. For the [Yb(H(2,2)-Me-3,2-HOPO)] complex, the q value we obtain of ca. 0.6 is slightly higher. However, since almost identical second sphere contributions should exist with this octadentate ligand topology, it is most likely that this value is similarly an overestimate, rather than an equilibrium involving a hydrated species, and we propose an eight coordinate q = 0 coordination geometry for the [Yb(H(2,2)-Me-3,2-HOPO)] complex. Notably, this result differs to that obtained12 for the isomeric H(2,2)-1,2-HOPO complex with Eu(III), where lifetime data clearly indicated a residual inner sphere water molecule (q = 1). The reason for the proposed differences can be rationalized as a combination of two factors; primarily due to the presence of four methyl groups in the 1' position of the Me-3,2-HOPO chromophores, which introduces additional steric crowding at the `open face' of the tetrapodal complex, and also the smaller effective ionic radius of the Yb(III) cation (CN = 8, 0.985 Å) compared to Eu(III) (CN = 8, 1.066 Å) may play a role.
For the Nd(III) complexes, the metal centre also has a much larger effective ionic radius (CN = 8, 1.109 Å) when compared with Yb(III). Using Faulkner's equation, we determine q values of ca. 1.0 and 0.7 for the 5LIO-Me-3,2-HOPO and H(2,2)-Me-3,2-HOPO Nd(III) complexes respectively, both of which are larger than the corresponding values with Yb(III). While these values should be interpreted with some caution, they no doubt reflect an improved solvent accessibility toward the larger metal. Moreover, while the increase in q for [Nd(H(2,2)-Me-3,2-HOPO)] is only marginal, the obtained q for [Nd(5LIO-Me-3,2-HOPO)2] is more than doubled, and this is accompanied by the previously mentioned concomitant switch in the order of luminescence lifetimes in aqueous solution. Hence, while [Yb(H(2,2)-Me-3,2-HOPO)] has a shorter lifetime than [Yb(5LIO-Me-3,2-HOPO)2] in aqueous solution, the opposite is true of the Nd(III) complexes. Evidently, the steric demands and rigidity of the octadentate [Yb(H(2,2)-Me-3,2-HOPO)] and [Nd(H(2,2)-Me-3,2-HOPO)] complexes can provide more adequate protection from non-radiative solvent deactivation, and effectively exclude the entry of water molecules to the inner coordination sphere of the metal. For the bis-tetradentate complexes, it would appear the more flexible ligands in combination with the larger Nd(III) metal ion can accommodate an equilibrium involving a hydrated [Nd(5LIO-Me-3,2-HOPO)2(H2O)] species.
Conclusions
The 1-methyl-3-hydroxy-pyridin-2-one (Me-3,2-HOPO) chelate group has proven to be a remarkably efficient sensitizer with the NIR emitting Ln(III) cations (Ln = Yb, Nd), displaying exceptional photophysical performance in terms of their overall quantum yield and luminescence lifetimes in aqueous solution. Furthermore, we have shown that, by analogy to the case for Eu(III), a detailed analysis of the NIR emission spectra from Yb(III) can also be useful in determining the site symmetry of the metal, as a result of readily observed crystal field splitting effects. In addition to their efficient NIR luminescence, these Me-3,2-HOPO based systems also demonstrate exceptional aqueous stabilities which will be of paramount importance in their further development should complexes such as these be used for in vivo applications, with pM values on the order of pYb ~ 21.9, no doubt reflecting the use of an exclusively oxygen containing donor set.
Supplementary Material
Table 1.
Summary of crystal data for the 5LIO-Me-3,2-HOPO ligand and corresponding Yb(III) complex.
| 5LIO-Me-3,2-HOPO | [Yb(5LIO-Me-3,2-HOPO)2] | |
|---|---|---|
| empirical formula | C18H26N4O9 | C39H40N8O19Yb |
| mol. wt. [g.mol−1] | 442.43 | 1105.88 |
| crystal system | Orthorhombic | Monoclinic |
| Space group | Pca21 | P21/b |
| a [Å] | 24.438(11) | 13.451 (2) |
| b[Å] | 9.722 (6) | 20.757 (3) |
| c[Å] | 8.925 (4) | 16.969 (3) |
| α[deg.] | 90 | 90 |
| β[deg.] | 90 | 101.126(2) |
| γ[deg.] | 90 | 90 |
| volume [Å3] | 2120.5 (19) | 4649.0 (13) |
| Z | 4 | 4 |
| density, ρ[g cm−3] | 1.386 | 1.58 |
| absorption coefficient, μ[mm−1] | 0.112 | 2.094 |
| F(000) | 936 | 2232 |
| crystal size [mm3] | 0.35 × 0.12 × 0.07 | 0.3 × 0.2 × 0.1 |
| Temperature [K] | 135 (2) | 104 (2) |
| Wavelength | 0.71073 | 0.71073 |
| θ range for data collection [deg] | 3.52 to 26.01 | 3.24 to 25.68 |
| limiting indices | −25 < h < 25, −1 < k <11, −10 < l < 10 | −16 < h < 16, −24 < k < 25, −11 < l < 20 |
| measured reflections | 1892 | 15656 |
| independent reflections | 1285 (Rint = 0.0245) | 15036 (Rint = 0.0122) |
| completeness to θ | 0.85* | 0.979 |
| refinement method | full matrix least squares on F2 | full matrix least squares on F2 |
| data / restraints /parameters | 1892/2/292 | 15656/1/1259 |
| goodness-of-fit on F2 | 1.001 | 1.035 |
| final R indices [I>2ρ(I)] | R1 = 0.0404, wR2 =0.0886 | R1 = 0.0253; wR2 =0.0642 |
| R indices (all data) | R1 = 0.0711, wR2 =0.1007 | R1 = 0.0271; wR2 =0.0651 |
| largest diff. peak and hole [eÅ −3] | 0.126 and −0.146 | 0.816 and −0.698 |
Data set incomplete due to crystal decomposition
Acknowledgements
This work was partially supported by the NIH (Grant HL69832) and supported by the Director, Office of Science, Office of Basic Energy Sciences, and the Division of Chemical Sciences, Geosciences, and Biosciences of the U.S. Department of Energy at LBNL under Contract No. DE-AC02-05CH11231. This technology is licensed to Lumiphore, Inc. in which some of the authors have a financial interest. Financial support was provided to C.J.J. and to M.S. by the German Research Foundation (DFG). The authors thank Dr. Kathleen Durkin for assistance with the TD-DFT calculations.
Footnotes
Supporting Information Available. X-ray crystallographic files (in CIF format), additional variable pH Yb(III) luminescence spectra (Fig. S1) and symmetry fits (Fig. S2), and quantum yield determinations (Fig S3 and S4). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Bünzli J-CG. Acc. Chem. Res. 2006;39:53–61. doi: 10.1021/ar0400894. [DOI] [PubMed] [Google Scholar]
- 2.Faulkner S, Beeby A, Dickins RS, Parker D, Williams JAG. J. Fluorescence. 1999;9:45–49. [Google Scholar]
- 3.Weissleder R, Ntziachristos V. Nat. Med. 2003;9:123–128. doi: 10.1038/nm0103-123. [DOI] [PubMed] [Google Scholar]
- 4.Faulkner S, Pope SJA, Burton-Pye BP. Appl. Spectrosc. Rev. 2005;40:1–31. [Google Scholar]
- 5.Frangioni JV. Curr. Opin. Chem. Biol. 2003;7:626–634. doi: 10.1016/j.cbpa.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 6.Aebischer A, Gumy F, Buenzli J-CG. Phys. Chem. Chem. Phys. 2009;11:1346–1353. doi: 10.1039/b816131c. [DOI] [PubMed] [Google Scholar]
- 7.Moore EG, Samuel APS, Raymond KN. Acc. Chem. Res. 2009;42:542–552. doi: 10.1021/ar800211j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ward MD. Coord. Chem. Rev. 2007;251:1663–1677. [Google Scholar]
- 9.Comby S, Imbert D, Chauvin A-S, Bunzli J-CG. Inorg. Chem. 2006;45:732–743. doi: 10.1021/ic0515249. [DOI] [PubMed] [Google Scholar]
- 10.Nonat A, Imbert D, Pecaut J, Giraud M, Mazzanti M. Inorg. Chem. 2009;48:4207–4218. doi: 10.1021/ic9000076. [DOI] [PubMed] [Google Scholar]
- 11.Moore EG, Xu J, Jocher CJ, Castro-Rodriguez I, Raymond KN. Inorg. Chem. 2008;47:3105–3118. doi: 10.1021/ic702144n. [DOI] [PubMed] [Google Scholar]
- 12.Moore EG, Jocher CJ, Xu J, Werner EJ, Raymond KN. Inorg. Chem. 2007;46:5468–5470. doi: 10.1021/ic700364t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moore EG, Seitz M, Raymond KN. Inorg. Chem. 2008;47:8571–8573. doi: 10.1021/ic801060x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moore EG, Szigethy G, Xu J, Palsson L-O, Beeby A, Raymond KN. Angew. Chem., Int. Ed. 2008;47:9500–9503. doi: 10.1002/anie.200802337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu J, Franklin SJ, Whisenhunt DW, Jr., Raymond KN. J. Amer. Chem. Soc. 1995;117:7245–7246. [Google Scholar]
- 16.Wagnon BK, Jackels SC. Inorg. Chem. 1989;28:1923–1927. [Google Scholar]
- 17.Xu J, Kullgren B, Durbin PW, Raymond KN. J. Med. Chem. 1995;38:2606–2614. doi: 10.1021/jm00014a013. [DOI] [PubMed] [Google Scholar]
- 18.SMART, Version 5.059: Area-Detector Software Package. Bruker Analytical X-ray Systems, Inc.; Madison, WI: 1999. [Google Scholar]
- 19.SAINT, Version 7.07B: SAX Area-Detector Integration Program. Siemens Industrial Automation, Inc.; Madison, WI: 2005. [Google Scholar]
- 20.XPREP, Version 6.12: Part of the SHELXTL Crystal Structure Determination Package. Bruker AXS Inc.; Madison, WI: 1995. [Google Scholar]
- 21.SADABS, Version 2.10: Siemens Area Detector Absorption Correction Program. University of Gottingen; Gottingen, Germany: 2005. [Google Scholar]
- 22.Farrugia LJ. J. Appl. Crystallogr. 1999;32:837–838. WinGX, 1.70.01. [Google Scholar]
- 23.Altomare A, Cascarano G, Giacovazzo C, Guagliardi A. J. Appl. Crystallogr. 1993;26:343–350. SIR92. [Google Scholar]
- 24.Sheldrick GM. Programs for Crystal Structure Analysis. Institut fur Anorganische Chemie der Universitat; Gottingen, Germany: 1998. SHELX97. [Google Scholar]
- 25.ORTEP3 for Windows. Farrugia LJ. J. Appl. Crystallogr. 1997;30:565. [Google Scholar]
- 26.Pierre VC, Botta M, Aime S, Raymond KN. Inorg. Chem. 2006;45:8355–8364. doi: 10.1021/ic061262q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Doble DMJ, Melchior M, O'Sullivan B, Siering C, Xu J, Pierre VC, Raymond KN. Inorg. Chem. 2003;42:4930–4937. doi: 10.1021/ic026240s. [DOI] [PubMed] [Google Scholar]
- 28.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, C., J. R., Montgomery JA, Jr., Vreven T, Kudin KN, B., J. C., Millam JM, Iyengar SS, Tomasi J, Barone V, M., B., Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, I., M., Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, L., X., Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, A., A. J., Cammi R, Pomelli C, Ochterski JW, Ayala PY, M., K., Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, M., D. K., Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, W., M. W., Gonzalez C, Pople JA. Gaussian 03, revision C.02. Gaussian, Inc.; Wallingford, CT: 2004. [Google Scholar]
- 29.Tsvirko MP, Meshkova SB, Venchikov VY, Bol'shoi DV. Opt. Spectrosc. (Engl. Transl.) 1999;87:866–870. [Google Scholar]
- 30.Meshkova SBT, Z. M., Bolshoy DV, Beltyukova SV, Tsvirko MP, Venchikov V. Ya. Acta Physica Polonica, A. 1999;95:983–990. [Google Scholar]
- 31.Xu J, Radkov E, Ziegler M, Raymond KN. Inorg. Chem. 2000;39:4156–4164. doi: 10.1021/ic000063i. [DOI] [PubMed] [Google Scholar]
- 32.Moore EG, Xu J, Jocher CJ, Werner EJ, Raymond KN. J. Am. Chem. Soc. 2006;128:10648–10649. doi: 10.1021/ja062597+. [DOI] [PubMed] [Google Scholar]
- 33.Porai-Koshits MA, Aslanov LA. Zhurnal Strukturnoi Khimii. 1972;13:266–276. [Google Scholar]
- 34.Kepert DL. Prog. Inorg. Chem. 1978;24:179–249. [Google Scholar]
- 35.Smith RM, Martell AE. Critical Stability Constants. Plenum Press; New York: 1976. [Google Scholar]
- 36.Dreuw A, Head-Gordon M. J. Am. Chem. Soc. 2004;126:4007–4016. doi: 10.1021/ja039556n. [DOI] [PubMed] [Google Scholar]
- 37.Cunningham KG, Newbold GT, Spring FS, Stark J. J. Chem. Soc. 1949:2091–2094. [Google Scholar]
- 38.Murray GMS, R. V., Peterson JR. Inorg. Chim. Acta. 1990;176:233–240. [Google Scholar]
- 39.Antic-Fidancev E, Jorma H, Lastusaari M. J. Phys.: Condens. Matter. 2003;15:863–876. [Google Scholar]
- 40.Samuel APS, Moore EG, Melchior M, Xu J, Raymond KN. Inorg. Chem. 2008;47:7535–7544. doi: 10.1021/ic800328g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Werts MHV, Verhoeven JW, Hofstraat JW. J. Chem. Soc., Perkin Trans. 2. 2000:433–439. [Google Scholar]
- 42.Beeby A, Clarkson IM, Dickins RS, Faulkner S, Parker D, Royle L, de Sousa AS, Williams JAG, Woods M. J. Chem. Soc., Perkin Trans. 2. 1999:493–503. [Google Scholar]
- 43.Faulkner S, Beeby A, Carrie M-C, Dadabhoy A, Kenwright AM, Sammes PG. Inorg. Chem. Commun. 2001;4:187–190. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.