

Abstract
BACKGROUND AND PURPOSE
Cerebral vasospasm is the persistent constriction of large conduit arteries in the base of the brain. This pathologically sustained contraction of the arterial myocytes has been attributed to locally elevated concentrations of vasoconstrictor agonists (spasmogens). We assessed the presence and function of KCNQ (Kv7) potassium channels in rat basilar artery myocytes, and determined the efficacy of Kv7 channel activators in relieving spasmogen-induced basilar artery constriction.
EXPERIMENTAL APPROACH
Expression and function of Kv7 channels in freshly isolated basilar artery myocytes were evaluated by reverse transcriptase polymerase chain reaction and whole-cell electrophysiological techniques. Functional responses to Kv7 channel modulators were studied in intact artery segments using pressure myography.
KEY RESULTS
All five mammalian KCNQ subtypes (KCNQ1-5) were detected in the myocytes. Kv currents were attributed to Kv7 channel activity based on their voltage dependence of activation (V0.5∼−34 mV), lack of inactivation, enhancement by flupirtine (a selective Kv7 channel activator) and inhibition by 10,10-bis(pyridin-4-ylmethyl)anthracen-9-one (XE991; a selective Kv7 channel blocker). XE991 depolarized the myocytes and constricted intact basilar arteries. Celecoxib, a clinically used anti-inflammatory drug, not only enhanced Kv7 currents but also inhibited voltage-sensitive Ca2+ currents. In arteries pre-constricted with spasmogens, both celecoxib and flupirtine were more effective in dilating artery segments than was nimodipine, a selective L-type Ca2+ channel blocker.
CONCLUSIONS AND IMPLICATIONS
Kv7 channels are important determinants of basilar artery contractile status. Targeting the Kv7 channels using flupirtine or celecoxib could provide a novel strategy to relieve basilar artery constriction in patients with cerebral vasospasm.
LINKED ARTICLES
To view two letters to the Editor regarding this article visit http://dx.doi.org/10.1111/j.1476-5381.2011.01454.x and http://dx.doi.org/10.1111/j.1476-5381.2011.01457.x
Keywords: cerebral vasospasm, basilar artery, KCNQ, flupirtine, celecoxib, membrane voltage, potassium channel, vasoconstriction
Introduction
Cerebral vasospasm, a sustained constriction of the arteries at the base of the brain, is a devastating consequence of subarachnoid hemorrhage (SAH). The affected arteries (primarily the basilar artery and arteries of the Circle of Willis) account for 10–39% of resistance to blood flow to the brain (Heistad and Konotos, 1983). Hence, spasm of these arteries results in a reduction of blood flow to the brain, leading to ischaemic neurological deficits and consequential high morbidity and mortality (Kassell et al., 1985). The condition is often diagnosed by angiographic narrowing of the basilar artery or altered basilar artery blood flow measured by transcranial Doppler ultrasonography.
The aetiology of vasospasm after SAH appears to be multifactorial, with spasmogens including 5-hydroxytryptamine (serotonin, 5-HT), endothelin (ET) and vasopressin [arginine8-vasopressin (AVP)] likely to be involved in the initiation and maintenance of the sustained constriction of arteries (Trandafir et al., 2004; Nishizawa and Laher, 2005). Vasospasm is produced by persistent contraction of the vascular smooth muscle cells (VSMC) in the arteries due to the sustained elevation of cytosolic Ca2+ concentrations ([Ca2+]cyt). The elevation of [Ca2+]cyt is mainly caused by increased influx of Ca2+ through voltage-sensitive Ca2+ channels (VSCC), which are activated by membrane depolarization (Harder et al., 1987; Zuccarello et al., 1996; Weyer et al., 2006).
Because the membrane voltage is regulated primarily by the K+ conductance (Faraci and Sobey, 1998), K+ channels are postulated as mediators in the pathogenesis of cerebral vasospasm and have therefore been identified as possible therapeutic targets to treat this condition (Wellman, 2006). K+ conductance was found to be reduced in basilar arteries after SAH (Harder et al., 1987; Jahromi et al., 2008a), and the dysfunction of 4-aminopyridine (4-AP)-sensitive Kv channels after SAH has been proposed as a mechanism contributing to cerebral vasospasm (Ishiguro et al., 2006; Jahromi et al., 2008a). However, none of the K+ channel subtypes previously found to be expressed in the cerebral vasculature has emerged as a successful target to treat cerebral vasospasm (Wellman, 2006). We recently demonstrated that K+ conductance through KCNQ (Kv7) channels (nomenclature follows Alexander et al., 2009) largely determines the resting membrane voltage in rat mesenteric artery myocytes (Mackie et al., 2008) and cultured rat aortic smooth muscle cells (Brueggemann et al., 2007; Mani et al., 2009). We also found that AVP, a potent vasoconstrictor, exerts its physiological effects on mesenteric arteries via suppression of Kv7 currents (Mackie et al., 2008). Here, we present evidence for the presence of this class of K+ channels in rat basilar artery myocytes and describe the results of functional studies that suggest that Kv7 channels contribute to the regulation of basilar artery tone and that clinically used Kv7 channel activators can attenuate spasmogen-induced basilar artery constriction. Hence, we propose that these drugs could be utilized as novel therapeutic agents to treat patients with cerebral vasospasm.
Methods
Animal procedures
All animal care and experimental procedures in this study were approved by the Institutional Animal Care and Use Committee, Loyola University Medical Center, Maywood, IL. Male Sprague-Dawley rats, 275–325 g, were killed by thoracotomy and excision of the heart while under 4% isoflurane anaesthesia. The brain was removed immediately and placed in ice-cold dissection solution [in mM: 145 NaCl, 4.7 KCl, 1.2 NaH2PO4, 1.2 MgSO4, 2 CaCl2, 2 pyruvic acid, 0.02 EDTA, 3 3-(N-morpholino) propanesulphonic acid (MOPS) and 5 d-glucose with 1% bovine serum albumin (BSA), pH 7.4 at 0°C, 300 mOsm·L−1]. The basilar artery was dissected free from the brain and used for pressure myography or for isolation of myocytes for patch-clamp recordings.
Isolation of basilar artery myocytes
The myocytes were isolated from the basilar artery by modifying the procedure described by Berra-Romani et al. (Berra-Romani et al., 2005). The dissected basilar artery was cut into two or three segments and placed in low-Ca2+ physiological saline solution (low-Ca2+ PSS, in mM): 140 NaCl, 5.36 KCl, 0.34 Na2HPO4, 0.44 K2HPO4, 10 HEPES, 1.2 MgCl2, 0.05 CaCl2 and 10 d-glucose, pH 7.2 at 37°C, 298 mOsm·L−1) for 30 min. The arterial segments were then subjected to enzymatic digestion in low-Ca2+ PSS containing (in mg·mL−1) 2 collagenase type XI; 0.16 elastase type IV and 2 BSA (fraction V, protease-free) at 37°C for 30 min. The digested segments were repeatedly washed with ice-cold low-Ca2+ PSS to remove the enzymes, and then the individual myocytes or clusters of myocytes were released from the segments by trituration using fire-polished Pasteur pipettes.
Electrophysiology
Individual myocytes were allowed to attach to glass coverslips and whole cell currents were measured by using a perforated patch configuration (by including 120 µg·mL−1 Amphotericin B in the internal solution) under voltage-clamp conditions. All experiments were performed at room temperature with continuous perfusion of bath solution as described previously (Mackie et al., 2008; Brueggemann et al., 2009). For recording the Kv7 currents, the bath solution contained (in mM): 140 NaCl, 5.36 KCl, 1.2 MgCl2, 2 CaCl2, 10 HEPES, 10 d-glucose, pH 7.3, 298 mOsm·L−1; the internal (pipette) solution contained (in mM): 135 KCl, 5 NaCl, 10 HEPES, 0.05 K2EGTA, 1 MgCl2, 20 d-glucose, pH 7.2, 298 mOsm·L−1. Kv7 currents were recorded in isolation by application of 5 s voltage steps from a −4 mV holding voltage to test voltages ranging from −84 mV to +16 mV. The bath solution contained GdCl3 (100 µM; sufficient to block L- and T-type Ca2+ channels, non-selective cation channels and to shift activation of 4-AP-sensitive KV currents to voltages positive to −20 mV; Supporting Information Figure S1) and spermine (100 µM; to inhibit the inwardly-rectifying K+ currents). Whole-cell K+ currents were digitized at 2 kHz and filtered at 1 kHz. The steady-state K+ currents recorded during the last 1000 ms (2000 points) of each voltage step were averaged and normalized to cell capacitance to obtain the current-voltage (I-V) relationship.
To record currents through voltage-sensitive Ca2+ channels, bath solution contained 10 mM Ba2+ as a charge carrier (in mM): 140 NaCl, 2.7 KCl, 10 BaCl2, 10 HEPES, pH 7.3, 298 mOsm·L−1 and internal solution contained (in mM): 135 CsCl, 10 HEPES, 10 Cs2EGTA, 2.5 MgCl2, 10 d-glucose, pH 7.2, 298 mOsm·L−1. A Cs2EGTA stock solution was prepared by titrating EGTA with CsOH to pH 7.2. Ba2+ currents were recorded in isolation by application of a 300 ms voltage step protocol from −90 mV holding voltage to test voltages ranging from −85.2 to +44.8 mV. Whole-cell voltage-sensitive Ba2+ currents were digitized at 10 kHz and filtered at 5 kHz, and the peak inward current was measured and normalized to cell capacitance.
Liquid junction potentials were calculated using Junction Potential Calculator provided by PCLAMP8 software and subtracted off-line. Current-voltage (I-V) curves for the K+ currents and voltage-sensitive Ba2+ currents (IBa) were derived after leak subtraction using a procedure described by Passmore et al. (2003). The voltage-dependence of channel activation of Kv7 currents was derived by calculating the conductance from the steady-state currents at each voltage (according to the equation G = I/(V–EK), where I, steady-state current; V, step voltage; EK reversal potential for potassium) and normalized to maximum conductance for each experiment (Wickenden et al., 2001). Normalized conductances were fitted by a Boltzmann function: G/Gmax = 1/[1 + exp(V0.5− Vm)/s], where G/Gmax is the fraction of maximal conductance, V0.5 is the voltage of half-maximal activation and s is the slope factor. Membrane voltages were recorded from individual myocytes or clusters of myocytes in current clamp (I = 0) mode. The effect of treatments on membrane voltage was determined by taking the mean of the values recorded over at least a 1 min period, prior to treatment, and at the end of treatment.
Pressure myography
The effects of various treatment conditions on basilar artery diameter were studied using a pressure myograph system (DMT-USA, Atlanta, GA, USA) as described previously (Henderson and Byron, 2007). The pipette contained physiological saline solution (in mM: 145 NaCl, 4.7 KCl, 1.2 NaH2PO4, 1.2 MgSO4, 2 CaCl2, 2 pyruvic acid, 0.02 EDTA, 3 MOPS and 5 d-glucose with 1% BSA, pH 7.4 at 37°C, 300 mOsm·L−1) and the bath contained the same solution without BSA. The artery segment was transferred to the pressure myograph system and secured to the glass pipettes using nylon sutures. By gradually increasing the pressure using a pressure column, the arteries were pressurized to 80 mmHg (the pressure that would be expected in the basilar artery in vivo) (Weyer et al., 2006). The bath solution was gradually warmed to 37°C. The viability of the vessel was confirmed by observation of a quick constrictor response to the depolarization induced by transient application of 60 mM KCl saline solution. The artery segment was then allowed to equilibrate, and arteries with diameters that were stable for at least 30 min were used in the study. Arteries that developed myogenic tone (a decrease in diameter coinciding with pressurization to 80 mmHg) or that failed to respond to KCl were discarded. The concentration of spasmogens was chosen based on the EC50 concentrations to constrict basilar artery (Nishimura, 1996; Mayhan, 1998; Katori et al., 2001). The vasoconstrictor/vasodilator responses were measured and the results are presented as changes in outer vessel diameter, in micrometres.
Polymerase chain reaction (PCR)
Reverse transcription PCR was used to detect the KCNQ gene products (Kv7 channel mRNA transcripts). Myocytes were freshly isolated from basilar artery segments by enzymatic digestion as for electrophysiological recordings and total RNA was isolated using RNeasy Kit (Qiagen, Valencia, CA, USA). PCR was performed essentially as described previously (Brueggemann et al., 2007). Primers were adapted from previous publications: KCNQ1–3 and KCNQ5 (Ohya et al., 2002) and KCNQ4 (Yeung et al., 2007) to correspond to rat sequences.
Data analysis
Data are presented as mean ± SEM. Data were analysed using Clampfit (Axon Instruments, Sunnyvale, CA, USA) and SigmaStat (Systat Software, Inc., Chicago, IL, USA) software programs. Paired Student's t-test was used for comparisons of parameters measured before and after treatments. Comparisons among multiple treatment groups were evaluated by analysis of variance (anova) followed by a Holm-Sidak post hoc test. Differences with P-value ≤0.05 were considered statistically significant.
Materials
All chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA) with the exception of the following. ET-1, amphotericin B and nimodipine were from Calbiochem (San Diego, CA, USA), BSA fraction V was from Boehringer Mannheim (Mannheim, Germany), celecoxib and rofecoxib were from LKT Laboratories, Inc. (St. Paul, MN, USA), flupirtine was from Tocris Cookson (Ellisville, MO, USA), 10,10-bis(pyridin-4-ylmethyl)anthracen-9-one (XE991) was from Ascent Scientific (Princeton, NJ, USA) and 2, 5-dimethyl-celecoxib was generously provided by Dr Axel H. Schönthal (University of Southern California, Los Angeles, CA, USA). Drugs were dissolved in DMSO (nimodipine, flupirtine, celecoxib, rofecoxib or dimethyl celecoxib) or water (XE991), and were used at dilutions of 1:1000 to 1:10 000 in the bath solution. The vehicle by itself did not have appreciable effects on currents or on membrane voltage.
Results
Expression of KCNQ genes in rat basilar arteries
Reverse transcriptase-PCR was used to evaluate the gene expression pattern of Kv7 channels in basilar artery myocytes. The mRNAs of all five mammalian KCNQ genes (KCNQ1-5, encoding Kv7.1- Kv7.5) were detected (Figure 1).
Figure 1.
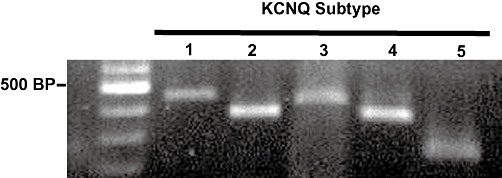
Kv7 channels are expressed in basilar artery myocytes. Reverse transcription polymerase chain reaction detection of KCNQ1 through KCNQ5 from mRNA extracted from basilar artery myocytes. Expected sizes of each reaction product are KCNQ1, 453 bp; KCNQ2, 372 bp; KCNQ3, 424 bp; KCNQ4, 359 bp; and KCNQ5, 240 bp. Molecular weight marker (100 base-pair ladder, New England Biolabs, Ipswich, MA, USA, is shown in the left lane).
Kv7 currents in basilar artery myocytes
To assess the presence of functional channels, we recorded Kv7 currents in basilar artery myocytes using patch-clamp electrophysiology under voltage-clamp conditions (Figure 2A). We recorded outwardly rectifying currents with a threshold for voltage-dependent activation <−60 mV and half-maximal activation (V0.5) at ∼−34 mV (Figure 2A, C and D). Addition of 10 µM flupirtine, a selective Kv7 channel activator, significantly enhanced the Kv7 currents at all voltages tested from −49.2 to +5.8 mV; at −20 mV, the voltage at which we observed near-maximal activation, currents more than doubled with the addition of flupirtine (0.04 ± 0.01 pA/pF before (control) compared with 0.09 ± 0.02 pA/pF after addition of flupirtine; Figure 2B, C). However there was no significant shift in the steady-state voltage-dependence of activation (V0.5 for flupirtine was −36.8 ± 0.8 mV compared with control −33.9 ± 1.5 mV; Figure 2D). XE991 (10 µM), a selective Kv7 channel blocker (Wang et al., 1998), effectively abolished the currents after wash out of flupirtine (Figure 2A, C) or in the presence of flupirtine (Supporting Information Figure S2).
Figure 2.
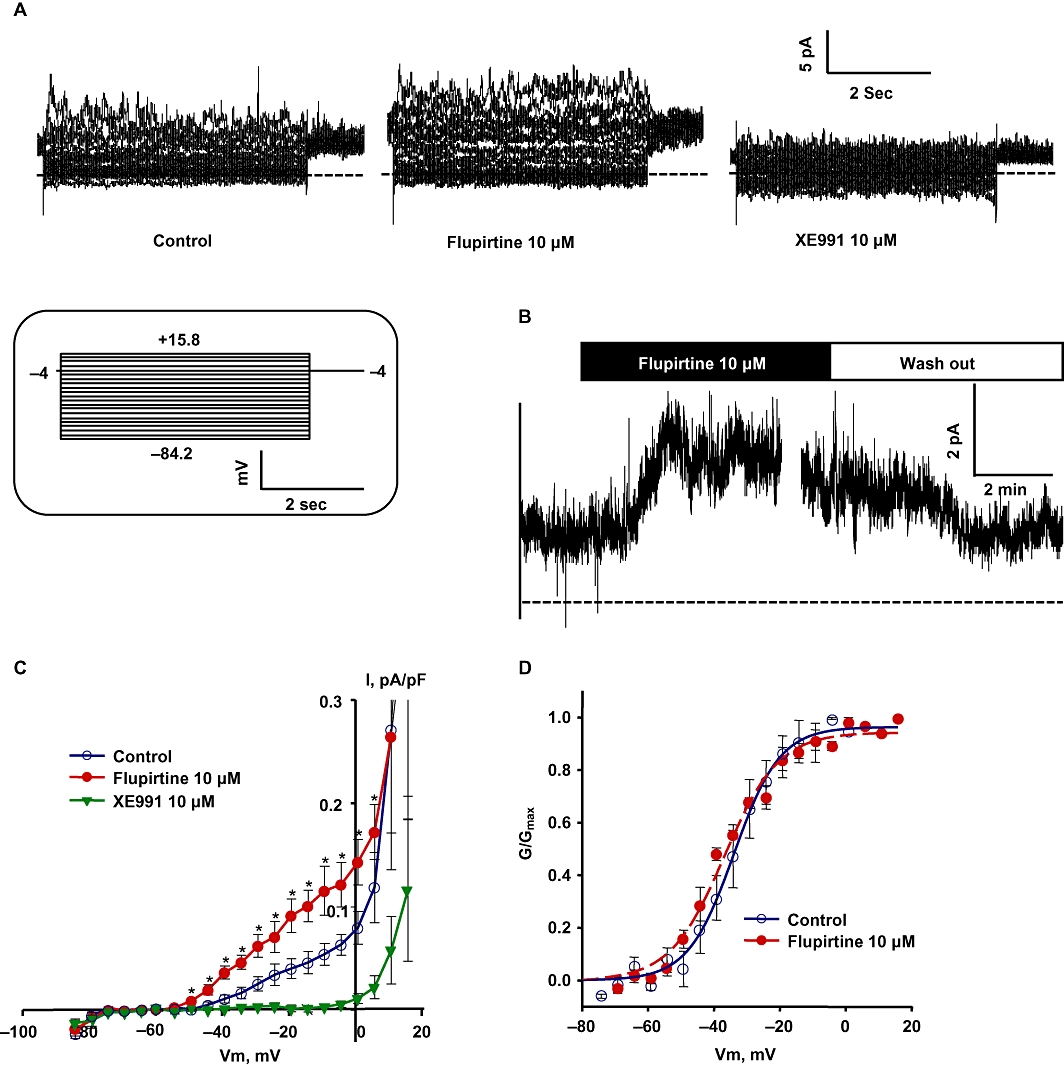
Kv7 currents in basilar artery myocytes. (A) Representative current traces recorded from a basilar artery myocyte (capacitance = 38.72 pF) under control conditions (left, untreated), treatment with 10 µM flupirtine (middle) and 10 µM XE991 (right; dotted lines indicate zero current). Inset shows the voltage protocol used to record the Kv7 currents. (B) Representative time course of Kv7 currents recorded at −20 mV in a single basilar artery myocyte before and during the application of 10 µM flupirtine. The currents were enhanced with the application of flupirtine and restored to control levels after wash out of flupirtine (bars above the trace indicate the duration of treatment or wash out). (C) Summarized I-V curves show the outwardly rectifying Kv7 currents and the response to application of 10 µM flupirtine and 10 µM XE991. Flupirtine significantly enhanced Kv7 currents at all tested voltages between −49.2 to +5.8 mV (n = 4 each, *P < 0.05, paired Student's t-test). (D) Normalized conductance plots fitted by a single Boltzmann function; control (voltage of half-maximal activation (V0.5) = −33.91 ± 1.46 mV; slope factor (s) = 6.98 ± 1.17 mV; 10 µM flupirtine (V0.5 = −36.8 ± 0.78 mV; s = 8.23 ± 0.64 mV, n = 4); G/Gmax, fraction of maximal conductance.
Contribution of Kv7 channels to membrane voltage
The Kv7 currents had a very negative threshold for voltage-dependent activation, suggesting that K+ conductance through Kv7 channels should contribute to maintenance of negative resting membrane voltages. To evaluate this possibility, we measured the effect of the Kv7 channel blocker XE991 on whole cell membrane voltage, in physiological concentrations of ions. Addition of 10 µM XE991 significantly depolarized the basilar artery myocytes (Figure 3A, B). To confirm that the depolarization of myocytes' membrane translates functionally into a constrictor response, we assessed if blockade of Kv7 channels would constrict the intact basilar artery using pressure myography. Application of 10 µM XE991 to pressurized basilar arteries produced a significant constrictor response (Figure 3C, D), which was almost completely reversed by addition of the L-type Ca2+ channel blocker nimodipine (2 µM, Figure 3C).
Figure 3.
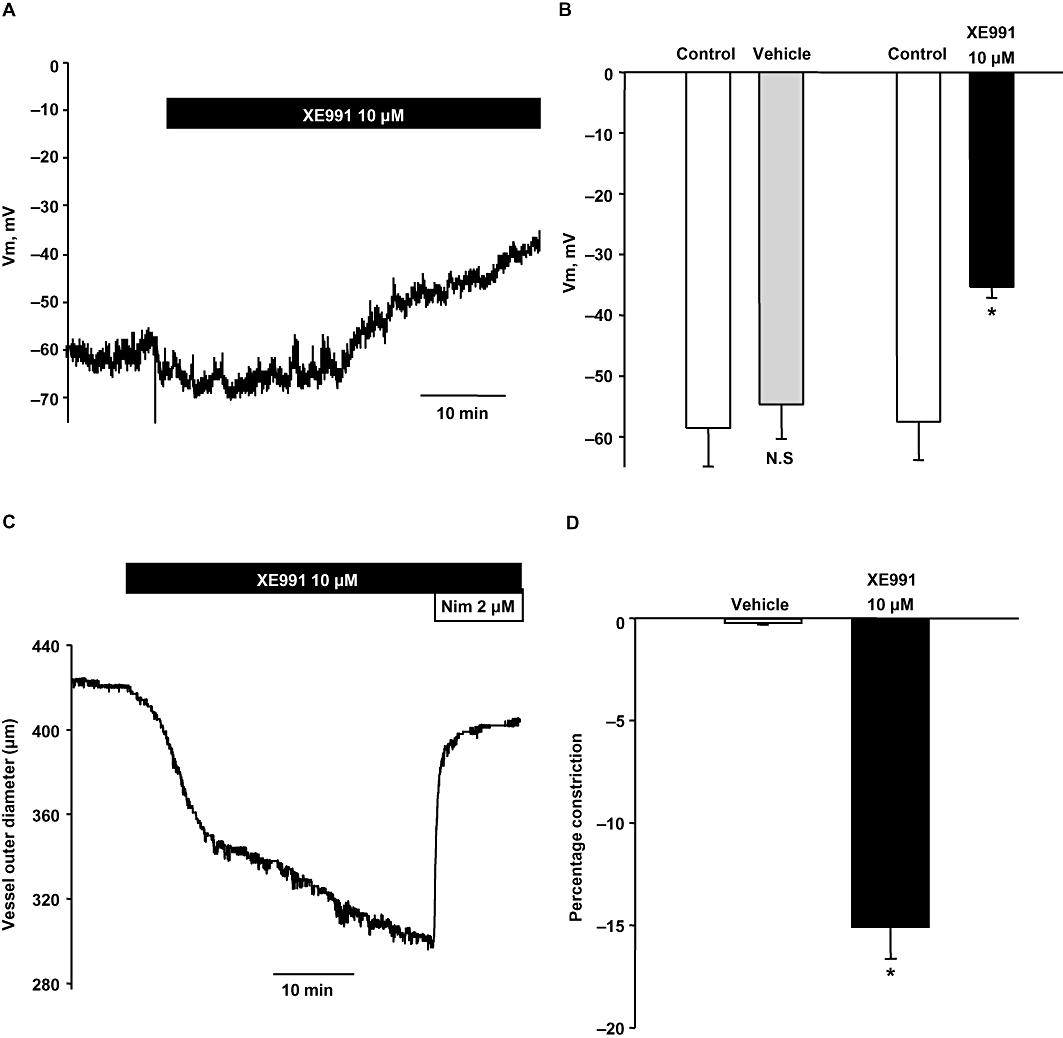
Kv7 currents determine resting membrane voltage in basilar artery myocytes and contractile status of basilar artery. (A) Representative time course of membrane voltage recorded in current-clamp mode from a basilar artery myocyte before and during the application of 10 µM XE991. (B) Mean membrane voltage values in basilar artery myocytes before (control) and during the addition of vehicle or 10 µM XE991. XE991 produced a significant membrane depolarization (n = 4, *P < 0.05, paired Student's ‘t’ test; N.S – Not significant). (C) Representative trace shows contraction produced in a pressurized basilar artery segment with the addition of 10 µM XE991. The constriction was reversed with the addition of 10 µM nimodipine. (D) Percentage change in the basilar artery diameter with the addition of 10 µM XE991 or vehicle. XE991 produced a significant constriction of basilar artery segments compared with vehicle control (n = 5, *P < 0.05, paired Student's t-test).
Effect of celecoxib on Kv7 and voltage-sensitive Ba2+ currents
We recently reported that celecoxib is a dual Kv7 channel activator and VSCC blocker in cultured rat aortic smooth muscle cells and rat mesenteric artery myocytes (Brueggemann et al., 2009). In accordance with our previous findings, we found here that celecoxib (10 µM) significantly enhanced the Kv7 currents at all voltages from −34.2 to +0.8 mV; at −20 mV, the currents more than doubled in amplitude (Figure 4). To record currents through voltage-sensitive Ca2+ channels, Ba2+ was used as a charge carrier (Figure 5A). Celecoxib (10 µM) inhibited IBa at all voltages from −20.2 to +39.8 mV and the peak inward currents were essentially abolished (Figure 5A, B).
Figure 4.
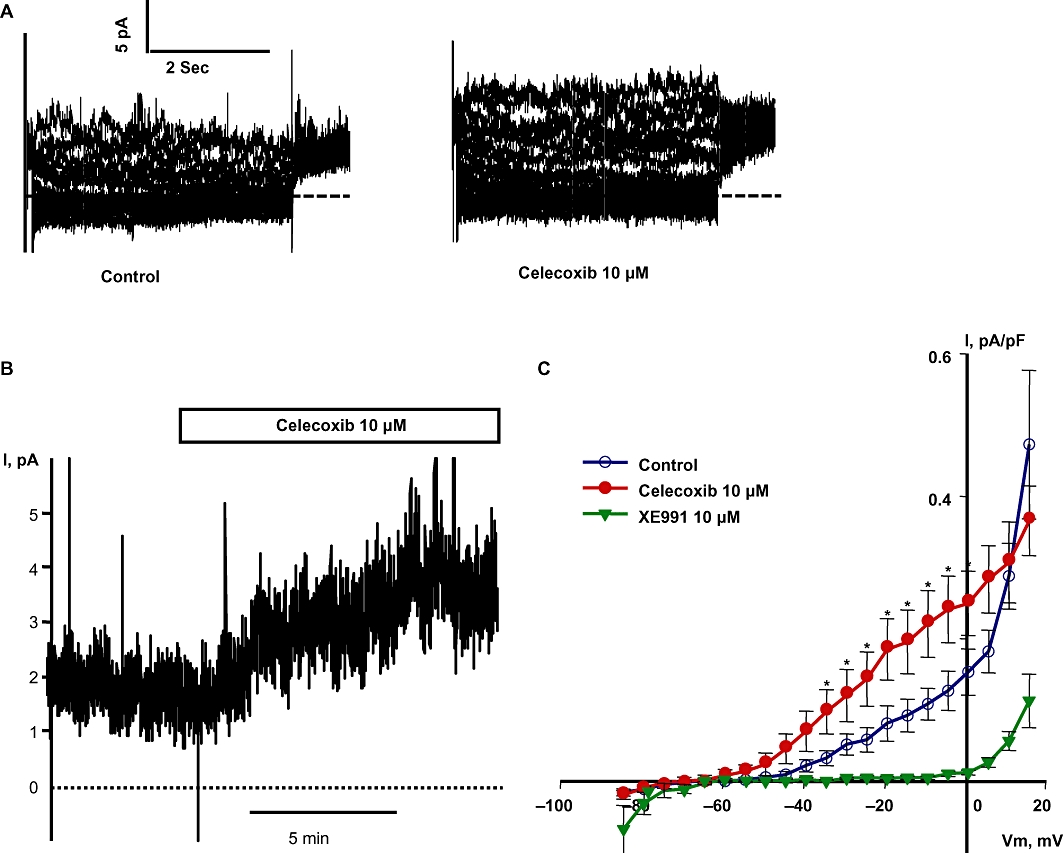
Celecoxib enhances Kv7 channel currents in basilar artery myocytes. (A) Representative Kv7 current traces before and during the application of 10 µM celecoxib. (B) Representative time course of Kv7 currents recorded at −20 mV in a single basilar artery myocyte before and during the application of 10 µM celecoxib. (C) Summarized I-V curve shows the Kv7 currents before and during application of 10 µM celecoxib. Celecoxib significantly enhanced the Kv7 currents at all tested voltages between −34.2 and +0.8 mV (n = 7, *P < 0.05, paired Student's t-test).
Figure 5.
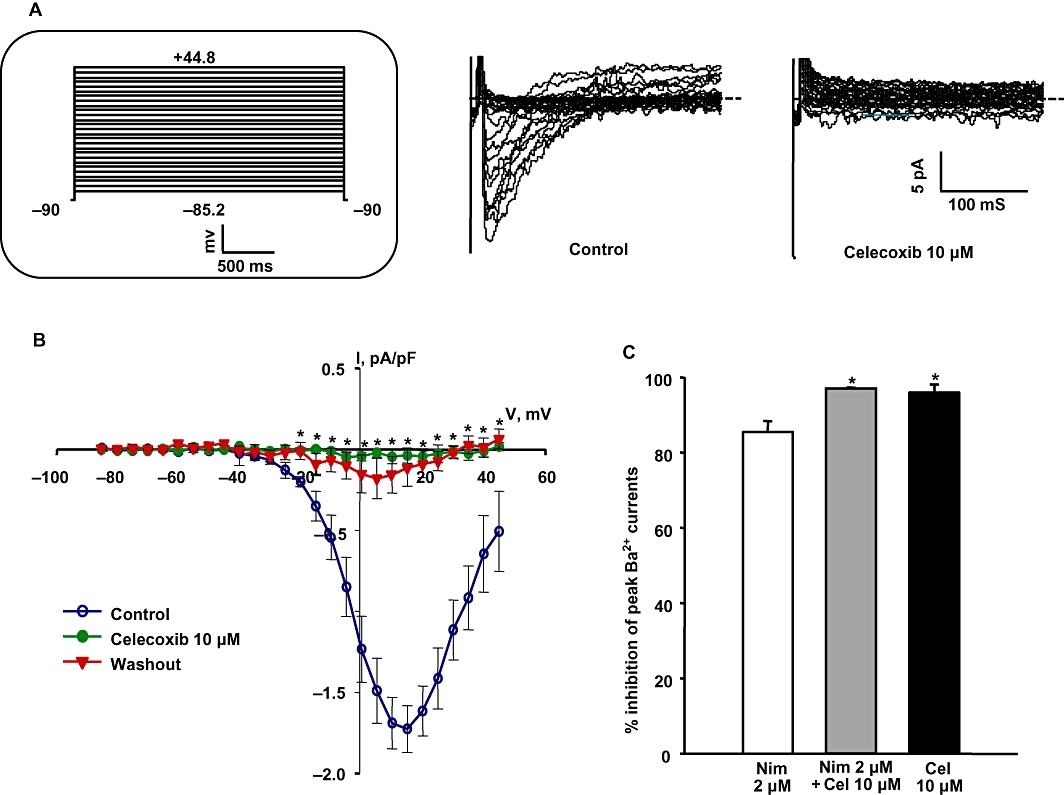
Celecoxib inhibits voltage-sensitive calcium currents in basilar artery myocytes. (A) Representative voltage-sensitive Ba2+ current (IBa) traces before and during the application of 10 µM celecoxib. Inset shows the schematic of the voltage protocol used to record IBa. (B) Summarized I-V curves show the effect of 10 µM celecoxib on IBa. Celecoxib significantly inhibited IBa at all tested voltages from −20.2 to +39.8 mV (n = 5, *P < 0.05, Student's t-test). (C) Percentage inhibition of peak IBa with the application of 2 µM nimodipine alone, 2 µM nimodipine + 10 µM celecoxib and 10 µM celecoxib alone. Celecoxib either alone or when added along with nimodipine produced significantly more inhibition of IBa compared with nimodipine alone (n = 5–6, *P < 0.05 using one-way anova followed by post hoc Holm-Sidak test).
Since nimodipine is the standard therapeutic agent used to treat patients with cerebral vasospasm, we compared the ability of nimodipine to inhibit IBa with that of celecoxib. The concentration of nimodipine used (2 µM) was found to be maximally effective in dilation of basilar artery segments pre-constricted with vasoconstrictor agonists (see Figure 7D). Celecoxib was significantly more effective in inhibiting IBa in basilar artery myocytes than was 2 µM nimodipine (Figure 5C).
Figure 7.
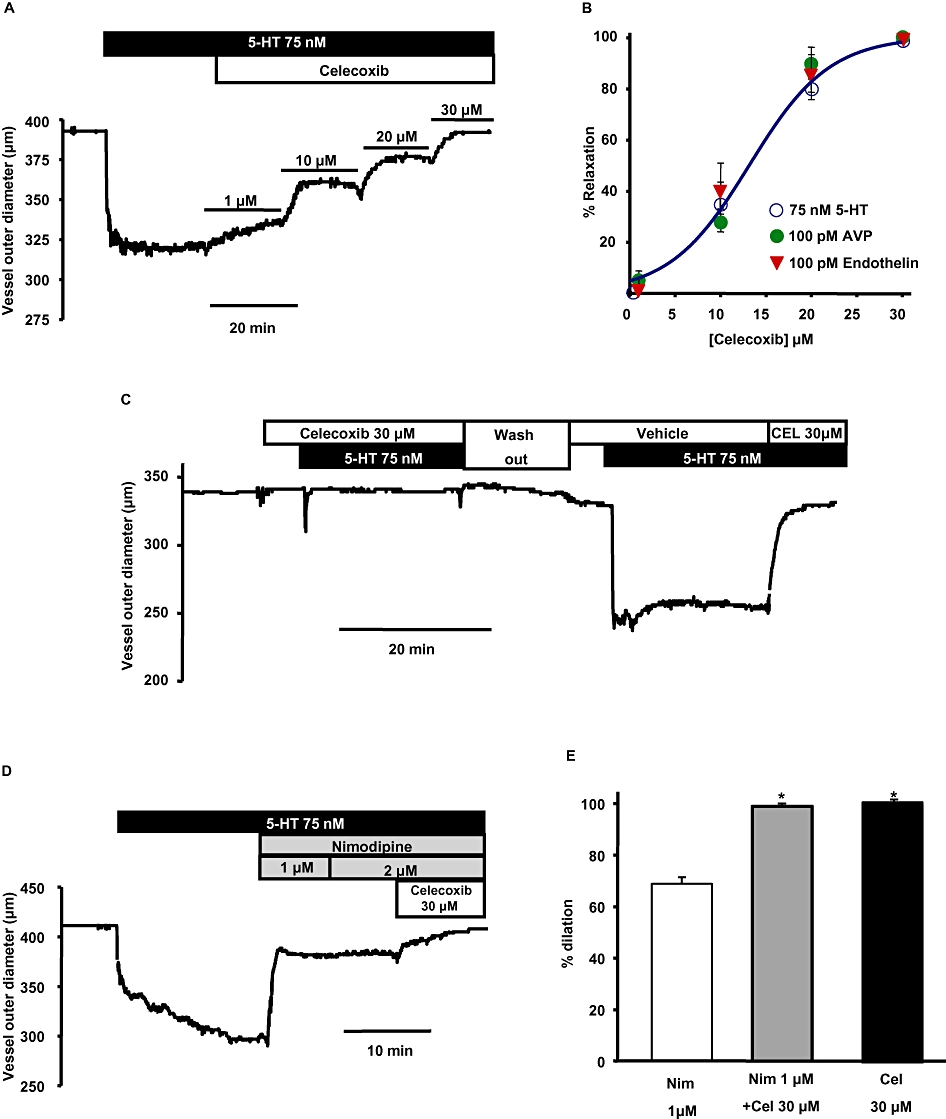
Celecoxib reverses the spasmogen-induced constriction in basilar artery more effectively than nimodipine. (A) Representative trace shows the concentration-dependent effect of celecoxib on a basilar artery segment pre-constricted with 75 nM 5-HT. (B) Mean concentration-response curves of the percentage relaxation produced by celecoxib in arteries pre-constricted with the spasmogens, 5-HT, AVP and ET-1. (C) Time-course shows 75 nM 5-HT was not able to constrict the artery in the presence of 30 µM celecoxib. However after wash out, 5-HT was able to constrict the same artery in the absence of celecoxib. Re-application of celecoxib completely reversed the constriction produced by 5-HT (representative of n = 3). (D) Representative time course trace shows the dilation produced by nimodipine and celecoxib when pre-constricted with 5-HT. Increasing the concentration of nimodipine above 1 µM did not produce additional dilation. However, addition of celecoxib produced additional dilation in the same artery. (E) Summary of the percentage dilation of basilar artery with the application of 2 µM nimodipine alone, 2 µM nimodipine + 10 µM celecoxib and 10 µM celecoxib alone. Celecoxib, either alone or when added along with nimodipine, produced significantly more dilation compared with nimodipine alone (n = 3–6, *P < 0.05 using one-way anova followed by post hoc Holm-Sidak test).
Effect of Kv7 channel activators on basilar artery constriction produced by spasmogens involved in cerebral vasospasm
To assess the functional consequences of Kv7 current enhancement, we studied the efficacy of flupirtine or celecoxib in reversing the constriction produced by spasmogens involved in the pathogenesis of cerebral vasospasm: ET-1, AVP and 5-HT. Flupirtine produced a concentration-dependent relaxation of basilar arteries pre-constricted with 100 pM ET-1, 100 pM AVP or 75 nM 5-HT (Figure 6A, B). The EC50 of relaxation produced by flupirtine was ∼40–60 µM (Figure 6B). Flupirtine (100 µM) did not reverse 5-HT-induced constriction in the presence of 10 µM XE991 (flupirtine produced 2.8 ± 2.5% dilation in the presence of XE991 compared with 97.4 ± 1.7% dilation in the absence of XE991). We also assessed whether flupirtine is more efficacious than nimodipine in dilating the basilar artery. Application of 100 µM flupirtine either alone or combined with nimodipine produced a significant additional dilation compared with nimodipine alone (Figure 6C, D).
Figure 6.
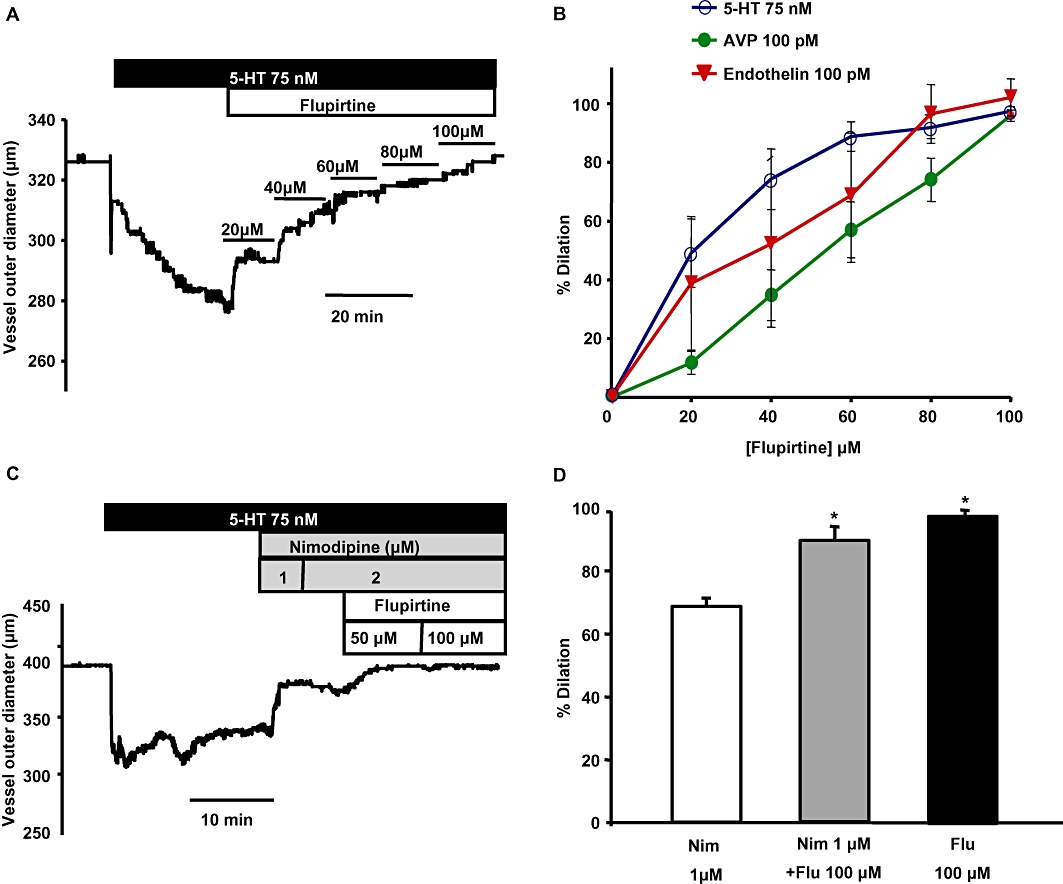
Flupirtine reverses the constriction induced by spasmogens in basilar artery. (A) Representative trace shows the concentration-dependent effect of flupirtine on a basilar artery segment pre-constricted with 75 nM 5-HT. (B) Mean concentration-response curves of the percentage relaxation produced by flupirtine in arteries pre-constricted with the spasmogens: 5-HT, AVP and ET-1. (C) Representative time course trace shows the dilation produced by nimodipine and flupirtine when an artery was pre-constricted with 5-HT. Increasing the concentration of nimodipine above 1 µM did not produce additional dilation. However, addition of flupirtine produced additional dilation in the same artery. (D) Summary of the percentage dilation of basilar artery with the application of 2 µM nimodipine alone, 2 µM nimodipine + 100 µM flupirtine and 100 µM flupirtine alone. Flupirtine either alone or when added along with nimodipine produced significantly more dilation compared with nimodipine alone (n = 3–6 *P < 0.05, one-way anova followed by post hoc Holm-Sidak test).
Celecoxib, the dual Kv7 channel activator and VSCC blocker, was a more potent dilator of the basilar artery compared with flupirtine (Figure 7). Basilar artery segments pre-constricted with ET-1, AVP or 5-HT were dilated by celecoxib in a concentration-dependent manner, with an EC50 of relaxation between 14 and 17 µM (Figure 7A, B). Celecoxib (30 µM), either alone or when combined with nimodipine (2 µM) produced significant additional dilation of basilar arteries compared with dilation induced by nimodipine alone (Figure 7D, E). We also sought to address the question of whether celecoxib could prevent the development of vasospasm when given before the constrictor agent. 5-HT was not able to produce constriction in basilar arteries that had been pre-treated with 30 µM celecoxib, but after wash out of both 5-HT and celecoxib, re-addition of 5-HT in the presence of the vehicle of celecoxib induced a robust constriction in the same artery (Figure 7C). The constriction produced by 5-HT in the presence of celecoxib was 0.11 ± 0.34% compared with 17.3 ± 1.7% produced in the presence of vehicle for celecoxib in the same arteries. The constriction produced by 5-HT alone was subsequently overcome by application of 30 µM celecoxib (Figure 7C).
Discussion
Our results provide clear functional evidence for the presence of Kv7 channels in rat basilar artery myocytes. K+ conductance through these channels contributes to resting membrane voltage and regulates the contractile status of basilar artery. Kv7 channel activators reverse the constrictor effects of spasmogens implicated in SAH-induced cerebral vasospasm.
Need for novel targets to treat cerebral vasospasm
Thus far, the only drug available to treat cerebral vasospasm is nimodipine, a selective L-type Ca2+ channel blocker, but it has limited efficacy. This may be due to the presence of heterogeneous Ca2+ channel subunits with varying sensitivities to nimodipine (Nikitina et al., 2007; Navarro-Gonzalez et al., 2009) and increased expression of nimodipine-insensitive T-type Ca2+ channels and R-type Ca2+ channels in basilar artery myocytes following SAH (Nikitina et al., 2010). Hence, a drug that inhibits the activation of all VSCC subtypes either directly or indirectly (e.g. by activation of K+ channels and prevention of the membrane depolarization that activates VSCC) would be expected to be more efficacious than nimodipine.
Kv7 channels are expressed and functional in rat basilar artery myocytes
Message transcripts of all the known KCNQ genes (KCNQ1-5, encoding Kv7.1-7.5 channels) were detected in rat basilar artery myocytes (Figure 1). Very recently, a quantitative assessment of the expression of KCNQ genes in rat basilar artery and middle cerebral artery myocytes by Zhong et al. also revealed the presence of all five KCNQ mRNAs, though the mRNAs of KCNQ2 and KCNQ3 were much less abundant than those of KCNQ1, KCNQ4 and KCNQ5 (Zhong et al., 2010). Kv7.1 channels are insensitive to drugs (e.g. flupirtine, retigabine and N-ethyl maleimide) that activate the other Kv7 channel subtypes (Gamper et al., 2005; Munro and Dalby-Brown, 2007), and Kv7.1 subunits do not form heteromers with other Kv7 channel subunits (Schwake et al., 2003). Considering that the whole cell Kv7 currents measured in basilar artery myocytes were robustly enhanced by flupirtine (Figure 2A–C), the contribution of outward K+ conductance through homomeric Kv7.1 channels is likely to be minimal. Hence, the functional Kv7 channels in basilar artery myocytes are likely to be predominantly constituted by homo- or hetero-tetramers of Kv7.4 and Kv7.5 channel subunits.
We isolated basilar artery myocyte Kv7 currents using whole cell voltage-clamp techniques (Figure 2). The KV currents recorded at test voltages between −60 mV and −20 mV can reasonably be attributed to Kv7 channel activity based on several observations: (i) the currents measured under our recording conditions were robustly enhanced by 10 µM flupirtine, a selective Kv7 channel activator; (ii) the currents were completely inhibited by 10 µM XE991, a selective Kv7 channel blocker; and (iii) the currents had electrophysiological characteristics expected of Kv7 currents (non-inactivating, voltage-dependent with a very negative threshold for activation and half-maximal activation (V0.5) of ∼−34 mV (Adams et al., 1982; Wickenden et al., 2001; Brueggemann et al., 2007; Mackie et al., 2008). Currents recorded at voltages more positive than −20 mV were not completely blocked by XE991, suggesting that at these more depolarized voltages we were recording a mix of currents, with contributions likely from other K+ channels, such as 4-AP-sensitive Kv channels and Ca2+-activated K+ channels (KCa) channels.
Kv7 channels are critical contributors to resting membrane voltage and basilar artery contractile status
The resting membrane voltage of basilar artery myocytes measured in the current study was −57.5 ± 6.3 mV, within the range reported in previous studies for basilar artery myocytes (Allen et al., 2002; Chrissobolis and Sobey, 2002; Jahromi et al., 2008a). The maximal density of K+ currents through Kv7 channels is small (<0.3 pA/pF, Figure 2C) compared with maximal current densities reported for 4-AP-sensitive-KV (36.9 pA/pF) and KCa channels (∼140 pA/pF) (Jahromi et al., 2008a,b;). However, Kv7 channels are likely to play an important role in determining the resting membrane voltage, more so than KV and KCa channels, as they are activated at more negative membrane voltages than 4-AP-sensitive KV and KCa channels [V0.5 for Kv7 currents is −34 mV (Figure 2D), compared with V0.5 of −1.3 mV for 4-AP-sensitive-KV currents (Jahromi et al., 2008a) and +86.8 mV for large-conductance KCa currents at 200 nM [Ca2+]cyt (Jahromi et al., 2008b)]. This is supported by the evidence that blockade of Kv7 channels with XE991 significantly depolarized the cell membrane (Figure 3A, B). This brought the membrane voltage from −58 mV, at which L-type VSCC have very low activity, to −35 mV, which is in the range of membrane voltage where VSCC activity increases in a steeply voltage-dependent manner (Figure 5B). Our results shown in Figure 3C support this hypothetical mechanism: XE991 robustly constricted the basilar artery and this effect was reversed by the L-type VSCC blocker nimodipine. Kv7 channels have a well-established role in stabilizing resting membrane voltages, and suppression of their activity is a common depolarizing stimulus in neurons and arterial myocytes (Adams et al., 1982; Mackie et al., 2008; Joshi et al., 2009). It remains to be determined whether suppression of Kv7 channel activity contributes substantially to the development of cerebral vasospasm after SAH.
Kv7 channel activators as candidates to treat cerebral vasospasm
The Kv7 channel activator flupirtine was able to reverse the constriction produced by the spasmogens involved in cerebral vasospasm, 5-HT, ET-1 and AVP (Figure 6). Furthermore, addition of flupirtine in the presence of nimodipine produced additional dilation. We speculate that flupirtine produced an additive vasodilatory effect by opposing the membrane depolarization produced by the spasmogens, and thereby preventing the activation of both nimodipine-sensitive and nimodipine-insensitive VSCC. Our results shown in Figure 5 are consistent with this idea in that we can detect Ba2+ currents in basilar artery myocytes that are activated by membrane depolarization, but not fully blocked by 2 µM nimodipine.
A 10-fold higher concentration of flupirtine was required in the vascular reactivity studies compared with patch-clamp studies. The discrepancy is likely, because, in intact arteries, depolarization of a small proportion of smooth muscle cells in response to addition of spasmogens leads to depolarization of the adjacent smooth muscle cells, which are connected by gap junctions. Therefore, the concentration of flupirtine required to oppose the concerted depolarization of VSMCs in a physiologically integrated arterial system is higher than in the single cell patch clamp studies. Conversely, a threefold lower concentration of spasmogen was required in vascular reactivity studies compared with patch-clamp studies (Mackie et al., 2008).
Celecoxib (Celebrex®) is marketed as a selective inhibitor of cyclooxygenase-2 (COX-2) and is widely prescribed to treat pain and inflammation. However, our present findings indicate that celecoxib is a robust Kv7 channel activator (like flupirtine) (Figure 4) as well as a VSCC blocker in basilar artery myocytes (more effective than 2 µM nimodipine; Figure 5). We recently demonstrated similar effects of celecoxib in mesenteric artery myocytes and provided evidence that these effects are apparent at concentrations of celecoxib that may be achieved with clinical therapy (Brueggemann et al., 2009). In contrast, the concentration of nimodipine (2 µM), which induced significantly less suppression of VSCC than 10 µM celecoxib did, is more than 1000-fold higher than concentrations achieved in cerebrospinal fluid after nimodipine administration in patients (Allen et al., 1983). Although the electrophysiological methods used may not reveal differences in drug effects that might occur under more physiological conditions, the pressure myography experiments are performed under much more physiological conditions and reveal a similar difference in efficacy between celecoxib and nimodipine. Though the mechanism(s) by which celecoxib modulates ion channels are not known, these effects are expected to influence the contractile status of basilar artery myocytes.
As expected from its dual ion channel effects, celecoxib very effectively reversed the constriction produced by the spasmogens (Figure 7A, B) and, like flupirtine, celecoxib was a more effective dilator of basilar artery than nimodipine (Figure 7D, E). The additional dilation produced by celecoxib could be due to two mechanisms: enhancement of Kv7 currents thereby limiting the voltage change necessary to activate all of the VSCCs, or Kv7 channel-independent inhibition of nimodipine-insensitive VSCC. Our results provide evidence for both of these mechanisms. The finding that flupirtine produces an additional dilation in the presence of nimodipine supports the former (Figure 6D), and that celecoxib produces additional inhibition of voltage-sensitive Ba2+ currents in the presence of 2 µM nimodipine supports the latter (Figure 5C).
The vasodilatory effects observed with celecoxib were independent of its ability to inhibit COX-2 as rofecoxib, a more potent inhibitor of COX-2 than celecoxib, did not induce vasodilatory responses, but dimethyl celecoxib, a structural analogue of celecoxib modified to eliminate COX-2 activity, also almost completely relaxed spasmogen pre-constricted artery segments (Supporting Information Figure S3; Brueggemann et al., 2009). We do not rule out the possibility of modulation of other ion channels or intracellular Ca2+ mobilization pathways by celecoxib that may also contribute in part to the vasodilatory responses observed here.
Direct Kv7 channel activators have already found their way to clinical trials to treat neuronal excitability disorders such as epilepsy and neuropathic pain (Miceli et al., 2008). Our findings suggest that, in addition to their direct neuroprotective effects (Boscia et al., 2006), these drugs with established safety profiles (Blackburn-Munro et al., 2005) can be readily adopted to prevent or limit the neurological deficits after SAH by reducing basilar artery vasospasm. Kv7 channel activators were also recently reported to reverse the pressure-induced constriction in resistance cerebral arteries (Zhong et al., 2010). Hence, the effect of Kv7 activators in improving the cerebral blood flow after SAH may extend beyond the ability of the drugs to dilate the conduit arteries like the basilar artery (Figure 6) that are primarily affected during the syndrome. In terms of potential utility in the treatment of SAH-induced cerebral vasospasm, celecoxib has additional effects that might also be beneficial. Celecoxib is a VSCC blocker, like nimodipine, and might therefore be expected to be more effective as a vasodilator than a drug like flupirtine that only activates Kv7 channels. It is increasingly recognized that an inflammatory component is associated with the development of cerebral vasospasm (Pradilla et al., 2010); the anti-inflammatory effects of celecoxib may therefore also be beneficial. Celecoxib could be an ideal drug to reduce the inflammation associated with SAH and simultaneously oppose the spasmogenic effects of locally elevated vasoconstrictor agonists. Preclinical trials to test the efficacy of these drugs to relieve basilar artery vasospasm in animal models of SAH are warranted.
In conclusion, this study provides the first evidence for the functional presence of Kv7 channels in basilar artery myocytes. Our findings suggest that K+ conductance through the Kv7 channels maintains hyperpolarized resting membrane voltages, preventing the activation of VSCC, and thereby regulating the contractile status of basilar artery. Flupirtine, a selective Kv7 channel activator or celecoxib, a dual Kv7 channel activator and VSCC blocker can reverse the constrictor effects of spasmogens implicated in SAH-induced cerebral vasospasm. These drugs could, therefore, be regarded as candidates for development of novel therapies for patients who have or may develop cerebral vasospasm.
Acknowledgments
This work was supported by grants from the National Institutes of Health (R01-HL089564 to KLB) and the American Heart Association (09PRE2260209 to BKM).
Glossary
Abbreviations
- 4-AP
4-aminopyridine
- AVP
arginine8-vasopressin
- ET
endothelin
- KCa
calcium-activated potassium channels
- KV channels
voltage-sensitive potassium channels
- SAH
subarachnoid hemorrhage
- VSMC
vascular smooth muscle cell
- XE991
10,10-bis(pyridin-4-ylmethyl)anthracen-9-one
Conflict of interest
The authors declare no conflict of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 A. Current-voltage curves show response of 4-AP-sensitive Kv currents to the addition of 100 µM Gd3+. The currents were isolated using protocol described previously (Mackie et al., 2008). B. Normalized conductance plots of 4-AP-sensitive-Kv currents fitted by a single Boltzmann function shows positive shift in activation of the currents with the addition of Gd3+ (half maximal activation of currents is shifted by approximately 15 mV). This enabled isolation of Kv7 currents at voltages ≤20 mV without contribution from 4-AP-sensitive Kv currents.
Figure S2 A. Current-voltage curves show that flupirtine significantly enhanced Kv7 currents at all tested voltages between -49.2 to +0.8 mV (n = 5 each, *P < 0.05, Paired Student's ‘t’ test, compared with control). XE991 (10 µM) abrogated Kv7 currents in the presence of 10 µM flupirtine (n = 5 each, #P < 0.05, Paired Student's ‘t’ test, compared with control).
Figure S3 A. Representative time course trace shows that 30 µM rofecoxib failed to dilate a basilar artery segment preconstricted with 75 nM 5-HT. Application of 30 µM celecoxib completely reversed the serotonin-induced constriction of the same artery. B. Representative time course trace shows that 30 µM dimethyl celecoxib completely reversed the serotonin-induced constriction of basilar artery. C. Summary of the percentage dilation produced by rofecoxib and dimethyl celecoxib (n = 3 each, *P < 0.05 using one-way anova followed by post hoc Holm-Sidak test).
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Adams PR, Brown DA, Constanti A. M-currents and other potassium currents in bullfrog sympathetic neurones. J Physiol. 1982;330:537–572. doi: 10.1113/jphysiol.1982.sp014357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC) Br J Pharmacol. (4th edn.) 2009;158(Suppl. 1):S1–S254. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen GS, Ahn HS, Preziosi TJ, Battye R, Boone SC, Boone SC, et al. Cerebral arterial spasm – a controlled trial of nimodipine in patients with subarachnoid hemorrhage. N Engl J Med. 1983;308:619–624. doi: 10.1056/NEJM198303173081103. [DOI] [PubMed] [Google Scholar]
- Allen T, Iftinca M, Cole WC, Plane F. Smooth muscle membrane potential modulates endothelium-dependent relaxation of rat basilar artery via myo-endothelial gap junctions. J Physiol. 2002;545:975–986. doi: 10.1113/jphysiol.2002.031823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berra-Romani R, Blaustein MP, Matteson DR. TTX-sensitive voltage-gated Na+ channels are expressed in mesenteric artery smooth muscle cells. Am J Physiol Heart Circ Physiol. 2005;289:H137–H145. doi: 10.1152/ajpheart.01156.2004. [DOI] [PubMed] [Google Scholar]
- Blackburn-Munro G, Dalby-Brown W, Mirza NR, Mikkelsen JD, Blackburn-Munro RE. Retigabine: chemical synthesis to clinical application. CNS Drug Rev. 2005;11:1–20. doi: 10.1111/j.1527-3458.2005.tb00033.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boscia F, Annunziato L, Taglialatela M. Retigabine and flupirtine exert neuroprotective actions in organotypic hippocampal cultures. Neuropharmacology. 2006;51:283–294. doi: 10.1016/j.neuropharm.2006.03.024. [DOI] [PubMed] [Google Scholar]
- Brueggemann LI, Moran CJ, Barakat JA, Yeh JZ, Cribbs LL, Byron KL. Vasopressin stimulates action potential firing by protein kinase C-dependent inhibition of KCNQ5 in A7r5 rat aortic smooth muscle cells. Am J Physiol Heart Circ Physiol. 2007;292:H1352–H1363. doi: 10.1152/ajpheart.00065.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brueggemann LI, Mackie AR, Mani BK, Cribbs LL, Byron KL. Differential effects of selective cyclooxygenase-2 inhibitors on vascular smooth muscle ion channels may account for differences in cardiovascular risk profiles. Mol Pharmacol. 2009;76:1053–1061. doi: 10.1124/mol.109.057844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrissobolis S, Sobey CG. Inhibitory effects of protein kinase C on inwardly rectifying K+- and ATP-sensitive K+ channel-mediated responses of the basilar artery. Stroke. 2002;33:1692–1697. doi: 10.1161/01.str.0000016966.89226.67. [DOI] [PubMed] [Google Scholar]
- Faraci FM, Sobey CG. Role of potassium channels in regulation of cerebral vascular tone. J Cereb Blood Flow Metab. 1998;18:1047–1063. doi: 10.1097/00004647-199810000-00001. [DOI] [PubMed] [Google Scholar]
- Gamper N, Li Y, Shapiro MS. Structural requirements for differential sensitivity of KCNQ K+ channels to modulation by Ca2+/calmodulin. Mol Biol Cell. 2005;16:3538–3551. doi: 10.1091/mbc.E04-09-0849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harder DR, Dernbach P, Waters A. Possible cellular mechanism for cerebral vasospasm after experimental subarachnoid hemorrhage in the dog. J Clin Invest. 1987;80:875–880. doi: 10.1172/JCI113146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heistad D, Konotos H. Cerebral circulation. In: Shepard J, Abboud F, editors. Handbook of Physiology. Section 2: The Cardiovascular System. Bethesad, MD: American Physiological Society; 1983. pp. 137–182. [Google Scholar]
- Henderson KK, Byron KL. Vasopressin-induced vasoconstriction: two concentration-dependent signaling pathways. J Appl Physiol. 2007;102:1402–1409. doi: 10.1152/japplphysiol.00825.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiguro M, Morielli AD, Zvarova K, Tranmer BI, Penar PL, Wellman GC. Oxyhemoglobin-induced suppression of voltage-dependent K+ channels in cerebral arteries by enhanced tyrosine kinase activity. Circ Res. 2006;99:1252–1260. doi: 10.1161/01.RES.0000250821.32324.e1. [DOI] [PubMed] [Google Scholar]
- Jahromi BS, Aihara Y, Ai J, Zhang ZD, Nikitina E, Macdonald RL. Voltage-gated K(+) channel dysfunction in myocytes from a dog model of subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2008a;28:797–811. doi: 10.1038/sj.jcbfm.9600577. [DOI] [PubMed] [Google Scholar]
- Jahromi BS, Aihara Y, Ai J, Zhang ZD, Weyer G, Nikitina E, et al. Preserved BK channel function in vasospastic myocytes from a dog model of subarachnoid hemorrhage. J Vasc Res. 2008b;45:402–415. doi: 10.1159/000124864. [DOI] [PubMed] [Google Scholar]
- Joshi S, Sedivy V, Hodyc D, Herget J, Gurney AM. KCNQ modulators reveal a key role for KCNQ potassium channels in regulating the tone of rat pulmonary artery smooth muscle. J Pharmacol Exp Ther. 2009;329:368–376. doi: 10.1124/jpet.108.147785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassell NF, Sasaki T, Colohan AR, Nazar G. Cerebral vasospasm following aneurysmal subarachnoid hemorrhage. Stroke. 1985;16:562–572. doi: 10.1161/01.str.16.4.562. [DOI] [PubMed] [Google Scholar]
- Katori E, Ohta T, Nakazato Y, Ito S. Vasopressin-induced contraction in the rat basilar artery in vitro. Eur J Pharmacol. 2001;416:113–121. doi: 10.1016/s0014-2999(01)00781-6. [DOI] [PubMed] [Google Scholar]
- Mackie AR, Brueggemann LI, Henderson KK, Shiels AJ, Cribbs LL, Scrogin KE, et al. Vascular KCNQ potassium channels as novel targets for the control of mesenteric artery constriction: based on studies in single cells, pressurized arteries and in vivo measurements of mesenteric vascular resistance. J Pharmacol Exp Ther. 2008;325:475–483. doi: 10.1124/jpet.107.135764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani BK, Brueggemann LI, Cribbs LL, Byron KL. Opposite regulation of KCNQ5 and TRPC6 channels contributes to vasopressin-stimulated calcium spiking responses in A7r5 vascular smooth muscle cells. Cell Calcium. 2009;45:400–411. doi: 10.1016/j.ceca.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayhan WG. Constrictor responses of the rat basilar artery during diabetes mellitus. Brain Res. 1998;783:326–331. doi: 10.1016/s0006-8993(97)01387-5. [DOI] [PubMed] [Google Scholar]
- Miceli F, Soldovieri MV, Martire M, Taglialatela M. Molecular pharmacology and therapeutic potential of neuronal Kv7-modulating drugs. Curr Opin Pharmacol. 2008;8:65–74. doi: 10.1016/j.coph.2007.10.003. [DOI] [PubMed] [Google Scholar]
- Munro G, Dalby-Brown W. Kv7 (KCNQ) channel modulators and neuropathic pain. J Med Chem. 2007;50:2576–2582. doi: 10.1021/jm060989l. [DOI] [PubMed] [Google Scholar]
- Navarro-Gonzalez MF, Grayson TH, Meaney KR, Cribbs LL, Hill CE. Non-L-type voltage-dependent calcium channels control vascular tone of the rat basilar artery. Clin Exp Pharmacol Physiol. 2009;36:55–66. doi: 10.1111/j.1440-1681.2008.05035.x. [DOI] [PubMed] [Google Scholar]
- Nikitina E, Zhang ZD, Kawashima A, Jahromi BS, Bouryi VA, Takahashi M, et al. Voltage-dependent calcium channels of dog basilar artery. J Physiol. 2007;580:523–541. doi: 10.1113/jphysiol.2006.126128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikitina E, Kawashima A, Takahashi M, Zhang ZD, Shang X, Ai J, et al. Alteration in voltage-dependent calcium channels in dog basilar artery after subarachnoid hemorrhage. J Neurosurg. 2010;113:870–880. doi: 10.3171/2010.2.JNS091038. [DOI] [PubMed] [Google Scholar]
- Nishimura Y. Characterization of 5-hydroxytryptamine receptors mediating contractions in basilar arteries from stroke-prone spontaneously hypertensive rats. Br J Pharmacol. 1996;117:1325–1333. doi: 10.1111/j.1476-5381.1996.tb16732.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishizawa S, Laher I. Signaling mechanisms in cerebral vasospasm. Trends Cardiovasc Med. 2005;15:24–34. doi: 10.1016/j.tcm.2004.12.002. [DOI] [PubMed] [Google Scholar]
- Ohya S, Asakura K, Muraki K, Watanabe M, Imaizumi Y. Molecular and functional characterization of ERG, KCNQ, and KCNE subtypes in rat stomach smooth muscle. Am J Physiol Gastrointest Liver Physiol. 2002;282:G277–G287. doi: 10.1152/ajpgi.00200.2001. [DOI] [PubMed] [Google Scholar]
- Passmore GM, Selyanko AA, Mistry M, Al-Qatari M, Marsh SJ, Matthews EA, et al. KCNQ/M currents in sensory neurons: significance for pain therapy. J Neurosci. 2003;23:7227–7236. doi: 10.1523/JNEUROSCI.23-18-07227.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradilla G, Chaichana KL, Hoang S, Huang J, Tamargo RJ. Inflammation and cerebral vasospasm after subarachnoid hemorrhage. Neurosurg Clin N Am. 2010;21:365–379. doi: 10.1016/j.nec.2009.10.008. [DOI] [PubMed] [Google Scholar]
- Schwake M, Jentsch TJ, Friedrich T. A carboxy-terminal domain determines the subunit specificity of KCNQ K+ channel assembly. EMBO Rep. 2003;4:76–81. doi: 10.1038/sj.embor.embor715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trandafir CC, Nishihashi T, Wang A, Murakami S, Ji X, Kurahashi K. Participation of vasopressin in the development of cerebral vasospasm in a rat model of subarachnoid haemorrhage. Clin Exp Pharmacol Physiol. 2004;31:261–266. doi: 10.1111/j.1440-1681.2004.03986.x. [DOI] [PubMed] [Google Scholar]
- Wang HS, Pan Z, Shi W, Brown BS, Wymore RS, Cohen IS, et al. KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel. Science. 1998;282:1890–1893. doi: 10.1126/science.282.5395.1890. [DOI] [PubMed] [Google Scholar]
- Wellman GC. Ion channels and calcium signaling in cerebral arteries following subarachnoid hemorrhage. Neurol Res. 2006;28:690–702. doi: 10.1179/016164106X151972. [DOI] [PubMed] [Google Scholar]
- Weyer GW, Jahromi BS, Aihara Y, Agbaje-Williams M, Nikitina E, Zhang ZD, et al. Expression and function of inwardly rectifying potassium channels after experimental subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2006;26:382–391. doi: 10.1038/sj.jcbfm.9600193. [DOI] [PubMed] [Google Scholar]
- Wickenden AD, Zou A, Wagoner PK, Jegla T. Characterization of KCNQ5/Q3 potassium channels expressed in mammalian cells. Br J Pharmacol. 2001;132:381–384. doi: 10.1038/sj.bjp.0703861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung SY, Pucovsky V, Moffatt JD, Saldanha L, Schwake M, Ohya S, et al. Molecular expression and pharmacological identification of a role for K(v)7 channels in murine vascular reactivity. Br J Pharmacol. 2007;151:758–770. doi: 10.1038/sj.bjp.0707284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong XZ, Harhun MI, Olesen SP, Ohya S, Moffatt JD, Cole WC, et al. Participation of KCNQ (Kv7) potassium channels in myogenic control of cerebral arterial diameter. J Physiol. 2010;588:3277–3293. doi: 10.1113/jphysiol.2010.192823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuccarello M, Bonasso CL, Lewis AI, Sperelakis N, Rapoport RM. Relaxation of subarachnoid hemorrhage-induced spasm of rabbit basilar artery by the K+ channel activator cromakalim. Stroke. 1996;27:311–316. doi: 10.1161/01.str.27.2.311. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.