

Abstract
Mitochondrial morphology and function are altered in intestinal epithelia during endotoxemia. However, it is unclear whether mitochondrial abnormalities occur in lung epithelial cells during acute sepsis or whether mitochondrial dysfunction corresponds with altered epithelial barrier function. Thus, we hypothesized that the intestinal epithelium is more susceptible to mitochondrial injury than the lung epithelium during acute sepsis and that mitochondrial dysfunction precedes impaired barrier function. Using a resuscitated feline model of Escherichia coli-induced sepsis, lung and ileal tissues were harvested after 6 h for histological and mitochondrial ultrastructural analyses in septic (n = 6) and time-matched controls (n = 6). Human lung epithelial cells (HLEC) and Caco-2 monolayers (n = 5) were exposed to ‘cytomix’ (TNFα: 40 ng/ml, IL-1β: 20 ng/ml, IFNγ: 10 ng/ml) for 24–72 h, and measurements of transepithelial electrical resistance (TER), epithelial permeability and mitochondrial membrane potential (ΔΨ) were taken. Lung epithelial morphology, mitochondrial ultrastructure and pulmonary gas exchange were unaltered in septic animals compared to matching controls. While histologically intact, ileal epithelia demonstrated marked mitochondrial ultrastructural damage during sepsis. Caco-2 monolayers treated with cytomix showed a significant decrease in mitochondrial ΔΨ within 24 h, which was associated with a progressive reduction in TER and increased epithelial permeability over the subsequent 48 h. In contrast, mitochondrial ΔΨ and epithelial barrier functions were preserved in HLEC following cytomix. These findings indicate that intestinal epithelium is more susceptible to mitochondrial damage and dysfunction than the lung epithelium in the context of sepsis. Early alterations in mitochondrial function portend subsequent epithelial barrier dysfunction.
Keywords: acute lung injury, feline, mitochondria, multiple organ failure, sepsis syndrome
Progressive organ dysfunction in the context of critical illness, also known as multiple organ dysfunction syndrome (MODS), most commonly caused by severe infections (sepsis), is the leading cause of mortality in critical care units (Tran et al. 1990; Vincent et al. 2006). A growing body of data indicates that altered cell metabolism, particularly mitochondrial, contributes to cell and organ dysfunction. The intestinal epithelium is particularly susceptible to mitochondrial damage (Hersch et al. 1990; Crouser et al. 1999) and impaired barrier function (Fink & Delude 2005) in the context of severe sepsis; however, little is known about the status of mitochondria in lung epithelium under these conditions. Furthermore, it is unclear whether lung epithelium fundamentally differs from intestinal epithelium in terms of susceptibility to the cytopathic events that characterize severe sepsis and the functional consequences thereof. In this regard, recent studies indicate that the barrier function of epithelial cells is sensitive to alterations in their bioenergetic status (Unno et al. 1996; Zheng & Cantley 2007).
Based upon human studies showing that intestinal epithelial cells undergo accelerated rates of programmed cell death during sepsis (Chang et al. 2005), we hypothesized that the intestinal epithelium is more susceptible to mitochondrial damage and dysfunction during severe acute bacterial sepsis than the lung epithelium and that consequent disruption of epithelial cell metabolism results in altered epithelial barrier function. A large animal model was employed for these investigations as it offers certain advantages over the prototypical rodent sepsis models, especially in the context of acute lung injury (ALI). Rodent models of sepsis are limited by the inability to control for potentially confounding physiological variables such as shock, hypoxaemia or acidosis, which are typically optimized in the human condition. In this regard, the intestinal epithelium is particularly sensitive to changes in systemic blood pressure and attendant ischaemia/reperfusion injury (Hotchkiss et al. 1999). Other advantages of large animal models include the ability to accurately assess lung gas exchange (e.g. during mechanical ventilation) and greater conformity to human anatomy (Ware 2008). An acute gram-negative bacteremia model [severe Escherichia coli bacteremia resulting in severe sepsis (without shock) and eventual organ failures and death (Schlag et al. 1992)] was chosen as it induces consistent dose-dependent pathology, such as is found in human sepsis, including alterations of the coagulation cascade (Taylor 1999) and lung and intestinal injury (Carraway et al. 1998; Welty-Wolf et al. 1998). This model is relevant to human bacteremia in the setting of gram-negative sepsis wherein the mortality is high, even with optimal treatment, and death occurs in a delayed fashion (Ziegler et al. 1991). In vitro analyses of lung and intestinal epithelium were performed in concert using a cocktail of pro-inflammatory cytokines (i.e. cytomix), which models the inflammatory milieu of sepsis and is known to alter the function of epithelial cells (Burke-Gaffney & Hellewell 1997; Wang et al. 2008).
The results of these investigations are interesting in that they show the intestinal epithelium to be more sensitive to ultrastructural damage, particularly mitochondrial, than the lung epithelium under conditions modelling acute severe sepsis. Furthermore, in vitro analyses indicate that loss of epithelial barrier function, as reflected by reduced transepithelial electrical resistance (TER) and increased permeability to small molecules, was preceded by a reduction in mitochondrial energy status, suggesting that mitochondrial performance could influence epithelial barrier function during acute inflammatory stress.
Materials and methods
Animal preparation
All experiments were approved by The Ohio State University Institutional Laboratory Animal Care and Use Committee, and all care and handling of the animals were in accord with National Institutes of Health guidelines. The experiments used an animal preparation that has been described previously (Crouser et al. 1999, 2002). Briefly, fasted, adult male cats were anesthetized with intramuscular ketamine hydrochloride (25 mg/kg) and intravenous sodium pentobarbital (10 mg/kg), intubated for mechanical ventilation and had a femoral artery and vein cannulated to enable continuous recording of systemic arterial pressure and arterial and venous blood sampling. Additional intravenous sodium pentobarbital (approximately 1–5 mg/kg) was provided, as needed, to maintain a proper level of sedation throughout the experimental time course. A blood gas analyser (ABL5; Radiometer America, Westlake, OH, USA) and a cooximeter (OSM3; Radiometer America), calibrated for feline blood, were used to assess ongoing blood gas and oxygenation status, enabling corrective measures as needed. Arterial pCO2 and pO2 were maintained within normal limits by adjusting the FIO2 and minute ventilation, as necessary. By administering intravenous bicarbonate, as required, arterial pH was normalized as well. Evaporative fluid loss was replenished with buffered isotonic saline (5–10 ml/kg/h), and rectal temperature was monitored and maintained at 39 °C.
Experimental protocol
After a 30-min stabilization period, baseline measurements were obtained, and animals were randomly assigned to receive either intravenous Escherichia coli [5.0 × 108 CFU/kg (in 20 ml), n = 6] or buffered isotonic saline vehicle (control, n = 6). Bacterial E. coli, serotype O86a:K61, was obtained from the American Type Culture Collection (33985™; Manassas, VA, USA), grown up and prepared as detailed previously (Welty-Wolf et al. 1998). The chosen acute dose proved to be sublethal for the experimental time period involved, yet evidence of acute organ injury typical of severe sepsis was demonstrated in the ileum, liver and heart within 2–4 h post-treatment consistent with that formerly described (Crouser et al. 1999, 2002, 2002; Joshi et al. 2006). In keeping with the results of Schlag et al. (1992), a dose of 1 × 107 CFU/kg E. coli failed to induce acute organ damage (data not shown). Approximately half of the E. coli dose was administered as an intravenous bolus over 1 min with the remaining half added to the intravenous drip and dispensed over the succeeding hour. Arterial pCO2 (35–40 mm Hg), pO2 (>100 mm Hg) and pH (7.3–7.4), paO2-to-FIO2 ratio (600 ± 50) and arterial pressure (>90 mm Hg), along with the relative ileal HbO2 content and blood volume, were kept within normal limits, as detailed elsewhere (Crouser et al. 1999, 2002). At 6 h post-treatment, the experiments were terminated, and tissue samples were harvested for histological and mitochondrial ultrastructural assessments.
Evaluation of ileal oxygen availability
Relative ileal HbO2 content and blood volume were measured at baseline and every 30 min thereafter using in vivo reflectance spectrophotometry as reported previously (Crouser et al. 1999, 2002). A diode-array UV–visible spectrophotometer (model 8452A; Hewlett Packard, Waldbronn, Germany) equipped with a remote reflectance fibre-optic probe (model RSA-HP-84F; Labsphere, North Sutton, NH, USA) was employed. Relative HbO2 contents were expressed as a percentage of the maximal measurable range in each animal, established by making similar measurements with the animal breathing an FIO2 of 1.0 at baseline (most oxygenated condition) and by excising the complete ileal segment at the end of the experiment (i.e. no further blood flow) and making measurements until tissue oxygen extraction had ended (least oxygenated condition) (Crouser et al. 1999). Relative changes in local tissue blood volume were expressed as a percentage of the same measurements made at baseline. At least four determinations of relative HbO2 content and blood volume were made at each time point by making measurements at randomly selected locations along the ileum.
Light and electron microscopy and evaluation of mitochondrial injury in the tissues
At 6 h post-treatment, lung and ileal tissue samples (approximately 0.5–2.0 mg) were excised and routinely processed for light and electron (Phillips CM-12 transmission electron microscope; Eindhoven, Holland) microscopy evaluation as discussed elsewhere (Crouser et al. 1999, 2002). For light microscopy, tissues were formalin-fixed for 72 h, blocked in paraffin, cut at 4 μm, de-paraffinized and then stained with haematoxylin and eosin. For electron microscopy, tissues were diced and fixed using 4% paraformaldehyde, 2.5% glutaraldehyde and 0.1 M sucrose in 0.1 M phosphate buffer (pH = 7.4). Specimens were then postfixed in 1% osmium tetroxide, dehydrated, embedded in Spurr media, cut, mounted on copper grids and stained with 2% uranyl acetate and Reynolds lead citrate.
Mitochondrial injury was assessed, as previously described (Crouser et al. 1999, 2002; Joshi et al. 2000), using a scoring system that is based upon the characteristic ultrastructural features of mitochondria attendant to the progressive stages of cellular injury (Trump et al. 1980). For each experiment, mitochondrial ultrastructure was evaluated at three randomly selected and widely separated areas for each tissue on each of two different grids. Two reviewers made the examinations in a blinded fashion, and the severity of ultrastructural injury was quantified by determining a composite score (based upon the scale of 0–5, as shown in Table 1), which represented all the mitochondria visualized within the high-power microscopy field. Thus, the reported mitochondrial injury score in each tissue for each experiment was derived from approximately 20 independent evaluations (i.e. approximately 120 independent evaluations for each tissue per group).
Table 1.
Mitochondrial injury scoring system based upon the stages of cellular injury*
| Cellular injury stage† | Mitochondrial injury score† | Mitochondrial characteristics |
|---|---|---|
| Stage I | 0.0 | Normal appearance |
| Stage II/III | 1.0 | Swelling of endoplasmic reticulum, minimal mitochondrial swelling |
| Stage IV‡ | 2.0 | Mild mitochondrial swelling |
| 3.0‡ | Moderate or focal high-amplitude swelling | |
| 4.0 | Diffuse high-amplitude swelling | |
| Stage V/VI | 5.0 | High-amplitude swelling with mitochondrial flocculent densities and/or calcifications |
Electron microscopy was used to visualize the extent of mitochondrial ultrastructural injury in the lungs and ileum, which was then quantitatively assessed (Scale of 0–5) based upon the cellular injury staging system described by Trump et al. (1980).
Stages and Scores do not directly correlate because indices of cellular injury other than mitochondrial changes were not taken into consideration during determinations of mitochondrial injury.
Irreversible cell damage is noted to occur during this stage, and mitochondrial injury scores >3.0 are predictive of ultimate cell death (Trump et al. 1980).
Feline Tumour Necrosis Factor α (TNFα) measurement
Serum from control and E. coli-treated animals was obtained at baseline and then again at 2, 4 and 6 h post-treatment. TNFα measurements were made by ELISA utilizing a mouse monoclonal capture antibody (Clone Mo199; Boehringer Mannheim, Indianapolis, IN, USA) and a rabbit polyclonal detection antibody to human TNFα. This assay is sensitive to TNFα concentrations ≥50 pg/ml and is highly specific for TNFα (Allen et al. 1992). Given that feline TNFα is known to have 90% homology with human TNFα, the ELISA specificity remains equally high for accurate quantification in feline serum samples as well.
Preparation of epithelial cell cultures
Human lungs were collected with approval from The Ohio State University Institutional Review Board and with patient consent. Normal, healthy adult donor lungs (n = 5) were obtained via open lung biopsy, and then primary human lung epithelial cells (HLEC) were isolated after enzymatic dissociation from the trachea, bronchi and bronchioles, seeded onto collagen-coated, semi-permeable membranes (0.6 cm2, Millicell-HA; Millipore, Billerica, MA, USA) and grown at an air–liquid interface, as previously described (Yamaya et al. 1992; Smith et al. 1996; Bao & Knoell 2006). Human lung epithelial cells were then cultured in a 50:50 mixture of Dulbecco's Modified Eagle media and Ham's F-12 media (D-MEM/F-12; Invitrogen Corp., Carlsbad, CA, USA), supplemented with 2% Ultroser G (BioSepra; Villeneuve, La Garenne, France) and multiple antibiotics [penicillin (100 U/ml), streptomycin (100 μg/ml), gentamycin (50 μg/ml), fluconazole (2 μg/ml) and amphotericin B (1.25 μg/ml)]. Similarly, Caco-2 cells were cultured in D-MEM with 20% foetal bovine serum (FBS) supplemented with penicillin (100 U/ml) and streptomycin (100 μg/ml) at 37 °C in a 5% CO2-humidified atmosphere. Forty-eight to 96 h after seeding, the epithelial cells formed a confluent culture with electrically tight junctions and a transepithelial resistance >800 Ω. The cells were then incubated with ‘cytomix’ (TNFα: 40 ng/ml, IL-1β: 20 ng/ml and IFNγ: 10 ng/ml) (Robbins et al. 1994) for 24–72 h at 37 °C in a 5% CO2-humidified environment. Human lung epithelial cells isolated from individual donors were cultured, treated and examined separately; pooling of cells across donors was never employed.
Determination of cell monolayer resistance and permeability
Human lung epithelial cells and Caco-2 cell layers, being fed exclusively from the basolateral aspect, were monitored until they excluded medium to the apical side. Cytomix was added to the basolateral side and incubated for the designated time periods. Barrier function, as reflected by the electrical resistance across the epithelial cell layer (Rt), was measured using a portable ohmmeter (Millipore). Epithelial barrier integrity was further evaluated by Lucifer yellow (MW = 457) diffusion. At each time point, Lucifer yellow (Millipore) was added to the donor (apical) chambers (final concentration: 0.1 mg/ml) to monitor membrane integrity. Aliquots (100 μl) were taken from the acceptor (basolateral) chambers and analysed in a fluorescent plate reader at excitation and emission wavelengths of 425 and 535 nm, respectively (Cytofluor II Fluorescence multiwell plate reader; PerSeptive Biosystems Inc., Foster City, CA, USA). A threshold value of ≥0.25%/h of Lucifer yellow flux was used to denote barriers that were abnormally permeable.
Evaluation of cellular mitochondrial membrane potential
To assess relative changes in mitochondrial membrane potential (ΔΨ) following cytomix treatment, HLEC and Caco-2 cells were harvested at 24 h post-treatment and washed with warm PBS. The cells were then resuspended in 1 ml warm PBS at a concentration of 1 × 106 and incubated with 2 μM JC-1 (Reers et al. 1991) using the MitoProbe™ JC-1 Assay kit (Invitrogen) at 37 °C, 5% CO2 for 20 min. Following a single wash with PBS, cells were resuspended and analysed by flow cytometry (Becton Dickinson FACS Calibur; BD Inc., Franklin Lakes, NJ, USA) using a 488 nm excitation wavelength with 519 nm (FITC-green) and 578 nm (PE-red) emission filters and photographed during fluorescence microscopy using a standard MF filter (Olympus BX40; Olympus America Inc., Melville, NY, USA). Fifty micromolar carbonyl cyanide 3-chlorophenylhydrazone (CCCP), an uncoupler of oxidative phosphorylation, at 37 °C for 5 min served as a positive control.
Assessment of apoptotic epithelial cell death
The presence of active caspase-3 was determined by the 7-amido-4-trifluoromethylcoumarin (AFC) assay using specific fluorosubstrates, as detailed previously (Coulter et al. 2002). For all AFC preparations, 3 × 106 cells were pelleted, washed with a magnesium sulphate-containing phosphate buffer and then lysed by multiple freeze–thaw cycles. Lysates were incubated with either DEVD-AFC [10% glycerol, 50 mM PIPES (pH 7.0), 1 mM EDTA, 1 mM DTT and 20 μM DEVD-AFC (Enzyme Systems Products, Livermore, CA, USA)] or YVAD-AFC [10% sucrose, 100 mM HEPES (pH 7.5), 0.1% CHAPS, 10 mM DTT, 20 μM YVAD-AFC (Enzyme Systems Products)]. The release of free AFC was determined using a Cytofluor 4000 fluorimeter (PerSeptive Biosystems Inc., Framingham, MA, USA) employing excitation and emission wavelengths of 400 and 505 nm, respectively.
Statistical analysis
The data were derived from a minimum of five independent experiments and were expressed as mean ± SEM, and statistical significance was based upon a value of P≤0.05. SigmaPlot 10.0 and SYSTAT 12.0 (Systat Software, Inc., Chicago, IL, USA) software were used to plot the data and carry out the statistical analyses, respectively. Comparisons of feline physiological parameters, TNFα levels and ileal mitochondrial injury scores were made using one-way analysis of variance (treatment) with repeated measures (time or replicates). Lung wet-to-dry ratios, the flow cytometry-derived red–green fluorescence intensity ratios, bicarbonate administration and lung mitochondrial injury scores were compared using the Student's t-test. Relative TER, epithelial permeability and relative caspase-3 activity measurements were analysed using two-way anova wherein the P-values of all pairwise comparisons were obtained using Tukey's multiple comparison method.
Results
Physiological parameters during the experimental protocol
In accordance with the experimental protocol, resuscitation was provided to maintain hemodynamic parameters, temperature and blood gas variables within a predetermined normal range. All physiological measurements remained relatively unchanged during the 6-h time period and did not differ significantly between the two groups (Figure 1). Mean arterial pressure, paO2/FIO2 and ileal oxygenation status were relatively constant throughout the experiment and did not vary between E. coli-treated and control groups.
Figure 1.
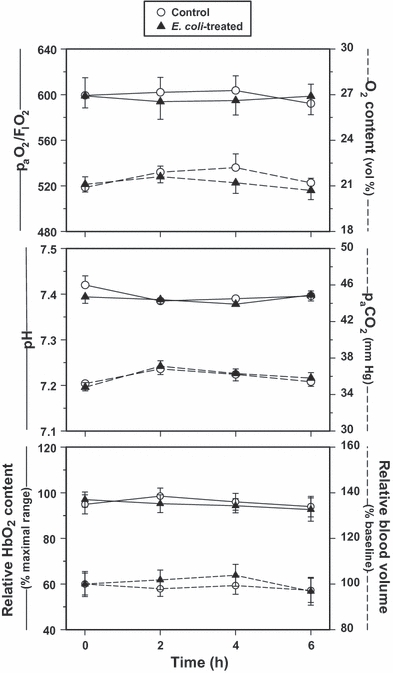
(Top) paO2-to-FIO2 ratio and arterial O2 content, (Middle) pH and paCO2 and (Bottom) relative ileal HbO2 content and blood volume of the Escherichia coli-treated and control groups as measured over time (values are mean ± SEM). All measurements were comparatively stable over the study period, indicating adequate resuscitation of the E. coli-treated animals and ample O2 availability in the ileal tissues throughout the 6-h experiment.
Unlike our previously established LPS model (Crouser et al. 1999, 2002, 2002; Joshi et al. 2006), only minor adjustments in minute ventilation, fluid resuscitation and bicarbonate administration were required through 6 h post-treatment in this bacteremia model. Positive end-expiratory pressure was never required, and vasoactive agents were never employed. There was no significant difference in the positive inspiratory pressure used (10–15 cm H2O, thus avoiding any confounding effects of mechanical injury related to ventilation itself), the inspiratory FIO2 (21%) and ventilatory frequency required, and the volume of fluid resuscitation needed over time between the groups. In addition, minimal supplemental bicarbonate administration was not different over the 6-h post-treatment period [11.6 ± 0.4 and 12.6 ± 1.1 mEq (NS), control vs. E. coli-treated, respectively]. However, these conditions did not hold true for longer experimental time courses. Evaluation much beyond 6 h was not conducted based upon the increasing mortality rates and enhanced difficulty in maintaining vital function status, resulting primarily from vascular leak-induced systemic hypotension and concomitant bicarbonate loss leading to acidosis, which could not be resolved through the stated resuscitative methods employed.
Arterial TNFα concentrations
Baseline TNFα levels were <200 pg/ml in the controls and remained low throughout the 6-h study period. As expected, TNFα levels increased significantly in the E. coli treatment group and peaked at 2 h post-treatment before returning to baseline values by 6 h (Figure 2).
Figure 2.
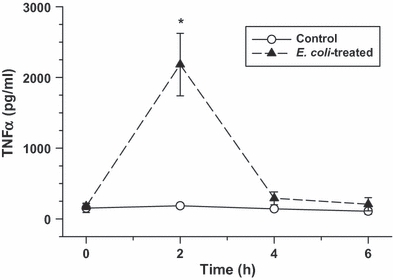
Arterial plasma TNFα levels as measured over time in the control and Escherichia coli -treated groups (values are mean ± SEM). Escherichia coli infusion caused a significant increase in TNFα levels that peaked at approximately 2 h post-treatment (*P<0.01, relative to baseline and time-matched control group).
Microscopic analyses of feline lung and ileal epithelia during sepsis
Lung tissue samples harvested 6 h post-treatment and processed for light microscopic analysis demonstrated an increased presence of septal neutrophilia following E. coli treatment (Figure 3, panels a and c). An average of 50 neutrophils per 100× visual field were observed in the lung tissues of the E. coli-treated group vs. <20 per 100× visual field in corresponding control tissues. Otherwise, no other differences in the appearance of the lung epithelia were noted between the groups. Lung wet-to-dry ratios were 5.3 ± 0.1 for controls and 6.2 ± 0.2 in the E. coli-treated group (P<0.05) reflecting mild lung oedema. In addition, there were no significant ultrastructural alterations of the lung epithelium, including mitochondria that appeared unchanged following E. coli treatment (Figure 3, panels b and d). Mitochondrial injury scores in the lung were 0.60 ± 0.09 and 0.65 ± 0.12 (NS) at 6 h post-treatment for the control and E. coli-treated groups, respectively. Histological analysis of the ileum was notable for retraction of intestinal villi in the E. coli-treated group; however, the mucosal layer remained essentially intact (Figure 4, panels a and c). Furthermore, ultrastructural analyses revealed high-amplitude mitochondrial swelling in the ileal epithelial cells of the E. coli-treated group relative to controls (Figure 4, panels b and d). Specifically, the mitochondrial injury score in the ileum at baseline was 0.63 ± 0.09. However, at 6 h post-treatment, the corresponding scores were 0.83 ± 0.11 (NS) and 3.43 ± 0.25 (P<0.01), respectively, for the control and E. coli-treated groups.
Figure 3.
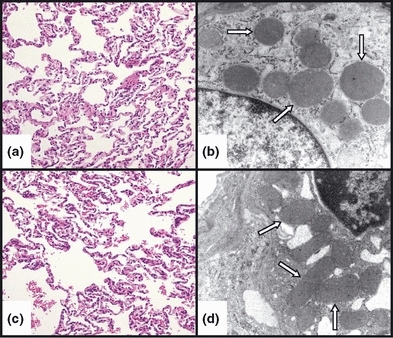
Representative light (LM) and electron (EM) photomicrographs, respectively, of control (a and b) and E. coli-treated (c and d) feline lung tissue. No significant difference was observed in the histological appearance of the alveolar epithelium or the epithelial mitochondrial ultrastructure between the control and E. coli-treated animals after 6 h. In addition, the mitochondria (white arrows) appeared intact and did not show the typical features of cytopathic injury, namely swelling and loss of cristae. (LM – staining: haematoxylin and eosin; original magnification: 20×, EM – staining: uranyl acetate and lead citrate; original magnification: 20,000×).
Figure 4.
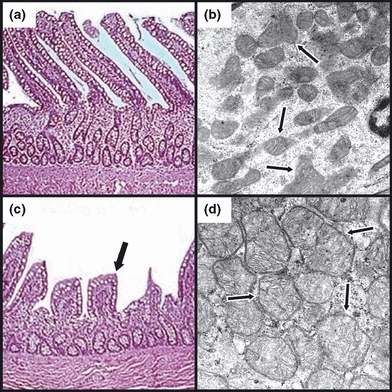
Representative light (LM) and electron (EM) photomicrographs, respectively, of control (a and b) and Escherichia coli-treated (c and d) feline ileal tissue. Escherichia coli-treated specimens (c) demonstrated retraction (black arrow) and occasional loss of villi compared to matching controls (a). In contrast to observations in the lungs, dramatic changes in mitochondrial morphology were evident in the ileum following E. coli treatment (d), evidenced by mitochondrial swelling with a loss of well-defined cristae (black arrows). (LM – staining: haematoxylin and eosin; original magnification: 20×, EM – staining: uranyl acetate and lead citrate; original magnification: 55,000×).
Transepithelial Electrical Resistance (TER) and permeability changes in response to putative sepsis mediators (cytomix)
In vitro analyses were performed to detect changes in TER in both lung (HLEC) and intestinal (Caco-2) epithelial monolayers over time in response to cytomix. TER was significantly reduced in HLEC monolayers at 24 h post-treatment relative to matched untreated controls but gradually returned towards baseline conditions becoming fully restored after 72 h (Figure 5). In contrast, Caco-2 monolayers demonstrated a progressive decline in TER over time becoming significant 48 h after cytomix exposure (Figure 5), corresponding with an ever-increasing permeability to small molecules (Figure 6).
Figure 5.
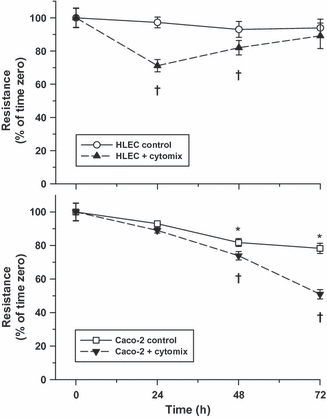
Transepithelial electrical resistance (TER) of human lung epithelial cells (HLEC) (Top) and Caco-2 (Bottom) monolayers exposed to control or cytomix-treated media as measured over time (values are mean ± SEM). Although a transitory drop in TER was observed in HLEC monolayers 24 h after cytomix treatment, resistance returned towards control levels thereafter. Conversely, TER fell progressively over time in Caco-2 monolayers, particularly following cytomix treatment (*P<0.05, compared to baseline; †P<0.05, relative to baseline and corresponding time-matched controls).
Figure 6.
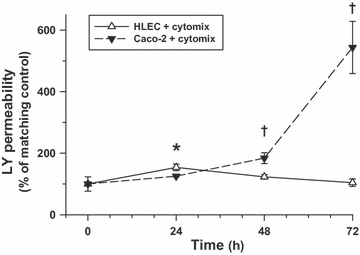
Epithelial permeability to Lucifer yellow of cytomix-treated human lung epithelial cells (HLEC) and Caco-2 monolayers over time (values are mean ± SEM). Following cytomix treatment, epithelial permeability was transiently elevated at 24 h post-treatment in HLEC monolayers compared to matching controls but normalized thereafter. By contrast, Caco-2 monolayers demonstrated a progressive increase in permeability beginning 48 h after cytomix treatment relative to matching controls (*P<0.05, compared to baseline; †P<0.05, relative to baseline and corresponding time-matched Caco-2 monolayers).
Mitochondrial membrane potential (ΔΨ) in HLEC and Caco-2 epithelial cells following cytomix
Relative changes in mitochondrial ΔΨ, as reflected by the alterations in the red–green fluorescence intensity ratio following exposure to the membrane potential-sensitive dye, JC-1, were determined in HLEC and Caco-2 epithelial cells 24 h after cytomix. No significant change in mitochondrial ΔΨ was observed in the lung epithelial cells. However, intestinal epithelial cell mitochondrial ΔΨ was dramatically diminished, consistent with a decreased red–green intensity ratio comparable with that induced by the positive control, CCCP (Figure 7).
Figure 7.
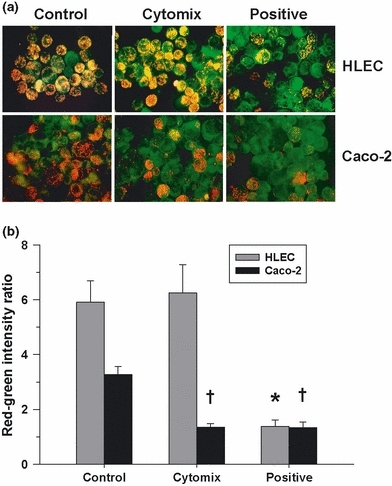
(a) Representative fluorescence photomicrographs of human lung epithelial cells (HLEC) and Caco-2 cells exposed to the membrane potential-sensitive dye, JC-1, 24 h after cytomix treatment or in time-matched controls. Orange–red fluorescence is indicative of cells having a normal elevated mitochondrial membrane potential (ΔΨ). Human lung epithelial cells appeared unchanged following cytomix, whereas Caco-2 cells fluoresced predominantly green, indicative of de-energization, and were comparable with carbonyl cyanide 3-chlorophenylhydrazone (CCCP)-treated (positive) cells. (b) Red–green fluorescence intensity ratio following flow cytometry of HLEC and Caco-2 cells exposed to JC-1 24 h post-treatment with cytomix or in time-matched controls (values are mean ± SEM). Unlike the HLEC cells, Caco-2 cells demonstrated a significant decrease in the red–green ratio related to the loss of ΔΨ similar to that observed following CCCP treatment (*†P<0.01, compared to matching controls).
Apoptosis following cytomix treatment
Increased programmed cell death, as reflected by an elevated caspase-3 activity, was evaluated over time in the HLEC and Caco-2 epithelial cells following cytomix treatment. No significant change in relative caspase-3 activity following cytomix was observed in the lung epithelial cells within 72 h post-treatment. However, enhanced apoptosis, indicated by an increased relative caspase-3 activity, was demonstrated in the intestinal epithelial cells 24 h after cytomix treatment but was unchanged from matching untreated controls thereafter (Figure 8).
Figure 8.
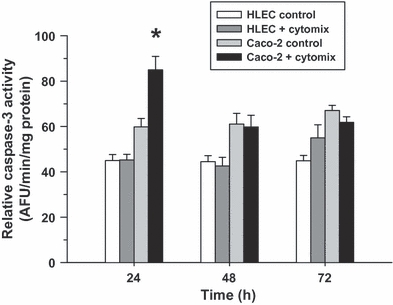
Relative caspase-3 activity in control and cytomix-treated human lung epithelial cells (HLEC) and Caco-2 cells over time (values are mean ± SEM). Although no change in activity was observed in the HLEC cells over time, a significant increase in activity was verified in the Caco-2 cells 24 h after cytomix treatment (*P<0.05, relative to matching controls) but not at later time points.
Discussion
These investigations show for the first time that lung epithelium, compared with intestinal epithelium, is resistant to mitochondrial injury in the context of acute severe sepsis. The in vivo studies showed that intestinal epithelial injury, characterized by altered mitochondrial morphology, is an early occurrence during severe bacteremic sepsis; however, the lung epithelium is preserved (Figures 3 and 4). In keeping with preserved lung morphology, pulmonary gas exchange was unchanged during the acute phase of sepsis (Figure 1). However, it was unclear from the in vivo investigations whether the relative resistance of the lung epithelium to sepsis relates to intrinsic cellular properties or is dependent upon other variables, such as differences in regional blood flow, inflammatory cell activation or the absence of bacteria (which is present in the intestinal lumen).
The in vitro model provided for a more sustained exposure to putative sepsis mediators while controlling for potentially confounding environmental variables, such as ambient oxygen tension, cytokine concentrations or regional bacterial populations. Under identical experimental conditions, cultured lung epithelial cells were shown to be more resistant to pro-inflammatory cytokine-mediated mitochondrial dysfunction compared with intestinal epithelial cells. Although mitochondrial damage can trigger cell death, changes in mitochondrial function (Figure 7) were not associated with significant cell death under our experimental conditions, as reflected in the relative caspase-3 activity (Figure 8), but did precede changes in barrier function, as indicated by the decreased TER (Figure 5) and increased permeability (Figure 6) to small molecules.
Certain organs are known to be highly susceptible to injury during critical illness. It has long been recognized that the intestinal epithelium is prone to damage during conditions modelling sepsis, even when controlling for blood flow (i.e. tissue oxygenation) (Crouser et al. 1999, 2002), and intestinal epithelial injury is believed to participate in the propagation of systemic inflammation by promoting the invasion of microorganisms or the translocation of bacterial products (Fink & Delude 2005). Likewise, lung epithelial integrity is thought to influence susceptibility to infection (i.e. barrier function) (van Kaam et al. 2004) and alveolar fluid accumulation in the context of ALI (Berthiaume & Matthay 2007). In keeping with the previous investigations performed in humans who had died from sepsis (Hotchkiss et al. 1999), this study demonstrated the ileum to be prone to acute damage; however, the lung was relatively resistant at least during the initial stage of sepsis. Moreover, the mitochondrial ultrastructural changes in the liver (not shown) and ileum (Figure 4), including high-amplitude swelling and loss of mitochondrial cristae integrity, conform to those described previously in these tissues in humans (Cowley et al. 1979; Watanabe et al. 2009) and animals (Crouser et al. 1999, 2002, 2002; Joshi et al. 2006) during severe sepsis or acute endotoxemia.
Although sepsis is a significant cause of ALI, the lung epithelial barrier was shown to resist injury, at least in the acute phase of severe bacteremic sepsis. Our findings are consistent with the previous studies, which found lung epithelial cells to be resistant to pro-inflammatory mediators (e.g. TNFα, IFNγ), unless the cells are rendered susceptible to injury by other factors, such as zinc depletion (Bao & Knoell 2006) or alcohol exposure (Brown et al. 2001). This would explain why a minority of patients presenting with sepsis develop severe ALI (Fein et al. 1983; Pittet et al. 1999; Vincent & Zambon 2006); whereas, the risk of ALI increases when a second risk factor is present, such as alcoholism (Moss et al. 1996), zinc depletion (Knoell et al. 2009) or ventilator-induced lung injury (ARDSnetwork, 2000; Frank et al. 2003). Presumably, there are a number of comorbid conditions contributing to sepsis-induced ALI in humans that are yet to be identified.
The incidence of intestinal epithelial damage and related changes in function during sepsis is difficult to determine because sensitive clinical diagnostic clinical tools are lacking. However, early postmortem examination of septic humans demonstrated that the intestinal mucosa is particularly prone to cytopathic changes (Hotchkiss et al. 1999; Berthiaume & Matthay 2007). This conforms to the common association of sepsis with altered gut motility and absorption (Johnston et al. 1996; Ritz et al. 2000; Overhaus et al. 2004) and supports the notion that the gut is the ‘motor’ of MODS, relating to epithelial damage, translocation of bacteria and bacterial products and the perpetuation of innate immune responses (Swank & Deitch 1996; Husain & Coopersmith 2003). Moreover, intestinal epithelial damage alters the capacity to provide nutrient support, thereby contributing to nutritional deficiencies in the setting of sepsis (Johnston et al. 1996; Ritz et al. 2000). Thus, further investigations into the mechanisms of intestinal epithelial injury are warranted.
Changes in intestinal barrier function are preceded by changes in the mitochondrial energy state (electrochemical gradient) in vitro. JC-1 is a fluorescent cationic dye that is rapidly sequestered within energized mitochondria, forming red-fluorescing aggregates. In contrast, when the mitochondria become de-energized, JC-1 remains in the cytoplasm in a monomeric form, which fluoresces green. Hence, the ratio of red to green fluorescence reflects the mitochondrial energization status (Huang et al. 2007). Using this sensitive approach, we observed that cytomix-treated gut epithelial cells exhibited mitochondrial de-energization within 24 h, whereas lung epithelial cells did not. Mitochondrial damage and dysfunction are well documented in various tissues, including the intestinal epithelium, in various models of sepsis (Cowley et al. 1979; Hersch et al. 1990; Crouser et al. 1999; Watts et al. 2004; Berthiaume & Matthay 2007; Watanabe et al. 2009). Mitochondrial mechanisms are believed to explain the high rate of intestinal epithelial apoptosis observed during sepsis (Coopersmith et al. 2002) and are expected to result in reduced ATP-dependent functions, including the maintenance of tight junctions and ion transport (Santa Cruz et al. 2005). Mitochondrial de-energization corresponded with subsequent alterations in intestinal epithelial barrier function, as reflected by reduced TER and increased permeability, such as is observed when epithelial cells are exposed to metabolic toxins (Santa Cruz et al. 2005). The dramatic increase in permeability to small molecules and reduction in TER observed in the Caco-2 cells 72 h after cytomix in the absence of significant ongoing cell death could be explained by the disruption of intercellular tight junctions.
Recent studies indicate that cellular bioenergetics strongly influences epithelial barrier functions, particularly with respect to tight junctions. Conditions favouring moderate levels of ATP depletion, such as sustained hypoxia or inhibition of glycolysis, are sufficient to promote altered intestinal epithelial permeability and disruption of tight junctions (Unno et al. 1996). Mechanistically, Zheng and Cantley recently demonstrated that epithelial tight junction assembly and disassembly is tightly regulated by AMP-activated protein kinase (AMPK). More specifically, the activity of AMPK is modulated by the cellular ATP/AMP ratio with lower ratios favouring disassembly of tight junctions (Zheng & Cantley 2007). Other possible mechanisms linking loss of epithelial barrier function to mitochondrial dysfunction include impaired ATP-dependent pump functions (Sugi et al. 2001) or increased mitochondrial oxidant production. With respect to the latter, oxidant stress is known to elicit remodelling of actin filaments leading to loss of barrier function (Boardman et al. 2004), and epithelial mitochondria are shown to produce higher levels of oxidants in response to putative sepsis mediators (Brown et al. 2001). Although altered ATP flux was not specifically addressed in these studies, the observed alterations in ultrastructure (in vivo) and function (in vitro) of intestinal mitochondria, the primary and most efficient source of ATP (compared to glycolysis), would predispose to ATP depletion such as is documented in other tissues during sepsis (Brealey et al. 2002). These bioenergetic mechanisms are likely modified by other factors known to regulate epithelial permeability, such as nitric oxide (Han et al. 2004), which could explain the observed transient reduction in HLEC resistance (Figure 5) and corresponding increase in HLEC permeability following cytomix treatment (Figure 6).
Additional investigations are needed to determine why the intestinal epithelium is more prone to damage and sustained dysfunction relative to the lung epithelium during sepsis/acute inflammation. Based upon the in vitro investigations, wherein the concentrations of cytokines, oxygen tension and other variables were carefully controlled, it is evident that intestinal epithelial injury is not merely explained by regional differences in oxygen tension (e.g. blood flow), inflammatory cell infiltration or the proximity to bacteria, which are variables that must be considered in vivo. Possible explanations for the greater sensitivity of the intestinal epithelium to sepsis and putative sepsis mediators (e.g. cytomix) include differences in oxidant/anti-oxidant balance or enhanced sensitivity to the effects of pro-inflammatory mediators such as IFNγ (Sugi et al. 2001) or TNFα (Fries et al. 2008).
There are several notable study limitations to be recognized. The large animal model, which requires continuous monitoring and resuscitation to normalize physiological variables, does not allow us to model subacute lung injury; however, this was not the goal of the in vivo investigations. We were primarily interested in determining whether the intestinal mucosa was more susceptible to injury than the lung epithelium in the context of ‘resuscitated’ sepsis, wherein variables such as blood gases and pH, blood pressure, oxygenation and body temperature were optimized, as is the standard in clinical practice. Maintaining these variables was part of the study design given that previous studies showed that ischaemia promotes epithelial barrier dysfunction (Sun et al. 1998) and mitochondrial damage (Madesh et al. 2000). Likewise, acidosis promotes mitochondrial swelling and related cellular de-energization (Kristian et al. 2001). The in vitro model (i.e. cytomix) does not exactly model conditions of sepsis but provides for reproducible exposure to the putative pro-inflammatory mediators of sepsis, which are known to contribute to cytopathic events. In particular, cytomix has been used extensively to model epithelial responses during acute inflammation (Han et al. 2004; Bastarache et al. 2007; Wang et al. 2008). Another potential limitation relates to the comparison of primary lung epithelial cell (HLEC) responses to those of an immortalized human intestinal epithelium cell line (Caco-2). Cultured primary human lung epithelial cells optimally represent the intact human lung epithelium. In lieu of primary gut epithelial cells, Caco-2 cells are considered to most closely approximate primary intestinal epithelium in terms of in vitro modelling (Sambuy et al. 2005; Shah et al. 2006), including preclinical pharmaceutical and toxicological testing (Meunier et al. 1995). Furthermore, when comparing the cytomix-induced responses of HLEC and Caco-2 cells to the epithelial pathology during gram-negative sepsis, the results are comparable in that both models show the intestinal epithelium to be more susceptible to injury, with evidence of early mitochondrial alterations.
In summary, this study confirmed that the intestinal epithelium is prone to injury during gram-negative bacteremia and that mitochondrial damage is an early manifestation. Under conditions modelling acute inflammation in vitro (i.e. cytomix), reduced mitochondrial transmembrane electrochemical gradient (de-energization) corresponded with subsequent impairment of intestinal epithelial barrier function. In contrast, the lung epithelium was resistant to both mitochondrial and barrier dysfunctions under these conditions. We speculate that mitochondrial dysfunction (e.g. impaired ATP production and/or increased ROS formation) may contribute to altered intestinal epithelial barrier function in the setting of sepsis. To the extent that changes in intestinal epithelial function equate with adverse clinical outcomes (e.g. bacterial translocation, impaired absorption of nutrients), it follows that cytoprotective strategies would hold promise for improving outcomes in sepsis. Indeed, recent studies suggest that interventions designed to protect intestinal epithelial mitochondria are beneficial in the context of sepsis (Coopersmith et al. 2002; Crouser et al. 2002). Future studies designed to determine why lung epithelium is relatively resistant to injury in the context of acute inflammation may lead to the discovery of novel therapeutic targets.
Acknowledgments
The authors wish to thank Edward P. Calomeni, Kathleen S. Wolken and the university's Campus Microscopy and Imaging Facility for their technical expertise and assistance in preparing the tissue and cell samples for light and electron microscopy analyses. In addition, the authors express appreciation to the Lifeline of Ohio for the lung donation procurements. National Institutes of Health (AI083912-01: EDC, HL086981-01: DLK and HL69899: RJF) and the OSU Medical Center Davis Developmental Grant (EDC).
References
- Allen JN, Herzyk DJ, Allen ED, Wewers MD. Human whole blood interleukin-1-b production: kinetics, cell source, and comparison with TNF-a. J. Lab. Clin. Med. 1992;119:538–546. [PubMed] [Google Scholar]
- ARDSnetwork. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. The Acute Respiratory Distress Syndrome Network. N. Engl. J. Med. 2000;342:1301–1308. doi: 10.1056/NEJM200005043421801. [DOI] [PubMed] [Google Scholar]
- Bao S, Knoell DL. Zinc modulates cytokine-induced lung epithelial cell barrier permeability. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006;291:L1132–L1141. doi: 10.1152/ajplung.00207.2006. [DOI] [PubMed] [Google Scholar]
- Bastarache JA, Wang L, Geiser T, et al. The alveolar epithelium can initiate the extrinsic coagulation cascade through expression of tissue factor. Thorax. 2007;62:608–616. doi: 10.1136/thx.2006.063305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthiaume Y, Matthay MA. Alveolar edema fluid clearance and acute lung injury. Respir. Physiol. Neurobiol. 2007;159:350–359. doi: 10.1016/j.resp.2007.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boardman KC, Aryal AM, Miller WM, Waters CM. Actin re-distribution in response to hydrogen peroxide in airway epithelial cells. J. Cell. Physiol. 2004;199:57–66. doi: 10.1002/jcp.10451. [DOI] [PubMed] [Google Scholar]
- Brealey D, Brand M, Hargreaves I, et al. Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet. 2002;360:219–223. doi: 10.1016/S0140-6736(02)09459-X. [DOI] [PubMed] [Google Scholar]
- Brown LA, Harris FL, Guidot DM. Chronic ethanol ingestion potentiates TNF-α-mediated oxidative stress and apoptosis in rat type II cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2001;281:L377–L386. doi: 10.1152/ajplung.2001.281.2.L377. [DOI] [PubMed] [Google Scholar]
- Burke-Gaffney A, Hellewell PG. Endogenous nitric oxide limits cytokine-induced damage of murine lung epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 1997;272:L707–L713. doi: 10.1152/ajplung.1997.272.4.L707. [DOI] [PubMed] [Google Scholar]
- Carraway MS, Welty-Wolf KE, Kantrow SP, et al. Antibody to E- and L-selectin does not prevent lung injury or mortality in septic baboons. Am. J. Respir. Crit. Care Med. 1998;157:938–949. doi: 10.1164/ajrccm.157.3.9707129. [DOI] [PubMed] [Google Scholar]
- Chang JX, Chen S, Ma LP, et al. Functional and morphological changes of the gut barrier during the restitution process after hemorrhagic shock. World J. Gastroenterol. 2005;11:5485–5491. doi: 10.3748/wjg.v11.i35.5485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coopersmith CM, Stromberg PE, Dunne WM, et al. Inhibition of intestinal epithelial apoptosis and survival in a murine model of pneumonia-induced sepsis. JAMA. 2002;287:1716–1721. doi: 10.1001/jama.287.13.1716. [DOI] [PubMed] [Google Scholar]
- Coulter KR, Doseff A, Sweeney P, et al. Opposing effect by cytokines on Fas-mediated apoptosis in A549 lung epithelial cells. Am. J. Respir. Cell Mol. Biol. 2002;26:58–66. doi: 10.1165/ajrcmb.26.1.4285. [DOI] [PubMed] [Google Scholar]
- Cowley RA, Mergner WJ, Fisher RS, Jones RT, Trump BF. The subcellular pathology of shock in trauma patients: studies using the immediate autopsy. Am. Surg. 1979;45:255–269. [PubMed] [Google Scholar]
- Crouser ED, Julian MW, Dorinsky PM. Ileal VO2-DO2 alterations induced by endotoxin correlate with severity of mitochondrial injury. Am. J. Respir. Crit. Care Med. 1999;160:1347–1353. doi: 10.1164/ajrccm.160.4.9810116. [DOI] [PubMed] [Google Scholar]
- Crouser ED, Julian MW, Blaho DV, Pfeiffer DR. Endotoxin-induced mitochondrial damage correlates with impaired respiratory activity. Crit. Care Med. 2002a;30:276–284. doi: 10.1097/00003246-200202000-00002. [DOI] [PubMed] [Google Scholar]
- Crouser ED, Julian MW, Joshi MS, et al. Cyclosporin A ameliorates mitochondrial ultrastructural injury in the ileum during acute endotoxemia. Crit. Care Med. 2002b;30:2722–2728. doi: 10.1097/00003246-200212000-00017. [DOI] [PubMed] [Google Scholar]
- Fein AM, Lippmann M, Holtzman H, Eliraz A, Goldberg SK. The risk factors, incidence and prognosis of ARDS following septicemia. Chest. 1983;83:40–42. doi: 10.1378/chest.83.1.40. [DOI] [PubMed] [Google Scholar]
- Fink MP, Delude RL. Epithelial barrier dysfunction: a unifying theme to explain the pathogenesis of multiple organ dysfunction at the cellular level. Crit. Care Clin. 2005;21:177–196. doi: 10.1016/j.ccc.2005.01.005. [DOI] [PubMed] [Google Scholar]
- Frank JA, Pittet JF, Lee H, Godzich M, Matthay MA. High tidal volume ventilation induces NOS2 and impairs cAMP-dependent air space fluid clearance. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003;284:L791–L798. doi: 10.1152/ajplung.00331.2002. [DOI] [PubMed] [Google Scholar]
- Fries W, Muja C, Crisafulli C, Cuzzocrea S, Mazzon E. Dynamics of enterocyte tight junctions: effects of experimental colitis and two different anti-TNF strategies. Am. J. Physiol. Gastrointest. Liver Physiol. 2008;294:G938–G947. doi: 10.1152/ajpgi.00469.2007. [DOI] [PubMed] [Google Scholar]
- Han X, Fink MP, Uchiyama T, Yang R, Delude RL. Increased iNOS activity is essential for pulmonary epithelial tight junction dysfunction in endotoxemic mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004;286:L259–L267. doi: 10.1152/ajplung.00187.2003. [DOI] [PubMed] [Google Scholar]
- Hersch M, Gnidec AA, Bersten AD, Troster M, Rutledge FS, Sibbald WJ. Histologic and ultrastructural changes in nonpulmonary organs during early hyperdynamic sepsis. Surgery. 1990;107:397–410. [PubMed] [Google Scholar]
- Hotchkiss RS, Swanson PE, Freeman BD, et al. Apoptotic cell death in patients with sepsis, shock and multiple organ dysfunction. Crit. Care Med. 1999;27:1230–1251. doi: 10.1097/00003246-199907000-00002. [DOI] [PubMed] [Google Scholar]
- Huang M, Camara AK, Stowe DF, Qi F, Beard DA. Quantitative analysis of mitochondrial membrane potential measurements with JC-1. FASEB J. 2007;21:946. [Google Scholar]
- Husain KD, Coopersmith CM. Role of intestinal epithelial apoptosis in survival. Curr. Opin. Crit. Care. 2003;9:159–163. doi: 10.1097/00075198-200304000-00013. [DOI] [PubMed] [Google Scholar]
- Johnston JD, Harvey CJ, Menzies IS, Treacher DF. Gastrointestinal permeability and absorptive capacity in sepsis. Crit. Care Med. 1996;24:1144–1149. doi: 10.1097/00003246-199607000-00013. [DOI] [PubMed] [Google Scholar]
- Joshi MS, Crouser ED, Julian MW, Schanbacher BL, Bauer JA. Digital imaging analysis for the study of endotoxin-induced mitochondrial ultrastructure injury. Anal. Cell. Pathol. 2000;21:41–48. doi: 10.1155/2000/201406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi MS, Julian MW, Huff JE, Bauer JA, Xia Y, Crouser ED. Calcineurin regulates myocardial function during acute endotoxemia. Am. J. Respir. Crit. Care Med. 2006;173:999–1007. doi: 10.1164/rccm.200411-1507OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Kaam AH, Lachmann RA, Herting E, et al. Reducing atelectasis attenuates bacterial growth and translocation in experimental pneumonia. Am. J. Respir. Crit. Care Med. 2004;169:1046–1053. doi: 10.1164/rccm.200312-1779OC. [DOI] [PubMed] [Google Scholar]
- Knoell DL, Julian MW, Bao S, et al. Zinc-deficiency increases organ damage and mortality in a murine model of polymicrobial sepsis. Crit. Care Med. 2009;37:1380–1388. doi: 10.1097/CCM.0b013e31819cefe4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristian T, Bernardi P, Siesjo BK. Acidosis promotes the permeability transition in energized mitochondria: implications for reperfusion injury. J. Neurotrauma. 2001;18:1059–1074. doi: 10.1089/08977150152693755. [DOI] [PubMed] [Google Scholar]
- Madesh M, Ramachandran A, Pulimood A, Vadranam M, Balasubramanian KA. Attenuation of intestinal ischemia/reperfusion injury with sodium nitroprusside: studies on mitochondrial function and lipid changes. Biochim. Biophys. Acta. 2000;1500:204–216. doi: 10.1016/s0925-4439(99)00107-6. [DOI] [PubMed] [Google Scholar]
- Meunier V, Bourrie M, Berger Y, Fabre G. The human intestinal epithelial cell line Caco-2; pharmacological and pharmacokinetic applications. Cell Biol. Toxicol. 1995;11:187–194. doi: 10.1007/BF00756522. [DOI] [PubMed] [Google Scholar]
- Moss M, Bucher B, Moore FA, Moore EE, Parsons PE. The role of chronic alcohol abuse in the development of acute respiratory distress syndrome in adults. JAMA. 1996;275:50–54. [PubMed] [Google Scholar]
- Overhaus M, Togel S, Pezzone MA, Bauer AJ. Mechanisms of polymicrobial sepsis-induced ileus. Am. J. Physiol. Gastrointest. Liver Physiol. 2004;287:G685–G694. doi: 10.1152/ajpgi.00359.2003. [DOI] [PubMed] [Google Scholar]
- Pittet D, Harbarth S, Suter PM, et al. Impact of immunomodulating therapy on morbidity in patients with severe sepsis. Am. J. Respir. Crit. Care Med. 1999;160:852–857. doi: 10.1164/ajrccm.160.3.9809033. [DOI] [PubMed] [Google Scholar]
- Reers M, Smith TW, Chen LB. J-aggregate formation of a carbocyanine as a quantitative fluorescent indicator of membrane potential. Biochemistry. 1991;30:4480–4486. doi: 10.1021/bi00232a015. [DOI] [PubMed] [Google Scholar]
- Ritz M, Fraser R, Tam W, Dent J. Impacts and patterns of disturbed gastrointestinal function in critically ill patients. Am. J. Gastroenterol. 2000;95:3044–3052. doi: 10.1111/j.1572-0241.2000.03176.x. [DOI] [PubMed] [Google Scholar]
- Robbins RA, Springall DR, Warren JB, et al. Inducible nitric oxide synthase is increased in murine lung epithelial cells by cytokine stimulation. Biochem. Biophys. Res. Commun. 1994;198:835–843. doi: 10.1006/bbrc.1994.1119. [DOI] [PubMed] [Google Scholar]
- Sambuy Y, De Angelis I, Ranaldi G, Scarino ML, Stammati A, Zucco F. The Caco-2 cell line as a model of the intestinal barrier: influence of cell culture and culture-related factors on Caco-2 cell functional characteristics. Cell Biol. Toxicol. 2005;21:1–26. doi: 10.1007/s10565-005-0085-6. [DOI] [PubMed] [Google Scholar]
- Santa Cruz V, Dugas TR, Kanz MF. Mitochondrial dysfunction occurs before transport of tight junction deficits in biliary epithelial cells exposed to bile from methylenedianiline-treated rats. Toxicol. Sci. 2005;84:129–138. doi: 10.1093/toxsci/kfi061. [DOI] [PubMed] [Google Scholar]
- Schlag G, Redl H, van Vuuren CJ, Davies J. Hyperdynamic sepsis in baboons: II. Relation of organ damage to severity of sepsis evaluated by a newly developed morphological scoring system. Circ. Shock. 1992;38:253–263. [PubMed] [Google Scholar]
- Shah P, Jogani V, Bagchi T, Misra A. Role of Caco-2 cell monolayers in prediction of intestinal drug absorption. Biotechnol. Prog. 2006;22:186–198. doi: 10.1021/bp050208u. [DOI] [PubMed] [Google Scholar]
- Smith JJ, Travis SM, Greenberg EP, Welsh MJ. Cystic fibrosis airway epithelia fail to kill bacteria because of abnormal airway surface fluid. Cell. 1996;85:229–236. doi: 10.1016/s0092-8674(00)81099-5. [DOI] [PubMed] [Google Scholar]
- Sugi K, Musch MW, Field M, Chang EB. Inhibition of Na+, K+-ATPase by interferon gamma down-regulates intestinal epithelial transport and barrier function. Gastroenterology. 2001;120:1393–1403. doi: 10.1053/gast.2001.24045. [DOI] [PubMed] [Google Scholar]
- Sun Z, Wang X, Deng X, et al. The influence of intestinal ischemia and reperfusion on bidirectional intestinal barrier permeability, membrane integrity, proteinase inhibitors, and cell death in rats. Shock. 1998;10:203–212. doi: 10.1097/00024382-199809000-00009. [DOI] [PubMed] [Google Scholar]
- Swank GM, Deitch EA. Role of the gut in multiple organ failure: bacterial translocation and permeability changes. World J. Surg. 1996;20:411–417. doi: 10.1007/s002689900065. [DOI] [PubMed] [Google Scholar]
- Taylor FB. Baboon model of E. coli sepsis: role of phospholipids microparticles in DIC. Sepsis. 1999;3:125–133. [Google Scholar]
- Tran DD, Groeneveld AB, van der Meulen J, Nauta JJ, Strack van Schijndel RJ, Thijs LG. Age, chronic disease, sepsis, organ failure, and mortality in a medical intensive care unit. Crit. Care Med. 1990;18:474–479. doi: 10.1097/00003246-199005000-00002. [DOI] [PubMed] [Google Scholar]
- Trump BF, Berezesky IK, Laiho KU, Osornio AU, Mergner WJ, Smith MW. The role of calcium in cell injury: a review. Scan. Electron Microsc. 1980;2:437–462. [PubMed] [Google Scholar]
- Unno N, Menconi MJ, Salzman AL, et al. Hyperpermeability and ATP depletion induced by chronic hypoxia or glycolytic inhibition in Caco-2BBe monolayers. Am. J. Physiol. Gastrointest. Liver Physiol. 1996;270:G1010–G1021. doi: 10.1152/ajpgi.1996.270.6.G1010. [DOI] [PubMed] [Google Scholar]
- Vincent J, Zambon M. Why do patients who have acute lung injury/acute respiratory distress syndrome die from multiple organ dysfunction syndrome? Implications for management. Clin. Chest Med. 2006;27:725–731. doi: 10.1016/j.ccm.2006.06.010. [DOI] [PubMed] [Google Scholar]
- Vincent JL, Sakr Y, Sprung CL, et al. Sepsis in European intensive care units: results of the SOAP study. Crit. Care Med. 2006;34:344–353. doi: 10.1097/01.ccm.0000194725.48928.3a. [DOI] [PubMed] [Google Scholar]
- Wang Q, Guo X, Wells-Byrum D, Noel G, Pritts TA, Ogle CK. Cytokine-induced epithelial permeability changes are regulated by the activation of the p38 mitogen-activated protein kinase pathway in cultured Caco-2 cells. Shock. 2008;29:531–537. doi: 10.1097/shk.0b013e318150737f. [DOI] [PubMed] [Google Scholar]
- Ware LB. Modeling human lung disease in animals. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008;294:L149–L150. doi: 10.1152/ajplung.00472.2007. [DOI] [PubMed] [Google Scholar]
- Watanabe E, Muenzer JT, Hawkins WG, et al. Sepsis induces extensive autophagic vacuolization in hepatocytes: a clinical and laboratory-based study. Lab. Invest. 2009;89:549–561. doi: 10.1038/labinvest.2009.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts JA, Kline JA, Thornton LR, Grattan RM, Brar SS. Metabolic dysfunction and depletion of mitochondria in hearts of septic rats. J. Mol. Cell. Cardiol. 2004;168:141–150. doi: 10.1016/j.yjmcc.2003.10.015. [DOI] [PubMed] [Google Scholar]
- Welty-Wolf KE, Carraway MS, Huang CT, Simonson SG, Kantrow SP, Piantadosi CA. Bacterial priming increases lung injury in gram-negative sepsis. Am. J. Respir. Crit. Care Med. 1998;158:610–619. doi: 10.1164/ajrccm.158.2.9704064. [DOI] [PubMed] [Google Scholar]
- Yamaya M, Finkbeiner WE, Chun SY, Widdicombe JH. Differentiated structure and function of cultures from human tracheal epithelium. Am. J. Physiol. 1992;262:L713–L724. doi: 10.1152/ajplung.1992.262.6.L713. [DOI] [PubMed] [Google Scholar]
- Zheng B, Cantley LC. Regulation of epithelial tight junction assembly and disassembly by AMP-activated protein kinase. Proc. Natl Acad. Sci. USA. 2007;104:819–822. doi: 10.1073/pnas.0610157104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler EJ, Fisher CJ, Sprung CL, et al. Treatment of gram-negative bacteremia and septic shock with HA-1A human monoclonal antibody against endotoxin. A randomized, double-blind, placebo-controlled trial. N. Engl. J. Med. 1991;324:429–436. doi: 10.1056/NEJM199102143240701. [DOI] [PubMed] [Google Scholar]