

Abstract
c-Jun N-terminal Kinases (JNKs) represent valuable targets in the development of new therapies. Present on the surface of JNK is a binding pocket for substrates and the scaffolding protein JIP1 in close proximity to the ATP binding pocket. We propose that bi-dentate compounds linking the binding energies of weakly interacting ATP and substrate mimetics could result in potent and selective JNK inhibitors. We describe here a bi-dentate molecule, 19, designed against JNK. 19 inhibits JNK kinase activity (IC50 = 18 nM; Ki = 1.5 nM) and JNK/substrate association in a displacement assay with a substrate peptide (compound 20; IC50 = 46 nM; Ki = 2 nM). Our data demonstrate that 19 targets for the ATP and substrate-binding sites on JNK concurrently. Finally, compound 19 not only inhibits JNK in a variety of cell-based experiments, but it elicits also in vivo activity where it is shown to improve glucose tolerance in diabetic mice.
INTRODUCTION
The c-Jun N-terminal kinases (JNKs) are a series of serine/threonine protein kinases belonging to the mitogen activated protein kinase (MAPK) family. In mammalian cells, three distinct genes encoding JNKs have been identified, JNK1, JNK2, and JNK3, and at least 10 different isoforms exist 1–3. JNK1, JNK2, and JNK3 share more than 90% amino acid sequence identity and the ATP pocket is >98% homologous. JNK1 and JNK2 are ubiquitously expressed, whereas JNK3 is most commonly found in the brain, cardiac muscle, and testis 2, 4. JNK activation in response to stimuli such as stress or cytokines results in activation of several transcription factors and cellular substrates implicated in inflammation, insulin signaling, mRNA stabilization, and cell proliferation and survival 3, 5–7. Because of the link between these pathways and the pathogenesis of diseases such as Parkinson’s and Alzheimer’s and inflammatory diseases, cancer, diabetes, atherosclerosis, and stroke, JNK inhibitors are expected to be useful therapeutic agents 1, 3, 8, 9.
JNK binds to substrates and scaffold proteins, such as JIP-1, that contain a D-domain, as defined by the consensus sequence R/K(2–3)X(1–6)L/I-X-L/I 10. A JIP1 D-domain peptide corresponding to amino acids 153–164, 20 (pepJIP1; sequence RPKRPTTLNLF; MW 1343), inhibits JNK activity in vitro and in cell while displaying extraordinary selectivity with negligible inhibition of the closely related MAP kinases p38 and Erk 11–13. The mechanism of this inhibition is thought to be due to competition of 20 with the D-domains of JNK substrates or upstream kinases 12, 14. In order to increase stability and increase cell permeability of 20, an all-D retro-inverso amino acid of compound 20 fused to the cell permeable HIV-TAT peptide, 11 (D-JNKI), was devised (sequence Ac-tdqsrpvqpflnlttprrprpprrrqrrkkrg-CONH2; MW = 3395) 15. 11 significantly decreases c-Jun phosphorylation by JNK when tested in cell, however, albeit very selective, inhibition studies suggest that 11 is only a modest JNK inhibitor 16. In comparison, the small molecule ATP mimetic, 21 (SP600125), is very potent in vitro but not very selective for JNK 17–19. Hence, most of the current efforts focus on optimization of 21 and other ATP mimetics for the design of JNK inhibitors 1, 9, 20.
Recently, using a combination of structure-based design guided by the X-ray structure of JNK1 in complex with 20 and 21, as well as NMR fragment-based drug discovery approaches 21, we proposed that by linking molecules that span these two sites we should be able to generate selective, high affinity bi-dentate JNK modulators. Indeed, we describe here a bi-dentate molecule with the aforementioned characteristics that functions as a JNK inhibitor both in vitro and in cell as well as exhibiting in vivo efficacy in a type 2 diabetes model.
RESULTS AND DISCUSSION
In the realm of drug discovery, fragment-based drug design approaches are becoming increasingly successful in tackling challenging targets, such as those involving protein-protein interactions 22. A common fragment-based drug design approach consists of designing bi-dentate compounds chemically linking two weakly interacting scaffolds that occupy adjacent pockets on the target’s surface (Figure 1a-c). In this case, the free energy of binding of the resulting bi-dentate compound with respect to those of the individual fragments can be expressed as:
Figure 1.

Fragment-based design and synthesis of bi-dentate JNK inhibitors. A) Schematic representation of the proposed approach overlaid on the surface representation of JNK1 in complex with 20 (RPKRPTTLNLF) and the ATP mimic 21 (PDB-ID 1UKI). The surface generated with MOLCAD50 and color coded according to cavity depth (blue, shallow; yellow, deep). B) Docked structure of 20 and 21 on the surface of JNK1. C) Docked structure of the bi-dentate compound 9 on the surface of JNK1. D) Scheme for the synthesis of 8 and the bi-dentate 9 (see methods for experimental details). E) In vitro JNK kinase activity inhibition by 9. F) Displacement of 20 from GST-JNK1 by 9 in absence (circles) and presence (squares) of a saturating amount of staurosporine (0.5 μM).
Where, R represents the Boltzman constant, T is the temperature of the system, ΔHA and ΔHB are the enthalpy of binding of fragments A and B respectively, ΔSAB represents the entropy loss upon binding of the bi-dentate compound, and KDA and KDB are the dissociation constants of the individual initial binders and E is the linking coefficient 23. The recently determined X-ray structure of JNK1 in complex with 20 and the ATP-mimic 21 24, reveals a close proximity between the ATP and the docking binding sites, suggesting the possibility of obtaining high affinity and selective compounds by designing appropriate bi-dentate molecules. Indeed, this approach has been successfully executed for a variety of other proteins and kinases 25–30. Hence, our hypothesis is that by tailoring a weak docking site binder to a weakly interacting ATP-mimic, it should be likewise possible to develop potent and selective inhibitors of JNK.
In order to define an optimal interacting docking peptide sequence for JNK we tested twenty peptide sequences derived from its putative substrates and scaffolding proteins, all presenting a D-domain consensus motif 10 (Supplementary Table S1). Each peptide was tested for its ability to displace 20 (residues 153–164) from JNK1 by using a Dissociation Enhanced Lanthanide Fluoro-Immuno Assay (DELFIA) platform. DELFIA is a heterogeneous assay whereby a biotin-linked 20 is adsorbed onto a streptavidin-coated plate followed by incubation with GST-JNK1. Detection of the 20/GST-JNK1 complex is facilitated by a highly fluorescent anti-GST Eu-antibody conjugate (Perkin–Elmer). Based on these data (Supplementary Table S1), a minimal peptide sequence, 10 (RPTTLNL), was identified as necessary and sufficient to displace full-length 20 in this assay. We are confident that inhibition is not at the level of either GST or streptavidin as 10 did not significantly displace a previously described and unrelated binding pair (SIAH1/phyllopod peptide) 31 in a similar DELFIA assay platform when tested up to 45μM.
The design of a weak ATP mimetic JNK inhibitor was based on the structure of 21 (Figure 1b) 24. Given that 21 was found to potently inhibit several protein kinases 17, 18, 20, in order to increase JNK selectivity and also facilitate subsequent synthetic efforts, we removed the keto group from 21. Furthermore, based on its docking mode, a convenient position to elongate the ATP-mimetic towards the docking site was identified in our previous work, by using a combination of structure-based design guided by NMR-relaxation measurements using paramagnetic spin-labeling approaches 21. These efforts resulted in the design of 8 (Figure 1d). The synthesis of 8 starts from the iodination of 1-azaindole with iodide and its protection with trimethylsilylethoxy methyl chloride, followed by a Suzuki coupling between 3 and 4-methoxycarbonylphenylboronic acid and attachment of the designed 3-carbon linker (Figure 1d).
8 is a relatively weak ATP mimetic, inhibiting JNK1 phosphorylation of the substrate ATF2 in the LanthaScreen™ Time-Resolved Fluorescence Resonance Energy Transfer (TR-FRET) based kinase activity assay (Invitrogen, Carlsbad, CA) with 14 μM IC50 value. Similarly, 10 was able to displace 20 in the DELFIA assay (IC50 25 μM) but was unable to inhibit JNK1 phosphorylation of the substrate ATF2 in the same TR-FRET based kinase activity assay at concentrations up to 25 μM, hence representing a fairly weakly interacting substrate binding scaffold. However, when linked to the indazole moiety of 8, the resulting compound 9 (Figure1d) was able to compete with 20 for JNK1 binding and inhibit JNK1 kinase activity with remarkable sub-nanomolar affinities (Table 1). In order to properly link compounds 8 and 10, two Gly residues were inserted as part of the linker based on molecular modeling and NMR-based considerations and the X-ray structure of the ternary complex, as described in our previous studies 21.
Table 1. JNK inhibition results.
In vitro activity data for reported JNK inhibitors.
| Molecule | ID (MW) | pepJIP Displacement | Kinase Inhibition |
|---|---|---|---|
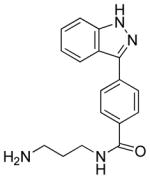
|
8 (294) | IC50 = >50 μM | IC50 = 14 μM |
| Ac-RPTTLNL-OH | 10 (856) | IC50 = 4.5 μM | IC50 = >50 μM |
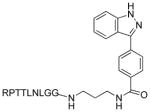
|
9 (1236) | IC50 = 0.9 nM | IC50 = 0.7 nM |
| Ac-tdqsrpvqpflnlttprrprpprrrqrrkkrg-OH | 11 (3994) | IC50 = ~ 10 μM | IC50 = >50μM |
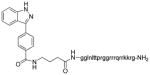
|
19 (2728) | IC50 = 46 nM Ki = 1.5 nM |
IC50 = 18 nM Ki = 2 nM |
Consistent with the bi-dentate binding mode of 9 to JNK1, the pre-incubation of JNK1 with an excess of ATP-mimetic staurosporine 32, hence eliminating one binding site, reduces the ability of 9 to compete with 20 binding to JNK1 by 600-fold (Figure 1f). As expected, under these circumstances, the IC50 for 9 was similar to that of the peptide alone (Table 1). Direct comparison of kinase inhibition properties of 9 (MW 1204) with 20 (MW 1343) clearly demonstrated an improvement of over 100-fold (Figure 1e). Hence, by linking the binding energy of a minimal 20 sequence with a modest 21 derived ATP mimetic we successfully produced a very potent bi-dentate molecule representing a new class of JNK inhibitors.
In order to confer further favorable pharmacological properties to this compound for in vivo studies, analogous to the clinical candidate 11 16, we produced an all-D retro-inverso version of 9 fused to the cell penetrating HIV-TAT sequence. As expected, the resulting compound, 19 (Table 1), efficiently competes with 20 for binding to JNK1 as well as strongly inhibiting JNK1 kinase activity (Figure 2a and b), with IC50 values of 18 and 46 nM respectively. Using the same in vitro kinase activity assay and the same ATF2 substrate, 19 was found to be inactive versus p38α at concentrations up to 100 μM, a member of the MAPK family with highest structural similarity to JNK thus demonstrating selectivity. Lineweaver-Burk analysis indicates that 19 is competitive with both ATP and ATF2 for binding to JNK1 as the data with an apparent K(i) of 2 and 1.5 nM respectively (Figure 2c and d). Consistent with the proposed bi-modal binding of 19 to JNK1, the data fit very well with both the mixed and the competitive inhibition modes and very poorly with either the non-competitive or the uncompetitive inhibition models (Supplementary Table S2).
Figure 2.

In vitro characterization of 19. A) Dose dependent displacement of biotinylated 20 from GST-JNK1 and B) JNK kinase activity inhibition by 19. C) and D) Lineweaver-Burk analysis with compound 19.
In an attempt to further profile the biological properties of 19, we compared its ability to function in the context of a complex cellular milieu with that of 11. For this analysis we employed a cell -based TR-FRET assay 33. In this assay, 19 was significantly more effective at inhibiting tumor necrosis factor-α (TNF-α) stimulated phosphorylation of c-Jun in B16-F10 melanoma cells (EC50 = 14 μM), while both 11 and 21 were significantly less effective under the same experimental conditions (Figure 3a). The cell-based system employed makes use of a GFP-c-Jun stable expression system. As a result, the levels of GFP-c-Jun in these cells are higher than endogenous levels. This could have an inflationary effect on the EC50 values obtained with this assay when testing substrate competitive compounds. Thus in an orthogonal assay we measured the ability of 19 to inhibit anisomycin stimulated endogenous c-Jun phosphorylation in HEK293T cells. Indeed, we found 19 to be significantly more effective in this system (EC50 = 2 μM; Figure 3b). To further profile 19 efficacy in cell as well as demonstrate its selectivity, we tested the ability of 19 to inhibit the release of cytokines from RAW 264.7 mouse macrophage cells in response to lipopolysaccharide (LPS). LPS stimulated secretion of TNF-α from macrophages is dependent on JNK activation 34 whereas IL-1β secretion from RAW 264.7 cells is known to be p38 dependent 35. We found that 19 was able to inhibit JNK dependent TNF-α release while p38α dependant IL-1β secretion was unaffected (Figure 3c). Taken together, these findings conclusively establish that 19 as a potent and selective dual substrate and ATP competitive JNK bi-dentate inhibitor able to function efficiently and selectively in a cellular context.
Figure 3.

Cell-based characterization of 19. A) TR-FRET analysis of c-Jun phosphorylation upon TNF-αstimulation of B16-F10 cells in the presence of increasing 19 (closed triangles), 21 (inverted triangles) and 11 (closed squares). B) Inhibitory effect of 19 on phospho-c-Jun upon anisomycin stimulation of HEK293T cell. C) Compounds 19 and 11 effect on TNF-α and IL-1β levels after 5 hours of exposure to LPS as compared to vehicle control. Results shown as percent of vehicle control ± S.D. (n = 3). Cytokine production was measured directly from cell culture medium by a sandwich immunoassay (Meso Scale Discovery).
JNK activation has been linked to the impaired glucose tolerance associated with type 2 diabetes 8, 36. Therefore we tested the ability of 19 to restore glucose tolerance in the type 2 diabetes mouse model NONcNZO10/LtJ37 (Jackson Labs, Bar Harbor, Maine 04609 USA), chosen because obesity in NONcNZO10/LtJ mice reflects most human obesities. For this analysis, glucose intolerant NONcNZO10/LtJ mice were injected intraperitoneally daily for four days with 25 mg/kg 19. The ability of mice to process glucose injected intraperitoneally was then measured. 19 was remarkably effective in restoring normoglycemia without inducing hypoglycemia compared to both the vehicle control and 11 (Figure 4a). The ability of 19 to improve glucose tolerance is consistent with its proposed function as an effective JNK inhibitor, while the observed shortcoming of compound 11 under our current experimental conditions can simply be a reflection of its limited potency against the target, as very recently corroborated by a comparative study 38.
Figure 4.

In vivo characterization of 19. A) Effect of 19 and 11 (25 mg/kg) on glucose tolerance in 26-week-old NONcNZO10/LtJ mice from Harlan (Jackson Labs, Bar Harbor, Maine 04609 USA). Closed diamonds, vehicle control; closed squares, 25 mg/kg 19; closed triangle, 25 mg/kg 11. ). p < 0.0001 for the comparison of compound 19 with control, by two-way ANOVA using all the measurements. B) Effect of 25 mg/kg 19 (open bars) on AST and ALT levels after Jo2 treatment as compared to vehicle control (filled bars). Data are mean ± SE for four mice. AST levels significantly different as compared to control mice p < 0.05. ALT levels significantly different as compared to control mice p < 0.02. C) Effect of 25 mg/kg 19 on phospho-c-Jun levels in C57/B6 mouse liver. Phospho-c-Jun levels measured by sandwich immunoassay (Meso Scale Discovery). Results shown as percent of vehicle control ± S.D. (n = 4). p < 0.0001 for the comparison of compound 19 with control, by unpaired t-test.
The link between hapototoxicity and JNK function has been established using JNK1−/− and JNK2−/− mice39–41. Thus, in order to extend our in vivo analysis of 19 we tested its ability to block Jo2-induced liver damage as measured by the release of liver enzymes alanine-aminotransferase (ALT) and aspartate aminotransferase (AST) into the blood. 19 effectively blunted the Jo2-induced elevation of AST and ALT levels relative to control animals (Figure 4b). Consistent with its proposed ability to effectively inhibit JNK function in vivo, 19 treatment resulted in significantly reduced levels of phosphorylated c-Jun in the liver of treated mice as compared to the control (Figure 4c).
In conclusion, by applying the principles of fragment-based drug discovery to the design of dual ATP- and substrate-competitive kinase inhibitors, we identified bi-dentate molecules with superior JNK inhibitory properties. We anticipate that this approach will find wide applications in the design and synthesis of other potent and selective bi-dentate kinase inhibitors. Regarding the reported bi-dentate compounds, given that 11 is currently a clinical candidate (under XG102 by Xigen Corp., Lausanne, CH) we can speculate that 19, having markedly improved biochemical and pharmacological properties and even reduced molecular weight over 11, could equally well enter further clinical investigations. Furthermore, our current efforts focused on the identification of small molecules 20 mimetics 42–45 could likewise lead, based on the reported results, to the design of additional bi-dentate compounds voided of the peptidyl nature.
Given the tremendous efforts of the past decade dedicated to the design of potent and selective kinase inhibitors for both therapeutics and basic cell biology studies, we are confident that the bi-dentate approach proposed will find wide applications in both the pharmaceutical and basic chemical biology arenas.
Materials and Methods
DELFIA Assay (dissociation enhanced lanthanide fluoro-immuno assay)
To each well of 96-well streptavidin-coated plates (Perkin-Elmer) 100 μL of a 100 ng/ml solution of biotin-labeled 20 (Biotin-lc-KRPKRPTTLNLF, where lc indicates a hydrocarbon chain of 6 methylene groups) was added. After 1 hr incubation and elimination of unbound biotin-20 by 3 washing steps, 87 μL of Eu-labeled anti-GST antibody solution (300 ng/ml; 1.9 nM), 2.5 μL DMSO solution containing test compound, and 10 μL solution of GST-JNK1 for a final protein concentration of 10 nM was added. After 1 hr incubation at 0 °C, each well was washed 5 times to eliminate unbound protein and the Eu-antibody if displaced by a test compound. Subsequently, 200 μL of enhancement solution (Perkin-Elmer) was added to each well and fluorescence measured after 10 min incubation (excitation wavelength, 340 nm; emission wavelength, 615 nm). Controls include unlabeled peptide and blanks receiving no compounds. Protein and peptide solutions were prepared in DELFIA buffer (Perkin-Elmer). Staurosporine (Calbiochem; San Diego, CA; Catalog # 569397) was included at 20-times molar equivalent GST-JNK1 and pre-incubated for 15 min on ice.
IN VITRO Kinase Assay
Assay platform from Invitrogen was utilized. The time-resolved fluorescence resonance energy transfer assay (TR-FRET) was performed in 384 well plates. Each well received JNK1 (0.8 nM), ATF2 (200 nM), and ATP (1 μM) in 50 mM HEPES, 10 mM MgCl 2, 1 mM EGTA and 0.01% Brij-35, pH 7.5 and test compounds. The kinase reaction was performed at room temperature for 1 hr. After this time, the terbium labeled antibody and EDTA were added into each well. After an additional hour incubation, the signal was measured at 520/495 nm emission ratio on a BMG Pherastar fluorescence plate reader.
Alternatively, 9 was kept as a 10 mM solution in 10% DMSO. Serial dilutions containing 1% DMSO were prepared and 9 was added at a ratio of 1:10 to each kinase reaction to obtain the indicated final concentrations. JNK kinase assays were performed with 50 ng per reaction of active JNK2α2 from Upstate/Millipore (Cat. No. 14–329) according to the recommendations of the manufacturer with the following changes: GST-c-Jun (1–79) was used as a substrate 1 ug per reaction 46. The kinase reactions were performed at 30 °C for 20 minutes without Brij-35, and were stopped by addition of 2x Laemmli loading buffer and boiling for 3 minutes. The proteins were then separated on a mini gel and transferred to a PVDF membrane by wet blot. The membranes were dried and exposed to film.
Phospho-c-Jun detection and quantification
All cell culture media and supplements were from Life Technologies. The B16-F10 murine melanocyte cell line was purchased from ATCC and maintained according to manufacturers recommendations. At 48 hr prior to measuring phospho-c-jun levels, cells were transduced with BacMam GFP–c-Jun (1–79). BacMam preparation and transductions were performed as previously reported47. Briefly, the cells were grown in 10 cm dishes to approximately 75% confluence. The transduction was performed by adding 10% vol/vol BacMam virus stock in combination with Trichstatin A at a final concentration of 0.5 mM. The cells were incubated for 24 hrs. BacMam GFP-c-Jun (1–79) transduction efficiency, as determined by fluorescence microscopy, exceeded in general 80% of the cell population. All transductions were performed at a signal saturating MOI (at least 500 IU/cell). Following the transduction the cells were trypsinized and plated in white tissue culture treated 384 well plates at a density of 25000 cell per well in 32 ml assay medium (Opti-MEM®, supplemented with 0.1% charcoal/dextran-treated FBS, 100 U/mL penicillin and 100 mg/mL streptomycin, 0.1 mM non-essential amino acids, 1 mM sodium pyruvate, 25 mM HEPES pH 7.3, and lacking phenol red). After overnight incubation, cells were pretreated for 60 min with compound (indicated concentrations) followed by 30 min of stimulation with 2 ng/ml of TNF-α. The medium was then removed by aspiration and the cells were lysed by adding 20 ml of lysis buffer (20 mM TRIS-HCl pH 7.6, 5 mM EDTA, 1% NP-40 substitute, 5 mM NaF, 150 mM NaCl, 1:100 protease and phosphatase inhibitor mix, SIGMA P8340 and P2850 respectively). The lysis buffer included 2 nM of the terbium labeled anti-pc-Jun (pSer73) detection antibody (Life Technologies). After allowing the assay to equilibrate for 1 hour at room temperature, TR-FRET emission ratios were determined on a BMG Pherastar fluorescence plate reader (excitation at 340 nm, emission 520 nm and 490 nm; 100 ms lag time, 200 ms integration time, emission ratio = Em520/Em 490).
Alternatively, HEK293T cells were maintained in DMEM and supplements (Invitrogen). Cells were seeded at 4000,000 cells per well in a 12-well plate and incubated with or without compound. After 16 hrs cells were treated with anisomycin (50–100 nM) (MP Biomedicals) for 5–10 minutes. Levels of phospho-c-Jun were measured using the phospho-c-Jun Whole Cell Lysate Kit (Meso Scale K151CGD) from Meso Scale Discovery (Gaithersburg, MD). The assay was performed according to the manufacturer’s instructions, with duplicates of 10 μg of total protein from either HEK293T or C57/B6 liver cells.
THP-1 Cell Assay for Inhibition of LPS-Induced TNF-α and IL-1β Production
THP-1 cells (ATCC TIB 202, ATCC, Rockville, MD) were maintained at 37 °C, 5% CO2 in 10% fetal bovine serum (FBS)/RPMI 1640 medium. The day of the assay, 2 × 106 cells were resuspended in 1 mL of 3% FBS/RPMI 1640 medium and plated in a 12-well plate. 19 and 11, 12.5 μM each, or DMSO vehicle was added to the cell mixture and allowed to preincubate for 60 min at 37 °C, 5% CO2, prior to stimulation with LPS (Sigma L6529, from E. coli serotype 055:B5; 1 μg/mL final). LPS stimulation was allowed to proceed for 5 hr at 37 °C, 5% CO2. TNF-α and IL-1β production was measured directly from cell culture medium by a commercially available sandwich immunoassay developed by Meso Scale Discovery (Meso Scale no. K15025B-1, Gaithersburg, MD). Levels of TNF-α and IL-1β in the cell culture medium were determined using a Meso Scale Discovery Sector Imager 2400 according to the manufacturer’s instructions.
Molecular Modeling
Computational docking studies were performed with GOLD 2.1 (The Cambridge Crystallographic Data Centre, Cambridge, UK) 48, 49 and analyzed with Sybyl (Tripos, St. Louis). Molecular surfaces were generated with MOLCAD 50. The X-ray coordinates of JNK1/20/21 (PDB-ID 1UKI) were used to dock the compounds. Peptide and 8 and bi-dentate 9 poses reported in Figure 1 of the manuscript correspond to those obtained directly from the X-ray coordinates.
Glucose Tolerance Test
26-week-old male NONcNZO10/LtJ mice from Harlan (Jackson Labs, Bar Harbor, Maine 04609 USA) were dosed intraperitoneally (i.p.) with 25 mg/kg of 19 and 11 daily for five days. Mice were fasted 16 hours before i.p. administration of 2 g/kg D-glucose. Blood samples were taken at designated time points and blood glucose levels were measured using a hand-held glucose meter (OneTouch Ultra, LifeScan, a Johnson & Johnson company, UK).
Liver injury
7-week-old female C57/B6 mice from Harlan were dosed i.p. with 25 mg/kg of 19, while control mice were be treated with the vehicle only (n = 4). One hour later, mice were injected i.p. with 0.2 μg/gram Jo-2 antibody (Fas/APO-1; BD Biosciences, Catalog # 554255). Serum and liver were collected 4 hours hence. AST and ALT levels in serum are determined using the IDEXX VetTest Chemistry Analyzer per manufacturer instructions.
Chemistry
All anhydrous solvents were commercially obtained and stored in Sure-seal bottles under nitrogen. All other reagents and solvents were purchased as the highest grade available and used without further purification. Thin-layer chromatography (TLC) analysis of reaction mixtures was performed using Merck silica gel 60 F254 TLC plates, and visualized using ultraviolet light. 1H NMR data were collected using a 300 MHz Varian instrument and recorded in deuteron-chloroform (CDCl3) or dimethyl sulfoxide-d6 (DMSO-d6). Chemical shifts (δ) are reported in parts per million (ppm) referenced to 1H (Me4Si at 0.00). Mass spectral data were acquired on a Shimadzu LCMS-2010EV for low resolution, and on an Agilent ESI-TOF for high resolution and low resolution. List of Abbreviations: equivalent (eqv), high performance liquid chromatography (HPLC), liquid chromatography/mass spectrometry (LC/MS), room temperature (rt). Purity of compounds was obtained in a HPLC Breeze from Waters Co. using an Atlantis T3 3μm 4.6 × 150 mm reverse phase column. All intermediate compounds were > 95% pure. Following the scheme reported in Figure 1: 1 (indazole) was commercially available, which was iodinated according to the reported procedures.
Synthesis of 19
18 was coupled with a peptide of D-amino acids on resin using standard peptide coupling conditions. After coupling reaction complete, resin was removed with the treatment of TFA. Final compound was purified by rev phase HPLC. The compound was dried and checked purity again with HPLC (purity was >93%) and analyzed with MALDI-mass. Please see supporting info file for HPLC trace and MALDI-Mass.
Synthesis of 3-iodo-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazole (3)
To a solution of 2 (1.22 g, 5 mmol) in DMF (10 mL) was added NaH (220 mg, 5.50 mmol) in three portions at 0 °C under nitrogen atmosphere. The reaction mixture was stirred at the same temperature for 30 min then SEM-Cl (0.9 mL, 5 mmol) was added dropwise to it. The resulting reaction mixture was stirred at 0 °C for 1 h then at room temperature for 4 h. The reaction mixture was quenched with cold water (100 mL) followed by extraction with ether (3 × 100 mL). The combined organic layers were washed with water (100 mL) and brine (100 mL), dried (MgSO4), and concentrated in vacuo. The residue was chromatographed over silica gel (5% ethyl acetate in hexane) to afford the colorless oil 3 (1.68 g, 90%), HPLC purity > 95%. 1H NMR (300 MHz, CDCl3) δ-0.07 (s, 9 H), 0.88 (t, J = 7.2 Hz, 2 H), 3.57 (t, J = 7.5 Hz, 2 H), 5.72 (s, 2 H), 7.27 (d, J = 8.2 Hz, 1 H), 7.44–7.58 (m, 3 H); HRMS calcd for C13H19IN2OSi 374.0311, found 374.0312.
Synthesis of methyl-4-(1-((2-(trimethylsilyl)ethoxy)methyl-1H-indazol-3yl)benzoate (4)
A mixture of 3 (374 mg, 1 mmol), 4-methoxycarbonylphenyl boronic acid (271 mg, 1.5 mmol), Pd(dppf)Cl2 (82 mg, 0.1 mmol), saturated aqueous Na2CO3 solution (4 mL), in ethanol (1 mL) and toluene (10 mL) was stirred at 80 °C for 12 h. Upon completion of the reaction (TLC), the reaction mixture was extracted with CH2Cl2 (3 × 50 mL). The combined organic layers were washed with water (50 mL), and brine (50 mL), dried (MgSO4), and concentrated in vacuo. The residue was chromatographed over silica gel (5 to 10% ethyl acetate in hexane) to yield the pure product 4 (295 mg, 77%), HPLC purity > 96%. 1H NMR (300 MHz, CDCl3) δ −0.05 (s, 9 H), 0.88 (t, J = 8.4 Hz, 2 H), 3.60 (t, J = 8.4 Hz, 2 H), 3.95 (s, 3 H), 5.81 (s, 2 H), 7.28 (t, J = 7.2 Hz, 1 H), 7.42 (t, J =7.5 Hz, 1 H), 7.62 (d, J = 8.4 Hz, 1 H), 7.85–8.22 (m, 5 H); EIMS m/z 383 (M+H)+, 325, 267, 265, 149, 121, 83; HRMS calcd for C21H27N2O3Si 383.1785, found 383.1784.
Synthesis of 4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazol-3-yl)benzoic acid (5)
To a solution of 4 (282 mg, 0.738 mmol) in THF (6 mL) and methanol (1 mL) was added LiOH solution (177 mg, 7.380 mmol) in water (2 mL). The resulting reaction mixture was stirred at room temperature for 18 h. The reaction mixture was acidified with 1 N HCl followed by extraction with CH2Cl2 (3 × 50 mL). The combined organic layers were dried (MgSO4) and concentrated in vacuo. The residue was chromatographed over silica gel (20 to 30% ethyl acetate in hexane) to afford the acid 5 (195 mg, 90%), HPLC purity > 95%. 1H NMR (300 MHz, CDCl3) δ-0.06 (s, 9 H), 0.92 (t, J = 8.4 Hz, 2 H), 3.64 (t, J = 8.4 Hz, 5.83 (s, 2 H), 7.32 (t, J = 7.2 Hz, 1H), 7.49 (t, J = 7.5 Hz, 1 H), 7.66 (d, J = 8.4 Hz, 1 H), 8.06 (d, J = 8.4 Hz, 1 H), 8.12 (d, J = 8.7 Hz, 2 H), 8.27 (d, J = 8.1 Hz, 2 H); EIMS m/z 369 (M+H)+, 339, 311, 251, 149, 121, 99, 55; HRMS calcd for C20H25N2O3Si 369.1629 (M+H), found 369.1627.
Synthesis of tert-butyl-3-(4-(1-((2-(trimethylsilyl)ethoxy)methyl-1H-indazol-3-yl)benzamido)propylcarbamate (6)
To a solution of 5 (155 mg, 0.421 mmol) in DMF (3 mL) were added EDC (96 mg, 0.505 mmol), HOBt (68 mg, 0.505 mmol), DIEA (0.19 mL, 1.052 mmol), and mono-Boc-1,3 –diamino propane (82 mg, 0.463 mmol). The reaction mixture was stirred at room temperature for 16 h. Upon completion the reaction mixture was diluted with water (40 mL) followed by extraction with ethyl acetate (3 × 40 mL). The combined organic layers were washed with saturated NaHCO3 solution (2 × 30 mL), water (3 × 30 mL), and brine (30 mL) successively, dried (MgSO4), and concentrated in vacuo. The residue was chromatographed over silica gel (50% ethyl acetate in hexane) to give the pure product 6 (175 mg, 79%), HPLC purity > 95%. 1H NMR (300 MHz, CDCl3) δ −0.063 (s, 9 H), 0.91 (t, J = 8.7 Hz, 2 H), 1.47 (s, 9 H), 1.74 (quintet, J = 5.7 Hz, 2 H), 3.29 (q, J = 6 Hz, 2 H), 3.55 (q, J = 6 Hz, 2 H), 3.63 (t, J = 8.4 Hz, 2 H), 4.95 (br s, 1 H, NH), 5.80 (s, 2 H), 7.30 (d, J = 7.2 Hz, 1 H), 7.47 (t, J = 7.2 Hz, 1 H), 7.63 (d, J = 8.4 Hz, 1 H), 7.98–8.12 (m, 5 H); HRMS calcd for C28H40N4O4Si 524.2819, found 524.2817.
Synthesis of tert-butyl-3-(4-(1H-indazol-3-yl)benzamido)propylcarbamate (7)
To a solution of 6 (76 mg, 0.141 mmol) in THF (5 mL) was added TBAF (0.7 mL, 1 M solution in THF) at room temperature. The reaction mixture was refluxed for 10 h. Upon completion the reaction mixture was partitioned between dichloromethane (40 mL) and water (30 mL). The organic layer was washed with water (30 mL) and brine (30 mL), dried (MgSO4), and concentrated in vacuo. The residue was chromatographed over silica gel (80% ethyl acetate in hexane) to give the pure product 7 (42 mg, 76%), HPLC purity > 95%. 1H NMR (300 MHz, DMSO-d6) δ 1.38 (s, 9 H), 1.65 (quintet, J = 6.6 Hz, 2 H), 3.01 (q, J = 6.3 Hz, 2 H), 3.30 (q, J = 6.3 Hz, 2 H), 6.83 (br s. NH), 7.24 (t, J = 7.8 Hz, 1 H), 7.42 (t, J = 6.9 Hz, 1 H), 7.62 (d, J = 8.4 Hz, 1 H),7.99 (d, J = 8.7 Hz, 2 H), 8.05–8.15 (m, 3 H), 8.52 (t, J = 5.4 Hz, 2 H), EIMS m/z 395 (M+H)+, 339, 295, 221, 83; HRMS calcd for C22H27N4O3 395.2078 (M+H), found 395.2077
Synthesis of N-(3-aminopropyl)-4-(1H-indazol-3-yl)benzamide (8)
To a solution of 7 (21 mg, 0.053 mmol) in CH2Cl2 (2 mL) was added TFA (0.5 mL). The resulting reaction mixture was stirred at room temperature for 2 h. TFA and dichloromethane were removed in vacuum to give 8. This compound was used for the next step without further purification.
Synthesis of N1-(1-(4-(1H-indazol-3-yl)phenyl-16-methyl-1,7,10,13-tetraoxo-2,6,9,12-tetraazaheptadecan-14-yl)-2(2-(2-(2-(1-(2-amino-5-guanidinopentanoyl)pyrolidine-2-carboxamido)-3-hydroxybutanamido)-3-hydroxybutanamido)-4-methylpentanamido)succinamide (9)
To a solution of 8 (15 mg, 0.051 mmol) in DMF (2 mL) were added EDC (10 mg, 0.051 mmol), HOBt (6 mg, 0.051 mmol), DIEA (0.5 mL), and Boc-Arg(Pbf)-Pro-Thr(otbu)-Thr(Otbu)-Leu-Asn(trt)-Leu-Gly-Gly-OH (70 mg, 0.042 mmol) at room temperature. The reaction mixture was stirred at 50 °C for 16 h. After completion of the reaction, DMF and DIEA were removed in vacuo to give the protected compound. The crude residue was directly treated with TFA (1 mL) and H2O (0.2 mL) for 3 h. The final product was obtained by the following HPLC purification: Atlantis Preparative T3 column (10 × 250 mm), acetonitrile-water system, RT = 5–34 minutes, yield: 35%, purity > 95%. 1H NMR (300 MHz, CD3OD) δ 0.74–0.79 (m, 12 H), 1.25–2.20 (m, 24 H), 2.42–2.82 (m, 2 H), 3.01–3.94 (m, 10 H), 4.10–4.65 (m, 8 H), 7.15 (br, NH), 7.34–7.45 (m, 4 H), 7.50 (br, NH), 7.61 (d, J = 8.4 Hz, 2 H), 7.74 (d, J = 7.8 Hz, 2 H), 7.89 (d, J = 8.4 Hz, 1 H), 7.95 (br, NH); EIMS m/z 1204 (M+H)+, 1051, 860, 602, 450, 295, 136, 130, 108; HRMS calculated for C56H86N17O13 1204.6585 (M+H), found 1204.6572.
Similarly, the synthesis of 19 was obtained by coupling methyl-4-(4-(1H-indazol-3-yl)benzamido) butanoate (analogue to 8 but with a free carboxylic acid in lieu of the free amine; Supplementary Material) was coupled with a peptide of D-amino acids on resin using standard peptide coupling conditions. After coupling reaction complete, resin was removed with the treatment of TFA. Final compound was purified by rev phase HPLC. The compound was dried and checked purity again with HPLC (purity was >93%) and analyzed with MALDI-mass (Supplementary Material).
Supplementary Material
Figure S1: Scheme for the synthesis of and analytical data for 19.
Figure S2: 9 analytical data.
Table S1: Peptide sequences and IC50 values relative to their ability to displace 20.
Table S2: R2 values for various types of inhibition by 19 at either substrate or ATP binding site.
Acknowledgments
Financial support is obtained by the National Institutes of Health, NIDDK branch, grant number R24DK080263. Small amounts of 19 for research purposes are available upon request.
Abbreviations list
- JNK
c-Jun N-terminal Kinase
- MAP
Mitogen Activated Protein
- MAPK
Mitogen Activated Protein Kinase
- NMR
Nuclear magnetic resonance spectroscopy
- DELFIA
Dissociation Enhanced Lanthanide Fluoro-Immuno Assay
- TR-FRET
Time-Resolved Fluorescence Resonance Energy Transfer
- ALT
Alanine-aminotransferase
- AST
aspartate aminotransferase
- TNF-α
Tumor Necrosis Factor-α
- LPS
Lipopolysaccharide
- IC50
Half Maximal Inhibitory Concentration
- EC50
Half Maximal Effective Concentration
- K(i)
Binding Affinity
- 1H NMR
1H Nuclear Magnetic Resonance Spectroscopy
- LCMS
Liquid Chromatography and Tandem Mass Spectrometry
- HPLC
High-Performance Liquid Chromatography
- MALDI
Matrix-Assisted Laser Desorption/Ionization
- FBS
Fetal Bovine Serum
- IL-1β
Interleukin 1, beta
- ERK
Extracellular signal-regulated kinase
- ATF
Cyclic AMP-dependent transcription factor
- DMSO
Dimethyl sulfoxide
- HIV
Human Immunodeficiency Virus
- HEK
Human Embryonic Kidney
References
- 1.Bogoyevitch MA. Therapeutic promise of JNK ATP-noncompetitive inhibitors. Trends Mol Med. 2005;11:232–239. doi: 10.1016/j.molmed.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 2.Gupta S, Barrett T, Whitmarsh AJ, Cavanagh J, Sluss HK, Derijard B, Davis RJ. Selective interaction of JNK protein kinase isoforms with transcription factors. Embo J. 1996;15:2760–2770. [PMC free article] [PubMed] [Google Scholar]
- 3.Manning AM, Davis RJ. Targeting JNK for therapeutic benefit: from junk to gold? Nat Rev Drug Discov. 2003;2:554–565. doi: 10.1038/nrd1132. [DOI] [PubMed] [Google Scholar]
- 4.Martin JH, Mohit AA, Miller CA. Developmental expression in the mouse nervous system of the p493F12 SAP kinase. Brain Res Mol Brain Res. 1996;35:47–57. doi: 10.1016/0169-328x(95)00181-q. [DOI] [PubMed] [Google Scholar]
- 5.Ip YT, Davis RJ. Signal transduction by the c-Jun N-terminal kinase (JNK)--from inflammation to development. Curr Opin Cell Biol. 1998;10:205–219. doi: 10.1016/s0955-0674(98)80143-9. [DOI] [PubMed] [Google Scholar]
- 6.Leppa S, Bohmann D. Diverse functions of JNK signaling and c-Jun in stress response and apoptosis. Oncogene. 1999;18:6158–6162. doi: 10.1038/sj.onc.1203173. [DOI] [PubMed] [Google Scholar]
- 7.Minden A, Karin M. Regulation and function of the JNK subgroup of MAP kinases. Biochim Biophys Acta. 1997;1333:F85–104. doi: 10.1016/s0304-419x(97)00018-8. [DOI] [PubMed] [Google Scholar]
- 8.Solinas G, Karin M. JNK1 and IKK{beta}: molecular links between obesity and metabolic dysfunction. FASEB J. 2010;24:2596–2611. doi: 10.1096/fj.09-151340. [DOI] [PubMed] [Google Scholar]
- 9.Bogoyevitch MA, Ngoei KR, Zhao TT, Yeap YY, Ng DC. c-Jun N-terminal kinase (JNK) signaling: recent advances and challenges. Biochim Biophys Acta. 2010;1804:463–475. doi: 10.1016/j.bbapap.2009.11.002. [DOI] [PubMed] [Google Scholar]
- 10.Bogoyevitch MA, Kobe B. Uses for JNK: the many and varied substrates of the c-Jun N-terminal kinases. Microbiol Mol Biol Rev. 2006;70:1061–1095. doi: 10.1128/MMBR.00025-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heo YS, Kim SK, Seo CI, Kim YK, Sung BJ, Lee HS, Lee JI, Park SY, Kim JH, Hwang KY, Hyun YL, Jeon YH, Ro S, Cho JM, Lee TG, Yang CH. Structural basis for the selective inhibition of JNK1 by the scaffolding protein JIP1 and SP600125. Embo J. 2004;23:2185–2195. doi: 10.1038/sj.emboj.7600212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dickens M, Rogers JS, Cavanagh J, Raitano A, Xia Z, Halpern JR, Greenberg ME, Sawyers CL, Davis RJ. A cytoplasmic inhibitor of the JNK signal transduction pathway. Science. 1997;277:693–696. doi: 10.1126/science.277.5326.693. [DOI] [PubMed] [Google Scholar]
- 13.Barr RK, Kendrick TS, Bogoyevitch MA. Identification of the critical features of a small peptide inhibitor of JNK activity. J Biol Chem. 2002;277:10987–10997. doi: 10.1074/jbc.M107565200. [DOI] [PubMed] [Google Scholar]
- 14.Ho DT, Bardwell AJ, Abdollahi M, Bardwell L. A docking site in MKK4 mediates high affinity binding to JNK MAPKs and competes with similar docking sites in JNK substrates. J Biol Chem. 2003;278:32662–32672. doi: 10.1074/jbc.M304229200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Borsello T, Clarke PG, Hirt L, Vercelli A, Repici M, Schorderet DF, Bogousslavsky J, Bonny C. A peptide inhibitor of c-Jun N-terminal kinase protects against excitotoxicity and cerebral ischemia. Nat Med. 2003;9:1180–1186. doi: 10.1038/nm911. [DOI] [PubMed] [Google Scholar]
- 16.Bonny C, Oberson A, Negri S, Sauser C, Schorderet DF. Cell-permeable peptide inhibitors of JNK: novel blockers of beta-cell death. Diabetes. 2001;50:77–82. doi: 10.2337/diabetes.50.1.77. [DOI] [PubMed] [Google Scholar]
- 17.Kim JA, Lee J, Margolis RL, Fotedar R. SP600125 suppresses Cdk1 and induces endoreplication directly from G2 phase, independent of JNK inhibition. Oncogene. 2010;29:1702–1716. doi: 10.1038/onc.2009.464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bain J, McLauchlan H, Elliott M, Cohen P. The specificities of protein kinase inhibitors: an update. Biochem J. 2003;371:199–204. doi: 10.1042/BJ20021535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, McLauchlan H, Klevernic I, Arthur JS, Alessi DR, Cohen P. The selectivity of protein kinase inhibitors: a further update. Biochem J. 2007;408:297–315. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Siddiqui MA, Reddy PA. Small molecule JNK (c-Jun N-terminal kinase) inhibitors. J Med Chem. 2010;53:3005–3012. doi: 10.1021/jm9003279. [DOI] [PubMed] [Google Scholar]
- 21.Vazquez J, De SK, Chen LH, Riel-Mehan M, Emdadi A, Cellitti J, Stebbins JL, Rega MF, Pellecchia M. Development of Paramagnetic Probes for Molecular Recognition Studies in Protein Kinases. J Med Chem. 2008;51:3460–3465. doi: 10.1021/jm800068w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shuker SB, Hajduk PJ, Meadows RP, Fesik SW. Discovering high-affinity ligands for proteins: SAR by NMR. Science. 1996;274:1531–1534. doi: 10.1126/science.274.5292.1531. [DOI] [PubMed] [Google Scholar]
- 23.Jahnke W, Blommers MJ, Fernandez C, Zwingelstein C, Amstutz R. Strategies for the NMR-based identification and optimization of allosteric protein kinase inhibitors. Chembiochem. 2005;6:1607–1610. doi: 10.1002/cbic.200500100. [DOI] [PubMed] [Google Scholar]
- 24.Murray BW, Bennett BL, Sasaki DT. Analysis of pharmacologic inhibitors of Jun N-terminal kinases. Methods Enzymol. 2001;332:432–452. doi: 10.1016/s0076-6879(01)32220-6. [DOI] [PubMed] [Google Scholar]
- 25.Enkvist E, Lavogina D, Raidaru G, Vaasa A, Viil I, Lust M, Viht K, Uri A. Conjugation of adenosine and hexa-(D-arginine) leads to a nanomolar bisubstrate-analog inhibitor of basophilic protein kinases. J Med Chem. 2006;49:7150–7159. doi: 10.1021/jm0605942. [DOI] [PubMed] [Google Scholar]
- 26.Hill ZB, Perera BG, Maly DJ. A chemical genetic method for generating bivalent inhibitors of protein kinases. J Am Chem Soc. 2009;131:6686–6688. doi: 10.1021/ja900871y. [DOI] [PubMed] [Google Scholar]
- 27.Lavogina D, Enkvist E, Uri A. Bisubstrate inhibitors of protein kinases: from principle to practical applications. ChemMedChem. 2010;5:23–34. doi: 10.1002/cmdc.200900252. [DOI] [PubMed] [Google Scholar]
- 28.Lee JH, Kumar S, Lawrence DS. Stepwise combinatorial evolution of Akt bisubstrate inhibitors. Chembiochem. 2008;9:507–509. doi: 10.1002/cbic.200700583. [DOI] [PubMed] [Google Scholar]
- 29.Pflug A, Rogozina J, Lavogina D, Enkvist E, Uri A, Engh RA, Bossemeyer D. Diversity of bisubstrate binding modes of adenosine analogue-oligoarginine conjugates in protein kinase a and implications for protein substrate interactions. J Mol Biol. 2010;403:66–77. doi: 10.1016/j.jmb.2010.08.028. [DOI] [PubMed] [Google Scholar]
- 30.Fischer PM. The design, synthesis and application of stereochemical and directional peptide isomers: a critical review. Curr Protein Pept Sci. 2003;4:339–356. doi: 10.2174/1389203033487054. [DOI] [PubMed] [Google Scholar]
- 31.Shah M, Stebbins JL, Dewing A, Qi J, Pellecchia M, Ronai ZA. Inhibition of Siah2 ubiquitin ligase by vitamin K3 (menadione) attenuates hypoxia and MAPK signaling and blocks melanoma tumorigenesis. Pigment Cell Melanoma Res. 2009;22:799–808. doi: 10.1111/j.1755-148X.2009.00628.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000;351:95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Robers MB, Horton RA, Bercher MR, Vogel KW, Machleidt T. High-throughput cellular assays for regulated posttranslational modifications. Anal Biochem. 2008;372:189–197. doi: 10.1016/j.ab.2007.09.012. [DOI] [PubMed] [Google Scholar]
- 34.Comalada M, Xaus J, Valledor AF, Lopez-Lopez C, Pennington DJ, Celada A. PKC epsilon is involved in JNK activation that mediates LPS-induced TNF-alpha, which induces apoptosis in macrophages. Am J Physiol Cell Physiol. 2003;285:C1235–1245. doi: 10.1152/ajpcell.00228.2003. [DOI] [PubMed] [Google Scholar]
- 35.Baldassare JJ, Bi Y, Bellone CJ. The role of p38 mitogen-activated protein kinase in IL-1 beta transcription. J Immunol. 1999;162:5367–5373. [PubMed] [Google Scholar]
- 36.Solinas G, Vilcu C, Neels JG, Bandyopadhyay GK, Luo JL, Naugler W, Grivennikov S, Wynshaw-Boris A, Scadeng M, Olefsky JM, Karin M. JNK1 in hematopoietically derived cells contributes to diet-induced inflammation and insulin resistance without affecting obesity. Cell Metab. 2007;6:386–397. doi: 10.1016/j.cmet.2007.09.011. [DOI] [PubMed] [Google Scholar]
- 37.Cho YR, Kim HJ, Park SY, Ko HJ, Hong EG, Higashimori T, Zhang Z, Jung DY, Ola MS, Lanoue KF, Leiter EH, Kim JK. Hyperglycemia, maturity-onset obesity, and insulin resistance in NONcNZO10/LtJ males, a new mouse model of type 2 diabetes. Am J Physiol Endocrinol Metab. 2007;293:E327–336. doi: 10.1152/ajpendo.00376.2006. [DOI] [PubMed] [Google Scholar]
- 38.Kaoud TS, Mitra S, Lee S, Taliaferro J, Cantrell M, Linse KD, Van Den Berg CL, Dalby KN. Development of JNK2-Selective Peptide Inhibitors That Inhibit Breast Cancer Cell Migration. ACS Chem Biol. 2011;6:658–666. doi: 10.1021/cb200017n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maeda S, Chang L, Li ZW, Luo JL, Leffert H, Karin M. IKKbeta is required for prevention of apoptosis mediated by cell-bound but not by circulating TNFalpha. Immunity. 2003;19:725–737. doi: 10.1016/s1074-7613(03)00301-7. [DOI] [PubMed] [Google Scholar]
- 40.Wang Y, Singh R, Lefkowitch JH, Rigoli RM, Czaja MJ. Tumor necrosis factorinduced toxic liver injury results from JNK2-dependent activation of caspase-8 and the mitochondrial death pathway. J Biol Chem. 2006;281:15258–15267. doi: 10.1074/jbc.M512953200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Malhi H, Gores GJ. Cellular and molecular mechanisms of liver injury. Gastroenterology. 2008;134:1641–1654. doi: 10.1053/j.gastro.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stebbins JL, De SK, Machleidt T, Becattini B, Vazquez J, Kuntzen C, Chen LH, Cellitti JF, Riel-Mehan M, Emdadi A, Solinas G, Karin M, Pellecchia M. Identification of a new JNK inhibitor targeting the JNK-JIP interaction site. Proc Natl Acad Sci U S A. 2008;105:16809–16813. doi: 10.1073/pnas.0805677105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.De SK, Chen V, Stebbins JL, Chen LH, Cellitti JF, Machleidt T, Barile E, Riel-Mehan M, Dahl R, Yang L, Emdadi A, Murphy R, Pellecchia M. Synthesis and optimization of thiadiazole derivatives as a novel class of substrate competitive c-Jun N-terminal kinase inhibitors. Bioorg Med Chem. 2010;18:590–596. doi: 10.1016/j.bmc.2009.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.De SK, Chen LH, Stebbins JL, Machleidt T, Riel-Mehan M, Dahl R, Chen V, Yuan H, Barile E, Emdadi A, Murphy R, Pellecchia M. Discovery of 2-(5-nitrothiazol-2-ylthio)benzo[d]thiazoles as novel c-Jun N-terminal kinase inhibitors. Bioorg Med Chem. 2009;17:2712–2717. doi: 10.1016/j.bmc.2009.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.De SK, Stebbins JL, Chen LH, Riel-Mehan M, Machleidt T, Dahl R, Yuan H, Emdadi A, Barile E, Chen V, Murphy R, Pellecchia M. Design, synthesis, and structure-activity relationship of substrate competitive, selective, and in vivo active triazole and thiadiazole inhibitors of the c-Jun N-terminal kinase. J Med Chem. 2009;52:1943–1952. doi: 10.1021/jm801503n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Caelles C, Morales M. Assays to measure stress-activated MAPK activity. Methods Mol Biol. 2004;282:145–156. doi: 10.1385/1-59259-812-9:145. [DOI] [PubMed] [Google Scholar]
- 47.Huwiler KG, Machleidt T, Chase L, Hanson B, Robers MB. Characterization of serotonin 5-hydroxytryptamine-1A receptor activation using a phospho-extracellular-signal regulated kinase 2 sensor. Anal Biochem. 2009;393:95–104. doi: 10.1016/j.ab.2009.06.018. [DOI] [PubMed] [Google Scholar]
- 48.Eldridge MD, Murray CW, Auton TR, Paolini GV, Mee RP. Empirical scoring functions: I. The development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes. J Comput Aided Mol Des. 1997;11:425–445. doi: 10.1023/a:1007996124545. [DOI] [PubMed] [Google Scholar]
- 49.Jones G, Willett P, Glen RC, Leach AR, Taylor R. Development and validation of a genetic algorithm for flexible docking. J Mol Biol. 1997;267:727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- 50.Teschner M, Henn C, Vollhardt H, Reiling S, Brickmann J. Texture mapping: a new tool for molecular graphics. J Mol Graph. 1994;12:98–105.k. doi: 10.1016/0263-7855(94)80074-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Scheme for the synthesis of and analytical data for 19.
Figure S2: 9 analytical data.
Table S1: Peptide sequences and IC50 values relative to their ability to displace 20.
Table S2: R2 values for various types of inhibition by 19 at either substrate or ATP binding site.