

Abstract
Expression of matrix metalloproteinases (MMPs), especially MMP9 correlates with blood–brain barrier (BBB) disruption during many neuroinflammatory diseases. During neurotropic coronavirus virus (JHMV) induced encephalomyelitis, MMP9 activity is restricted to neutrophils. Furthermore, myeloid cell depletion implicated MMP9 in facilitating leukocyte central nervous system (CNS) infiltration via loss of BBB integrity. The requirement of MMP9 in BBB disruption was thus assessed in JHMV infected MMP9 deficient (MMP9−/−) mice. Depletion of neutrophils reduced CNS accumulation of monocytes and T cells, albeit without affecting overall pathogenesis. By contrast, infected MMP9−/− mice revealed no differences in CNS leukocyte infiltration, composition or localization, consistent with BBB disruption similar to wild‐type (WT) mice. Unimpaired T cell mediated virus control supported an unexpectedly redundant role of MMP9 in promoting leukocyte access to the brain parenchyma. Although MMP9 deficiency did not expand the overall limited pattern of MMP expression during JHMV infection, it coincided with MMP3 upregulation. MMP3 expression remained largely confined to astrocytes, similar to WT mice. These data demonstrate that neutrophil‐derived MMP9 is not the sole mediator facilitating parenchymal leukocyte entry via BBB disruption during viral encephalomyelitis. Moreover, significantly enhanced MMP3 expression by astrocytes in infected MMP9−/− mice suggests an active role of resident cells in participating and potentially collaborating with infiltrating cells in regulating BBB permeability. Overall, these results highlight the complexity of targeting individual MMPs as a strategy to regulate inflammation. © 2011 Wiley‐Liss, Inc.
Keywords: coronavirus, central nervous system (CNS), leukocytes, matrix metalloproteinase (MMP), astrocytes
INTRODUCTION
Leukocyte recruitment into the central nervous system (CNS) parenchyma is highly regulated by the blood–brain barrier (BBB), limiting invasion of cell‐associated pathogens as well as immune‐mediated neuronal dysregulation and tissue damage (Bechmann et al.,2007). However, leukocyte entry into the CNS parenchyma is essential to protect the host following CNS infection. Among key factors controlling BBB permeability are matrix metalloproteinases (MMPs), which belong to a large family of endoproteinases primarily involved in turnover and remodeling of the extracellular matrix (Candelario‐Jalil et al.,2009). CNS infections, degenerative diseases, autoimmune inflammation, as well as stroke are all associated with upregulation of MMPs, which degrade components of the basement membrane and extracellular matrix, thus facilitating leukocyte invasion (Hartung and Kieseier,2000; Parks et al.,2004; Rosell and Lo,2008; Wang et al.,2008). Intracranial MMP injection into naïve mice supports the role of MMPs in increasing BBB permeability and CNS leukocyte migration (Anthony et al.,1998). Conversely, inhibition of MMPs decreases BBB breakdown, limits parenchymal leukocyte entry and ameliorates clinical symptoms (Liedtke et al.,1998; Toft‐Hansen et al.,2006; Tran et al.,1998). Furthermore, broad‐spectrum MMP inhibitors retain leukocytes in the perivascular space supporting a role for MMPs in degrading the glia limitans (Toft‐Hansen et al.,2006).
MMP9 is the most prominent in promoting BBB disruption among the various MMPs associated with CNS inflammation. Its upregulation coincides with loss of BBB integrity after lipopolysaccharide injection, during experimental autoimmune encephalitis (EAE), meningitis, viral encephalitis, as well as trauma‐induced neuroinflammation (Dubois et al.,1999; Mun‐Bryce and Rosenberg,1998; Noble et al.,2002; Paul et al.,1998; Sporer et al.,1998; Wang et al.,2008). Disease activity in multiple sclerosis patients further coincides with high MMP9 serum levels (Opdenakker et al.,2003). MMP9 most likely functions by disrupting the glia limitans, based on its ability to cleave dystroglycan, an anchoring component of astrocyte endfeet, and its high affinity for Laminins 1 and 2, two major components of the glia limitans (Agrawal et al.,2006; Sixt et al.,2001). Although these data implicate MMP9 in promoting leukocyte access into the CNS parenchyma, an essential role for MMP9 in mediating disruption of the glia limitans has not been demonstrated.
Characterization of discrete roles of individual MMPs is complicated by the wide variation of expression patterns, kinetics, substrate, and cell specificity in different inflammatory settings. The present study used a viral encephalomyelitis model characterized by a highly limited MMP expression pattern (Zhou et al.,2005) to define the role of MMP9 in BBB disruption at the glia limitans. Infections with both lethal and sublethal neurotropic JHM strains of mouse hepatitis virus (JHMV) induce only three MMPs: MMP9 is produced by inflammatory neutrophils (Savarin et al.,2010; Zhou et al.,2003). MMP3 expression is largely restricted to astrocytes (Zhou et al.,2003,2005). Finally, MMP12 is produced by both CNS resident cells and infiltrating leukocytes (Zhou et al.,2005). Compared to the sublethal infection, the lethal infection is characterized by significantly increased neutrophil inflammation as well as MMP expression; combined neutrophil/monocyte ablation abrogated CNS MMP9 expression and BBB disruption (Zhou et al.,2003). Anti‐CXCR2 treatment to block CNS neutrophil migration during the sublethal infection also reduced CNS leukocyte accumulation, impaired viral control, and accelerated mortality (Hosking et al.,2009). Together, these data supported a protective role of neutrophils in promoting BBB disruption via MMP9 release.
To identify MMP9 as the key neutrophil component enhancing BBB disruption during sublethal JHMV infection, lymphocyte access and viral control were compared in MMP9 deficient (MMP9−/−) and neutrophil depleted mice. Sublethal infection is characterized by early CNS neutrophil and monocyte recruitment, followed by T and B cell infiltration, both critical for virus control (Bergmann et al.,2006). Neutrophil depletion reduced infiltrating monocytes and T cells by ∼50%–60%, but did not affect clinical disease or viral control. Surprisingly, infected MMP9−/− mice exhibited identical BBB permeability, CNS leukocyte recruitment and distribution as wild‐type (WT) mice. Analysis of potential MMP dysregulation in the absence of MMP9 revealed a marked, transient increase specifically of MMP3 in the CNS. Similar to WT controls, MMP3 expression was restricted to astrocytes, suggesting that enhanced focal MMP3 activity at the parenchymal side of the BBB contributes to BBB disruption and may compensate for the absence of MMP9. Overall, these results demonstrate that neither neutrophils nor MMP9 are essential in mediating protective lymphocyte access to the CNS parenchyma during sublethal viral encephalomyelitis. Limited MMP activation in this model further implicates compensatory or alternative mechanisms breaching BBB permeability.
MATERIALS AND METHODS
Mice, Virus, and Poly I:C
Homozygous C57BL/6‐MMP9 deficient (MMP9−/−) mice (Dubois et al.,1999) backcrossed for 12 generations to C57BL/6 mice were provided by Dr. Robert Fairchild, Cleveland Clinic. C57BL/6 mice were purchased from the National Cancer Institute (Frederick, MS). All mice were used at 6–7 weeks of age and injected intracranially (i.c.) in the left hemisphere with 30 μL of endotoxin‐free Dulbecco's phosphate‐buffered saline (PBS) containing 250 plaque‐forming units (PFU) of the glia tropic 2.2v‐1 monoclonal antibody (mAb)‐derived variant of mouse hepatitis virus strain JHM (Fleming et al.,1986). Clinical disease severity was graded daily as described (Fleming et al.,1986): 0, healthy; 1, hunched back; 2, partial hind limb paralysis or inability to maintain the upright position; 3, complete hind limb paralysis; 4, moribund or dead. For innate immune activation mice were injected i.c. with 200 μg poly (I:C) (high molecular weight) (Invivogen, San Diego, CA) in 30 μL endotoxin‐free PBS. Brains were removed at 2, 6, and 12 h post injection (p.i.), snap frozen in liquid nitrogen for gene expression analysis. Neutrophils were depleted by intraperitoneal (i.p.) injection of 250 μg of anti‐Ly6G (clone 1A8) every other day, starting 2 days prior to infection until Day 7 p.i. Control mice were injected with 250 μg of irrelevant mAb (clone GL113). Virus titers were determined from clarified brain homogenates by plaque assay on DBT cell monolayers as described (Lin et al.,1997; Stohlman et al.,2008). All procedures were carried out in compliance with Institutional Animal Care and Use Committee approved protocols.
Isolation of CNS Derived Cells and Flow Cytometry
After perfusion with PBS, brains were homogenized individually in Dulbecco's PBS using Tenbroeck tissue homogenizers. Supernatants for virus titer determination were collected after centrifugation at 400 × g for 7 min at 4°C and CNS derived cells were isolated using percoll gradients (Pharmacia, Uppsala, Sweden) as previously described (Savarin et al.,2010). After isolation, cells were washed in RPMI 1640‐HEPES medium prior to analysis. Following pre‐incubation with mouse serum and anti‐mouse CD16/CD32 mAb (clone 2.4G2, BD PharMingen, San Diego, CA), cells were stained for surface markers in 0.1% bovine serum albumin (BSA) in PBS for 30 min on ice with FITC‐, PE‐, PerCP‐, or APC‐ conjugated mAb (all from BD PharMingen except when indicated), including anti‐CD45 (clone Ly‐5), CD4 (clone GK1.5), CD8 (clone 53‐6.7), CD11b (clone m1/70), F4/80 (Serotec, Raleigh, NC), and Ly6G (clone 1A8). Samples were analyzed using a FACS Calibur flow cytometer and CellQuest Software (BD Biosciences, Mountain View, CA).
Individual CNS derived cell populations were purified from the CNS of infected mice (n = 4) as described (Phares et al.,2010). Briefly, brains and spinal cords were finely minced, digested in 0.25% Trypsin for 30 min at 37°C, and trypsin activity inhibited by adding RPMI 1640‐HEPES supplemented with 20% newborn calf serum. Cells were collected by centrifugation, washed, and enriched using percoll gradients. Cells were stained with APC‐CD45 mAb to separate glial cells (CD45−), microglia (CD45lo) and infiltrating leukocytes (CD45hi) using a FACS Aria (BD Biosciences) cell sorter. Purified cells were resuspended in TRIzol reagent (Invitrogen, Carlsbad, CA) for subsequent gene expression analysis.
Zymography
Unfractionated cells purified from the CNS were resuspended in lysis buffer (1% Triton X‐100, 300 mM NaCl, 50 mM tris(hydroxymethyl)aminomethane (Tris), pH 7.4) and lysates from 2.5 × 105 cells separated on 10% acrylamide gels containing 1% gelatin (Bio‐Rad, Hercules, CA). Following electrophoresis, gels were consecutively placed in 1X renaturing buffer (Bio‐Rad) for 30 min at room temperature, 1X developing buffer (Bio‐Rad) for 20 min at room temperature, and then overnight at 37°C. Gels were stained with 0.25% Coomassie brilliant blue R‐250 (Bio‐Rad) and destained with the destain solution (Bio‐Rad).
Gene Expression Analysis
Individual brains were frozen in liquid nitrogen and stored at −80°C. RNA was extracted with TRIzol reagent (Invitrogen) according to the manufacturer's instructions. cDNA was synthesized using 2 μg of RNA, SuperScript II Reverse Transcriptase (Invitrogen) with oligo(dT)12–18. Quantitative real‐time PCR was performed using the SYBR green kit (Roche, Basel, Switzerland), a LightCycler (Roche) and the following primer sets: MMP2: F: 5′‐TTCCCTAAGCTCATCGCAGACT‐3′, R: 5′‐CACGCTCTTGAGACTTTGGTTCT‐3′; MMP3: F: 5′‐TTTAAAGGAAATCAGTTCTGGGCTATA‐3′, R: 5′‐CGA TCTTCTTCACGGTTGCA‐3′; MMP7: F: 5′‐TGGCT TCGAAGGAGAGATC‐3′, R: 5′‐CGAAGGCATGACCTAG AGTGTTC‐3′; MMP12: F: 5′‐GGAGCTCACGGAGACTTC AACT‐3′, R: 5′‐CCTTGAATACCAGGTCCAGGATA‐3′; MMP14: F: 5′‐TAAGCACTGGGTGTTTGACGAA‐3′, R: 5′‐CCCTCGGCCAAGCTCCT‐3′; TIMP1: F: 5′‐CCAGAG CCGTCACTTTGCTT‐3′, R: 5′‐AGGAAAAGTAGACAGTG TTCAGGCTT‐3′; TNF: F: 5′‐GCCACCACGCTCTTCTG TCT‐3′, R: 5′‐GGTCTGGGCCATAGAACTGATG‐3′; IL‐1β: F: 5′‐GACGGCACACCCACCCT‐3′, R: 5′‐AAACCG TTTTTCCATCTTCTTCTTT‐3′; Neutrophil elastase: F: 5′‐AGAGGCGTGGAGGTCATTTCT‐3′, R: 5′‐GGGCTGC TGACATGACGAA‐3′; Ubiquitin: F: 5′‐TGGCTATTAATT ATTCGGTCTGCAT‐3′, R: 5′‐GCAAGTGGCTAGAGTGCA GAGTAA‐3′. Taqman primers and 2X TaqMan fast master mix (Applied Biosystems, Carlsbad, CA) were used to assessed Cathepsin G and MMP8 mRNA levels. mRNA expression was compared relative to ubiquitin mRNA and converted to a linearized value using the following formula: [1.8e(Ctubiquitin−Ctgenex)] × 105 (Zhou et al.,2005).
Blood–Brain Barrier Permeability
Ten minutes before sacrifice, mice were injected i.p. with 100 μL of sodium fluorescein (NaF; 100 mg/mL) (Sigma, St Louis, MO) diluted in PBS. Following perfusion with PBS, brains were removed and homogenized in 4 mL PBS using Tenbroeck tissue homogenizers. Supernatants were collected following centrifugation at 400 × g for 7 min at 4°C and treated with an equal volume of 15% trichloroacetic acid. Samples were centrifuged at 10,000 × g for 10 min. NaOH was added to a final concentration of 1 M. Fluorescence was determined at 530 nm using a Spectramax M2 microplate reader (Molecular Devices, Sunnyvale, CA) and compared using a NaF standard curve.
Immunohistology
Leukocyte distribution at the BBB was determined as described (Savarin et al.,2010). Briefly, mice were perfused with PBS followed by 4% paraformaldehyde. Brains and spinal cords were then stored in cryoprotection solution until 30‐μm sections were prepared using a microtome. Before staining, sections were treated with 1% Triton X‐100 for 30 min followed by blocking solution (1% BSA and 10% normal goat serum in PBS) for 30 min at RT. Sections were stained with rabbit anti‐mouse laminin (Cedarlane Laboratories, ON, Canada) and rat anti‐mouse CD45 Ab (Serotec) overnight at 4°C. Alexa Fluor 594 goat anti‐rabbit (Invitrogen) and biotinylated anti‐rat Ab (Vector Laboratories, Burlingame, CA) were added for 1 h at RT, followed by streptavidin FITC (Vector Laboratories) for 1 h at RT. To visualize MMP3 and Glial Fibrillary Acidic Protein (GFAP), mice were perfused with ice‐cold PBS, brains dissected, frozen in OCT and stored at −80°C. 10‐μm sections were fixed for 5 min in acetone at −20°C. After incubation in blocking solution for 30 min, sections were incubated overnight at 4°C with goat anti‐mouse MMP3 (Santa Cruz Biotechnology, Santa Cruz, CA) and mouse anti‐mouse GFAP (Sigma). For immunofluorescence, Alexa Fluor 488 goat anti‐mouse (Invitrogen) and Rhodamine donkey anti‐goat (Jackson ImmunoResearch, West Grove, PA) secondary antibodies were added for 1 h at RT. Sections were mounted with Vectashield mounting medium with 4′‐6‐Diamidino‐2‐phenylindole (DAPI) (Vector Laboratories). For immunohistochemistry, biotinylated anti‐goat (Jackson ImmunoResearch) secondary antibody was added for 1 h at RT. Sections were incubated with ABC peroxidase kit (Vector Laboratories) for 1 h at RT and revealed with diaminobenzidine. Tissue sections were analyzed using a Leica DM4000B microscope (Leica, Microsystems, Wetzlar, Germany).
Statistical Analyses
Statistical differences were calculated using a two‐tailed unpaired Student's t test or the Mann‐Whitney nonparametric test. P values <0.05 were considered as significant.
RESULTS
Decreased CNS Inflammation in Neutrophil Depleted Mice
Sustained BBB integrity coincident with the absence of MMP9 expression in neutropenic mice infected with lethal JHMV suggested that neutrophil‐secreted MMP9 disrupts the BBB (Zhou et al.,2003). However, the rapid mortality after lethal JHMV infection prevented an assessment of altered BBB permeability specifically on CNS T cell recruitment. In addition, a subsequent study using sublethal JHMV infection revealed a role of monocytes in promoting BBB disruption at the glia limitans (Savarin et al.,2010). To more precisely characterize the role of neutrophil‐derived MMP9 in enhancing CNS leukocyte access during sublethal JHMV infection, mice were treated with anti‐Ly6G mAb to selectively deplete neutrophils (Daley et al.,2008). Neutrophil depletion did not affect disease severity or mortality compared to control animals (Fig. 1A and data not shown). Clinical symptoms during sublethal JHMV infection are largely determined by lymphocyte infiltration and antiviral effector function, rather than magnitude of viral replication (Ireland et al.,2009; Kapil et al.,2009; Savarin et al.,2008). The similar disease course therefore suggested that neutrophils expressing MMP9 were redundant in promoting CNS leukocyte infiltration. Analysis of total numbers of CD45hi cells within the CNS showed that overall leukocyte infiltration was not affected by neutrophil depletion at Days 3 and 5 p.i. (Fig. 1B). However, at the peak of inflammation (Day 7 p.i.) and subsequently, infiltrating cells were reduced by ∼50% (Fig. 1B). In control mice CNS neutrophil recruitment peaked at Day 3 p.i. and decreased between Days 5–7 p.i., but comprised an overall small fraction of infiltrating cells (Fig. 1C). Very few neutrophils were detected at any time in the CNS of anti‐Ly6G treated mice confirming depletion efficiency (Fig. 1C). Neutrophil depletion resulted in decreased macrophages (CD45hiF4/80+), CD4+ and CD8+ T cell populations at Day 7 p.i., although no differences were evident prior to peak leukocyte accumulation (Fig. 1C). By Day 10 p.i., macrophages were decreased relative to Day 7 p.i. in both groups reaching similar overall levels. By contrast, both CD4+ and CD8+ T cell subsets remained reduced in neutropenic mice relative to controls, with differences waning by Day 14 p.i. (Fig. 1C). Despite decreased T cell numbers between Days 7 and 10 p.i., viral replication was controlled below the level of detection by Day 14 p.i. similar to neutrophil sufficient mice (data not shown). These data indicate that neutrophils promote, but are not essential for T cell migration across the BBB or viral control during sublethal JHMV infection.
Figure 1.

Neutrophil depletion reduces CNS leukocyte accumulation. A: Disease severity was scored daily (n = 60 per group) after JHMV i.c. infection using the following grades: 0, healthy; 1, hunched back or ruffled fur; 2, partial hind limb paralysis or inability to maintain the upright position; 3, complete hind limb paralysis; 4, moribund or dead. Numbers of bone marrow‐derived infiltrating leukocytes (CD45hi) (B), neutrophils (Ly6G+), macrophages (F4/80+), CD4+, and CD8+ T cells (C) were determined in the CNS of control and neutrophil‐depleted mice between Day 3 and 14 p.i. using flow cytometry. Data represent the mean (±SEM) of six mice per group per time point from two separate experiments. *P < 0.05.
MMP9 Deficiency does not Affect Disease Severity or Leukocyte Infiltration
Prepackaged MMP9 is localized exclusively in neutrophils during JHMV infection (Savarin et al.,2010; Zhou et al.,2005). Consistent with the burst of neutrophil accumulation at Day 3 in WT mice (Fig. 1C), MMP9 activity was prominent within the CNS at Day 3 p.i., but declined by Day 5 p.i. (Fig. 2A). To demonstrate that MMP9 is the major protease facilitating leukocyte extravasation into the CNS parenchyma, JHMV pathogenesis was compared in syngeneic MMP9−/− and WT mice. Both the groups exhibited clinical signs of encephalitis, characterized by hunched posture and ruffled fur, at Day 6 p.i. and subsequently progressed to hind limb paralysis (Fig. 2B); survival rates were also not altered (data not shown). The absence of MMP9 therefore had no overt consequences on clinical manifestations or disease progression, similar to neutrophil‐depleted mice.
Figure 2.
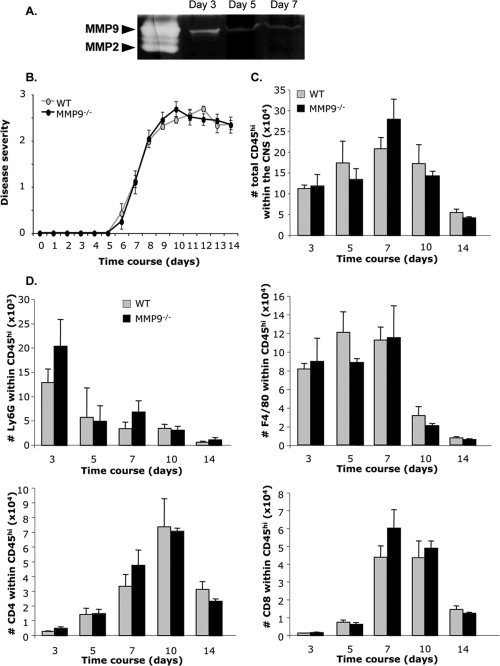
Similar disease severity and CNS inflammation in WT and MMP9−/− mice. A: MMP9 activity was analyzed by zymography in the CNS of infected WT mice at days 3, 5, and 7 p.i. B: Clinical symptoms were monitored daily in WT (n = 80) and MMP9−/− (n = 80) mice using the grades described in Fig. 1. Total numbers of inflammatory leukocytes (CD45hi) (C), neutrophils (Ly6G+), macrophages (F4/80+), CD4+, and CD8+ T cells (D) were analyzed by flow cytometry in the CNS of WT and MMP9−/− mice. Data represent the means (±SEM) of nine mice per group per time point from three separate experiments.
To assess whether MMP9 deficiency reduced CNS leukocyte accumulation to the same extent as neutrophil depletion, CD45hi inflammatory cells and their composition were compared to WT mice. Unexpectedly, CD45hi cells in the CNS were similar in infected MMP9−/− compared to WT mice throughout Days 3–14 p.i. (Fig. 2C). CNS accumulation of neutrophils (Ly6G+), macrophages (F4/80+), CD4+, and CD8+ T cells was also not affected by MMP9 deficiency. Neutrophils peaked at Day 3 p.i. and decreased similarly in both groups (Fig. 2D), whereas macrophages only decreased to low levels at Day 10, as T cells accumulated (Fig. 2D). Both T cell subsets peaked at similar levels between Days 7 and 10 p.i. in WT and MMP9−/− mice. No differences in total leukocytes, as well as subsets in the CNS of MMP9−/− mice suggested MMP9 is not essential in disrupting BBB integrity.
MMP9 Deficiency does not Alter BBB Disruption or Virus Clearance
To confirm the apparently redundant role of MMP9 in disrupting the BBB during the course of viral encephalitis, mice were injected with NaF at distinct times following infection (Fig. 3A). At Day 3 p.i., the peak of neutrophil accumulation (Fig. 2D), the loss of BBB integrity was similar comparing both groups. No differences were detected at later time points p.i. and BBB function was restored to a similar degree by Day 10 p.i. in both groups (Fig. 3A). BBB disruption was thus not correlated to MMP9 activity during sublethal JHMV infection.
Figure 3.
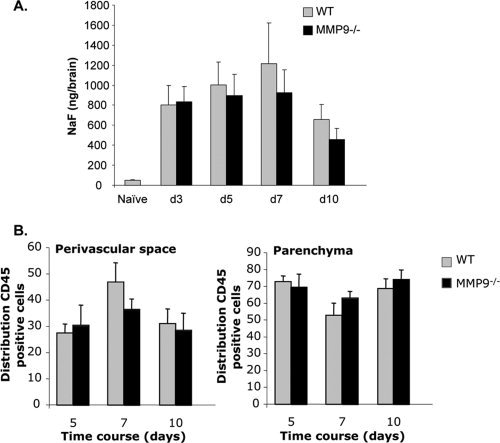
BBB disruption and CNS leukocyte distribution are identical in MMP9−/− mice and controls. A: BBB permeability was assessed by NaF. Data represent the average NaF amount per brain (±standard deviations) from nine mice per time point from three individual experiments. B: Leukocyte distribution in the perivascular space versus parenchyma was quantified in brain sections of infected WT and MMP9−/− mice using anti‐CD45 and anti‐laminin Abs. Ten pictures per animal were analyzed between days 5 and 10 p.i. Data represent the mean (±standard errors) of three animals per group per time point.
Trafficking through the BBB is a two‐step process that requires migration from the blood to the perivascular space and subsequently into the parenchyma (Owens et al.,2008). MMPs enhance leukocyte penetration through the endothelial cell tight junction, as well as through the glia limitans (Toft‐Hansen et al.,2006). Neither flow cytometry nor NaF measurements distinguish a specific role of MMP9 in this two‐step process. To assess whether the absence of MMP9 had more subtle effects on leukocyte localization within the BBB, the distribution of inflammatory cells was quantified following laminin staining to visualize the two basement membranes that define the perivascular space (Savarin et al.,2010). CNS leukocyte accumulation was too sparse at Day 3 p.i. for quantitative analysis. At Day 5 p.i. the majority of infiltrating cells were already localized in the parenchyma in both groups of mice (Fig. 3B). Furthermore, no difference in leukocyte distribution within the perivascular space versus parenchyma was detected comparing WT and MMP9−/− mice at Days 7 and 10 p.i. (Fig. 3B). Sublethal JHMV primarily infects glial cells and requires T cell interaction with infected cells to diminish viral replication (Bergmann et al.,2001; Savarin et al.,2008). Similar parenchymal T cell entry was thus further verified by monitoring viral control as a measure of T cell function. The peak, magnitude, and decline of infectious virus were all identical in both groups (data not shown), supporting the concept that MMP9 deficiency did not interfere with anti‐viral T cell activity. Overall, these results demonstrate that MMP9 does not affect leukocyte entry into the CNS parenchyma during viral encephalitis.
Transient MMP3 Increase in MMP9−/− Mice
The apparent redundancy of MMP9 for leukocyte access to the CNS parenchyma contrasted with indirect evidence that neutrophil‐derived MMP9 contributes to BBB disruption during JHMV infection (Hosking et al.,2009; Zhou et al.,2003). It also contradicted decreased CNS leukocyte recruitment after neutrophil depletion (see Fig. 1) and suggested alternative neutrophil derived proteases contribute to BBB breakdown. In addition to MMP9, neutrophil granules contain a variety of proteases involved in endothelial cell interactions, vascular permeability and anti‐microbial activity (DiStasi and Ley,2009; Hager et al.,2010). Thus, neutrophil elastase, MMP8 and Cathepsin G, all upregulated during neuroinflammatory diseases (DiStasi and Ley,2009; Hager et al.,2010; Toft‐Hansen et al.,2004), were examined as potential alternative mediators of BBB disruption in absence of MMP9. Neutrophil elastase was upregulated at Days 3 and 5 p.i. in WT mice, when neutrophils peak in the CNS (Fig. 4A); however, no increase in mRNA expression was observed in MMP9−/− mice. Moreover, MMP8 and Cathepsin G mRNA levels both remained very low relative to naïve mice and compared to neutrophil elastase or other MMPs. A slight but not significant MMP8 increase was observed in MMP9−/− mice. Although these data suggested that neutrophil‐derived proteases were not dysregulated in the absence of MMP9 (Fig. 4A), higher levels of prepackaged proteases remains possible.
Figure 4.
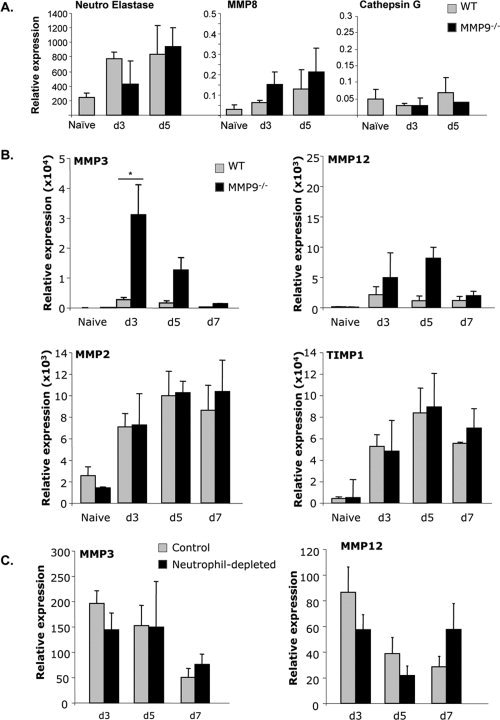
MMP3 mRNA is increased in MMP9−/− mice. A: Neutrophil elastase, MMP8, and Cathepsin G mRNA expression in brains were analyzed by real time PCR at the peak of CNS neutrophil infiltration (Day 3 and 5 p.i.) of WT and MMP9−/− mice B. MMP3, MMP12, MMP2, and TIMP1 mRNA expression was analyzed by real time PCR in the brain of naïve, WT, and MMP9−/− mice. *P < 0.05 C. MMP3 mRNA expression was analyzed in the brain of control and neutrophil‐depleted mice between Day 3 and 7 p.i. Data represent the average mRNA expression (±standard deviations) relative to ubiquitin of three mice per group per time point and are representative of two separate experiments.
We next determined if up‐regulation of other members of the MMP family compensated for the absence of MMP9. Unlike other inflammatory models, e.g., EAE (Pagenstecher et al.,1998), JHMV infection only induces a restricted number of MMPs, namely MMP3 and MMP12 (Zhou et al.,2005). Therefore, MMP3 and MMP12 were initially monitored as compensatory candidates for MMP9. MMP3 mRNA expression was increased greater than 10‐fold in MMP9−/− compared to WT mice at Day 3 p.i., coincident with peak neutrophil recruitment (Fig. 4B). Although levels remained significantly elevated at Day 5 p.i., they declined to similar levels as in WT mice after Day 7 p.i. By contrast, no differences in CNS MMP3 mRNA levels were observed comparing neutrophil‐depleted and control mice at Days 3, 5, and 7 p.i. (Fig. 4C). MMP12 mRNA expression was also elevated in the absence of MMP9, most prominently at Day 5 p.i. (Fig. 4B). Similar to MMP3 however, no significant variations in MMP12 mRNA expression were observed comparing neutropenic with control mice (Fig. 4C). The only MMP inhibitor, tissue inhibitor of metalloproteinase 1 (TIMP‐1), induced during JHMV infection (Zhou et al.,2005), was also identical in WT and MMP9−/− mice. Expression of MMP2, MMP7, and MMP14, upregulated during other neuroinflammatory disorders (Hartung and Kieseier,2000; Rosenberg,2002), was also assessed to determine if the absence of MMP9 diversified the pattern of MMP expression. MMP2 mRNA was constitutively expressed in the CNS of naïve mice and increased to similar levels in infected WT and MMP9−/− mice (Fig. 4B). However, as previously described (Zhou et al.,2005), MMP2 enzymatic activity could not be detected in the CNS after JHMV infection suggesting post‐transcriptional regulation (Fig. 2A). In addition, neither MMP7 nor MMP14 mRNA were upregulated in the CNS of either group following infection. Finally, expression of ADAM12 and ADAM17, proteases related to the MMP family (Seals and Courtneidge,2003), also remained undetectable in the CNS of infected WT and MMP9−/− mice (data not shown). Thus, MMP9 deficiency did not alter the restricted MMP expression pattern induced by JHMV infection.
These data implicate MMP3 as a potential candidate that compensates for the absence of MMP9 activity in promoting leukocyte entry into the CNS parenchyma during JHMV infection. However, in WT mice MMP3 expression by astrocytes is parenchymal (Zhou et al.,2005), whereas MMP9 is likely to act on the endothelium and/or glia limitans, posing a physical discrepancy. To address whether MMP3 expression remains restricted to astrocytes in the absence of MMP9, MMP3 mRNA levels were determined in purified populations of CNS infiltrating leukocytes (CD45hi), microglia (CD45lo) and glial (CD45neg) cells. Elevated MMP3 in total CNS RNA from MMP9−/− mice was recapitulated primarily in the CD45− CNS resident population containing astrocytes, in which MMP3 mRNA was already detectable at Day 3 p.i. and increased further at Day 5 p.i. (Fig. 5A); in WT mice MMP3 was not detected until Day 5 p.i. and levels were substantially lower. At Day 5 p.i. MMP3 mRNA was also elevated in microglia (CD45lo) and infiltrating leukocytes (CD45hi) derived from MMP9−/− mice (Fig. 5A), albeit at lower levels compared to the astrocyte enriched population. MMP3 protein expression by astrocytes was confirmed by immunofluorescence. At Day 3 p.i. all MMP3 positive cells in infected MMP9−/− mice co‐expressed GFAP (Fig. 5B), supporting MMP3 expression primarily by astrocytes, similar to WT mice (Zhou et al.,2005). Moreover, higher MMP3 mRNA levels did not correlate with an increased number of cells expressing MMP3 but rather enhanced expression per cell (Fig. 5C). As astrocytes are integral components of the glia limitans, their significantly elevated MMP3 production may contribute to disruption of the basement membrane in MMP9−/− mice.
Figure 5.
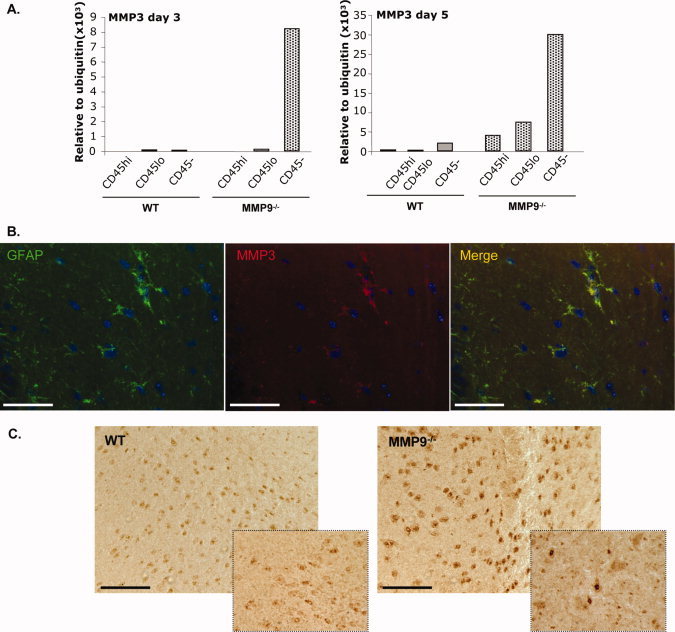
MMP3 is expressed by astrocytes. A: MMP3 mRNA expression was analyzed in FACS purified populations of glial cells (CD45−), microglia (CD45lo) and CNS infiltrating leukocytes (CD45hi) isolated from WT and MMP9−/− mice at Day 3 and 5 p.i. B: MMP3 and GFAP co‐expression in brain sections from MMP9−/− mice at Day 3 p.i. Scale bar, 25 μm. C: MMP3 expression was analyzed by immunohistochemistry in WT and MMP9−/− mice at Day 3 p.i. and at 4× higher magnification in the inset. Scale bar = 100 μm.
Mechanisms of MMP3 upregulation in the absence of MMP9 remain elusive. A direct effect of MMP9 in down‐regulating MMP3 was ruled out as abrogation of MMP9 via neutrophil depletion in WT mice did not upregulate MMP3. Early MMP3 upregulation following infection suggested the innate immune response might thus provide the induction signal. This is supported by IL‐1β and TNF mediated induction of MMP3 and MMP9 in cultured astrocytes and microglia (Crocker et al.,2006; Gottschall and Yu,1995). However, similar magnitude and kinetics of TNF and IL‐1β mRNA expression in the CNS of infected WT and MMP9−/− mice (Fig. 6A) did not support a direct relationship between these proinflammatory cytokines and MMP3. To investigate a correlation between MMP induction and proinflammatory signals in vivo in the absence of infection and inflammatory cell recruitment, innate responses in the CNS were initiated by injection of WT mice with poly (I:C), as a viral RNA mimic. Although TNF and IL‐1β mRNA were specifically upregulated by poly (I:C) as early as 2 h post injection, expression was transient and returned to baseline levels by 6 h (Fig. 6B). By contrast, MMP3 mRNA upregulation did not reach peak levels until 12 h. MMP12 mRNA levels were already induced at 6 h and remained constant until 12 h. These data suggest that neither TNF, IL‐1β nor innate immune signals are primary factors inducing MMP3 upregulation in vivo. Overall, the data support alternative or additional factors distinct from neutrophil‐secreted proteases, TNF and IL‐1β, underlying dysregulated MMP3 expression in the absence of MMP9 during viral encephalitis.
Figure 6.
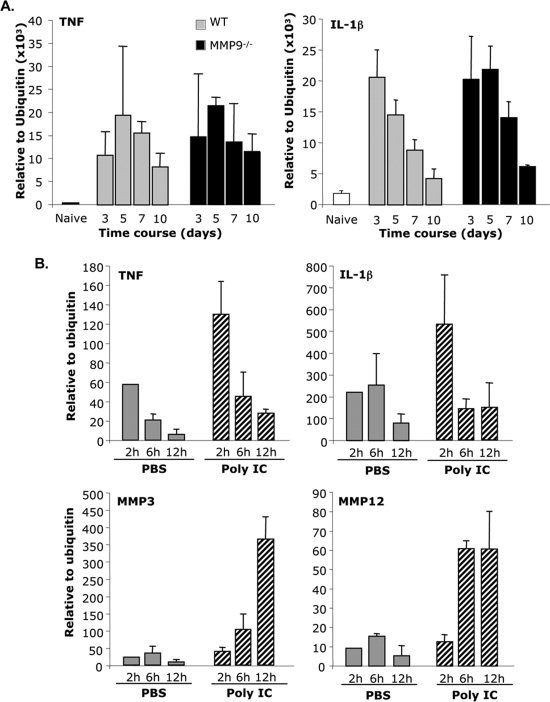
MMPs induction does not correlate with TNF and IL‐1β expression. A: Kinetics of TNF and IL‐1β mRNA expression was compared between WT and MMP9−/− mice. B: mRNA expression of TNF, IL‐1β, MMP3, and MMP12 was analyzed at 2, 6, and 12 h after poly (I:C) injection. Data represent the average mRNA expression (±standard deviations) relative to ubiquitin of 3–4 mice per group per time point.
DISCUSSION
Among various MMPs upregulated during neuroinflammatory disorders, MMP9 has been a consistent candidate for promoting BBB disruption, including in the JHMV infection model (Hosking et al.,2009; Zhou et al.,2003). However, rapid mortality following neutrophil/monocyte ablation during lethal JHMV infection preceded peak lymphocyte accumulation (Zhou et al.,2003), leaving the role of neutrophils and MMP9 on T cell migration through the BBB inconclusive. Using a sublethal virus variant, the present study shows that neutrophil depletion decreased CNS leukocyte recruitment by ∼50%, predominantly at the peak of inflammation. Although macrophage and T cell infiltration were both reduced in neutropenic mice, similar virus clearance and disease severity as in control mice suggested that T cells accessing the CNS parenchyma are sufficient to exert effective anti‐viral responses. By contrast, a similar decrease in CNS macrophage and T cell recruitment after blocking ELR+ CXC chemokine to limit CXCR2‐mediated neutrophil migration resulted in increased mortality and delayed virus clearance (Hosking et al.,2010). CXCR2 expression on CNS resident cells (Omari et al.,2005,2006) or a role of CXCR2 in oligodendrocyte protection during JHMV infection (Hosking et al.,2010) may explain this discrepancy. Altogether, our data suggest that while neutrophils participate in leukocyte accumulation in the CNS, they are not essential to protect the host from JHMV infection. Monocytes have also been implicated in facilitating T cell migration through the glia limitans at early time points during JHMV infection (Savarin et al.,2010). Neutrophils and monocytes are the first cells to access the CNS and are both a reservoir of proteases. We propose cooperation between these two innate cells in breaching the BBB early following infection. By releasing granule‐sequestrated proteases, neutrophils may induce initial BBB disruption, which is amplified by monocytes to enhance leukocyte migration through the glia limitans. This scenario is supported by the transient retention of leukocytes, including CD4+ and CD8+ T cells, in the absence of monocytes.
In contrast to neutropenic mice, MMP9−/− mice infected with JHMV exhibited no difference in virus clearance, disease severity or CNS inflammation compared to WT mice, indicating that MMP9 is redundant in promoting leukocyte migration through the BBB. Similar clinical disease and CNS inflammation were also observed in MMP9−/− and WT mice during EAE (Dubois et al.,1999). The absence of MMP9 may thus be overcome by additional neutrophil derived proteases contributing to degrading BBB components (Hager et al.,2010). Indeed, neutrophil elastase compensates for MMP9 deficiency in a model of experimental peritonitis (Kolaczkowska et al.,2009). However, neutrophil elastase, MMP8 and Cathepsin G, were not significantly upregulated in JHMV infected MMP9−/− compared to WT mice, suggesting that neutrophil‐derived proteases did not compensate for MMP9 deficiency at the BBB. Potential compensation by other MMPs in MMP9−/− mice is difficult to test during EAE due to the wide range of MMPs upregulated in this model (Rosenberg,2002). However, upregulation of the limited array of MMPs characteristic of JHMV infection was conserved in MMP9−/− mice. Moreover, enhanced MMP3 expression specifically in astrocytes further demonstrated that the source of MMPs also remained unaltered in MMP9−/− mice. As astrocytes endfeet are part of the glia limitans (Bechmann et al.,2007), MMP3 release from the parenchymal side may exert similar degrading effects as MMP9 released from the luminal side of the perivascular space. A role for MMP3 in regulating BBB permeability has previously been suggested using a lipopolysaccharide‐induced neuroinflammatory model (Gurney et al.,2006). However, our data are distinct in suggesting that MMP9 activity at the luminal side of the BBB may be compensated by MMP3 expressed by astrocytes at the abluminal side. Unfortunately, to our knowledge there are no in vivo specific MMP3 inhibitors available to test this notion. It is also noted that the absence of MMP upregulation at the mRNA level does not rule out compensatory mechanisms by other MMPs, as MMP activity may be enhanced by post‐translational cleavage. However, as noted above, activity measurements of individual MMPs are difficult due to the limited assays currently available. Nevertheless, our data emphasize an important role of astrocytes in maintaining BBB integrity, especially at the glia limitans.
The signaling pathway controlling MMP3 expression in the absence of MMP9 remains unclear. Our in vivo data suggested that the pro‐inflammatory cytokines IL‐1β and TNF are insufficient to induce MMP3, contrasting their effect on glial cell cultures (Crocker et al.,2006; Gottschall and Yu,1995). Moreover, neutrophil depletion did not increase MMP3 in WT mice, suggesting MMP9 does not directly regulate MMP3. A similar discrepancy was observed during EAE comparing young and adult MMP9−/− mice. Whereas adult MMP9−/− mice were indistinguishable from WT mice, young MMP9−/− mice (≤4 weeks of age) were resistant to EAE (Dubois et al.,1999). These data suggest that compensatory mechanisms may occur developmentally, when MMPs play an important role in CNS development. Indeed, MMP9 expression during early postnatal development is an important factor regulating myelin formation (Larsen et al.,2006).
In conclusion, our data show that neutrophils participate in BBB disruption during JHMV infection, but are not essential in protecting the host from infection. Moreover, the specific role of neutrophil‐derived MMP9 in BBB breakdown, especially at the glia limitans, remains unclear due to potential alternative/compensatory pathways dependent on MMP3. These data emphasizes the difficulty of targeting MMPs to alter CNS inflammation due to their complex regulation and dual roles (beneficial vs. detrimental).
Acknowledgements
The authors thank Kate Koch and Sherry Yu for their technical assistance and Jennifer Powers for FACS purification.
REFERENCES
- Agrawal S,Anderson P,Durbeej M,van Rooijen N,Ivars F,Opdenakker G,Sorokin LM. 2006. Dystroglycan is selectively cleaved at the parenchymal basement membrane at sites of leukocyte extravasation in experimental autoimmune encephalomyelitis. J Exp Med 203: 1007–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anthony DC,Miller KM,Fearn S,Townsend MJ,Opdenakker G,Wells GM,Clements JM,Chandler S,Gearing AJ,Perry VH. 1998. Matrix metalloproteinase expression in an experimentally‐induced DTH model of multiple sclerosis in the rat CNS. J Neuroimmunol 87: 62–72. [DOI] [PubMed] [Google Scholar]
- Bechmann I,Galea I,Perry VH. 2007. What is the blood‐brain barrier (not)? Trends Immunol 28: 5–11. [DOI] [PubMed] [Google Scholar]
- Bergmann CC,Lane TE,Stohlman SA. 2006. Coronavirus infection of the central nervous system: Host‐virus stand‐off. Nat Rev Microbiol 4: 121–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergmann CC,Marten NW,Hinton DR,Parra B,Stohlman SA. 2001. CD8 T cell mediated immunity to neurotropic MHV infection. Adv Exp Med Biol 494: 299–308. [DOI] [PubMed] [Google Scholar]
- Candelario‐Jalil E,Yang Y,Rosenberg GA. 2009. Diverse roles of matrix metalloproteinases and tissue inhibitors of metalloproteinases in neuroinflammation and cerebral ischemia. Neuroscience 158: 983–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crocker SJ,Milner R,Pham‐Mitchell N,Campbell IL. 2006. Cell and agonist‐specific regulation of genes for matrix metalloproteinases and their tissue inhibitors by primary glial cells. J Neurochem 98: 812–823. [DOI] [PubMed] [Google Scholar]
- Daley JM,Thomay AA,Connolly MD,Reichner JS,Albina JE. 2008. Use of Ly6G‐specific monoclonal antibody to deplete neutrophils in mice. J Leuk Biol 83: 64–70. [DOI] [PubMed] [Google Scholar]
- DiStasi MR,Ley K. 2009. Opening the flood‐gates: How neutrophil‐endothelial interactions regulate permeability. Trends Immunology 30: 547–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubois B,Masure S,Hurtenbach U,Paemen L,Heremans H,van den Oord J,Sciot R,Meinhardt T,Hammerling G,Opdenakker G, et al. 1999. Resistance of young gelatinase B‐deficient mice to experimental autoimmune encephalomyelitis and necrotizing tail lesions. J Clin Invest 104: 1507–1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming JO,Trousdale MD,el‐Zaatari FA,Stohlman SA,Weiner LP. 1986. Pathogenicity of antigenic variants of murine coronavirus JHM selected with monoclonal antibodies. J Virol 58: 869–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottschall PE,Yu X. 1995. Cytokines regulate gelatinase A, B (matrix metalloproteinase 2 and 9) activity in cultured rat astrocytes. J Neurochem 64: 1513–1520. [DOI] [PubMed] [Google Scholar]
- Gurney KJ,Estrada EY,Rosenberg GA. 2006. Blood‐brain barrier disruption by stromelysin‐1 facilitates neutrophil infiltration in neuroinflammation. Neurobiol Dis 23: 87–96. [DOI] [PubMed] [Google Scholar]
- Hager M,Cowland JB,Borregaard N. 2010. Neutrophil granules in health and disease. J Intern Med 268: 25–34. [DOI] [PubMed] [Google Scholar]
- Hartung HP,Kieseier BC. 2000. The role of matrix metalloproteinases in autoimmune damage to the central and peripheral nervous system. J Neuroimmunol 107: 140–147. [DOI] [PubMed] [Google Scholar]
- Hosking MP,Liu L,Ransohoff RM,Lane TE. 2009. A protective role for ELR+ chemokines during acute viral encephalomyelitis. PLoS Pathog 5: e1000648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosking MP,Tirotta E,Ransohoff RM,Lane TE. 2010. CXCR2 signaling protects oligodendrocytes and restricts demyelination in a mouse model of viral‐induced demyelination. PLoS One 5: e11340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ireland DD,Stohlman SA,Hinton DR,Kapil P,Silverman RH,Atkinson RA,Bergmann CC. 2009. RNase L mediated protection from virus induced demyelination. PLoS Pathog 5: e1000602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapil P,Atkinson R,Ramakrishna C,Cua DJ,Bergmann CC,Stohlman SA. 2009. Interleukin‐12 (IL‐12), but not IL‐23, deficiency ameliorates viral encephalitis without affecting viral control. J Virol 83: 5978–5986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolaczkowska E,Grzybek W,van Rooijen N,Piccard H,Plytycz B,Arnold B,Opdenakker G. 2009. Neutrophil elastase activity compensates for a genetic lack of matrix metalloproteinase‐9 (MMP‐9) in leukocyte infiltration in a model of experimental peritonitis. J Leukoc Biol 85: 374–381. [DOI] [PubMed] [Google Scholar]
- Larsen PH,DaSilva AG,Conant K,Yong VW. 2006. Myelin formation during development of the CNS is delayed in matrix metalloproteinase‐9 and ‐12 null mice. J Neurosci 26: 2207–2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liedtke W,Cannella B,Mazzaccaro RJ,Clements JM,Miller KM,Wucherpfennig KW,Gearing AJ,Raine CS. 1998. Effective treatment of models of multiple sclerosis by matrix metalloproteinase inhibitors. Ann Neurol 44: 35–46. [DOI] [PubMed] [Google Scholar]
- Lin MT,Stohlman SA,Hinton DR. 1997. Mouse hepatitis virus is cleared from the central nervous systems of mice lacking perforin‐mediated cytolysis. J Virol 71: 383–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mun‐Bryce S,Rosenberg GA. 1998. Gelatinase B modulates selective opening of the blood‐brain barrier during inflammation. Am J Physiol 274 (5 Part 2): R1203–R1211. [DOI] [PubMed] [Google Scholar]
- Noble LJ,Donovan F,Igarashi T,Goussev S,Werb Z. 2002. Matrix metalloproteinases limit functional recovery after spinal cord injury by modulation of early vascular events. J Neurosci 22: 7526–7535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omari KM,John G,Lango R,Raine CS. 2006. Role for CXCR2 and CXCL1 on glia in multiple sclerosis. Glia 53: 24–31. [DOI] [PubMed] [Google Scholar]
- Omari KM,John GR,Sealfon SC,Raine CS. 2005. CXC chemokine receptors on human oligodendrocytes: Implications for multiple sclerosis. Brain 128 (Part 5): 1003–1015. [DOI] [PubMed] [Google Scholar]
- Opdenakker G,Nelissen I,Van Damme J. 2003. Functional roles and therapeutic targeting of gelatinase B, chemokines in multiple sclerosis. Lancet Neurol 2: 747–756. [DOI] [PubMed] [Google Scholar]
- Owens T,Bechmann I,Engelhardt B. 2008. Perivascular spaces and the two steps to neuroinflammation. J Neuropathol Exp Neurol 67: 1113–1121. [DOI] [PubMed] [Google Scholar]
- Pagenstecher A,Stalder AK,Kincaid CL,Shapiro SD,Campbell IL. 1998. Differential expression of matrix metalloproteinase and tissue inhibitor of matrix metalloproteinase genes in the mouse central nervous system in normal and inflammatory states. Am J Pathol 152: 729–741. [PMC free article] [PubMed] [Google Scholar]
- Parks WC,Wilson CL,Lopez‐Boado YS. 2004. Matrix metalloproteinases as modulators of inflammation and innate immunity. Nat Rev Immunol 4: 617–629. [DOI] [PubMed] [Google Scholar]
- Paul R,Lorenzl S,Koedel U,Sporer B,Vogel U,Frosch M,Pfister HW. 1998. Matrix metalloproteinases contribute to the blood–brain barrier disruption during bacterial meningitis. Ann Neurol 44: 592–600. [DOI] [PubMed] [Google Scholar]
- Phares TW,Marques CP,Stohlman SA,Hinton DR,Bergmann CC. 2010. Factors supporting intrathecal humoral responses following viral encephalomyelitis. J Virol 85: 2589–2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosell A,Lo EH. 2008. Multiphasic roles for matrix metalloproteinases after stroke. Curr Opin Pharmacol 8: 82–89. [DOI] [PubMed] [Google Scholar]
- Rosenberg GA. 2002. Matrix metalloproteinases in neuroinflammation. Glia 39: 279–291. [DOI] [PubMed] [Google Scholar]
- Savarin C,Bergmann CC,Hinton DR,Ransohoff RM,Stohlman SA. 2008. Memory CD4+ T‐cell‐mediated protection from lethal coronavirus encephalomyelitis. J Virol 82: 12432–12440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savarin C,Stohlman SA,Atkinson R,Ransohoff RM,Bergmann CC. 2010. Monocytes regulate T cell migration through the glia limitans during acute viral encephalitis. J Virol 84: 4878–4888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seals DF,Courtneidge SA. 2003. The ADAMs family of metalloproteases: Multidomain proteins with multiple functions. Genes Dev 17: 7–30. [DOI] [PubMed] [Google Scholar]
- Sixt M,Engelhardt B,Pausch F,Hallmann R,Wendler O,Sorokin LM. 2001. Endothelial cell laminin isoforms, laminins 8 and 10, play decisive roles in T cell recruitment across the blood‐brain barrier in experimental autoimmune encephalomyelitis. J Cell Biol 153: 933–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sporer B,Paul R,Koedel U,Grimm R,Wick M,Goebel FD,Pfister HW. 1998. Presence of matrix metalloproteinase‐9 activity in the cerebrospinal fluid of human immunodeficiency virus‐infected patients. J Infect Dis 178: 854–857. [DOI] [PubMed] [Google Scholar]
- Stohlman SA,Hinton DR,Parra B,Atkinson R,Bergmann CC. 2008. CD4 T cells contribute to virus control and pathology following central nervous system infection with neurotropic mouse hepatitis virus. J Virol 82: 2130–2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toft‐Hansen H,Buist R,Sun XJ,Schellenberg A,Peeling J,Owens T. 2006. Metalloproteinases control brain inflammation induced by pertussis toxin in mice overexpressing the chemokine CCL2 in the central nervous system. J Immunol 177: 7242–7249. [DOI] [PubMed] [Google Scholar]
- Toft‐Hansen H,Nuttall RK,Edwards DR,Owens T. 2004. Key metalloproteinases are expressed by specific cell types in experimental autoimmune encephalomyelitis. J Immunol 173: 5209–5218. [DOI] [PubMed] [Google Scholar]
- Tran EH,Hoekstra K,van Rooijen N,Dijkstra CD,Owens T. 1998. Immune invasion of the central nervous system parenchyma and experimental allergic encephalomyelitis, but not leukocyte extravasation from blood, are prevented in macrophage‐depleted mice. J Immunol 161: 3767–3775. [PubMed] [Google Scholar]
- Wang P,Dai J,Bai F,Kong KF,Wong SJ,Montgomery RR,Madri JA,Fikrig E. 2008. Matrix metalloproteinase 9 facilitates West Nile virus entry into the brain. J Virol 82: 8978–8985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J,Marten NW,Bergmann CC,Macklin WB,Hinton DR,Stohlman SA. 2005. Expression of matrix metalloproteinases and their tissue inhibitor during viral encephalitis. J Virol 79: 4764–4773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J,Stohlman SA,Hinton DR,Marten NW. 2003. Neutrophils promote mononuclear cell infiltration during viral‐induced encephalitis. J Immunol 170: 3331–3336. [DOI] [PubMed] [Google Scholar]