

Abstract
Nitric oxide (NO) produced by the endothelium is an important protective molecule in the vasculature. It is generated by the enzyme endothelial NO synthase (eNOS). Similar to all NOS isoforms, functional eNOS transfers electrons from nicotinamide adenine dinucleotide phosphate (NADPH), via the flavins flavin adenine dinucleotide and flavin mononucleotide in the carboxy-terminal reductase domain, to the heme in the amino-terminal oxygenase domain. Here, the substrate L-arginine is oxidized to L-citrulline and NO. Cardiovascular risk factors such as diabetes mellitus, hypertension, hypercholesterolaemia or cigarette smoking reduce bioactive NO. These risk factors lead to an enhanced production of reactive oxygen species (ROS) in the vessel wall. NADPH oxidases represent major sources of this ROS and have been found upregulated in the presence of cardiovascular risk factors. NADPH-oxidase-derived superoxide avidly reacts with eNOS-derived NO to form peroxynitrite (ONOO-). The essential NOS cofactor (6R-)5,6,7,8-tetrahydrobiopterin (BH4) is highly sensitive to oxidation by this ONOO-. In BH4 deficiency, oxygen reduction uncouples from NO synthesis, thereby converting NOS to a superoxide-producing enzyme. Among conventional drugs, compounds interfering with the renin-angiotensin-aldosterone system and statins can reduce vascular oxidative stress and increase bioactive NO. In recent years, we have identified a number of small molecules that have the potential to prevent eNOS uncoupling and, at the same time, enhance eNOS expression. These include the protein kinase C inhibitor midostaurin, the pentacyclic triterpenoids ursolic acid and betulinic acid, the eNOS enhancing compounds AVE9488 and AVE3085, and the polyphenolic phytoalexin trans-resveratrol. Such compounds enhance NO production from eNOS also under pathophysiological conditions and may thus have therapeutic potential.
Keywords: NADPH oxidase; oxidative stress; (6R-)5,6,7,8-tetrahydrobiopterin; midostaurin; betulinic acid; ursolic acid; trans-resveratrol
Introduction
The signalling molecule nitric oxide (NO) regulates vital functions such as neurotransmission or vascular tone (via activation of soluble guanylyl cyclase), gene transcription and mRNA translation (via iron-responsive elements), and post-translational modifications of proteins via ADP-ribosylation (Förstermann et al., 1994). However, NO can also react with superoxide anion (O2-·), forming the potent cytotoxin peroxynitrite (ONOO-). ONOO- causes oxidative damage, nitration and S-nitrosylation of biomolecules including proteins, lipids and DNA (Ridnour et al., 2004).
Three isoforms of NO synthase (NOS; EC 1.14.13.39) have been found in mammals. Neuronal ‘n’NOS (or NOS I) is a low-output enzyme that is constitutively expressed in neurons and some other cell types. Inducible ‘i’NOS (or NOS II) is a high-output enzyme, whose expression can be induced by cytokines and other agents in almost any cell type. Endothelial ‘e’NOS (or NOS III) is also a low-output enzyme that is constitutively expressed in endothelial cells and few other cell types. The nature of nNOS and eNOS as low-output enzymes and iNOS as a high-output enzyme depends not so much on the conversion rate of the different isozymes, but rather reflects the short-lasting, pulsatile, Ca2+-activated NO production of nNOS and eNOS versus the continuous, Ca2+-independent NO production by iNOS. Whereas eNOS-derived NO is a protective principle in the vasculature (see below), the large amounts of NO produced by iNOS are mainly responsible for the fall in blood pressure in septic shock (MacMicking et al., 1995; Liu and Huang, 2008). iNOS-derived NO may also contribute to vascular peroxynitrite formation (Upmacis et al., 2007) and has been shown to be proatherogenic (Kuhlencordt et al., 2001).
eNOS, an enzyme of major importance in the vasculature
In blood vessels, eNOS is the predominant NOS isoforms; it is responsible for most of the NO produced in this tissue (Schwarz et al., 1999; Li and Förstermann, 2000b). NO dilates blood vessels by directly stimulating soluble guanylyl cyclase and increasing cyclic GMP in smooth muscle cells (Rapoport et al., 1983; Förstermann et al., 1986; Ignarro et al., 1986). NO released towards the lumen of a blood vessel is a potent inhibitor of platelet aggregation and adhesion to the vascular wall (Alheid et al., 1987; Busse et al., 1987; Radomski et al., 1987). NO can also inhibit leukocyte adhesion to the vessel wall by either interfering with the ability of the leukocyte adhesion molecule CD11/CD18 to bind to the endothelial cell surface or by suppressing CD11/CD18 expression on leukocytes (Kubes et al., 1991; Arndt et al., 1993). Leukocyte adherence is an early event in the development of atherosclerosis and therefore, NO may protect against the onset of atherogenesis. Furthermore, NO has been shown to inhibit DNA synthesis, mitogenesis and proliferation of vascular smooth muscle cells (Garg and Hassid, 1989; Nakaki et al., 1990; Hogan et al., 1992; Nunokawa and Tanaka, 1992). These antiproliferative effects are mostly mediated by cGMP (Garg and Hassid, 1989; Nakaki et al., 1990; Nunokawa and Tanaka, 1992; Southgate and Newby, 1990). The inhibition of platelet aggregation and adhesion protects smooth muscle from exposure to platelet-derived growth factor(s). Therefore, NO also prevents a later step in atherogenesis, fibrous plaque formation. Finally, eNOS is probably essential for the function of endothelial progenitor cells involved in vascular repair (Aicher et al., 2003; Sasaki et al., 2006).
It is not surprising that a loss of function of such an enzyme in vascular disease has major pathophysiologic consequences. Pharmacological approaches to maintain or restore eNOS functionality (ability of the eNOS enzyme to produce NO) are thus warranted.
Normal function of eNOS and regulation of enzyme activity
Functional NOS enzymes are homodimers. The C-terminal reductase domain of one monomer [that binds nicotinamide adenine dinucleotide phosphate (NADPH), flavin mononucleotide (FMN) and flavin adenine dinucleotide (FAD)] is linked to the N-terminal oxygenase domain of the other monomer. This oxygenase domain carries a prosthetic heme group. The oxygenase domain also binds (6R-)5,6,7,8-tetrahydrobiopterin (BH4), molecular oxygen and the substrate L-arginine (Crane et al., 1998; Alderton et al., 2001). All NOS isozymes catalyse flavin-mediated electron transfer from the C-terminally bound NADPH to the heme on the N-terminus. Calmodulin (CaM), upon calcium-induced binding to the NOS, increases both the electron transfer within the reductase domain (from NADPH to the flavins) and also the electron transfer from the reductase domain to the heme center in the oxygenase domain (Hemmens and Mayer, 1998). At the heme, the electrons are used to reduce and activate O2. In order to synthesize NO, the enzyme needs to cycle twice. In a first step, NOS hydroxylates L-arginine to Nω-hydroxy-L-arginine (which remains largely bound to the enzyme). In a second step, NOS oxidizes N-hydroxy-L-arginine to citrulline and NO (Noble et al., 1999; Stuehr et al., 2001).
eNOS synthesizes NO in a pulsatile, Ca2+/CaM-dependent manner with eNOS activity markedly increasing when intracellular Ca2+ rises. However, eNOS can be activated by stimuli that do not produce sustained increases in intracellular Ca2+, but still induce a long-lasting release of NO. Such best established stimulus is the shear stress of the flowing blood, which can increase enzyme activity at resting Ca2+ levels. This activation is mediated by phosphorylation of the enzyme (Fleming and Busse, 2003). The eNOS protein can be phosphorylated on several serine, threonine and tyrosine residues; however, major changes in enzyme function have been reported for the phosphorylation of serine 1177 (activation) and threonine 495 (inhibition) in the human eNOS sequence (Fleming, 2010). These mechanisms, together with eNOS expression levels and eNOS functionality, are determining factors of vascular NO production.
Cardiovascular risk factors lead to an enhanced production of reactive oxygen species (ROS) in the vascular wall
Many cardiovascular diseases are associated with increased levels of ROS in the vessel wall (Förstermann, 2008) (Figure 1). Several enzyme systems can potentially produce these ROS; however, four enzyme systems seem to predominate. These include the NADPH oxidases, xanthine oxidase, enzymes of the mitochondrial respiratory chain and uncoupled eNOS (Mueller et al., 2005). Of these, NADPH oxidases seem to be the predominant source of ROS in the vasculature (Förstermann, 2008).
Figure 1.
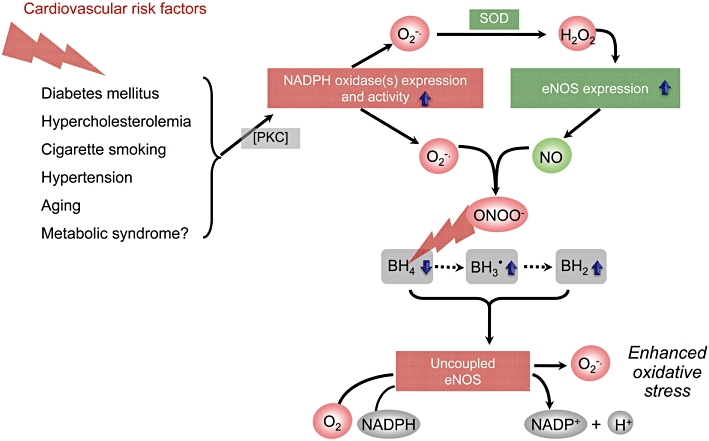
Potential mechanisms by which risk factors for atherosclerosis and cardiovascular disease lead to endothelial nitric oxide synthase (eNOS) uncoupling and endothelial dysfunction. In many types of vascular disease, nicotinamide adenine dinucleotide phosphate (NADPH) oxidases are upregulated in the vascular wall and generate superoxide (O2-·). At least in diabetes and angiotensin II-induced hypertension the increased NADPH oxidase expression and activity seem to be a consequence of PKC activation. NADPH oxidases derived O2-· can be dismutated (or reduced) by superoxide dismutase (SOD) (or superoxide reductase). The resulting hydrogen peroxide (H2O2) can increase the expression eNOS, and, indeed, eNOS expression is increased in most types of vascular disease. The products of NADPH oxidases and eNOS, O2-· and nitric oxide (NO), respectively, rapidly recombine to form peroxynitrite (ONOO-). This can oxidize the essential cofactor of eNOS (6R-)5,6,7,8-tetrahydrobiopterin (BH4) to trihydrobiopterin radical (BH3·). BH3· can be converted to the quinonoid 6,7-[8H]-H2-biopterin, which is non-enzymatically rearranged to 7,8-dihydrobiopterin (BH2). As a consequence, oxygen (O2) reduction by eNOS is uncoupled from NO formation, and a functional eNOS is converted into a dysfunctional O2-·-generating enzyme that contributes to vascular oxidative stress. The enhanced eNOS expression (see above) aggravates the situation.
Different isoforms of O2-·-producing NADPH oxidase exist in the vascular wall. They are expressed in endothelial and smooth muscle cells, as well as the adventitia. Cardiovascular risk factors increase the expression and/or activity of these enzymes, leading to increased ROS production. Evidence for an activation of NADPH oxidase in the vasculature has been provided in animal models of hypertension [such as angiotensin II infusion (Rajagopalan et al., 1996; Fukui et al., 1997) or spontaneously hypertensive rats (SHR) (Morawietz et al., 2001; Li et al., 2006)] and for different forms of diabetes mellitus (Hink et al., 2001). Also experimental hypercholesterolaemia is associated with an activation of NADPH oxidase (Warnholtz et al., 1999). In atherosclerotic arteries, there is evidence for increased expression of the NADPH oxidase subunits gp91phox (Nox2) and Nox4, both of which can contribute to increased oxidative stress (Sorescu et al., 2002).
In hypercholesterolaemia, both systemic and local renin angiotensin systems may be activated. Angiotensin-converting enzyme activity, as well as local angiotensin II concentrations are increased in atherosclerotic plaques (Diet et al., 1996; Ohishi et al., 1997), and the inflammatory cells present in the atherosclerotic vessel wall are capable of producing large amounts of angiotensin II. The stimulating effects of angiotensin II on the activity of NADPH oxidases suggest that an activated renin angiotensin system could cause increased vascular O2-· production and thus vascular dysfunction (Griendling et al., 2000). In vessels from hypercholesterolaemic animals (Vergnani et al., 2000) and in platelets from hypercholesterolaemic patients (Nickenig et al., 1999), the angiotensin II receptor subtype AT1 has been found to be upregulated, probably in response to low-density lipoprotein (Nickenig et al., 1997). Thus, studies in laboratory animals and man have provided evidence for a stimulation of the renin angiotensin system in atherosclerosis and for a (subsequent) activation of NADPH oxidases in the vascular wall. In certain types of pathophysiology, other enzymes may also become important sources of ROS.
One such enzyme is xanthine oxidase (Landmesser et al., 2007). However, its role in cardiovascular disease is controversial. Whereas some investigators reported an improvement of endothelial dysfunction in hypercholesterolaemic and diabetic patients with xanthine oxidase inhibitors such as oxypurinol and allopurinol (Cardillo et al., 1997; Butler et al., 2000), others failed to show an effect with allopurinol (O'Driscoll et al., 1999).
Increased ROS formation in the vascular wall reduces bioactive NO by promoting NO inactivation (by reaction of superoxide with NO forming peroxynitrite) and by decreasing NO production (eNOS uncoupling, see below).
eNOS itself can be a source of superoxide
As described above, a functional eNOS transfers electrons from NADPH, via the flavins FAD and FMN in the carboxy-terminal reductase domain, to the heme in the amino-terminal oxygenase domain, where the substrate L-arginine is oxidized to L-citrulline and NO. The flow of electrons within NOS is tightly regulated, and if disturbed the chemical reduction of oxygen and the generation of NO are uncoupled and O2-· is generated from the oxygenase domain (Stuehr et al., 2001).
eNOS uncoupling has been seen in vitro in endothelial cells treated with low-density lipoprotein (Pritchard et al., 1995), in peroxynitrite-treated rat aorta (Laursen et al., 2001), and in isolated blood vessels from animals with pathophysiological conditions such as SHR (Cosentino and Luscher, 1998), stroke-prone spontaneously hypertensive rats (Kerr et al., 1999), angiotensin II-induced hypertension (Mollnau et al., 2002), deoxycorticosterone acetate (DOCA)-salt hypertension (Landmesser et al., 2003), streptozotocin-induced diabetes (Hink et al., 2001) or nitroglycerin tolerance (Münzel et al., 2000). eNOS uncoupling has also been observed in patients with endothelial dysfunction due to diabetes mellitus (Heitzer et al., 2000b), essential hypertension (Higashi et al., 2002), hypercholesterolaemia (Stroes et al., 1997) and in chronic smokers (Heitzer et al., 2000a).
eNOS uncoupling may have major consequences on endothelial function. On the one hand, it reduces or abolishes formation, and on the other hand, it boosts pre-existing oxidative stress.
Oxidative stress produced by NADPH oxidase induces eNOS uncoupling
Due to the enhanced oxidative stress (see above), an increased degradation of NO by its reaction with O2-· is likely to occur in vascular disease. This, in turn, can lead to eNOS uncoupling by ONOO- and thus endothelial dysfunction.
There is a growing body of evidence that vascular NADPH oxidase plays a crucial role in the phenomenon of eNOS uncoupling. The important hint came from experiments with NADPH oxidase (p47phox) knockout animals (Landmesser et al., 2003). When hypertension was induced in normal mice with a combination of the mineralocorticoid DOCA and salt, these animals showed an increased production of vascular ROS. This was significantly reduced by the NOS inhibitor NG-nitro-L-arginine methyl ester (L-NAME), demonstrating a marked contribution of uncoupled eNOS to oxidative stress in vascular tissue. p47phox knockout animals showed much less oxidative stress upon DOCA-salt treatment, and levels of ROS could no longer be reduced with L-NAME (Landmesser et al., 2003).
These findings demonstrate that NADPH oxidase-derived ROS can indeed represent the trigger leading to eNOS uncoupling, and that uncoupled eNOS significantly contributes to oxidative stress (Landmesser et al., 2003).
Protein kinase C (PKC) is involved in some types of endothelial dysfunction
In some types of vascular disease, PKC activation is involved in the induction of oxidative stress. Vascular PKC activity has been found to be elevated in models of diabetes and angiotensin II-induced hypertension (Honing et al., 1998; Hink et al., 2001; Mollnau et al., 2002). In these models, the increased NADPH oxidase expression and activity in the vascular wall, the enhanced O2-· generation and the eNOS uncoupling seem to be a consequence of PKC activity. PKC inhibitors reduced O2-· production, inhibited upregulation of NADPH oxidase in vivo and reversed eNOS uncoupling. Thus, increases in the expression and activity of NADPH oxidases are at least, in part, PKC-dependent. PKC activation also leads to an enhanced eNOS expression (Li et al., 1998; Hink et al., 2001), which, if the enzyme becomes uncoupled, aggravates the pathophysiological situation.
Oxidation of BH4 can trigger eNOS uncoupling
NO and L-citrulline production by eNOS in endothelial cells correlates closely with the intracellular concentration of BH4 (Werner-Felmayer et al., 1993; Rosenkranz-Weiss et al., 1994). In isolated arteries (Cosentino and Katusic, 1995) or rats in vivo (Yamashiro et al., 2002), a BH4 depletion produced endothelial dysfunction in a short period of time (Figure 1). In isolated aortas from pre-hypertensive SHR, BH4 supplementation diminished the NOS-dependent generation of O2-· (Cosentino and Luscher, 1998). Administration of BH4 restored endothelial function in animal models of diabetes (Pieper, 1997) and insulin-resistance (Shinozaki et al., 2000), as well as in patients with hypercholesterolaemia (Stroes et al., 1997), diabetes mellitus (Heitzer et al., 2000b), essential hypertension (Higashi et al., 2002) and in chronic smokers (Heitzer et al., 2000a).
Intracellular BH4 levels depend on the balance of its de novo synthesis and its degradation/oxidation. BH4 is one of the most potent naturally occurring reducing agents. It is therefore reasonable to hypothesize that oxidative stress may lead to excessive oxidation and depletion of BH4 (Milstien and Katusic, 1999; Laursen et al., 2001). As oxidative stress occurs in cardiovascular pathophysiology (see above), oxidation of BH4 may be the common cause of eNOS dysfunction in these situations. In agreement with this concept, BH4 levels have been found decreased in the aorta from insulin-resistant rats (Shinozaki et al., 1999), in plasma of SHR compared with age-matched WKY rats (Hong et al., 2001), in aorta of hypercholesterolaemic apolipoprotein E- (apoE)-knockout mice (Laursen et al., 2001) and in DOCA-salt-treated hypertensive rats (Landmesser et al., 2003). Conversely, an infusion of the eNOS cofactor BH4 can restore eNOS functionality, as demonstrated by studies in chronic smokers (Heitzer et al., 2000a), oral glucose challenge (Ihlemann et al., 2003), diabetics (Heitzer et al., 2000b), hypercholesterolaemic patients (Stroes et al., 1997) and hypertensive individuals (Higashi et al., 2002).
It is particularly ONOO-, the direct reaction product of NO and O2-·, that oxidizes BH4 to BH3· radical. BH3· can be re-reduced to BH4 by NOS itself or non-enzymatically when enough reducing equivalents such as vitamin C are available (Kuzkaya et al., 2003; Werner et al., 2003). BH3· radical can also be disproportionate to the quinonoid 6,7-[8H]-H2-biopterin, which again can be reduced by vitamin C back to BH4 (Werner et al., 2003; Heller et al., 2006).
Therapeutic effects of enhancing eNOS expression and preventing eNOS uncoupling
Strategies to increase eNOS protein without a concomitant augmentation of endothelial BH4 levels may lead to eNOS uncoupling, enhanced oxidative stress and progression of vascular diseases. Therefore, compounds that increase eNOS protein levels are only beneficial when guaranteeing eNOS functionality (Li and Forstermann, 2009a). In the past, we have found compounds that maintain eNOS functionality in disease, and at the same time, upregulate expression of the enzyme.
Compounds AVE9488 and AVE3085
Two small-molecular-weight eNOS transcription enhancers, 4-fluoro-N-indan-2-yl-benzamide (AVE9488) and 2,2-difluoro-benzo[1,3]dioxole-5-carboxylic acid indan-2-ylamide (AVE3085), have been identified in a high throughput screening (Wohlfart et al., 2008). These compounds stimulate eNOS transcription in endothelial cells in vitro and in vascular tissues in vivo. Importantly, treatment of apoE knockout mice with AVE9488 enhances vascular BH4 levels and reverses eNOS uncoupling. In apoE knockout mice, but not in eNOS-knockout mice, treatment with AVE9488 reduces cuff-induced neointima formation. A 12-week treatment with AVE9488 or AVE3085 reduces atherosclerotic plaque formation in apoE knockout mice, but not in apoE/eNOS-double knockout mice (Wohlfart et al., 2008). Moreover, AVE9488 reverses impaired functional activity of endothelial progenitor cells from patients with ischaemic cardiomyopathy (Sasaki et al., 2006), and improves cardiac remodeling and heart failure after experimental myocardial infarction (Fraccarollo et al., 2008). Despite the promising results in the above in vivo experiments, the long-term therapeutic benefit of AVE9488 is not (yet) known.
The protein kinase C inhibitor midostaurin
As mentioned above, PKC activation is involved in the induction of oxidative stress in several types of vascular disease. The ROS H2O2 in turn, enhances eNOS expression (Cai et al., 2001). This may be an attempt of the organism to compensate for the reduced NO bioactivity. However, this compensation mechanism is often futile, because the eNOS enzyme becomes or remains uncoupled under pathological conditions. In fact, the upregulation of eNOS expression makes the situation even worse, because an uncoupled eNOS aggravates oxidative stress.
Midostaurin (4′-N-benzoyl staurosporine, CGP41251, PKC-412) is a glycosidic indolocarbazole analog of staurosporine that, via PKC inhibition, reduces NADPH oxidase expression (Li et al., 2006). As a consequence of reduced oxidative stress, midostaurin increases BH4 levels and reverses eNOS uncoupling in SHR and in atherosclerosis-prone apoE knockout mice (Li et al., 2005, 2006).
However, based on the above mechanisms, inhibition of PKC also leads to a normalization of eNOS expression (Hink et al., 2001). The result is a functional, NO-producing eNOS enzyme, yet at normal expression levels (Hink et al., 2001). These effects are seen at low doses of midostaurin, used to inhibit PKC. At higher doses, however, midostaurin not only reverses eNOS uncoupling (via PKC inhibition), but also upregulates eNOS expression by a PKC-independent mechanism (Li and Forstermann, 2000a; Li et al., 2005, 2006). The result is a NO-mediated vasodilation and a reduction in blood pressure (Li et al., 2005, 2006).
Unfortunately, the therapeutic usefulness of the PKC inhibitor is limited by significant systemic toxicity in vivo.
Betulinic acid and ursolic acid
Recently, we have described vascular effects of two pentacyclic triterpenoid acids that occur in various plants: betulinic acid (Steinkamp-Fenske et al., 2007b) and ursolic acid (Steinkamp-Fenske et al., 2007a). Both compounds are secondary plant metabolites widespread in fruit peel, leaves and stem bark. They are also important components of oriental and traditional medicine herbs widely distributed all over the world (Ovesna et al., 2004; Jäger et al., 2009). So far, both betulinic acid (Steinkamp-Fenske et al., 2007b) and ursolic acid are mainly known for their anti-tumour activity (Ovesna et al., 2004; Mullauer et al., 2010). We found that these triterpenoid acids upregulate eNOS expression, and at the same time, reduce NADPH oxidase expression in human endothelial cells through PKC-independent mechanisms (Steinkamp-Fenske et al., 2007a,b;) (Figure 2). The triterpenoids thus have the potential to reverse eNOS uncoupling. In addition, betulinic acid also enhances eNOS enzymatic activity by phosphorylation of eNOS at serine 1177 and dephosphorylation of eNOS at threonine 495 (N. Hohmann et al., unpubl. data). Both compounds are devoid of prominent in vivo toxicity (at least in rodents) (Jäger et al., 2009; Mullauer et al., 2010). Their therapeutic potential in cardiovascular disease needs to be further investigated in in vivo studies.
Figure 2.
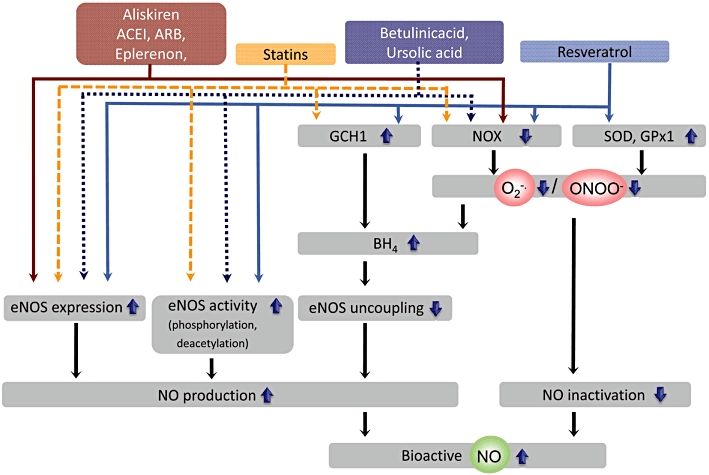
Therapeutic effects of enhancing endothelial nitric oxide synthase (eNOS) expression and preventing eNOS uncoupling. The renin inhibitor aliskiren, angiotensin-converting enzyme inhibitors (ACEI), angiotensin II receptor 1 blockers (ARB), as well as the selective aldosterone antagonist eplerenone enhance the expression of eNOS. In addition, these drugs prevent eNOS uncoupling by downregulating nicotinamide adenine dinucleotide phosphate oxidase (NOX) expression and activity, and by preventing (6R-)5,6,7,8-tetrahydrobiopterin (BH4) oxidation (see text). Statins (3-hydroxy-3-methylgulutaryl-coenzyme A [HMG-CoA] reductase inhibitors) stabilize eNOS mRNA, downregulate NOX and increase BH4 biosynthesis by upregulating GTP cyclohydrolase 1 (GCH1). Betulinic acid and ursolic acid are compounds that enhance eNOS expression and, at the same time, downregulate NOX expression. Trans-resveratrol stimulates the expression of eNOS. It also enhances enzyme activity by increasing phosphorylation at serine 1177 and by activating the protein deacetylase SIRT1, which in turn deacetylates eNOS at lysines 496 and 506 (in the calmodulin-binding domain), thereby stimulating eNOS activity. In addition, it upregulates antioxidant enzymes (such as superoxide dismutases (SOD) and glutathione peroxidase 1 (GPx1) and downregulates NOX. This leads to a reduction of peroxynitrite (ONOO-)-mediated BH4 oxidation and NO inactivation by superoxide O2-·. Trans-resveratrol also stimulates the expression of GCH1. The enhanced nitric oxide (NO) bioactivity resulting from increased NO production and reduced NO inactivation is likely to mediate/contribute to the vasoprotective effects of the aforementioned compounds.
Trans-resveratrol
Trans-resveratrol (3,5,4′-trihydroxy-trans-stilbene) is a polyphenolic phytoalexin found in red grapes and several other plants (Li and Forstermann, 2009). Trans-resveratrol has been shown to prevent or slow the progression of a wide variety of diseases including cancer and cardiovascular diseases (Bradamante et al., 2004). Trans-resveratrol also extends the lifespan of various organisms from yeast to vertebrates (Baur and Sinclair, 2006). This is true despite its low bioavailability. Trans-resveratrol can accumulate in tissues resulting in a ∼30-fold enrichment over serum concentrations. In addition, in vivo concentrations of bioactive metabolites can be more than 10 times higher than the native compound (Baur and Sinclair, 2006). No significant in vivo toxicity has been reported for trans-resveratrol (Cottart et al., 2010) which allows the use of the compound at high doses.
We have previously demonstrated that trans-resveratrol increases eNOS expression in human endothelial cells (Wallerath et al., 2002) (Figure 2). In addition, others have shown that trans-resveratrol stimulates a complicated signalling pathway in endothelial cells that increases the interaction between oestrogen receptor-α, caveolin-1 and c-Src, and stimulates phosphorylation of caveolin-1, c-Src and eNOS (at serine 1177) (Klinge et al., 2008). This results in an increased NO production by eNOS (Klinge et al., 2008) (Figure 2). Furthermore, eNOS has been shown to co-localize (and co-precipitate) with the protein deacetylase SIRT1 (silent mating type information regulation 2 homolog 1) (Mattagajasingh et al., 2007). Trans-resveratrol can activate SIRT1, which in turn, deacetylates eNOS at lysines 496 and 506 in the calmodulin-binding domain, thereby stimulating eNOS activity and increasing endothelial NO production (Mattagajasingh et al., 2007) (Figure 2).
Recent data from our own laboratory demonstrate that trans-resveratrol can also reverse eNOS uncoupling (Xia et al., 2010). As a polyphenolic compound, trans-resveratrol has been shown to scavenge several types of radicals (including hydroxyl, superoxide and metal-induced radicals (Bradamante et al., 2004). However, the direct antioxidant effect of trans-resveratrol is poor; the protective effects of trans-resveratrol against oxidative injury are likely to be attributed mostly to the upregulation of endogenous cellular antioxidant system, rather than its direct ROS scavenging activity. Treatment with trans-resveratrol leads to an upregulation of superoxide dismutases 1 (SOD1), SOD2, SOD3, glutathione peroxidase 1 and catalase in the hypercholesterolaemic apoE-knockout mice (Xia et al., 2010), as well as in cultured human endothelial cells (Spanier et al., 2009; Xia et al., 2010) (Figure 2). At the same time, the expression and activity of NADPH oxidases are downregulated. As a result, peroxynitrite levels and BH4 oxidation are reduced by trans-resveratrol (Xia et al., 2010). Importantly, trans-resveratrol also increases BH4 levels by upregulating GTP cyclohydrolase 1 (GCH1), the rate-limiting enzyme for BH4 biosynthesis (Xia et al., 2010) (Figure 2). The resulting reversal of eNOS uncoupling, along with the enhanced expression levels (Wallerath et al., 2002) and enzymatic activity (eNOS phosphorylation and eNOS deacetylation) (Li and Forstermann, 2009), is likely to contribute to the protective effects of trans-resveratrol.
Statins
Statins (3-hydroxy-3-methylgulutaryl-coenzyme A reductase inhibitors) are a group of lipid-lowering drugs used in the prevention and treatment of cardiovascular disease. Although it is widely accepted that most of the clinical benefit obtained with statins is a direct result of their lipid-lowering properties, these agents appear to display additional cholesterol-independent or pleiotropic effects on various aspects of cardiovascular disease (Liao, 2002). These include improvement of endothelial function, stabilization of atherosclerotic plaques, inhibition of oxidative stress and inflammation, and reduction of thrombogenic responses (Liao and Laufs, 2005). These beneficial effects of statins are, in part, mediated by an effect on eNOS because they can be inhibited by eNOS inhibitors (John et al., 1998) and are absent in eNOS-deficient mice (Landmesser et al., 2004).
Statins increase the expression of eNOS by a post-transcriptional mechanism involving inhibition of geranylgeranylation of Rho GTPase, and stabilization of eNOS mRNA (Laufs and Liao, 1998; Laufs et al., 1998a) (Figure 2). Statins can also enhance eNOS activity by decreasing caveolin abundance (Feron et al., 2001) and by post-translational mechanisms involving activation of the phosphatidylinositol 3-kinase/Akt pathway (Kureishi et al., 2000) (Figure 2).
In addition, several statins inhibit endothelial O2-· formation by reducing the expression and/or activity of NADPH oxidase and by preventing the isoprenylation of p21 Rac, which is critical for the assembly of NADPH oxidase (Wagner et al., 2000) (Figure 2). Extracellular SOD activity was more than doubled by simvastatin. Simvastatin treatment also increased the number of functionally active endothelial progenitor cells (Landmesser et al., 2005).
Statins have also been shown to increase GCH1 mRNA expression in endothelial cells and to elevate intracellular BH4 levels (Hattori et al., 2003) (Figure 2). In streptozotocin-induced diabetic rats, atorvastatin normalizes endothelial function, reduces oxidative stress by inhibiting vascular NADPH oxidases and prevents eNOS uncoupling by an upregulation of GCH1 (Wenzel et al., 2008).
The aforementioned effects may be responsible for part of the anti-atherogenic action of statins (Nissen et al., 2006; Patel et al., 2007).
Drugs interfering with the renin–angiotensin–aldosterone system
The renin–angiotensin–aldosterone system is upregulated in the vasculature of atherosclerotic vessels. Angiotensin II and aldosterone both promote endothelial dysfunction and atherosclerosis (Imanishi et al., 2008a). Angiotensin II activates NADPH oxidases via AT1 stimulation (Griendling et al., 2000). In addition, the AT1 receptor is upregulated in vitro by low-density lipoprotein (Nickenig et al., 1997). Accordingly, drugs interfering with the renin–angiotensin–aldosterone system decrease vascular oxidative stress and improve bioavailability of vascular NO by various mechanisms.
The renin inhibitor aliskiren increases eNOS expression, enhances eNOS phosphorylation at serine 1177 (thereby increasing activity), decreases NADPH oxidase expression, augments vascular BH4 levels and restores eNOS uncoupling in Watanabe heritable hyperlipidaemic rabbits (Imanishi et al., 2008b) (Figure 2). The anti-atherosclerotic effect of aliskiren (Verma and Gupta, 2008) is comparable with the AT1 receptor blocker (ARB) valsartan (Imanishi et al., 2008b) or irbesartan (Nussberger et al., 2008). Combination therapy of aliskiren and valsartan had an additive effect on endothelial function, BH4 content, NO release and plaque volume reduction (Imanishi et al., 2008b).
Angiotensin-converting enzyme inhibitors (ACEI) and ARB have indirect antioxidant effects by preventing the activation NADPH oxidase (Mancini et al., 1996; Warnholtz et al., 1999; Wassmann et al., 2002; Klingbeil et al., 2003) (Figure 2). In addition, they can also increase the activity of extracellular SOD (SOD3) (Hornig et al., 2001). ACEI significantly reduce cardiovascular events in patients with established coronary artery disease or at high risk for the disease (Bauersachs and Fraccarollo, 2008). ARB can improve eNOS functionality; losartan restored glomerular NO production by increasing GCH1 protein expression and elevating BH4 bioavailability in diabetic rats (Satoh et al., 2008).
Eplerenone, a selective aldosterone antagonist, has been shown to attenuate atherosclerosis in cholesterol-fed monkeys (Takai et al., 2005). Imanishi et al. investigated the effect of eplerenone and enalapril, alone or in combination, on atherosclerotic changes in genetically hyperlipidaemic rabbits (Imanishi et al., 2008a). Both eplerenone and enalapril reduce NADPH oxidase activity, elevate vascular BH4 levels (and thus limit eNOS uncoupling), and enhance eNOS expression and NO bioavailability (Figure 2). Eplerenone also increases eNOS phosphorylation at serine 1177. Both drugs decrease atherosclerotic plaque formation and the combination leads to an additive reduction (Imanishi et al., 2008a).
These multiple pleiotropic effects of compounds interfering with the renin–angiotensin–aldosterone system may make important contributions to the therapeutic benefit of such drugs.
Conclusions
The pathophysiological causes of oxidative stress are likely to involve changes in a number of different enzyme systems; most importantly, there is an upregulation of NADPH oxidases and eNOS. Together they lead to an increased production of ONOO-. This conveys oxidative damage to eNOS and/or its cofactor BH4, leading to ‘uncoupling’ of the enzyme. As consequence, an increased production of ROS by uncoupled eNOS is likely to contribute significantly to vascular oxidative stress and endothelial dysfunction. Several drugs in clinical use have pleiotropic actions that improve endothelial function, and novel pharmacological approaches to prevent or reverse endothelial dysfunction are being investigated.
Acknowledgments
Original work from our own laboratory contributing to this review was supported by the Collaborative Research Center SFB 553 and by grant LI-1042/1–1 from the DFG (Deutsche Forschungsgemeinschaft), Bonn, Germany
Glossary
Abbreviations
- ACEI
angiotensin-converting enzyme inhibitor
- ARB
AT1 receptor blocker
- BH4
(6R-)5,6,7,8-tetrahydrobiopterin
- DOCA
deoxycorticosterone acetate
- eNOS
endothelial nitric oxide synthase
- GCH1
guanosine-5′-triphosphate cyclohydrolase 1
- GPx1
glutathione peroxidase 1
- NO
nitric oxide
- Nox
homolog protein of the nicotinamide adenine dinucleotide phosphate oxidase subunit gp91phox
- O2-·
superoxide anion
- ONOO-
peroxynitrite
- ROS
reactive oxygen species
- SHR
spontaneously hypertensive rats
- SOD
superoxide dismutase
Conflicts of interest
None.
References
- Aicher A, Heeschen C, Mildner-Rihm C, Urbich C, Ihling C, Technau-Ihling K, et al. Essential role of endothelial nitric oxide synthase for mobilization of stem and progenitor cells. Nat Med. 2003;9:1370–1376. doi: 10.1038/nm948. [DOI] [PubMed] [Google Scholar]
- Alderton WK, Cooper CE, Knowles RG. Nitric oxide synthases: structure, function and inhibition. Biochem J. 2001;357(Pt 3):593–615. doi: 10.1042/0264-6021:3570593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alheid U, Frölich JC, Förstermann U. Endothelium-derived relaxing factor from cultured human endothelial cells inhibits aggregation of human platelets. Thromb Res. 1987;47:561–571. doi: 10.1016/0049-3848(87)90361-6. [DOI] [PubMed] [Google Scholar]
- Arndt H, Smith CW, Granger DN. Leukocyte-endothelial cell adhesion in spontaneously hypertensive and normotensive rats. Hypertension. 1993;21:667–673. doi: 10.1161/01.hyp.21.5.667. [DOI] [PubMed] [Google Scholar]
- Bauersachs J, Fraccarollo D. More NO – no more ROS: combined selective mineralocorticoid receptor blockade and angiotensin-converting enzyme inhibition for vascular protection. Hypertension. 2008;51:624–625. doi: 10.1161/HYPERTENSIONAHA.107.106625. [DOI] [PubMed] [Google Scholar]
- Baur JA, Sinclair DA. Therapeutic potential of resveratrol: the in vivo evidence. Nat Rev Drug Discov. 2006;5:493–506. doi: 10.1038/nrd2060. [DOI] [PubMed] [Google Scholar]
- Bradamante S, Barenghi L, Villa A. Cardiovascular protective effects of resveratrol. Cardiovasc Drug Rev. 2004;22:169–188. doi: 10.1111/j.1527-3466.2004.tb00139.x. [DOI] [PubMed] [Google Scholar]
- Busse R, Luckhoff A, Bassenge E. Endothelium-derived relaxant factor inhibits platelet activation. Naunyn Schmiedebergs Arch Pharmacol. 1987;336:566–571. doi: 10.1007/BF00169315. [DOI] [PubMed] [Google Scholar]
- Butler R, Morris AD, Belch JJ, Hill A, Struthers AD. Allopurinol normalizes endothelial dysfunction in type 2 diabetics with mild hypertension. Hypertension. 2000;35:746–751. doi: 10.1161/01.hyp.35.3.746. [DOI] [PubMed] [Google Scholar]
- Cai H, Davis ME, Drummond GR, Harrison DG. Induction of endothelial NO synthase by hydrogen peroxide via a Ca(2+)/calmodulin-dependent protein kinase II/janus kinase 2-dependent pathway. Arterioscler Thromb Vasc Biol. 2001;21:1571–1576. doi: 10.1161/hq1001.097028. [DOI] [PubMed] [Google Scholar]
- Cardillo C, Kilcoyne CM, Cannon RO, 3rd, Quyyumi AA, Panza JA. Xanthine oxidase inhibition with oxypurinol improves endothelial vasodilator function in hypercholesterolemic but not in hypertensive patients. Hypertension. 1997;30(1 Pt 1):57–63. doi: 10.1161/01.hyp.30.1.57. [DOI] [PubMed] [Google Scholar]
- Cosentino F, Katusic ZS. Tetrahydrobiopterin and dysfunction of endothelial nitric oxide synthase in coronary arteries. Circulation. 1995;91:139–144. doi: 10.1161/01.cir.91.1.139. [DOI] [PubMed] [Google Scholar]
- Cosentino F, Luscher TF. Tetrahydrobiopterin and endothelial function. Eur Heart J. 1998;19(Suppl. G):G3–G8. [PubMed] [Google Scholar]
- Cottart CH, Nivet-Antoine V, Laguillier-Morizot C, Beaudeux JL. Resveratrol bioavailability and toxicity in humans. Mol Nutr Food Res. 2010;54:7–16. doi: 10.1002/mnfr.200900437. [DOI] [PubMed] [Google Scholar]
- Crane BR, Arvai AS, Ghosh DK, Wu C, Getzoff ED, Stuehr DJ, et al. Structure of nitric oxide synthase oxygenase dimer with pterin and substrate. Science. 1998;279:2121–2126. doi: 10.1126/science.279.5359.2121. [DOI] [PubMed] [Google Scholar]
- Diet F, Pratt RE, Berry GJ, Momose N, Gibbons GH, Dzau VJ. Increased accumulation of tissue ACE in human atherosclerotic coronary artery disease. Circulation. 1996;94:2756–2767. doi: 10.1161/01.cir.94.11.2756. [DOI] [PubMed] [Google Scholar]
- Feron O, Dessy C, Desager JP, Balligand JL. Hydroxy-methylglutaryl-coenzyme A reductase inhibition promotes endothelial nitric oxide synthase activation through a decrease in caveolin abundance. Circulation. 2001;103:113–118. doi: 10.1161/01.cir.103.1.113. [DOI] [PubMed] [Google Scholar]
- Fleming I. Molecular mechanisms underlying the activation of eNOS. Pflugers Arch. 2010;459:793–806. doi: 10.1007/s00424-009-0767-7. [DOI] [PubMed] [Google Scholar]
- Fleming I, Busse R. Molecular mechanisms involved in the regulation of the endothelial nitric oxide synthase. Am J Physiol Regul Integr Comp Physiol. 2003;284:R1–12. doi: 10.1152/ajpregu.00323.2002. [DOI] [PubMed] [Google Scholar]
- Förstermann U. Oxidative stress in vascular disease: causes, defense mechanisms and potential therapies. Nat Clin Pract Cardiovasc Med. 2008;5:338–349. doi: 10.1038/ncpcardio1211. [DOI] [PubMed] [Google Scholar]
- Förstermann U, Mülsch A, Böhme E, Busse R. Stimulation of soluble guanylate cyclase by an acetylcholine-induced endothelium-derived factor from rabbit and canine arteries. Circ Res. 1986;58:531–538. doi: 10.1161/01.res.58.4.531. [DOI] [PubMed] [Google Scholar]
- Förstermann U, Closs EI, Pollock JS, Nakane M, Schwarz P, Gath I, et al. Nitric oxide synthase isozymes. Characterization, purification, molecular cloning, and functions. Hypertension. 1994;23(6 Pt 2):1121–1131. doi: 10.1161/01.hyp.23.6.1121. [DOI] [PubMed] [Google Scholar]
- Fraccarollo D, Widder JD, Galuppo P, Thum T, Tsikas D, Hoffmann M, et al. Improvement in left ventricular remodeling by the endothelial nitric oxide synthase enhancer AVE9488 after experimental myocardial infarction. Circulation. 2008;118:818–827. doi: 10.1161/CIRCULATIONAHA.107.717702. [DOI] [PubMed] [Google Scholar]
- Fukui T, Ishizaka N, Rajagopalan S, Laursen JB, Capers Q, Taylor WR, et al. p22phox mRNA expression and NADPH oxidase activity are increased in aortas from hypertensive rats. Circ Res. 1997;80:45–51. doi: 10.1161/01.res.80.1.45. [DOI] [PubMed] [Google Scholar]
- Garg UC, Hassid A. Nitric oxide-generating vasodilators and 8-bromo-cyclic guanosine monophosphate inhibit mitogenesis and proliferation of cultured rat vascular smooth muscle cells. J Clin Invest. 1989;83:1774–1777. doi: 10.1172/JCI114081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griendling KK, Sorescu D, Ushio-Fukai M. NAD(P)H oxidase: role in cardiovascular biology and disease. Circ Res. 2000;86:494–501. doi: 10.1161/01.res.86.5.494. [DOI] [PubMed] [Google Scholar]
- Hattori Y, Nakanishi N, Akimoto K, Yoshida M, Kasai K. HMG-CoA reductase inhibitor increases GTP cyclohydrolase I mRNA and tetrahydrobiopterin in vascular endothelial cells. Arterioscler Thromb Vasc Biol. 2003;23:176–182. doi: 10.1161/01.atv.0000054659.72231.a1. [DOI] [PubMed] [Google Scholar]
- Heitzer T, Brockhoff C, Mayer B, Warnholtz A, Mollnau H, Henne S, et al. Tetrahydrobiopterin improves endothelium-dependent vasodilation in chronic smokers : evidence for a dysfunctional nitric oxide synthase. Circ Res. 2000a;86:E36–E41. doi: 10.1161/01.res.86.2.e36. [DOI] [PubMed] [Google Scholar]
- Heitzer T, Krohn K, Albers S, Meinertz T. Tetrahydrobiopterin improves endothelium-dependent vasodilation by increasing nitric oxide activity in patients with Type II diabetes mellitus. Diabetologia. 2000b;43:1435–1438. doi: 10.1007/s001250051551. [DOI] [PubMed] [Google Scholar]
- Heller R, Werner-Felmayer G, Werner ER. Antioxidants and endothelial nitric oxide synthesis. Eur J Clin Pharmacol. 2006;62(Suppl 13):21–28. [Google Scholar]
- Hemmens B, Mayer B. Enzymology of nitric oxide synthases. Methods Mol Biol. 1998;100:1–32. doi: 10.1385/1-59259-749-1:1. [DOI] [PubMed] [Google Scholar]
- Higashi Y, Sasaki S, Nakagawa K, Fukuda Y, Matsuura H, Oshima T, et al. Tetrahydrobiopterin enhances forearm vascular response to acetylcholine in both normotensive and hypertensive individuals. Am J Hypertens. 2002;15(4 Pt 1):326–332. doi: 10.1016/s0895-7061(01)02317-2. [DOI] [PubMed] [Google Scholar]
- Hink U, Li H, Mollnau H, Oelze M, Matheis E, Hartmann M, et al. Mechanisms underlying endothelial dysfunction in diabetes mellitus. Circ Res. 2001;88:E14–E22. doi: 10.1161/01.res.88.2.e14. [DOI] [PubMed] [Google Scholar]
- Hogan M, Cerami A, Bucala R. Advanced glycosylation endproducts block the antiproliferative effect of nitric oxide. Role in the vascular and renal complications of diabetes mellitus. J Clin Invest. 1992;90:1110–1115. doi: 10.1172/JCI115928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong H-J, Hsiao G, Cheng T-H, Yen M-H. Supplemention with tetrahydrobiopterin suppresses the development of hypertension in spontaneously hypertensive rats. Hypertension. 2001;38:1044–1048. doi: 10.1161/hy1101.095331. [DOI] [PubMed] [Google Scholar]
- Honing ML, Morrison PJ, Banga JD, Stroes ES, Rabelink TJ. Nitric oxide availability in diabetes mellitus. Diabetes Metab Rev. 1998;14:241–249. doi: 10.1002/(sici)1099-0895(1998090)14:3<241::aid-dmr216>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- Hornig B, Landmesser U, Kohler C, Ahlersmann D, Spiekermann S, Christoph A, et al. Comparative effect of ace inhibition and angiotensin II type 1 receptor antagonism on bioavailability of nitric oxide in patients with coronary artery disease: role of superoxide dismutase. Circulation. 2001;103:799–805. doi: 10.1161/01.cir.103.6.799. [DOI] [PubMed] [Google Scholar]
- Ignarro LJ, Harbison RG, Wood KS, Kadowitz PJ. Activation of purified soluble guanylate cyclase by endothelium-derived relaxing factor from intrapulmonary artery and vein: stimulation by acetylcholine, bradykinin and arachidonic acid. J Pharmacol Exp Ther. 1986;237:893–900. [PubMed] [Google Scholar]
- Ihlemann N, Rask-Madsen C, Perner A, Dominguez H, Hermann T, Kober L, et al. Tetrahydrobiopterin restores endothelial dysfunction induced by an oral glucose challenge in healthy subjects. Am J Physiol Heart Circ Physiol. 2003;285:H875–H882. doi: 10.1152/ajpheart.00008.2003. [DOI] [PubMed] [Google Scholar]
- Imanishi T, Ikejima H, Tsujioka H, Kuroi A, Kobayashi K, Muragaki Y, et al. Addition of eplerenone to an angiotensin-converting enzyme inhibitor effectively improves nitric oxide bioavailability. Hypertension. 2008a;51:734–741. doi: 10.1161/HYPERTENSIONAHA.107.104299. [DOI] [PubMed] [Google Scholar]
- Imanishi T, Tsujioka H, Ikejima H, Kuroi A, Takarada S, Kitabata H, et al. Renin inhibitor aliskiren improves impaired nitric oxide bioavailability and protects against atherosclerotic changes. Hypertension. 2008b;52:563–572. doi: 10.1161/HYPERTENSIONAHA.108.111120. [DOI] [PubMed] [Google Scholar]
- Jäger S, Trojan H, Kopp T, Laszczyk M, Scheffler A. Pentacyclic triterpene distribution in various plants – rich sources for a new group of multi-potent plant extracts. Molecules. 2009;14:2016–2031. doi: 10.3390/molecules14062016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John S, Schlaich M, Langenfeld M, Weihprecht H, Schmitz G, Weidinger G, et al. Increased bioavailability of nitric oxide after lipid-lowering therapy in hypercholesterolemic patients: a randomized, placebo-controlled, double-blind study. Circulation. 1998;98:211–216. doi: 10.1161/01.cir.98.3.211. [DOI] [PubMed] [Google Scholar]
- Kerr S, Brosnan MJ, McIntyre M, Reid JL, Dominiczak AF, Hamilton CA. Superoxide anion production is increased in a model of genetic hypertension : role of the endothelium. Hypertension. 1999;33:1353–1358. doi: 10.1161/01.hyp.33.6.1353. [DOI] [PubMed] [Google Scholar]
- Klingbeil AU, John S, Schneider MP, Jacobi J, Handrock R, Schmieder RE. Effect of AT1 receptor blockade on endothelial function in essential hypertension. Am J Hypertens. 2003;16:123–128. doi: 10.1016/s0895-7061(02)03154-0. [DOI] [PubMed] [Google Scholar]
- Klinge CM, Wickramasinghe NS, Ivanova MM, Dougherty SM. Resveratrol stimulates nitric oxide production by increasing estrogen receptor alpha-Src-caveolin-1 interaction and phosphorylation in human umbilical vein endothelial cells. FASEB J. 2008;22:2185–2197. doi: 10.1096/fj.07-103366. [DOI] [PubMed] [Google Scholar]
- Kubes P, Suzuki M, Granger DN. Nitric oxide: an endogenous modulator of leukocyte adhesion. Proc Natl Acad Sci U S A. 1991;88:4651–4655. doi: 10.1073/pnas.88.11.4651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlencordt PJ, Chen J, Han F, Astern J, Huang PL. Genetic deficiency of inducible nitric oxide synthase reduces atherosclerosis and lowers plasma lipid peroxides in apolipoprotein E-knockout mice. Circulation. 2001;103:3099–3104. doi: 10.1161/01.cir.103.25.3099. [DOI] [PubMed] [Google Scholar]
- Kureishi Y, Luo Z, Shiojima I, Bialik A, Fulton D, Lefer DJ, et al. The HMG-CoA reductase inhibitor simvastatin activates the protein kinase Akt and promotes angiogenesis in normocholesterolemic animals. Nat Med. 2000;6:1004–1010. doi: 10.1038/79510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzkaya N, Weissmann N, Harrison DG, Dikalov S. Interactions of peroxynitrite, tetrahydrobiopterin, ascorbic acid, and thiols: implications for uncoupling endothelial nitric-oxide synthase. J Biol Chem. 2003;278:22546–22554. doi: 10.1074/jbc.M302227200. [DOI] [PubMed] [Google Scholar]
- Landmesser U, Dikalov S, Price SR, McCann L, Fukai T, Holland SM, et al. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J Clin Invest. 2003;111:1201–1209. doi: 10.1172/JCI14172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landmesser U, Engberding N, Bahlmann FH, Schaefer A, Wiencke A, Heineke A, et al. Statin-induced improvement of endothelial progenitor cell mobilization, myocardial neovascularization, left ventricular function, and survival after experimental myocardial infarction requires endothelial nitric oxide synthase. Circulation. 2004;110:1933–1939. doi: 10.1161/01.CIR.0000143232.67642.7A. [DOI] [PubMed] [Google Scholar]
- Landmesser U, Bahlmann F, Mueller M, Spiekermann S, Kirchhoff N, Schulz S, et al. Simvastatin versus ezetimibe: pleiotropic and lipid-lowering effects on endothelial function in humans. Circulation. 2005;111:2356–2363. doi: 10.1161/01.CIR.0000164260.82417.3F. [DOI] [PubMed] [Google Scholar]
- Landmesser U, Spiekermann S, Preuss C, Sorrentino S, Fischer D, Manes C, et al. Angiotensin II induces endothelial xanthine oxidase activation: role for endothelial dysfunction in patients with coronary disease. Arterioscler Thromb Vasc Biol. 2007;27:943–948. doi: 10.1161/01.ATV.0000258415.32883.bf. [DOI] [PubMed] [Google Scholar]
- Laufs U, Liao JK. Post-transcriptional regulation of endothelial nitric oxide synthase mRNA stability by Rho GTPase. J Biol Chem. 1998;273:24266–24271. doi: 10.1074/jbc.273.37.24266. [DOI] [PubMed] [Google Scholar]
- Laufs U, La Fata V, Plutzky J, Liao JK. Upregulation of endothelial nitric oxide synthase by HMG CoA reductase inhibitors. Circulation. 1998a;97:1129–1135. doi: 10.1161/01.cir.97.12.1129. [DOI] [PubMed] [Google Scholar]
- Laursen JB, Somers M, Kurz S, McCann L, Warnholtz A, Freeman BA, et al. Endothelial regulation of vasomotion in apoE-deficient mice: implications for interactions between peroxynitrite and tetrahydrobiopterin. Circulation. 2001;103:1282–1288. doi: 10.1161/01.cir.103.9.1282. [DOI] [PubMed] [Google Scholar]
- Li H, Forstermann U. Structure-activity relationship of staurosporine analogs in regulating expression of endothelial nitric-oxide synthase gene. Mol Pharmacol. 2000a;57:427–435. doi: 10.1124/mol.57.3.427. [DOI] [PubMed] [Google Scholar]
- Li H, Förstermann U. Nitric oxide in the pathogenesis of vascular disease. J Pathol. 2000b;190:244–254. doi: 10.1002/(SICI)1096-9896(200002)190:3<244::AID-PATH575>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Li H, Forstermann U. Prevention of atherosclerosis by interference with the vascular nitric oxide system. Curr Pharm Des. 2009a;15:3133–3145. doi: 10.2174/138161209789058002. [DOI] [PubMed] [Google Scholar]
- Li H, Forstermann U. Resveratrol: a multifunctional compound improving endothelial function. Editorial. Cardiovasc Drugs Ther. 2009b;23:425–429. doi: 10.1007/s10557-009-6209-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Oehrlein SA, Wallerath T, Ihrig-Biedert I, Wohlfart P, Ulshöfer T, et al. Activation of protein kinase C alpha and/or epsilon enhances transcription of the human endothelial nitric oxide synthase gene. Mol Pharmacol. 1998;53:630–637. doi: 10.1124/mol.53.4.630. [DOI] [PubMed] [Google Scholar]
- Li H, Hergert SM, Schafer SC, Brausch I, Yao Y, Huang Q, et al. Midostaurin upregulates eNOS gene expression and preserves eNOS function in the microcirculation of the mouse. Nitric Oxide. 2005;12:231–236. doi: 10.1016/j.niox.2005.04.001. [DOI] [PubMed] [Google Scholar]
- Li H, Witte K, August M, Brausch I, Gödtel-Armbrust U, Habermeier A, et al. Reversal of eNOS uncoupling and upregulation of eNOS expression lowers blood pressure in hypertensive rats. J Am Coll Cardiol. 2006;47:2536–2544. doi: 10.1016/j.jacc.2006.01.071. [DOI] [PubMed] [Google Scholar]
- Liao JK. Beyond lipid lowering: the role of statins in vascular protection. Int J Cardiol. 2002;86:5–18. doi: 10.1016/s0167-5273(02)00195-x. [DOI] [PubMed] [Google Scholar]
- Liao JK, Laufs U. Pleiotropic effects of statins. Annu Rev Pharmacol Toxicol. 2005;45:89–118. doi: 10.1146/annurev.pharmtox.45.120403.095748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu VW, Huang PL. Cardiovascular roles of nitric oxide: a review of insights from nitric oxide synthase gene disrupted mice. Cardiovasc Res. 2008;77:19–29. doi: 10.1016/j.cardiores.2007.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacMicking JD, Nathan C, Hom G, Chartrain N, Fletcher DS, Trumbauer M, et al. Altered responses to bacterial infection and endotoxic shock in mice lacking inducible nitric oxide synthase. Cell. 1995;81:641–650. doi: 10.1016/0092-8674(95)90085-3. [DOI] [PubMed] [Google Scholar]
- Mancini GB, Henry GC, Macaya C, O'Neill BJ, Pucillo AL, Carere RG, et al. Angiotensin-converting enzyme inhibition with quinapril improves endothelial vasomotor dysfunction in patients with coronary artery disease. The TREND (Trial on Reversing ENdothelial Dysfunction) Study. Circulation. 1996;94:258–265. doi: 10.1161/01.cir.94.3.258. [DOI] [PubMed] [Google Scholar]
- Mattagajasingh I, Kim CS, Naqvi A, Yamamori T, Hoffman TA, Jung SB, et al. SIRT1 promotes endothelium-dependent vascular relaxation by activating endothelial nitric oxide synthase. Proc Natl Acad Sci U S A. 2007;104:14855–14860. doi: 10.1073/pnas.0704329104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milstien S, Katusic Z. Oxidation of tetrahydrobiopterin by peroxynitrite: implications for vascular endothelial function. Biochem Biophys Res Commun. 1999;263:681–684. doi: 10.1006/bbrc.1999.1422. [DOI] [PubMed] [Google Scholar]
- Mollnau H, Wendt M, Szocs K, Lassegue B, Schulz E, Oelze M, et al. Effects of angiotensin II infusion on the expression and function of NAD(P)H oxidase and components of nitric oxide/cGMP signaling. Circ Res. 2002;90:E58–E65. doi: 10.1161/01.res.0000012569.55432.02. [DOI] [PubMed] [Google Scholar]
- Morawietz H, Weber M, Rueckschloss U, Lauer N, Hacker A, Kojda G. Upregulation of vascular NAD(P)H oxidase subunit gp91phox and impairment of the nitric oxide signal transduction pathway in hypertension. Biochem Biophys Res Commun. 2001;285:1130–1135. doi: 10.1006/bbrc.2001.5312. [DOI] [PubMed] [Google Scholar]
- Mueller CF, Laude K, McNally JS, Harrison DG. Redox mechanisms in blood vessels. Arterioscler Thromb Vasc Biol. 2005;25:274–278. doi: 10.1161/01.ATV.0000149143.04821.eb. [DOI] [PubMed] [Google Scholar]
- Mullauer FB, Kessler JH, Medema JP. Betulinic acid, a natural compound with potent anticancer effects. Anticancer Drugs. 2010;21:215–227. doi: 10.1097/CAD.0b013e3283357c62. [DOI] [PubMed] [Google Scholar]
- Münzel T, Li H, Mollnau H, Hink U, Matheis E, Hartmann M, et al. Effects of long-term nitroglycerin treatment on endothelial nitric oxide synthase (NOS III) gene expression, NOS III-mediated superoxide production, and vascular NO bioavailability. Circ Res. 2000;86:E7–E12. doi: 10.1161/01.res.86.1.e7. [DOI] [PubMed] [Google Scholar]
- Nakaki T, Nakayama M, Kato R. Inhibition by nitric oxide and nitric oxide-producing vasodilators of DNA synthesis in vascular smooth muscle cells. Eur J Pharmacol. 1990;189:347–353. doi: 10.1016/0922-4106(90)90031-r. [DOI] [PubMed] [Google Scholar]
- Nickenig G, Sachinidis A, Michaelsen F, Bohm M, Seewald S, Vetter H. Upregulation of vascular angiotensin II receptor gene expression by low-density lipoprotein in vascular smooth muscle cells. Circulation. 1997;95:473–478. doi: 10.1161/01.cir.95.2.473. [DOI] [PubMed] [Google Scholar]
- Nickenig G, Baumer AT, Temur Y, Kebben D, Jockenhovel F, Bohm M. Statin-sensitive dysregulated AT1 receptor function and density in hypercholesterolemic men. Circulation. 1999;100:2131–2134. doi: 10.1161/01.cir.100.21.2131. [DOI] [PubMed] [Google Scholar]
- Nissen SE, Nicholls SJ, Sipahi I, Libby P, Raichlen JS, Ballantyne CM, et al. Effect of very high-intensity statin therapy on regression of coronary atherosclerosis: the ASTEROID trial. JAMA. 2006;295:1556–1565. doi: 10.1001/jama.295.13.jpc60002. [DOI] [PubMed] [Google Scholar]
- Noble MA, Munro AW, Rivers SL, Robledo L, Daff SN, Yellowlees LJ, et al. Potentiometric analysis of the flavin cofactors of neuronal nitric oxide synthase. Biochemistry. 1999;38:16413–16418. doi: 10.1021/bi992150w. [DOI] [PubMed] [Google Scholar]
- Nunokawa Y, Tanaka S. Interferon-gamma inhibits proliferation of rat vascular smooth muscle cells by nitric oxide generation. Biochem Biophys Res Commun. 1992;188:409–415. doi: 10.1016/0006-291x(92)92400-r. [DOI] [PubMed] [Google Scholar]
- Nussberger J, Aubert JF, Bouzourene K, Pellegrin M, Hayoz D, Mazzolai L. Renin inhibition by aliskiren prevents atherosclerosis progression: comparison with irbesartan, atenolol, and amlodipine. Hypertension. 2008;51:1306–1311. doi: 10.1161/HYPERTENSIONAHA.108.110932. [DOI] [PubMed] [Google Scholar]
- O'Driscoll JG, Green DJ, Rankin JM, Taylor RR. Nitric oxide-dependent endothelial function is unaffected by allopurinol in hypercholesterolaemic subjects. Clin Exp Pharmacol Physiol. 1999;26:779–783. doi: 10.1046/j.1440-1681.1999.03125.x. [DOI] [PubMed] [Google Scholar]
- Ohishi M, Ueda M, Rakugi H, Naruko T, Kojima A, Okamura A, et al. Enhanced expression of angiotensin-converting enzyme is associated with progression of coronary atherosclerosis in humans. J Hypertens. 1997;15:1295–1302. doi: 10.1097/00004872-199715110-00014. [DOI] [PubMed] [Google Scholar]
- Ovesna Z, Vachalkova A, Horvathova K, Tothova D. Pentacyclic triterpenoic acids: new chemoprotective compounds. Minireview. Neoplasma. 2004;51:327–333. [PubMed] [Google Scholar]
- Patel TN, Shishehbor MH, Bhatt DL. A review of high-dose statin therapy: targeting cholesterol and inflammation in atherosclerosis. Eur Heart J. 2007;28:664–672. doi: 10.1093/eurheartj/ehl445. [DOI] [PubMed] [Google Scholar]
- Pieper GM. Acute amelioration of diabetic endothelial dysfunction with a derivative of the nitric oxide synthase cofactor, tetrahydrobiopterin. J Cardiovasc Pharmacol. 1997;29:8–15. doi: 10.1097/00005344-199701000-00002. [DOI] [PubMed] [Google Scholar]
- Pritchard KA, Jr, Groszek L, Smalley DM, Sessa WC, Wu M, Villalon P, et al. Native low-density lipoprotein increases endothelial cell nitric oxide synthase generation of superoxide anion. Circ Res. 1995;77:510–518. doi: 10.1161/01.res.77.3.510. [DOI] [PubMed] [Google Scholar]
- Radomski MW, Palmer RM, Moncada S. The anti-aggregating properties of vascular endothelium: interactions between prostacyclin and nitric oxide. Br J Pharmacol. 1987;92:639–646. doi: 10.1111/j.1476-5381.1987.tb11367.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopalan S, Kurz S, Munzel T, Tarpey M, Freeman BA, Griendling KK, et al. Angiotensin II-mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation. Contribution to alterations of vasomotor tone. J Clin Invest. 1996;97:1916–1923. doi: 10.1172/JCI118623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapoport RM, Draznin MB, Murad F. Endothelium-dependent relaxation in rat aorta may be mediated through cyclic GMP-dependent protein phosphorylation. Nature. 1983;306:174–176. doi: 10.1038/306174a0. [DOI] [PubMed] [Google Scholar]
- Ridnour LA, Thomas DD, Mancardi D, Espey MG, Miranda KM, Paolocci N, et al. The chemistry of nitrosative stress induced by nitric oxide and reactive nitrogen oxide species. Putting perspective on stressful biological situations. Biol Chem. 2004;385:1–10. doi: 10.1515/BC.2004.001. [DOI] [PubMed] [Google Scholar]
- Rosenkranz-Weiss P, Sessa WC, Milstien S, Kaufman S, Watson CA, Pober JS. Regulation of nitric oxide synthesis by proinflammatory cytokines in human umbilical vein endothelial cells. Elevations in tetrahydrobiopterin levels enhance endothelial nitric oxide synthase specific activity. J Clin Invest. 1994;93:2236–2243. doi: 10.1172/JCI117221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki K, Heeschen C, Aicher A, Ziebart T, Honold J, Urbich C, et al. Ex vivo pretreatment of bone marrow mononuclear cells with endothelial NO synthase enhancer AVE9488 enhances their functional activity for cell therapy. Proc Natl Acad Sci U S A. 2006;103:14537–14541. doi: 10.1073/pnas.0604144103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh M, Fujimoto S, Arakawa S, Yada T, Namikoshi T, Haruna Y, et al. Angiotensin II type 1 receptor blocker ameliorates uncoupled endothelial nitric oxide synthase in rats with experimental diabetic nephropathy. Nephrol Dial Transplant. 2008;23:3806–3813. doi: 10.1093/ndt/gfn357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz PM, Kleinert H, Forstermann U. Potential functional significance of brain-type and muscle-type nitric oxide synthase I expressed in adventitia and media of rat aorta. Arterioscler Thromb Vasc Biol. 1999;19:2584–2590. doi: 10.1161/01.atv.19.11.2584. [DOI] [PubMed] [Google Scholar]
- Shinozaki K, Kashiwagi A, Nishio Y, Okamura T, Yoshida Y, Masada M, et al. Abnormal biopterin metabolism is a major cause of impaired endothelium-dependent relaxation through nitric oxide/O2- imbalance in insulin-resistant rat aorta. Diabetes. 1999;48:2437–2445. doi: 10.2337/diabetes.48.12.2437. [DOI] [PubMed] [Google Scholar]
- Shinozaki K, Nishio Y, Okamura T, Yoshida Y, Maegawa H, Kojima H, et al. Oral administration of tetrahydrobiopterin prevents endothelial dysfunction and vascular oxidative stress in the aortas of insulin-resistant rats. Circ Res. 2000;87:566–573. doi: 10.1161/01.res.87.7.566. [DOI] [PubMed] [Google Scholar]
- Sorescu D, Weiss D, Lassegue B, Clempus RE, Szocs K, Sorescu GP, et al. Superoxide production and expression of nox family proteins in human atherosclerosis. Circulation. 2002;105:1429–1435. doi: 10.1161/01.cir.0000012917.74432.66. [DOI] [PubMed] [Google Scholar]
- Southgate K, Newby AC. Serum-induced proliferation of rabbit aortic smooth muscle cells from the contractile state is inhibited by 8-Br-cAMP but not 8-Br-cGMP. Atherosclerosis. 1990;82:113–123. doi: 10.1016/0021-9150(90)90150-h. [DOI] [PubMed] [Google Scholar]
- Spanier G, Xu H, Xia N, Tobias S, Deng S, Wojnowski L, et al. Resveratrol reduces endothelial oxidative stress by modulating the gene expression of superoxide dismutase 1 (SOD1), glutathione peroxidase 1 (GPx1) and NADPH oxidase subunit (Nox4) J Physiol Pharmacol. 2009;60(Suppl 4):111–116. [PubMed] [Google Scholar]
- Steinkamp-Fenske K, Bollinger L, Voller N, Xu H, Yao Y, Bauer R, et al. Ursolic acid from the Chinese herb danshen (Salvia miltiorrhiza L.) upregulates eNOS and downregulates Nox4 expression in human endothelial cells. Atherosclerosis. 2007a;195:e104–e111. doi: 10.1016/j.atherosclerosis.2007.03.028. [DOI] [PubMed] [Google Scholar]
- Steinkamp-Fenske K, Bollinger L, Xu H, Yao Y, Horke S, Forstermann U, et al. Reciprocal regulation of endothelial nitric-oxide synthase and NADPH oxidase by betulinic acid in human endothelial cells. J Pharmacol Exp Ther. 2007b;322:836–842. doi: 10.1124/jpet.107.123356. [DOI] [PubMed] [Google Scholar]
- Stroes E, Kastelein J, Cosentino F, Erkelens W, Wever R, Koomans H, et al. Tetrahydrobiopterin restores endothelial function in hypercholesterolemia. J Clin Invest. 1997;99:41–46. doi: 10.1172/JCI119131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuehr D, Pou S, Rosen GM. Oxygen reduction by nitric-oxide synthases. J Biol Chem. 2001;276:14533–14536. doi: 10.1074/jbc.R100011200. [DOI] [PubMed] [Google Scholar]
- Takai S, Jin D, Muramatsu M, Kirimura K, Sakonjo H, Miyazaki M. Eplerenone inhibits atherosclerosis in nonhuman primates. Hypertension. 2005;46:1135–1139. doi: 10.1161/01.HYP.0000184640.81730.22. [DOI] [PubMed] [Google Scholar]
- Upmacis RK, Crabtree MJ, Deeb RS, Shen H, Lane PB, Benguigui LE, et al. Profound biopterin oxidation and protein tyrosine nitration in tissues of ApoE-null mice on an atherogenic diet: contribution of inducible nitric oxide synthase. Am J Physiol Heart Circ Physiol. 2007;293:H2878–H2887. doi: 10.1152/ajpheart.01144.2006. [DOI] [PubMed] [Google Scholar]
- Vergnani L, Hatrik S, Ricci F, Passaro A, Manzoli N, Zuliani G, et al. Effect of native and oxidized low-density lipoprotein on endothelial nitric oxide and superoxide production : key role of L-arginine availability. Circulation. 2000;101:1261–1266. doi: 10.1161/01.cir.101.11.1261. [DOI] [PubMed] [Google Scholar]
- Verma S, Gupta MK. Aliskiren improves nitric oxide bioavailability and limits atherosclerosis. Hypertension. 2008;52:467–469. doi: 10.1161/HYPERTENSIONAHA.108.114488. [DOI] [PubMed] [Google Scholar]
- Wagner AH, Kohler T, Ruckschloss U, Just I, Hecker M. Improvement of nitric oxide-dependent vasodilatation by HMG-CoA reductase inhibitors through attenuation of endothelial superoxide anion formation. Arterioscler Thromb Vasc Biol. 2000;20:61–69. doi: 10.1161/01.atv.20.1.61. [DOI] [PubMed] [Google Scholar]
- Wallerath T, Deckert G, Ternes T, Anderson H, Li H, Witte K, et al. Resveratrol, a polyphenolic phytoalexin present in red wine, enhances expression and activity of endothelial nitric oxide synthase. Circulation. 2002;106:1652–1658. doi: 10.1161/01.cir.0000029925.18593.5c. [DOI] [PubMed] [Google Scholar]
- Warnholtz A, Nickenig G, Schulz E, Macharzina R, Brasen JH, Skatchkov M, et al. Increased NADH-oxidase-mediated superoxide production in the early stages of atherosclerosis: evidence for involvement of the renin-angiotensin system. Circulation. 1999;99:2027–2033. doi: 10.1161/01.cir.99.15.2027. [DOI] [PubMed] [Google Scholar]
- Wassmann S, Hilgers S, Laufs U, Bohm M, Nickenig G. Angiotensin II type 1 receptor antagonism improves hypercholesterolemia-associated endothelial dysfunction. Arterioscler Thromb Vasc Biol. 2002;22:1208–1212. doi: 10.1161/01.atv.0000022847.38083.b6. [DOI] [PubMed] [Google Scholar]
- Wenzel P, Daiber A, Oelze M, Brandt M, Closs E, Xu J, et al. Mechanisms underlying recoupling of eNOS by HMG-CoA reductase inhibition in a rat model of streptozotocin-induced diabetes mellitus. Atherosclerosis. 2008;198:65–76. doi: 10.1016/j.atherosclerosis.2007.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner ER, Gorren AC, Heller R, Werner-Felmayer G, Mayer B. Tetrahydrobiopterin and nitric oxide: mechanistic and pharmacological aspects. Exp Biol Med. 2003;228:1291–1302. doi: 10.1177/153537020322801108. [DOI] [PubMed] [Google Scholar]
- Werner-Felmayer G, Werner ER, Fuchs D, Hausen A, Reibnegger G, Schmidt K, et al. Pteridine biosynthesis in human endothelial cells. Impact on nitric oxide-mediated formation of cyclic GMP. J Biol Chem. 1993;268:1842–1846. [PubMed] [Google Scholar]
- Wohlfart P, Xu H, Endlich A, Habermeier A, Closs EI, Hubschle T, et al. Antiatherosclerotic effects of small-molecular-weight compounds enhancing endothelial nitric-oxide synthase (eNOS) expression and preventing eNOS uncoupling. J Pharmacol Exp Ther. 2008;325:370–379. doi: 10.1124/jpet.107.128009. [DOI] [PubMed] [Google Scholar]
- Xia N, Daiber A, Habermeier A, Closs EI, Thum T, Spanier G, et al. Resveratrol reverses endothelial nitric-oxide synthase uncoupling in apolipoprotein E knockout mice. J Pharmacol Exp Ther. 2010;335:149–154. doi: 10.1124/jpet.110.168724. [DOI] [PubMed] [Google Scholar]
- Yamashiro S, Kuniyoshi Y, Arakaki K, Miyagi K, Koja K. The effect of insufficiency of tetrahydrobiopterin on endothelial function and vasoactivity. Jpn J Thorac Cardiovasc Surg. 2002;50:472–477. doi: 10.1007/BF02919638. [DOI] [PubMed] [Google Scholar]