

Abstract
Acute hypercalcemia inhibits plasma renin activity. How this occurs is unknown. We hypothesized that acute hypercalcemia inhibits plasma renin activity via the calcium-sensing receptor due to parathyroid hormone-mediated increases in renal cortical interstitial calcium via TRPV5. To test our hypothesis, acute in vivo protocols were run in sodium-restricted, anesthetized rats. TRPV5 messenger RNA expression was measured with real-time quantitative reverse-transcriptase polymerase chain reaction. Acute hypercalcemia significantly decreased plasma renin activity by 37% from 32.0 ± 3.3 to 20.3 ± 2.6 ng Ang I/ml/hr (p<0.001). Acute hypercalcemia also significantly increased renal cortical interstitial calcium by 38% (1.73±0.06 mmol/L) compared to control values (1.25±0.05 mmol/L, p<0.001). Plasma renin activity did not decrease in hypercalcemia in the presence of a calcium-sensing receptor antagonist, Ronacaleret (22.8±4.3 vs. 21.6±3.6 ng Ang I/ml/hr). Increasing plasma calcium did not decrease plasma renin activity in parathyroidectomized rats (22.5 ± 2.6 vs. 22.0 ± 3.0 ng Ang I/ml/hr). Parathyroidectomized rats were unable to increase their renal cortical interstitial calcium in response to hypercalcemia (1.01±0.11 mmol/L). Acutely replacing plasma parathyroid hormone levels did not modify the hypercalcemic inhibition of plasma renin activity in parathyroid-intact rats (39.1±10.9 vs. 16.3±3.2 ng Ang I/ml/hr, p<0.05). Renal cortical TRPV5 messenger RNA expression decreased by 67% in parathyroidectomized (p<0.001) compared to intact rats. Our data suggest that acute hypercalcemia inhibits plasma renin activity via the calcium-sensing receptor due to parathyroid hormone-mediated increases in renal cortical interstitial calcium via TRPV5.
Keywords: Renin, calcium, calcium-sensing receptor, blood pressure, parathyroid hormone, renal cortical interstitial calcium, TRPV5
INTRODUCTION
Renin secretion has a unique relationship with extracellular calcium (Ca) in that increased extracellular Ca inhibits renin release both in vitro and in vivo. In vitro, increased extracellular Ca inhibits renin release from juxtaglomerular (JG) cells by inhibition of adenylyl cyclase 5 and stimulation of phosphodiesterase-1C, thus decreasing cyclic adenosine monophosphate (cAMP), the stimulatory second messenger for renin release.1-4 In vivo, acute hypercalcemia also decreases renin secretion.5-7 However, how hypercalcemia inhibits renin secretion is unknown.
It is possible that hypercalcemia inhibits renin by acting on the calcium-sensing receptor (CaSR). The CaSR is ubiquitously expressed and senses changes in extracellular Ca, transducing these changes into intracellular signaling.8 In vivo, the CaSR is integral for maintaining homeostatic control of plasma Ca. It does so primarily by decreasing parathyroid hormone (PTH) secretion and renal Ca reabsorption in response to hypercalcemia.8 Recently, we have found that the renin-secreting JG cells express the CaSR, and that stimulating the CaSR decreases renin release in vitro9 and plasma renin activity (PRA) in vivo.10 However, due to the ubiquitous expression of the CaSR, it is unknown how increases in plasma Ca could be translated into CaSR-mediated inhibition of renin secretion. The basolateral surface of the JG cells is bathed in the renal cortical interstitium.11 Thus, in addition to increased plasma Ca, increased renal cortical interstitial Ca could directly stimulate the CaSR to inhibit renin secretion. Increasing plasma Ca has been shown to increase renal cortical interstitial Ca.12 However, whether increased renal cortical interstitial Ca inhibits renin secretion is unknown.
PTH is an 84 amino-acid peptide released from the chief cells of the parathyroid gland. PTH increases plasma Ca, primarily by stimulating Ca resorption from bone and Ca reabsorption in the kidney. PTH stimulates the renal Ca reabsorption, predominantly in the distal tubule via TRPV5, the distal tubule epithelial Ca transporter.13 PTH increases the expression of TRPV5 and also increases TRPV5 Ca transport.14,15 These actions of PTH translate into increases in renal cortical interstitial Ca.12 As such, hypercalcemia may inhibit renin secretion via PTH-mediated increases in renal cortical interstitial Ca through TRPV5.
With these considerations, we hypothesized that acute hypercalcemia inhibits PRA via the CaSR due to PTH-mediated increases in renal cortical interstitial Ca via TRPV5.
METHODS
Acute in vivo preparation
Male Sprague-Dawley rats were placed on a 0.05% NaCl (Harlan-Teklad, Madison, WI) diet for 10 days prior to in vivo experimentation. The low NaCl diet was used to stimulate PRA.5 Stimulation of basal PRA using dietary sodium restriction was used to amplify the possible inhibition of PRA in response to hypercalcemia. Furthermore, it has been reported that high Ca infusions inhibit elevated PRA more efficaciously than basal PRA.5,7 Rats were fasted over night prior to being anesthetized with 125 mg/kg body weight thiobutabarbitol (Inactin, Sigma, St. Louis, MO) and placed on heating pad for the duration of the experiment. Rats were then given a tracheotomy using PE-240 tubing (Clay/Adams Becton Dickinson, Parsipanny, NJ). The femoral vein was catheterized using PE-50 tubing and a maintenance infusion of 10 μl/min of 0.9% NaCl was given. The femoral artery was catheterized with PE-50 tubing attached to a Statham pressure transducer (Viggo-Spectramed, Oxnard, CA), and connected through an iWorx 118 A to D Signal Processor to a computer using iWorx Labscribe 2.065 data acquisition software (iWorx, Dover, NH) for continuous monitoring of mean arterial pressure (MAP). The pressure transducers were calibrated using a digital, mercury-free “traceable” manometer (Fisher Scientific, Pittsburgh, PA). The peritoneal cavity was opened with a midline incision, and the intestines were wrapped with wet gauze and gently moved under the right abdominal wall to expose the left kidney. A dialysis catheter, consisting of a single 1-cm strand of dialysis tubing (Hemoflow F-8, Fresenius, Waltham, MA) fused to two sections of PE-20 tubing using nail polish (Revlon, New York, NY), was inserted under the renal capsule in the renal cortical interstitium. The dialysis catheter was perfused at a rate of 2 μl/min (see in situ dialysis methods below). At the completion of the surgery, the rat received a 1.0 ml bolus of 6% heat-inactivated bovine serum albumin (BSA, Sigma, St. Louis, MO). Any blood withdrawn was replaced with an equal volume bolus of 6% BSA i.v. Rats were allowed to recover from the surgery for 1 hour. Before the commencement of experimental manipulations, blood for basal PRA, PTH and plasma Ca were withdrawn at a volume of 300 μl each. Rats were subjected to various experimental manipulations (listed below) for 90 minutes before blood for PRA, PTH and plasma Ca were withdrawn again. Rats were then euthanized via pneumothorax and aortic transaction. The left kidney was removed for the inspection of any anatomical abnormalities, including ischemia, swelling and hydronephrosis. Rats with visible visceral trauma were excluded from analyses. All procedures were approved by the Henry Ford Health System IACUC committee, which is AALAC approved, and adhere to the guiding principles in the care and use of experimental animals in accordance with the National Institute of Health (NIH) guidelines.
In situ microdialysis
Our methods of in situ microdialysis are based on the work of R.D. Bukoski.12 During each experimental period, 3 sequential infusions of salines with different concentrations of Ca were pumped through the dialysis tubing at a rate of 2 μl/min. These salines consisted of 0.9% NaCl with varying concentrations of Ca (0.3 mmol/L, 1.0 mmol/L, 2.2 mmol/L). Each saline was infused through the dialysis tubing over 30 minutes. Only the effluent during the last 20 minutes was collected, in order to allow for the clearing of the dead space in the tubing.
The concentration of Ca for infusions and their paired effluent collections were measured using a NOVA-8 (NOVA Biomedical, Waltham, MA) electrolyte analyzer. To determine the concentration of Ca in the cortical interstitium, the concentration of Ca in the infused saline and its paired effluent were plotted on a graph, as previously described.12 The Ca concentration infused was plotted on the X-axis and the paired difference between the effluent Ca concentration and the infusion Ca concentration was plotted on the Y-axis. A line of best fit was generated, and the X-intercept (or point of zero flux) determined the interstitial Ca concentration.12
Parathyroidectomy (PTX)
Parathyroidectomies were performed as described previously,10 with the exception that rats were used for in vivo protocols 48-72 hrs after the completion of the surgery. PTX rats were placed on the 0.05% NaCl diet for 7-8 days prior to the PTX surgery, identical to rats in the other protocols, and were maintained on 0.05% NaCl diet for the 48-72 hr recovery period prior to any acute experimentation. PTX was confirmed by undetectable plasma PTH levels.
Plasma Renin Activity (PRA)
PRA was analyzed from 300 μl of femoral venous blood. Blood was centrifuged at 16000 x g for 6 min at 4° C and the plasma was aspirated and stored at −20° C until PRA was determined. PRA was analyzed by generation of angiotensin I (Ang I/ hr/min) using a Gamma Coat RIA kit (DiaSorin, Stillwater, MN) according to the manufacturer’s instructions.
Plasma parathyroid hormone (PTH) and ionized Ca
Measurements of plasma PTH and Ca were performed on femoral venous blood samples. Plasma PTH was measured using a commercial rat PTH immunoassay kit (Alpco Diagnostics, Salem, NH) according to the manufacturer’s instructions. Plasma ionized Ca was measured using a NOVA-8 electrolyte analyzer (Nova Biomedical, Waltham, MA).
Real-Time Quantitative Reverse-Transcriptase Polymerase Chain Reaction (RT-PCR)
Real time RT-PCR for TRPV5 was performed by quantitative real-time RT-PCR using a SYBR green method. Custom rat-specific primers from TIB Molbiol (Adelphia, NJ) were used for all PCR reactions. The primer sequences for TRPV5 are; forward: 5′-tgtgagccatttgtaggtcag-3′, reverse: 5′-gaggttgtgggaacttcga-3′. Real time RT-PCR was performed as follows: 1 μg of DNAse-treated total RNA sample was reverse transcribed using random primers and Omniscript reverse transcriptase (Qiagen, Valencia, CA) in a total volume of 20 μl for 1 hr at 37 °C followed by an inactivation step of 95 °C for 5 min. 2 μl of the reverse transcription reaction was then amplified in a Roche version 2.0 lightcycler PCR instrument (Roche, Indianapolis, IN) using SYBR green dye (SA Biosciences, Frederick, MD) and specific primers. Reactions were set up in a final volume of 20 μl, which contained 2 μl of sample, 1 μM each of both the primers and 10 μl of 2x SYBR green PCR mix. After an initial “hot start” at 95°C for 10 min, amplification occurred by denaturation at 95°C for 15s, annealing at 58°C for 45 sec, and extension at 72°C for 1 min for a total of 30-40 cycles. At the end of PCR cycling, melting curve analyses were performed. A relative quantitation method [ΔΔCt]16 was used to evaluate expression of TRPV5. RT-PCR of GAPDH was used for normalization of all data.
Statistics
Single changes from baseline to post-treatment were analyzed using a Student’s paired t-test. Repeated changes from baseline to post-treatment were analyzed using a One-way, Repeated-Measures ANOVA with a Student-Newman-Keuls test for post-hoc analyses. Intergroup analyses were performed using a One-way ANOVA with a Student-Newman-Keuls test for post-hoc analyses. Values p < 0.05 were considered statistically significant. All data are presented as mean ± 1 SEM. For the purpose of simplicity, a single asterisk is used to denote statistical significance in figures, regardless of p-value. Actual p-values are provided in the text.
In vivo protocols
Protocol 1: NaCl Control
(n=16) Rats were instrumented as described in the acute in vivo preparation section. Rats received a control 0.9% NaCl i.v. infusion for the duration of the experiment.
Protocol 2: High-Ca
(n=17) Rats received an i.v. infusion of 0.3 mg Ca/kg bodyweight/min (dissolved in 0.9% NaCl, pH 7.4) over the duration of the experimental period. Calcium lactate pentahydrate (Sigma, St. Louis, MO) was the Ca salt used for the infusion to avoid increasing plasma chloride.
Protocol 3a: High-Ca infusion+CaSR inhibition (Ronacaleret)
(n=8) The calcilytic Ronacaleret was used to inhibit the CaSR. Ronacaleret has a half-life of 4-5 hours in vivo.17 Ronacaleret was generously provided by GlaxoSmithKline, Molecular Discovery Research, Research Triangle Park, N.C. Rats received the same high-Ca infusion as above, but also received the CaSR antagonist Ronacaleret. Ronacaleret was given as a 1 mg/kg bodyweight i.v. bolus in 300 μl 0.9% NaCl every 30 minutes, commencing with the beginning of the high-Ca infusion. As such, each rat received three 1 mg/kg doses of Ronacaleret during the experiment.
Protocol 3b: Ronacaleret control
(n=5) Rats were maintained on 0.05% NaCl chow for 10 days before being anesthetized and instrumented identically to rats from the High-Ca in vivo protocols. Rats were given a bolus of 1 mg/kg i.v. Ronacaleret after the removal of blood for basal PRA, plasma PTH and Ca measurements. Blood for PRA, PTH and plasma Ca measurements were taken again 30 minutes post-Ronacaleret.
Protocol 4a: High-Ca+parathyroidectomy (PTX)
(n=8) PTX was performed as described previously.10 Rats were used 48-72 hours post-PTX for acute protocols. Rats received the same high-Ca infusion as protocol 2, as described above.
Protocol 4b: Extended High-Ca+parathyroidectomy (PTX)
(n=4) In 4 PTX rats from protocol 4a we continued the high-Ca i.v. infusion for an additional 90 minutes after the removal of blood for the measurement of experimental PRA, Ca and PTH values. During this additional 90 minute i.v. infusion, renal cortical interstitial Ca was measured again. At the end of the additional 90 minute i.v. infusion, blood was withdrawn for a third determination of PRA, plasma Ca and plasma PTH.
Protocol 5: High-Ca +PTH replacement infusion
(n=8) After the withdrawal of blood for the determination of basal PRA, plasma PTH and Ca, rats received a 200 ng/kg i.v. bolus of rat PTH 1-84 (Bachem, Torrance, CA) delivered in a 300 μl bolus of 0.9% saline. During the i.v. high-Ca infusion, rats were concomitantly infused with 20 ng/kg/min of rat PTH 1-84, i.v. These rats were parathyroid-intact, not PTX. This was done to examine the differences between the chronic and acute effects of PTH on the hypercalcemic inhibition of PRA.
Protocol 6: Effect of PTX on TRPV5 expression
PTX (n=3) or sham (n=4) rats were anesthetized with Inactin (125mg/kg) 72 hours after recovering from their respective surgeries. The right femoral artery was catheterized and 1 ml of blood was withdrawn for plasma Ca and PTH quantification. The left kidney was exposed via a mid-ventral incision, excised and decapsulated before cortical tissue was harvested on an ice-cold Lucite block. Cortical tissue was minced in ice-cold Tri-Reagent (Molecular Research Center, Inc., Cincinnati, OH) before being centrifugated at 12,000 g for 10 min at 4° C. The supernatant was collected and snap frozen in liquid nitrogen and stored at −80° C until extraction of RNA.
RESULTS
In vivo protocols
Protocol 1: NaCl Control
Time controls were run without any change in Ca delivery. Data for protocol 1 are summarized in Figure 1a, Figure 1b and Table 1. The control i.v. NaCl infusion had no effect on PRA, plasma Ca, plasma PTH or MAP. Renal cortical interstitial Ca was 1.25±0.05 mmol/L.
Figure 1.
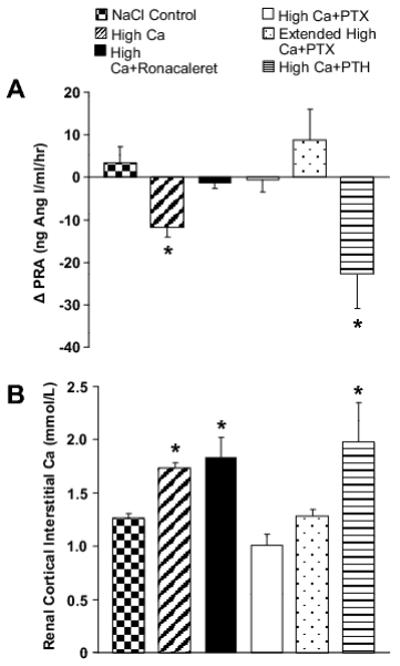
a The effects of CaSR, PTX and acute PTH replacement on Ca-mediated PRA inhibition. Increasing plasma Ca decreased PRA by 37%. High plasma Ca did not decrease PRA in the presence of CaSR inhibition (Ronacaleret) or in PTX rats. Increasing plasma Ca decreased PRA by 58% in rats receiving acute PTH replacement. * = p<0.05. See text for specific p-values. Data are presented as mean ± 1 SEM.
Figure 1b The effects of high plasma Ca on renal cortical interstitial Ca. Increasing plasma Ca increased renal cortical interstitial Ca 38% above i.v. NaCl control values. Renal cortical interstitial Ca was also increased by 47% in the presence of high plasma Ca and CaSR antagonism (Ronacaleret) compared to NaCl controls. Renal cortical interstitial Ca in PTX rats receiving the i.v. high-Ca infusion did not differ from i.v. NaCl controls, even in the presence of the extended i.v. high-Ca infusion. Renal cortical interstitial Ca was elevated 58% over i.v. NaCl control values in rats that received the i.v. high-Ca infusion with PTH replacement. * = p<0.05 versus NaCl control. See text for specific p-values. Data are presented as mean ± 1 SEM.
Table 1. The effects of various I.V. infusions on experimental parameters.
Parameters measured in each of the various protocols. Rats were maintained on a low-NaCl diet for 10 days prior to being anesthetized and administered the I.V. treatment denoted in the column on the left.
| Treatment | Basal PRA (ng Ang I/ml/hr) |
Experimental PRA (ng Ang I/ml/hr) |
Basal Plasma Ca (mmol/L) |
Experimental Plasma Ca (mmol/L) |
Basal PTH (pg/ml) |
Experimental PTH (pg/ml) |
Basal MAP (mm Hg) |
Experimental MAP (mm Hg) |
|---|---|---|---|---|---|---|---|---|
|
NaCl I.V.
Control |
26.7±4.6 | 30.2±4.9 | 1.12±0.04 | 1.18±0.02 | 104.8±9.8 | 90.3±8.7 | 106±3 | 107±3 |
|
High-Ca I.V.
Infusion |
32.0±3.3 | 20.3±2.6* | 1.19±0.04 | 1.91±0.07* | 171.3±32.9 | 4.7±4.7* | 106±2 | 117±2* |
|
High-Ca I.V.
Infusion+ Ronacaleret |
22.8±4.3 | 21.6±3.6 | 1.36±0.03 | 1.77±0.09* | 80.4±19.9 | 34.1±23.6 | 97 ±2 | 108±2* |
|
Ronacaleret
Control |
35.0±11.3 | 27.2±15.1 | 1.17±0.04 | 1.27±0.04* | 198.1±5.8 | 129.2±25.0 | 104±4 | 105±5 |
|
High-Ca I.V.
Infusion+ PTX |
22.5±2.6 | 22.0±3.0 | 0.62±0.04† | 1.50±0.06* | 0±0 | 0±0 | 104±3 | 123±4* |
|
Extended
High-Ca I.V. Infusion+ PTX |
21.9±3.7 | 30.5±5.7 | 0.69±0.06† | 1.94±0.19*‡ | 0±0 | 0±0 | 103±2 | 122±4* |
|
High-Ca I.V.
Infusion+ PTH |
39.1±10.9 | 16.3±3.2* | 1.19±0.05 | 1.81±0.10* | 161.4±54.8 | 542.8±250.9 | 112±3 | 128±4* |
= p<0.05 versus paired basal value.
= p<0.05 versus basal i.v. high-Ca value.
= p<0.05 versus i.v. high-Ca infusion + PTX. See text for specific p-values. Data are presented as mean ± 1 SEM.
Protocol 2: High-Ca infusion
The effect of hypercalcemia was tested by increasing the concentration of Ca infused into the circulation. The i.v. high-Ca infusion significantly decreased PRA by 37% (p<0.001, Figure 1a, Table 1). Plasma Ca significantly increased (p<0.001), while plasma PTH significantly decreased (p<0.001, Table 1). Furthermore, the i.v. high-Ca infusion significantly increased MAP (p<0.001, Table 1). Renal cortical interstitial Ca was significantly elevated compared to the rats treated with the control i.v. infusion from protocol 1 (1.73±0.06 mmol/L, p<0.001, Figure 1b).
Protocol 3a: High-Ca +CaSR inhibition (Ronacaleret)
To determine if the response to hypercalcemia was mediated by the CaSR, we used the calcilytic Ronacaleret to block the CaSR. PRA did not change from basal levels in rats that received the i.v. high-Ca infusion and Ronacaleret (Figure 1a and Table 1). Plasma Ca significantly increased (p<0.01). However, plasma PTH did not significantly decrease from basal levels, indicating that the administration of Ronacaleret was effective at inhibiting the CaSR (Table 1). As before, MAP increased in the presence of the i.v. high-Ca infusion and Ronacaleret (p<0.001, Table 1), and renal cortical interstitial Ca was elevated compared to the NaCl control i.v. infusion (1.84±0.18 mmol/L, p<0.01, Figure 1b.).
Protocol 3b: Ronacaleret control
To determine any effect of the CaSR blocker alone, we ran time controls without hypercalcemia. 1 mg/kg Ronacaleret had no effect on PRA or PTH 30 min after treatment. Plasma Ca increased 30 minutes after Ronacaleret administration (p<0.01, Table 1) indicating the administration of Ronacaleret was successful.
Protocol 4a: High-Ca+parathyroidectomy (PTX)
To determine the role of PTH in the hypercalcemia-mediated inhibition of PRA, we repeated our i.v. high-Ca infusion in PTX rats. Basal plasma Ca was decreased in PTX rats (p<0.001). When receiving the i.v. high-Ca infusion, PRA did not significantly decrease (Figure 1a, Table 1), while plasma Ca significantly increased (p<0.001), although the final plasma Ca was less than other groups receiving the high-Ca infusion (p<0.01). Furthermore, MAP increased significantly (p<0.001). Renal cortical interstitial Ca was not significantly different than the control NaCl rats (1.01±0.11 mmol/L, Figure 1b).
Protocol 4b: Extended High-Ca+parathyroidectomy (PTX)
Because the i.v. high-Ca infusion did not sufficiently increase plasma Ca in PTX rats (protocol 4a), we continued the infusion for an additional 90 minutes in 4 PTX rats to determine if further increasing plasma Ca could decrease PRA. As summarized in Table 1, plasma Ca increased further (p<0.05). PRA remained unaffected by the extended i.v. high-Ca infusion (Figure 1a, Table 1). MAP was still elevated compared to control values (p<0.05). As before, renal cortical interstitial Ca remained low and unchanged, even in the presence of further elevated plasma Ca (1.27±0.07 mmol/L, Figure 1b).
To determine if PTX decreases basal PRA, we compared the basal PRA of our PTX rats to the basal PRA of all other rats used in the i.v. high-Ca infusion protocol (parathyroid-intact). Basal PRA in parathyroid intact rats was 30.3±2.6 ng Ang I/ml/hr (n=54), which was not significantly different from PTX rats (22.5±2.6 ng Ang I/ml/hr, n=8).
Protocol 5: High-Ca +PTH replacement infusion
Acute hypercalcemia decreases PTH, and PTH may stimulate renin secretion.18,19 As such, we tested whether the decrease in PRA from acute hypercalcemia was due to the acute decrease in plasma PTH. To test this, we infused PTH with the i.v. high-Ca infusion in parathyroid-intact rats. PRA significantly decreased when rats were infused with both i.v. high-Ca and PTH (p<0.05, Figure 1a, Table 1). Both plasma Ca (p<0.001) and MAP (p<0.001) increased, while plasma PTH levels did not significantly change from basal levels (Table 1). Renal cortical interstitial Ca was increased compared to rats receiving the i.v. NaCl control infusion (1.98±0.38 mmol/L, p<0.01, Figure 1b.). Our data exclude the possibility that acute reductions in plasma PTH by the high-Ca infusion are directly causing the decrease in PRA.
Protocol 6: Effect of PTX on TRPV5 expression
To determine the cause of the impaired renal cortical interstitial Ca response in PTX rats to hypercalcemia, we measured the effects of PTX on the expression of the distal tubule Ca transporter, TRPV5. Plasma Ca was significantly lower in PTX compared to sham-operated rats (0.78±0.03 mmol/L vs. 1.16±0.03 mmol/L, p<0.001). PTH was undetectable in the PTX group and 114.73±14.6 ng/ml in the sham-operated group (p<0.001), indicating the PTX was successful. As seen in figure 2, renal cortical TRPV5 messenger RNA (mRNA) expression was significantly less in PTX rats compared to sham-operated (0.33±0.08 vs. 1.00±0.05 fold expression, respectively, p<0.001).
Figure 2.
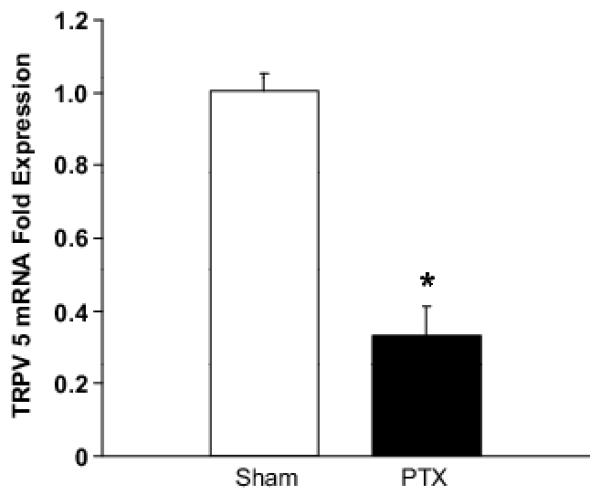
The effect of PTX on renal cortical TRPV5 mRNA expression as measured with real-time quantitative RT-PCR. TRPV5 mRNA expression was decreased by 67% in PTX rats. * = p<0.05 versus sham control. See text for specific p-values. Data are presented as mean ± 1 SEM.
DISCUSSION
We have shown that acutely increasing plasma Ca decreases PRA, and that inhibiting the CaSR completely eliminated the hypercalcemia-mediated inhibition of PRA. PTX also completely eliminated the hypercalcemia-mediated decrease in PRA, even when plasma Ca was elevated further for an additional 90 minutes. Noticeably, PTX totally eliminated the change in renal cortical interstitial Ca seen with hypercalcemia. While the i.v. high-Ca infusion decreased plasma PTH, acutely replacing plasma PTH with the i.v. high-Ca infusion did not impair the hypercalcemic inhibition of PRA. TRPV5 mRNA expression was decreased in PTX rats. These data support the hypothesis that acute hypercalcemia inhibits PRA via the CaSR due to PTH-mediated increases in renal cortical interstitial Ca through TRPV5. These data are consistent with the notion of a direct inhibitory effect of Ca on the JG cell.1-4,9
PRA is the ability of renin to generate angiotensin I from angiotensinogen endogenous to the plasma and is used as a marker of renin secretion. PRA represents renin synthesized and secreted from the renal cortical JG cells, as PRA from bilaterally-nephrectomized animals decreases to undetectable levels.20,21 PRA has been shown to be inversely related to the plasma Ca concentration. Similar to our results, acutely increasing the plasma Ca concentration in vivo decreases PRA.5-7 However, a mechanism of how this occurs is completely unknown. It should be noted that the increase in plasma Ca seen in our protocols, as well as previous experiments examining renin in hypercalcemia, are superphysiological.5,7 However, the levels of ionized plasma Ca that we obtain with our i.v. high-Ca infusion (~1.8-1.9 mmol/L) are quantitatively similar to ionized plasma Ca levels take from patients with hypercalcemia of malignancy or hyperparathyroidism (1.72 mmol/L).22
We have previously shown that JG cells in rats and mice, both in vitro and in vivo, contain CaSR.9,10 The CaSR is a seven-transmembrane domain receptor responsible for homeostatic regulation of plasma Ca.8 It was first described in the parathyroid gland where it acts by decreasing PTH secretion and increasing urinary Ca excretion in response to hypercalcemia.8 We have also reported that activation of the CaSR using a calcimimetic in vivo decreases PRA.10 Thus, we hypothesized that the CaSR should also mediate the inhibition of PRA by high plasma Ca. To test this, we used a calcilytic antagonist of the CaSR (Ronacaleret) during hypercalcemia, and found that blocking the CaSR completely eliminated the inhibition of PRA by high plasma Ca (and elevated cortical interstitial Ca). This result confirms that high plasma Ca inhibits PRA by acting on the CaSR.
As a positive control, we also measured the effect of Ronacaleret on the hypercalcemia-mediated inhibition of PTH. The CaSR is also integral in regulating PTH secretion,8 and calcilytics block both parathyroid gland as well as JG cell CaSR. High plasma Ca negatively feeds back on the parathyroid chief cells to decrease plasma PTH via the CaSR.8,23 This is illustrated in our results, as we observed that acute hypercalcemia significantly decreased circulating plasma PTH, and this decrease was blunted by CaSR antagonism using Ronacaleret.
PTH is an 84 amino acid peptide secreted from the chief cells of the parathyroid gland. PTH increases plasma Ca by stimulating Ca resorption from bone and also by stimulating renal Ca reabsorption. PTH stimulates renal Ca transport primarily in the distal tubule by increasing the expression and activation of the epithelial Ca transporter, TRPV5.13-15 To test whether acute hypercalcemia inhibits PRA via PTH-mediated increases in renal cortical interstitial Ca, we repeated the high-Ca infusion in PTX rats. We found that acute hypercalcemia did not decrease PRA in PTX rats. Furthermore, we found that renal cortical interstitial Ca did not increase with the i.v. high-Ca infusion in PTX rats. However, due to a low basal plasma Ca from the PTX, the high-Ca infusion did not increase plasma Ca to an equivalent level seen in other groups receiving the i.v. high-Ca infusion. To control for this, we extended the i.v. high-Ca infusion in 4 of the PTX rats in an attempt to further increase plasma and renal cortical interstitial Ca. The extended high-Ca infusion increased plasma Ca levels to similar levels seen in other groups with hypercalcemia. However, this extended high-Ca infusion still failed to either decrease PRA or increase renal cortical interstitial Ca in the PTX rats. These data demonstrate that acute hypercalcemia inhibits PRA via PTH-mediated increases in renal cortical interstitial Ca.
An important question raised by our experiments is: how could a PTX impair the increase in renal cortical interstitial Ca in response to hypercalcemia? PTH stimulates Ca reabsorption by increasing TRPV5-mediated Ca transport and TRPV5 expression.13-15 As such, we anticipated that our PTX rats could not increase their renal cortical interstitial Ca because of low TRPV5 expression. To test this, we measured renal cortical TRPV5 mRNA expression in PTX rats and sham controls. As expected, cortical TRPV5 mRNA expression was significantly decreased in PTX rats compared to sham-operated animals. This indicates that the inability of PTX rats to increase renal cortical interstitial Ca in response to hypercalcemia is in part due to decreased TRPV5 expression.
We also sought to control for many possible confounding factors in our experiment. PTH has also been reported to stimulate PRA.18,19 It is possible that this may be due to a direct effect of PTH on the JG cell, as PTH receptors are found in isolated glomeruli with attached vessels.24 As such, acute hypercalcemia could decrease PRA by acutely decreasing plasma PTH, decreasing the stimulatory effect of PTH on renin secretion. To test if this was the case, we repeated the i.v. high-Ca infusion in parathyroid intact rats with a concomitant i.v. PTH infusion to keep plasma PTH from decreasing. Even when plasma PTH was maintained at physiologic levels in acute hypercalcemia, PRA still decreased. This indicates that acute hypercalcemia does not decrease PRA through acute decreases in PTH.
Changes in blood pressure regulate PRA through the baroreceptor mechanism,25 such that increases in blood pressure decrease PRA. Hypercalcemia increases blood pressure,26 ostensibly by a direct action on L-type voltage-gated Ca channels on the vascular smooth muscle.27 As such, we measured the effects of our high-Ca infusion on MAP. The high-Ca infusion increased MAP, as expected. However, it is unlikely that this increase in MAP affected PRA, as it also increased in the Ronacaleret and PTX groups, which experienced no change in PRA. As such, it appears that high plasma Ca does not decrease PRA through increased MAP.
Since the PTX eliminated the response of PRA to hypercalcemia, we examined whether the chronic PTX lowered basal PRA. We compared the basal PRA values from our PTX rats versus the basal PRA values from all parathyroid-intact rats. Basal PRA values were not significantly lower in PTX rats versus controls, indicating that the loss of PRA inhibition was not due to a low basal PRA value. As an additional control, we measured the effect of Ronacaleret on basal PRA and found that CaSR blockade had no effect. Thus, Ronacaleret does not stimulate PRA, per se, but inhibits its decline in response to hypercalcemia-mediated CaSR stimulation.
One potential criticism of our work is that our basal PRA values are highly variable. This is due to the low-NaCl diet our rats were placed on, to elevate basal PRA levels and increase the likelihood that we would see an inhibition of PRA by high plasma Ca. This practice is consistent with previous experiments in the literature.5,7 Additionally, the basal PRA level in the rats receiving the i.v. high-Ca infusion (protocol 2) appears higher than other groups. However, there is no statistical difference between these basal PRA levels, and furthermore, they do not affect our results. When we take rats from protocol 2 with numerically identical baselines to other groups, the i.v. high-Ca infusion still decreases PRA from 21.2±1.6 to 12.3±1.2 ng Ang I/ml/hr (p<0.001, n=9). Our findings are identical for protocol 5; when we take rats with a numerically identical baseline, the i.v. high-Ca infusion with PTH replacement still decreased PRA from 24.2±4.3 to 12.7±2.8 ng Ang I/ml/hr (p<0.05, n=6). As such, apparent, but not actual, baseline PRA differences do not affect our PRA results.
In summary, our data address a 30-year old question in renal physiology: how does acute hypercalcemia inhibit PRA in vivo?5 The answer appears to be that hypercalcemia inhibits PRA by acting on the CaSR. These data are consistent with a direct effect of renal cortical interstitial Ca acting on the basolateral membrane of the JG cell. The cascade downstream of JG cell CaSR activation, involving inhibition of adenylyl cyclases and stimulation of phosphodiesterases, has already been described in detail.1-4,9 Plasma PTH is necessary for the inhibition of PRA by hypercalcemia by mediating the increase in renal interstitial Ca in response to hypercalcemia via renal cortical TRPV5 expression. Our data do not support the idea that hypercalcemia decreases PRA by acutely decreasing (a renin-stimulating effect of) plasma PTH. All of these data help support the growing body of evidence that the CaSR is an integral component in regulating renin.
Perspectives
How do the calcium-mediated changes in renin secretion translate into normal physiology or pathophysiology? The extent of hypercalcaemia we produce is large but not supraphysiologic, as they are similar to ionized plasma Ca levels found in patients with hypercalcaemia of malignancy or hyperparathyroidism.22 Clinically, acute hypercalcaemia is common in patients with rhabdomyolysis during the diuretic phase of acute renal injuries.28 Interestingly, PRA also decreases significantly during the diuretic phase in these patients.29 As such, the hypercalcaemia may contribute to this decline in PRA, though this remains to be tested. But what about the effects of less dramatic changes in calcium? While acute hypercalcaemia evokes a mild pressor response, chronic calcium supplementation has been shown to be antihypertensive. Epidemiologic data from a number of human studies over the past 20 years have reported an inverse relationship between blood pressure and calcium intake.30 A survey of animal models of hypertension lists 80 studies in normotensive and various models of hypertensive rats in which dietary calcium supplementation decreased blood pressure as much as 56 mmHg.31 In these studies, the mechanism for this antihypertensive influence dietary calcium is not known, and PRA’s have not been consistently reported. However, a chronic calcium-mediated lowering of renin or diminishing the response of renin to stimulation by retarding adenylyl cyclase-V activity via increased CaSR activation11 might reduce the role of renin in maintaining blood pressure. However, these interesting possibilities are yet to be tested.
Acknowledgement
The authors would like to acknowledge Dr. Robert Marquis, from GlaxoSmithKline, who generously provided us with the Ronacaleret used in these experiments.
Sources of Funding: This research was supported by funding from the National Institutes of Health (NIH) from grants F30DK084654-02 and PPG 5PO1HL090550-02. Mr. Atchison is a member of the Wayne State University School of Medicine MD/PhD program.
Footnotes
Disclosures: There are no conflicts or disclosures to report.
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Ortiz-Capisano MC, Ortiz PA, Harding P, Garvin JL, Beierwaltes WH. Decreased intracellular calcium stimulates renin release via calcium-inhibitable adenylyl cyclase. Hypertension. 2007;49:162–169. doi: 10.1161/01.HYP.0000250708.04205.d4. [DOI] [PubMed] [Google Scholar]
- 2.Ortiz-Capisano MC, Ortiz PA, Harding P, Garvin JL, Beierwaltes WH. Adenylyl cyclase isoform v mediates renin release from juxtaglomerular cells. Hypertension. 2007;49:618–624. doi: 10.1161/01.HYP.0000255172.84842.d2. [DOI] [PubMed] [Google Scholar]
- 3.Grunberger C, Obermayer B, Klar J, Kurtz A, Schweda F. The calcium paradox on of renin release: calcium suppresses renin exocytosis by inhibition of calcium-dependent adenylate cyclases AC5 and AC6. Circ Res. 2006;99:1197–1206. doi: 10.1161/01.RES.0000251057.35537.d3. [DOI] [PubMed] [Google Scholar]
- 4.Ortiz-Capisano MC, Liao TD, Ortiz PA, Beierwaltes WH. Calcium-dependent phosphodiesterase 1C inhibits renin release from isolated juxtaglomerular cells. Am J Physiol -Regul Integr Comp Physiol. 2009;297:R1469–1476. doi: 10.1152/ajpregu.00121.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kotchen TA, Mauli KI, Luke R, Rees D, Flamenbaum W. Effect of acute and chronic calcium administration on plasma renin. J Clin Invest. 1974;54:1279–86. doi: 10.1172/JCI107873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Watkins BE, Davis JO, Lohmeier TE, Freeman RH. Intrarenal site of action of calcium on renin secretion in dogs. Circ Res. 1976;39:847–853. doi: 10.1161/01.res.39.6.847. [DOI] [PubMed] [Google Scholar]
- 7.Kotchen TA, Maull KI, Kotchen JM, Luke RG. Effect of calcium gluconate infusion on renin in the dog. J Lab Clin Med. 1977;89:181–189. [PubMed] [Google Scholar]
- 8.Brown EM, MacLeod RJ. Extracellular calcium sensing and extracellular calcium signaling. Physiol Rev. 2001;81:239–297. doi: 10.1152/physrev.2001.81.1.239. [DOI] [PubMed] [Google Scholar]
- 9.Ortiz-Capisano MC, Ortiz PA, Garvin JL, Harding P, Beierwaltes WH. Expression and function of the calcium-sensing receptor in juxtaglomerular cells. Hypertension. 2007;50:737–743. doi: 10.1161/HYPERTENSIONAHA.107.095158. [DOI] [PubMed] [Google Scholar]
- 10.Atchison DK, Ortiz-Capisano MC, Beierwaltes WH. Acute activation of the calcium-sensing receptor inhibits plasma renin activity in vivo. Am J Physiol -Regul Integr Comp Physiol. 2010;299:R1020–1026. doi: 10.1152/ajpregu.00238.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beierwaltes WH. The role of calcium in the regulation of renin secretion. Am J Physiol Renal Physiol. 2010;298:F1–F11. doi: 10.1152/ajprenal.00143.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mupanomunda MM, Tian B, Ishioka N, Bukoski RD. Renal interstitial Ca2+ Am J Physiol Renal Physiol. 2000;278:F644–F649. doi: 10.1152/ajprenal.2000.278.4.F644. [DOI] [PubMed] [Google Scholar]
- 13.Hoenderop JG, Nilius B, Bindels RJ. Calcium reabsorption across epithelia. Physiol Rev. 2005;85:373–422. doi: 10.1152/physrev.00003.2004. [DOI] [PubMed] [Google Scholar]
- 14.van Abel M, Hoenderop JG, Van der Kemp AW, Friedlaender MM, van Leeuwen JP, Bindels RJ. Coordinated control of renal Ca(2+) transport proteins by parathyroid hormone. Kidney Int. 2005;4:1708–1721. doi: 10.1111/j.1523-1755.2005.00587.x. [DOI] [PubMed] [Google Scholar]
- 15.de Groot T, Lee K, Langeslag M, Xi Q, Jalink K, Bindels RJ, Hoenderop JG. Parathyroid hormone activates TRPV5 via PKA-dependent phosphorylation. J Am Soc Nephrol. 2009;20:1693–1704. doi: 10.1681/ASN.2008080873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Winer J, Jung CK, Shackel I, Williams PM. Development and validation of real-time quantitative reverse transcriptase-polymerase chain reaction for monitoring gene expression in cardiac myocytes in vitro. Anal Biochem. 1999;270:41–49. doi: 10.1006/abio.1999.4085. [DOI] [PubMed] [Google Scholar]
- 17.Roux S. New treatment targets in osteoporosis. Joint Bone Spine. 2010;3:222–228. doi: 10.1016/j.jbspin.2010.02.004. [DOI] [PubMed] [Google Scholar]
- 18.Smith JM, Mouw DR, Vander AJ. Effect of parathyroid hormone on plasma renin activity and sodium excretion. Am J Physiol. 1979;236:311–319. doi: 10.1152/ajprenal.1979.236.3.F311. [DOI] [PubMed] [Google Scholar]
- 19.Kovács L, Góth MI, Szabolcs I, Dohán O, Ferencz A, Szilágyi G. The effect of surgical treatment on secondary hyperaldosteronism and relative hyperinsulinemia in primary hyperparathyroidism. Eur J Endocrinol. 1998;138:543–547. doi: 10.1530/eje.0.1380543. [DOI] [PubMed] [Google Scholar]
- 20.Oates HF, Fretten JA, Stokes GS. Dissappearance rate of circulating renin after bilateral nephrectomy in the rat. Clin Exp Pharmacol Physiol. 1974;1:547–549. doi: 10.1111/j.1440-1681.1974.tb00576.x. [DOI] [PubMed] [Google Scholar]
- 21.Katz SA, Opsahl JA, Lunzer MM, Forbis LM, Hirsch AT. Effect of bilateral nephrectomy on active renin, angiotensinogen, and renin glycoforms in plasma and myocardium. Hypertension. 1997;30:259–266. doi: 10.1161/01.hyp.30.2.259. [DOI] [PubMed] [Google Scholar]
- 22.Wills MR, Lewin MR. Plasma calcium fractions and the protein-binding of calcium in normal subjects and in patients with hypercalcaemia and hypocalcaemia. J Clin Pathol. 1971;9:856–866. doi: 10.1136/jcp.24.9.856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grant FD, Conlin PR, Brown EM. Rate and concentration dependence of parathyroid hormone dynamics during stepwise changes in serum ionized calcium in normal humans. J Clin Endocrinol Metab. 1990;71:370–378. doi: 10.1210/jcem-71-2-370. [DOI] [PubMed] [Google Scholar]
- 24.Yang T, Hassan S, Huang YG, Smart AM, Briggs JP, Schnermann JB. Expression of PTHrp, PTH/PTHrP receptor, and Ca(2+)-sensing receptor mRNA along the rat nephron. Am J Physiol. 1997;272:F751–758. doi: 10.1152/ajprenal.1997.272.6.F751. [DOI] [PubMed] [Google Scholar]
- 25.Skinner SL, McCubin JW, Page IH. Control of renin secretion. Circ Res. 1964;15:64–76. doi: 10.1161/01.res.15.1.64. [DOI] [PubMed] [Google Scholar]
- 26.Suzuki T, Aoki K. Hypertensive effects of calcium infusion in subjects with normotension and hypertension. J Hypertens. 1998;6:1003–1008. doi: 10.1097/00004872-198812000-00008. [DOI] [PubMed] [Google Scholar]
- 27.Berl T, Levi M, Ellis M, Chaimovitz C. Mechanism of acute hypercalcemic hypertension in the conscious rat. Hypertension. 1985;7:923–930. doi: 10.1161/01.hyp.7.6.923. [DOI] [PubMed] [Google Scholar]
- 28.Akmal M, Bishop JE, Telfer N, Norman AW, Massry SG. Hypocalcemia and hypercalcemia in patients with rhabdomyolysis with and without acute renal failure. J Clin Endocrinol Metab. 1986;63:137–142. doi: 10.1210/jcem-63-1-137. [DOI] [PubMed] [Google Scholar]
- 29.Tu WH. Plasma renin activity in acute tubular necrosis and other renal diseases associated with hypertension. Circulation. 1965;31:686–695. doi: 10.1161/01.cir.31.5.686. [DOI] [PubMed] [Google Scholar]
- 30.McCarron DA. Epidemiological evidence and clinical trials of dietary calcium’s effect on blood pressure. Contrib Nephrol. 1991;90:2–10. doi: 10.1159/000420115. [DOI] [PubMed] [Google Scholar]
- 31.Hatton DC, Scrogin KE, Levine D, Feller D, McCarron DA. Dietary calcium modulates blood pressure through alpha 1-adrenergic receptors. Am J Physiol. 1993;264:F234–F238. doi: 10.1152/ajprenal.1993.264.2.F234. [DOI] [PubMed] [Google Scholar]