

Abstract
BACKGROUND AND PURPOSE
Inhibition of the human cardiac Na+ channel (hNav1.5) can prolong the QRS complex and has been associated with increased mortality in patients with underlying cardiovascular disease. The safety implications of blocking hNav1.5 channels suggest the need to test for this activity early in drug discovery in order to design out any potential liability. However, interpretation of hNav1.5 blocking potency requires knowledge of how hNav1.5 block translates into prolongation of the QRS complex.
EXPERIMENTAL APPROACH
We tested Class I anti-arrhythmics, other known QRS prolonging drugs and drugs not reported to prolong the QRS complex. Their block of hNav1.5 channels (as IC50 values) was measured in an automated electrophysiology-based assay. These IC50 values were compared with published reports of the corresponding unbound (free) plasma concentrations attained during clinical use (fCmax) to provide an IC50 : fCmax ratio.
KEY RESULTS
For 42 Class I anti-arrhythmics and other QRS prolonging drugs, 67% had IC50 : fCmax ratios <30. For 55 non-QRS prolonging drugs tested, 72% had ratios >100. Finally, we determined the relationship between the IC50 value and the free drug concentration associated with prolongation of the QRS complex in humans. For 37 drugs, QRS complex prolongation was observed at free plasma concentrations that were about 15-fold lower than the corresponding IC50 at hNav1.5 channels.
CONCLUSIONS AND IMPLICATIONS
A margin of 30- to 100-fold between hNav1.5 IC50 and fCmax appears to confer an acceptable degree of safety from QRS prolongation. QRS prolongation occurs on average at free plasma levels 15-fold below the IC50 at hNav1.5 channels.
LINKED ARTICLE
This article is commented on by Gintant et al., pp. 254–259 of this issue. To view this commentary visit http://dx.doi.org/10.1111/j.1476-5381.2011.01433.x
Keywords: Nav1.5 channels, QRS complex, prolongation, ECG, drug safety
Introduction
Drug-induced changes to the electrocardiogram (ECG) are a key concern to both the pharmaceutical industry (Pollard et al., 2008) and regulators (ICH S7A and B, ICH E14: Anonymous, 2000; 2005a,b;). This concern largely resulted from the promiscuity of the hERG K+ channel, and its relationship to drug-induced QT interval prolongation and Torsade de Pointes (TdP). However, drug block of cardiac ion channels other than hERG is beginning to receive some consideration (Chen et al., 2009). As a result, pharmaceutical companies and contract research organizations have employed screening against a panel of cardiac ion channels as one of the earliest steps of preclinical cardiac safety assessment (Wible et al., 2008; Chen et al., 2009).
The cardiac Na+ channel (hNav1.5, encoded by the SCN5a gene; nomenclature follows Alexander et al., 2009) is one such safety target. This channel is primarily responsible for the depolarization of atrial and ventricular myocytes. Blockade of the Na+ current decreases the rate of depolarization, which in turn slows the velocity of excitation conduction. If the slowing of conduction is large enough, this can be measured as prolongation of the QRS complex on the ECG. Indeed, many drugs that block the cardiac Na+ channel are associated with QRS prolongation: these include Class I anti-arrhythmics, which block the cardiac Na+ channel as the primary mechanism of action such as flecainide and propafenone, as well as members of several other classes of non-cardiovascular drugs (e.g. tricyclic antidepressants, antipsychotics, anticonvulsants and antimalarials; Madias, 2008). Furthermore, an analysis of the FDA's Adverse Events Reporting System showed that there were 1194 clinical QRS adverse events reported, attributed to more than 500 drugs, between November 1997 and November 2007 (Valentin, 2010).
The safety implications of blockade of the cardiac Na+ channel and QRS prolongation remain a matter of debate. Genetic evidence suggests that loss of hNav1.5 channel activity is associated with numerous cardiac disorders and is often linked with potentially fatal arrhythmias (Tan et al., 2003). Furthermore, pharmacological inhibition of the cardiac Na+ channel may also trigger arrhythmias and can be associated with an increased mortality rate (Echt et al., 1991). A prolonged QRS duration has also been linked with an increase in mortality – in patients with right bundle branch block, for every 10 ms increase in QRS duration, the risk of death rises by 26.6% (Adesanya et al., 2008). Despite this, the link between QRS prolongation and arrhythmias in healthy individuals is not well understood (Seger, 2006). Nonetheless, the possible safety concerns of cardiac Na+ channel blockade make it prudent to identify and eliminate this liability early in the drug discovery process. To this end, we have previously described a hNav1.5 channel assay based on the IonWorks™ planar patch technology (Harmer et al., 2008).
Interpretation of the potency of a compound to block hNav1.5 channels requires knowledge of how hNav1.5 channel block translates into prolongation of the QRS complex. To explore this relationship, we determined the blocking potencies (IC50 values) at hNav1.5 channels for three categories of drugs: (i) Class I anti-arrhythmics; (ii) other drugs associated with QRS complex prolongation; and (iii) drugs not reported to prolong the QRS complex. Safety margins for these drugs were calculated by comparing the values of IC50 at hNav1.5 channels with published reports of the maximum unbound (i.e. free) plasma concentrations attained during clinical use (fCmax). We hope that these data will provide evidence for setting provisional safety margins, and aid in the interpretation of potency data for block of hNav1.5 channels, obtained during safety screening, early in the drug discovery process.
Methods
hNav1.5 IonWorks™ assay
Blocking potencies at hNav1.5 channels (as IC50) were determined using the IonWorks™ assay previously described (Harmer et al., 2008). This assay is able to determine hNav1.5 channel blocking potencies that are in good agreement with those determined using conventional patch clamp, as well as well as predicting Na+ channel effects in more integrated systems such as the cardiac Purkinje fibre action potential. Briefly, currents were recorded from hNav1.5 channel-expressing CHO cells using the Population Patch mode of the IonWorks™ device. Experiments were conducted at room temperature. Pre- and post-compound hNav1.5 currents were evoked by a single voltage train consisting of a 15 s period holding at −90 mV, a 160 ms step to −100 mV (to obtain an estimate of leak current), a 100 ms step back to −90 mV, followed by a series of eight pulses to 0 mV for 50 ms from a holding potential of −90 mV applied at 3 Hz. In between the pre- and post-compound voltage pulses there was no clamping of the membrane potential. Currents were leak-subtracted based on the estimate of current evoked during the step to −100 mV at the start of the voltage pulse protocol. The degree of inhibition of the hNav1.5 current for each well was assessed by dividing the post-scan hNav1.5 current by the respective pre-scan hNav1.5 current for the eighth test pulse. Each compound plate was laid out in 12 columns to enable 10 8-point non-cumulative concentration–effect curves to be constructed; the remaining two columns on the plate were taken up with vehicle (final concentration 0.33% dimethyl sulphoxide), to define the assay baseline, and a supra-maximal blocking concentration of flecainide (final concentration 316 µM) to define the 100% inhibition level. A full concentration-response curve to flecainide was also obtained for each plate to compare to historical data as a quality control measure. Data were normalized to vehicle and 100% blocking levels and the resulting data fitted to the Hill equation using a custom-written Origin-based fitting program (Origin 7.5; OriginLab Corporation, Northampton, MA, USA). The IC50 values quoted are the geometric mean of at least two independent experiments, with each test concentration being tested in a minimum of six wells.
Drugs
Redfern et al. (2003) have previously described in detail an approach to defining safety margins between the IC50, at hERG channels, and the drug free plasma concentrations and risk of generating TdP. We therefore adopted a similar approach with respect to the safety margins between block of hNav1.5 channels and drug-induced QRS complex prolongation. Specifically, we have compared hNav1.5 channel blocking potency data for a range of drugs with known propensity to prolong the QRS complex and compared this to their therapeutic plasma concentrations, as the IC50 : fCmax ratio in order to establish safety margins.
A list of 132 drugs that are known or suspected to inhibit hNav1.5 channels was initially compiled. These drugs were sourced either from Sigma-Aldrich (Dorset, UK), Apin Chemicals (Oxford, UK), Tocris Cookson (Bristol, UK) or the AstraZeneca compound collection (Macclesfield, UK).
Assays and data collection
These 132 drugs were then tested in the hNav1.5 IonWorks™ assay over eight half-log10 spaced concentrations. Drugs were tested to the limit of their solubility, up to a maximum test concentration of 100 µM (solubility was assessed by visual inspection). Those compounds that inhibited hNav1.5 channels by <40% did not have an IC50 calculated, and were not analysed further. Information on the remaining 98 drugs was collected and analysed, as described below, and tabulated.
Human plasma protein binding data and therapeutic plasma concentrations were primarily obtained from Redfern et al. (2003) and the BIOPRINT™ database (Krejsa et al., 2003). Schulz and Schmoldt's (2003) study was also used as a source of therapeutic plasma concentration data, although this publication generally lists trough values at steady state. Other literature sources used are referenced in the tables. Where available, the highest value for the effective therapeutic plasma concentration, or the highest plasma concentration achieved in humans during normal clinical use, has been used for each drug. For the purposes of this paper, this value has been referred to as the Cmax (maximum clinical concentration). The unbound (free) Cmax value was calculated using the plasma protein binding value, and is referred to as fCmax.
The MEDLINE (PubMed) database was searched for each drug using QRS as an additional search term. The drugs were divided into three categories: (i) Class I antiarrhythmics; (ii) other drugs associated with QRS complex prolongation; and (iii) drugs not reported to prolong the QRS complex (based on a search of the MEDLINE database). A drug was deemed to have changed the QRS complex if the authors concluded that the QRS complex change was noteworthy and drug-related; the magnitude of QRS complex change was not taken into account. For each of the 98 drugs, the ratio of IC50 at hNav1.5 channels to fCmax was calculated and plotted on a logarithmic scale. For each drug, the literature was also searched for studies where drug-induced QRS complex prolongation was measured alongside plasma drug concentrations. When possible, the plasma concentration associated with the lowest significant or noteworthy change in QRS complex duration is quoted.
The incidence of false positives/negatives was calculated for a given IC50 : fCmax ratio as follows: Class I anti-arrhythmics and other known drugs associated with QRS complex prolongation were classified as positives, and drugs not reported to prolong the QRS complex were classified as negatives. Then, a specific margin was selected (e.g. 30-fold). A positive compound with an IC50 : fCmax ratio above 30 was classified as a false positive, a negative compound with an IC50 : fCmax ratio below 10 was classified as a false negative. This approach was taken for a series of IC50 : fCmax ratios.
Results
An initial set of 132 drugs was tested in the hNav1.5 channel assay in order to determine IC50 values. The drugs listed below were tested up to the limit of their solubility, up to a maximum test concentration of 100 µM. These drugs were either inactive in the hNav1.5 assay (i.e. below the normal baseline activity for the assay), or did not inhibit the hNav1.5 channel sufficiently in order to fit a concentration–effect curve and determine an IC50 value:
Alfuzosin, amantadine, amiloride, anthralin, atenolol (S), benzocaine, berberine chloride, betahistine, chlorotrianisene, ciprofloxacin, clobetasol, dofetilide, epirubicin, ethotoin, fampridine, fenoterol, fluorometholone, gabapentin, gabexate, gatifloxacin, ipriflavone, levetiracetam, levocetirizine, levofloxacin, mephenytoin, montelukast, norethindrone, orlistat, phenacemide, d-sotalol, sparfloxacin, tiapride Cl, tocainide HCl and zonisamide. As these drugs were deemed to be inactive at hNav1.5 channels, they were considered outside the scope of this study and were excluded from further analysis.
Data for the remaining 98 drugs tested in this study are listed in Tables 1, 2 and 3. Table 1 lists Class I anti-arrhythmics. Table 2 lists drugs that are associated with prolongation of the QRS complex. Table 3 lists drugs that have not been reported to cause prolongation of the QRS complex. Each shows the hNav1.5 channel IC50 value, molecular weight, plasma protein binding, fCmax range and the log10 (hNav1.5 channel IC50 : highest fCmax). Tables 1 and 2 also show, where data are available, free drug concentrations determined in patients or volunteers where a change in the QRS complex duration has also been measured. The log10 of the ratio of IC50 to the mean free drug concentration associated with a QRS change (IC50 : [QRS]) is also shown. The data contained within the tables formed the basis for further analysis described below. A more comprehensive version of these tables can be found in the supplementary data (Tables S1–S3).
Table 1.
Class I anti-arrhythmics
| Drug | MW | PPB % | Plasma concentration range (nM) | hNav1.5 IC50 (µM) | Log10 IC50 : fCmax | [QRS]free (nM) | Log10 mean IC50 : [QRS]free |
|---|---|---|---|---|---|---|---|
| Ajmaline | 326.4 | 49 | 65–17 103 | 43.2 | 0.48 | 24 510 | 0.2 |
| Aprindine | 322.5 | 90 | 239–620 | 1.1 | 0.23 | 211; 192 | 0.7 |
| Cibenzoline | 262.4 | 55 | 976–1837 | 10.4 | 0.75 | 900 | 1.1 |
| Disopyramide | 339.5 | 69 | 742–6470 | 302.3 | 1.7 | 1950; 3327; 1950; 1756 | 2.1 |
| Encainide | 352.5 | 75 | 213–816 | 11.2 | 1.1 | 139; 202; 171; 348 | 1.7 |
| Flecainide | 414.3 | 57 | 753–840 | 5.8 | 0.84 | 265; 525; 559; 229 | 1.2 |
| Lidocaine | 234.3 | 67 | 7112–12 802 | 30.9 | 0.40 | 2077; 1963; 4552 | 1.1 |
| Lorcainide | 370.9 | 85 | 162 | 2.3 | 1.2 | 227; 113; 120 | 1.2 |
| Mexiletine | 179.3 | 67 | 1869–4129 | 25.7 | 0.79 | 1121; 9718 | 0.9 |
| Moricizine | 427.5 | 88 | 281 | 1.2 | 0.64 | 253; 393 | 0.6 |
| Phenytoin | 252.3 | 87 | 4360–7631 | 120.6 | 1.2 | ||
| Pilsicainide | 272.4 | 27 | 2687 | 51.1 | 1.3 | 1290; 1451; 6503; 16 661 | 1.1 |
| Procainamide | 235.3 | 16 | 30 340–54 186 | 2050.0 | 1.8 | 35 695; 53 185; 26 771; 56 397; 29 270 | 1.7 |
| Propafenone | 341.5 | 90 | 241–879 | 1.2 | 0.13 | 88; 29; 1171; 81; 291 | 0.9 |
| Quinidine | 324.4 | 87 | 1566–3237 | 9.7 | 0.48 | 916; 1018; 1174; 1057 | 1.0 |
MW, molecular weight; PPB %, human plasma protein binding; [QRS]free (nM), the free plasma concentration associated with QRS complex prolongation in human – more than one value may be quoted, see supplemental data for more details. Log10 mean IC50 : [QRS] = log10 of (hNav1.5 IC50 divided by [QRS]free (nM). Plasma concentration range is the free (unbound) value. See supplemental tables for additional information and references.
Table 2.
Drugs associated with QRS complex prolongation
| Drug | MW | PPB % | Plasma concentration range (nM) | hNav1.5 IC50 (µM) | Log10 IC50 : fCmax | [QRS]free (nM) | Log10 mean IC50 : [QRS]free |
|---|---|---|---|---|---|---|---|
| Amiodarone | 645.3 | 98 | 0.5–62 | 4.8 | 1.9 | 121; 47; 62; 39 | 1.9 |
| Amitriptyline | 277.4 | 95 | 4.9–49 | 1.6 | 1.5 | 208; 160 | 1.2 |
| Amodiaquine | 355.9 | 95 | 25–58 | 9.7 | 2.2 | 58 | 2.2 |
| Amoxapine | 313.8 | 90 | 16–191 | 5.8 | 1.5 | 287 | 1.3 |
| Bupivacaine | 288.4 | 96 | 32–312 | 4.3 | 1.1 | 190; 328 | 1.2 |
| Bupropion | 239.7 | 84 | 67–95 | 76.2 | 2.9 | 659 | 2.1 |
| Carbamazepine | 236.3 | 74 | 7061–13 034 | 152.0 | 1.1 | 57 493; 52 617 | 0.4 |
| Chloroquine | 319.9 | 58 | 660 | 36.5 | 1.7 | 16 900; 3848 | 0.7 |
| Citalopram | 324.4 | 80 | 46–123 | 14.7 | 2.1 | 1196 | 1.1 |
| Desethylamodiaquine | 327.8 | 86 | 353 | 6.9 | 0.44 | ||
| Desipramine | 266.4 | 81 | 27–357 | 2.4 | 0.82 | 123 | 1.3 |
| Diltiazem | 414.5 | 86 | 1091 | 14.2 | 1.1 | ||
| Diphenhydramine | 255.4 | 92 | 23–34 | 16.4 | 2.7 | 776 | 1.3 |
| Dolasetron | 324.4 | 72 | 637 | 79.7 | 2.1 | ||
| Fluoxetine | 309.3 | 94 | 6.5–93 | 4.9 | 1.7 | 167 | 1.5 |
| Imipramine | 280.4 | 90 | 73–128 | 3.6 | 1.4 | 43; 74; 53 | 1.8 |
| Lamotrigine | 256.1 | 56 | 14 249–24 327 | 63.4 | 0.42 | 25 721 | 0.3 |
| Maprotiline | 277.4 | 89 | 10–238 | 2.8 | 1.1 | 83 | 1.5 |
| Mesoridazine | 386.6 | 95 | 129 | 7.2 | 1.7 | 2069 | 0.5 |
| Nortriptyline | 263.4 | 94 | 23–36 | 2.0 | 1.7 | 23 | 1.9 |
| Perhexiline | 277.5 | 90 | 216 | 3.4 | 1.2 | ||
| Procaine | 236.3 | 6 | 39 778–39 778 | 84.6 | 0.33 | 40 574 | 0.3 |
| Quinine | 324.4 | 92 | 1834 | 17.8 | 1.0 | 2682; 995; 1336 | 1.1 |
| Risperidone | 410.5 | 89 | 2–2130 | 20.2 | 1.0 | 287 | 1.8 |
| Ropivacaine | 274.4 | 94 | 394 | 11.2 | 1.5 | 481 | 1.4 |
| Thioridazine | 370.6 | 98 | 84–979 | 3.5 | 0.55 | 262 | 1.1 |
| Venlafaxine | 277.4 | 27 | 395–1053 | 90.1 | 1.9 | 14 473 | 0.8 |
MW, molecular weight; PPB %, human plasma protein binding; [QRS]free (nM), the free plasma concentration associated with QRS complex prolongation in human – more than one value may be quoted, see supplemental data for more details. Log10 mean IC50 : [QRS] = Log10 of (hNav1.5 IC50 divided by [QRS]free) (nM). Plasma concentration range is the free (unbound) value. See supplemental tables for additional information and references.
Table 3.
Drugs not associated with QRS complex prolongation
| Drug | MW | PPB % | Plasma concentration range (nM) | hNav1.5 IC50 (µM) | Log10 IC50 : fCmax |
|---|---|---|---|---|---|
| Ambroxol | 378.1 | 90 | 23 | 15.2 | 2.8 |
| Astemizole | 458.6 | 97 | 0.04–3.5 | 2.3 | 2.8 |
| Bepridil HCl | 366.6 | 100 | 5.1–33 | 1.9 | 1.8 |
| Bromopride | 344.3 | 40 | 105 | 41.4 | 2.6 |
| Buspirone | 385.5 | 91 | 0.9–1.0 | 125.4 | 5.1 |
| Chloramphenicol | 323.1 | 51 | 150 48–27 689 | 215.0 | 1.1 |
| Chlorpromazine | 318.9 | 96 | 25–65 | 2.8 | 1.6 |
| Cinnarizine | 368.5 | 91 | 67 | 1.0 | 1.2 |
| Cisapride | 466.0 | 0 | 4.9 | 4.2 | 2.9 |
| Clemastine | 343.9 | N/A | 4.7 | 4.0 | 2.9 |
| Clomipramine | 314.9 | 98 | 16–30 | 2.6 | 1.9 |
| Clozapine | 326.8 | 95 | 49–92 | 11.6 | 2.1 |
| Cyproheptadine | 287.4 | 96 | 6.0–7.0 | 6.1 | 2.9 |
| Desloratadine | 310.8 | 85 | 1.9 | 14.1 | 3.9 |
| Diazepam | 284.8 | 98 | 29–180 | 73.2 | 2.6 |
| Dipyridamole | 504.6 | 99 | 26–34 | 19.3 | 2.7 |
| Domperidone | 425.9 | 92 | 3.8–19 | 5.6 | 2.5 |
| Ebastine | 469.7 | 0 | 5.1 | 1.3 | 2.4 |
| Exemestane | 296.4 | 90 | 2.7 | 71.3 | 4.4 |
| Fenfluramine | 231.3 | 36 | 76–166 | 47.0 | 2.5 |
| Flunarizine | 404.5 | 93 | 5.5–34 | 2.6 | 1.9 |
| Fluphenazine | 437.5 | 90 | 0.5–0.9 | 8.0 | 3.9 |
| Haloperidol | 375.9 | 91 | 3.6–5.0 | 2.3 | 2.7 |
| Hydroxyzine | 374.9 | 93 | 19–22 | 32.8 | 3.2 |
| Ketoconazole | 531.4 | 99 | 113–709 | 44.9 | 1.8 |
| Lofexidine | 259.1 | 85 | 984 | 27.2 | 1.4 |
| Loperamide | 477.1 | 97 | 0.15–0.2 | 2.9 | 4.1 |
| Loratadine | 382.9 | 98 | 1.0–1.4 | 33.6 | 4.4 |
| Memantine | 179.3 | 45 | 256 | 92.6 | 2.6 |
| Mepivacaine | 246.4 | 77 | 373–817 | 81.4 | 2.0 |
| Mifepristone | 429.6 | 98 | 50–92 | 25.8 | 2.4 |
| Moxifloxacin | 401.4 | 42 | 6083 | 206.7 | 1.5 |
| Nicardipine | 479.5 | 99 | 2.3–2.6 | 4.3 | 3.2 |
| Nifedipine | 346.3 | 97 | 7.7–10 | 33.5 | 3.5 |
| Nilvadipine | 385.4 | 98 | 0.2–0.6 | 2.3 | 3.6 |
| Olanzapine | 312.4 | 93 | 1.8–6.7 | 39.0 | 3.8 |
| Oxatomide | 426.6 | 91 | 7.4 | 1.0 | 2.1 |
| Pimozide | 461.6 | 99 | 0.2–0.4 | 1.7 | 3.6 |
| Prilocaine HCl | 220.3 | 29 | 974–6491 | 72.6 | 1.9 |
| Propranolol | 259.4 | 93 | 32–80 | 7.5 | 2.0 |
| Protriptyline | 263.4 | 92 | 91 | 3.1 | 1.5 |
| Pyrimethamine | 248.7 | 86 | 154 | 24.6 | 2.2 |
| Quinacrine | 400.0 | 80 | 247 | 20.2 | 1.9 |
| Reserpine | 608.7 | 96 | 0.1 | 10.5 | 4.9 |
| Riluzole | 234.2 | 96 | 66–256 | 17.6 | 1.8 |
| Salmeterol | 415.6 | 96 | 0.14–0.1 | 7.2 | 4.7 |
| Sertindole | 441.0 | 99 | 1.6–1600 | 8.1 | 5.6 |
| Sertraline | 306.2 | 98 | 9.3–33 | 4.3 | 2.1 |
| Tacrine | 198.3 | 55 | 23–36 | 47.1 | 3.1 |
| Terfenadine | 471.7 | 97 | 0.3–0.6 | 2.0 | 3.5 |
| Terguride (S−) | 340.5 | 70 | 2.0 | 22.1 | 4.0 |
| Trifluoperazine | 407.5 | 99 | 0.04–0.2 | 7.4 | 4.5 |
| Trimipramine | 294.4 | 95 | 4.8–42 | 2.7 | 1.8 |
| Verapamil | 454.6 | 95 | 11–81 | 9.3 | 2.1 |
| Ziprasidone | 412.9 | 99 | 1.5–2.2 | 170.0 | 4.9 |
MW, molecular weight; PPB %, human plasma protein binding. Plasma concentration range is the free (unbound) value. See supplemental tables for additional information and references.
Figure 1 shows examples of IC50 : fCmax ratio calculations for two drugs. For each drug, the graphs show the concentration–effect curve for blockade of hNav1.5 channels, as well as the highest fCmax value quoted in the tables. Verapamil (not associated with QRS prolongation) has an IC50 : fCmax ratio of 115, whereas propafenone (a QRS prolonging Class I drug) has an IC50 : fCmax ratio of 1.3.
Figure 1.
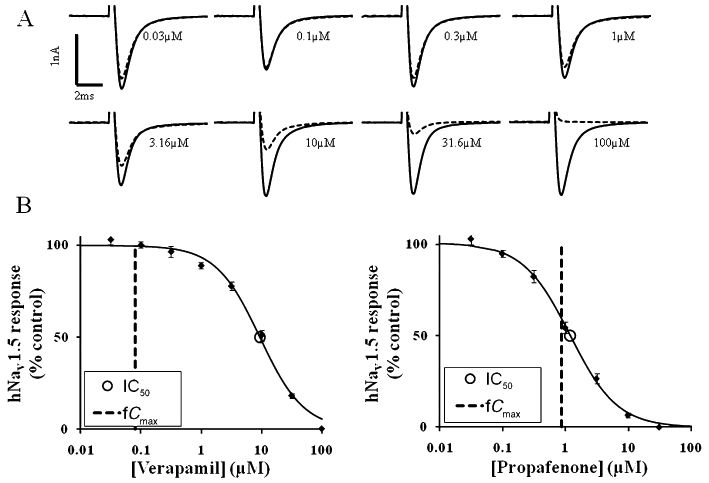
IC50 : fCmax ratio calculations for verapamil and propafenone. (A) Typical hNav1.5 current recordings generated using an IonWorks™-based assay. Each individual recording shows data from a single well in response to vehicle (0.33% dimethyl sulphoxide, solid line) followed by a single concentration of verapamil (dotted line). Each recording is taken from the eighth pulse to 0 mV from a holding potential of −90 mV. (B) Graphs show concentration–effect curves for hNav1.5 channels generated using the same assay. The half maximal inhibitory concentration (IC50) and maximal free plasma concentration in human (fCmax) values are also plotted. For propafenone, the hNav1.5 IC50 is 1.2 µM and fCmax is 0.9 µM, giving a ratio (safety margin) of 1.3. For verapamil, the hNav1.5 IC50 is 9.3 µM and the fCmax is 0.08 µM, giving a ratio of 115.
The same ratios were calculated for all 98 drugs, and the data are presented graphically in Figure 2. The data in Figure 2 are divided into the three drug categories. For category 1 drugs, the IC50 : fCmax ratios ranged from 1.3 to 57.4 (mean = 13, n = 15). The majority (87%) of the Class I drugs had an IC50 : fCmax ratio of less than 30. For category 2 drugs, margins ranged from 2.1 to 800 (mean = 85, n = 27). When categories 1 and 2 were combined, the majority of drugs (67%) had IC50 : fCmax ratios of less than 30-fold. For category 3 drugs, the margins ranged from 13 to 406 050 (mean = 16 193, n = 55). The majority of category 3 drugs (72%) had ratios greater than 100.
Figure 2.
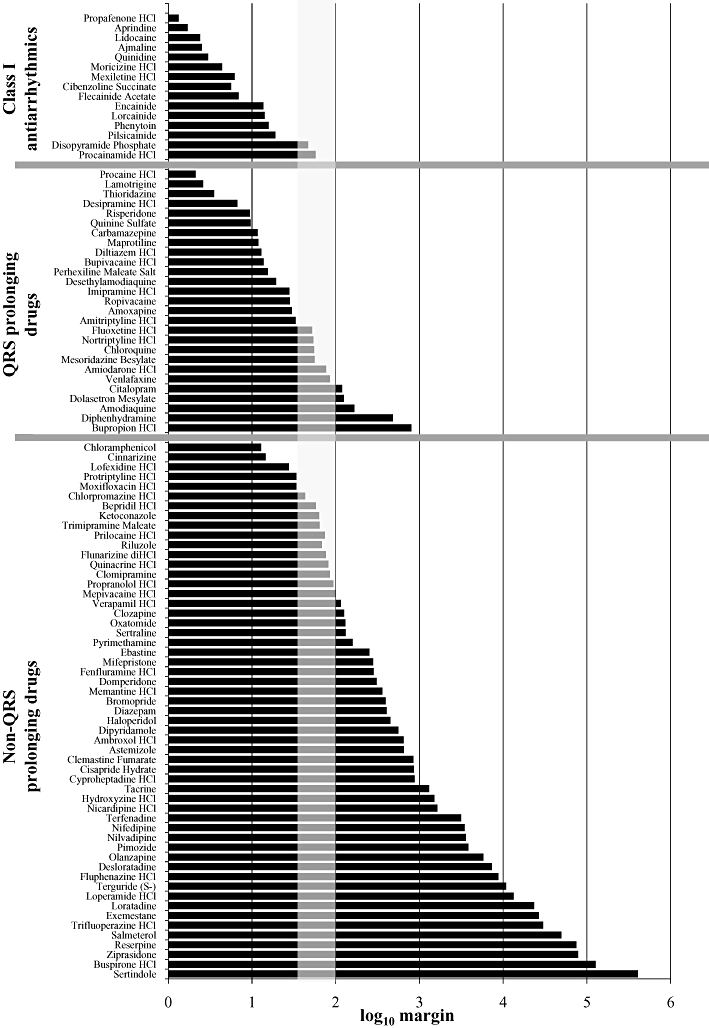
Log10 of the IC50 : fCmax ratios. IC50 values at hNav1.5 channels were generated using a IonWorks™-based assay. The fCmax values are derived from published values of the maximum plasma concentration achieved in a clinical setting and the plasma protein binding value in humans. The vertical grey box indicates where ratios between 30 and 100 would fall. For source data, refer to Tables 1–3.
In order to further characterize the relationship between the fCmax and hNav1.5 channel IC50 values, we carried out two separate analyses. Initially, we conducted a crude statistical analysis by fitting a Gaussian distribution to the frequency plots for the fCmax and IC50 values for the 98 drugs tested in this study. The frequency/distribution curves are plotted in Figure 3A. The separation between the two distribution means is 109 ± 1. It is worth noting that the distribution for the hNav1.5 IC50 values is quite narrow when compared to the drug fCmax values.
Figure 3.
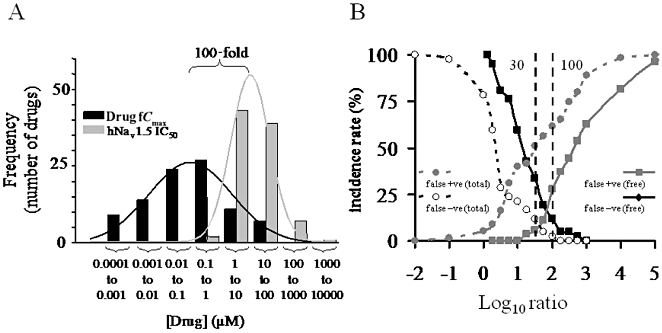
Analysis of fCmax, hNav1.5 IC50 and derived ratios. (A) Distribution curves for fCmax and IC50 values for the drugs analysed in this study. The distribution means [expressed as log10 of (drug) ] were −6.6 ± 0.1 (R2 = 0.95) for fCmax data and −4.5 ± 0.03 (R2 = 0.99) for the IC50 data. (B) Analysis of false positive (false +ve) and false negative (false −ve) rates versus IC50 : fCmax ratios. Class I anti-arrhythmics (Na+ channel blockers) and other known QRS-prolonging drugs were classified as positive, and drugs not known to prolong QRS were classified as negative. The false positive/negative incidence was calculated for ratios using total and unbound (free) Cmax values. A ratio of 30−100 has the optimal false positive/negative incidence for free drug concentration, as indicated by the dotted line.
The data were then pooled into two groups: Class I antiarrhythmics and other drugs associated with QRS complex prolongation were classified as positives, and drugs not reported to prolong the QRS complex were classified as negatives. The false positive/false negative incidence was then calculated for a range of IC50 : fCmax ratios [shown in Figure 3B as log10 (ratio) ]. This analysis was conducted using both total and unbound (free) Cmax values. The results of this analysis are plotted in Figure 3B. When only the free drug concentrations were taken into account, the point where the false positive/false negative incidence was equal occurred within the 30 to 100 ratio range. When total concentrations were taken into account, this point occurred between ratios of 1 and 10 [log10 (ratio) 0 to 1].
Finally, we attempted to determine the relationship between the hNav1.5 IC50 value and the free drug concentration that is associated with prolongation of the QRS complex in humans ([QRS]free). The IC50 : [QRS]free ratio (therapeutic window) was calculated for each drug where data were available. The results of this analysis for 37 drugs are presented in Figure 4. The IC50 : [QRS]free therapeutic window for these drugs ranged from 1.8 to 165 (mean = 15). It should be noted that for some drugs, the QRS data were derived from case studies that often involved drug overdose. Also, as noted previously, there is a paucity of drug-induced QRS clinical data in the literature on which to conduct this kind of analysis.
Figure 4.
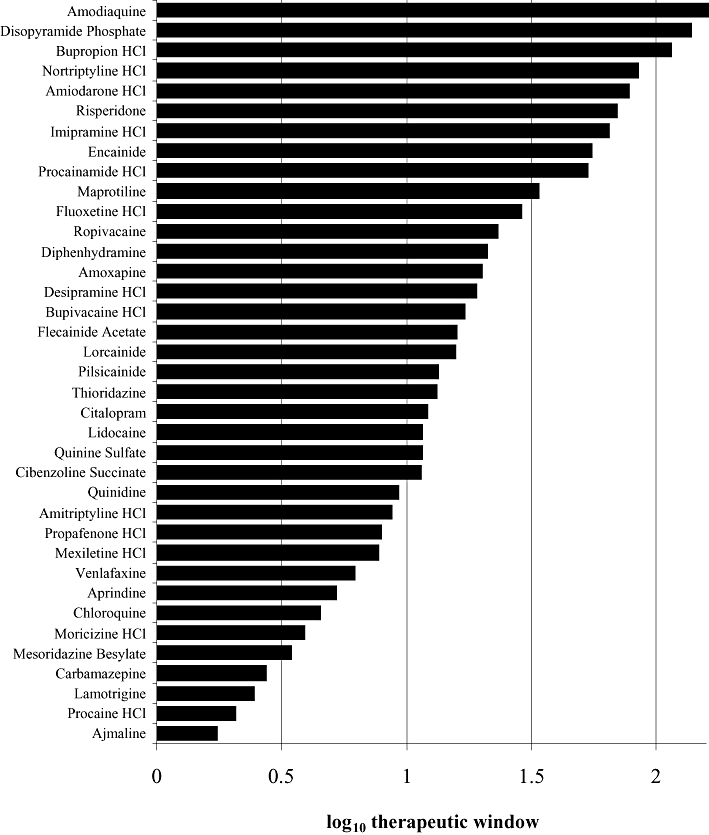
Log10 of (hNav1.5 IC50 : [QRS]free), shown as therapeutic window. IC50 data were generated using the IonWorks™-based assay. [QRS]free is the published value of mean lowest free plasma concentration associated with prolongation of the QRS complex in humans. For source data, refer to Tables 1 and 2.
Discussion
The importance of preclinical assessment of blocking activity at hNav1.5 channels
Drug block of the cardiac Na+ channel can be associated with intracardiac conduction delay, which in turn is manifested as prolongation of the QRS complex on the ECG (Delk et al., 2007). Furthermore, ventricular tachycardia and other arrhythmias are considered to contribute to the morbidity and mortality associated with Na+ channel blocking drugs (Thanacoody and Thomas, 2005; Delk et al., 2007). However, Seger (2006) has suggested that the true incidence and aetiology of ventricular arrhythmias is unknown: ‘There is no evidence that the arrhythmias that occur in (Na+ channel blocking drug) poisoning prior to the development of a failing heart are a result of non-uniform conduction slowing.’ Despite this, a drug-induced change to the ECG, such as QRS prolongation, remains an undesirable property for new chemical entities. For example, QRS prolongation has been identified as a marker for increased mortality (Horwich et al., 2003; Kalahasti et al., 2003; Breidthardt et al., 2007; Adesanya et al., 2008). Therefore, it seems prudent to screen out liability to block hNav1.5 channels early in a drug discovery programme, where a medium-throughput hNav1.5 channel assay could generate data in a time frame that could influence medicinal chemistry design–make–test cycle (Harmer et al., 2008).
Pharmaceutical regulators have also highlighted the importance of drug-induced ECG changes – it is a requirement to perform a thorough ECG assessment before a candidate drug enters the final stage of clinical development (ICH E14 guidelines: Anonymous, 2005a). The primary aim of the thorough ECG assessment is to determine the effects of the candidate drug on the QTc interval as a surrogate marker for the cardiac arrhythmia TdP. Although QTc prolongation is recognized as an imperfect marker for TdP risk, the potential for sudden death and the difficulty in predicting such a rare arrhythmia has resulted in such prolongation becoming a major focus for drug safety (Darpo, 2010). If we have learnt anything from the hERG/QT/TdP story, it is that the best way to minimize uncertainty over the link between QT prolongation and TdP is to avoid having QT interval prolongation in the first place, by early application of the hERG screen (Pollard et al., 2010; Valentin et al., 2010).
An analogous situation exists for block of hNav1.5 channels and drug-induced QRS changes: the relationship between QRS prolongation and arrhythmia is poorly defined. Nonetheless, given the choice between two otherwise equal new drugs, one that does not widen QRS and one that does, the former would be the rational choice. Furthermore, given the association between QRS prolongation and mortality, and the potential for drug-induced arrhythmia, it remains a sensible precaution to remove any activity at hNav1.5 channels before compounds reach human volunteers.
In this study, we have avoided making any link between potency as a blocker of hNav1.5 channels and arrhythmia risk, as this is a challenging task beyond the scope of this study. However, it is worth pointing out that preclinical cardiac risk assessment would also include integrating information from other ion channel assays (hERG, L-type Ca2+ channel, etc.), as well as other in vitro and in vivo cardiovascular assays. Taken together, the integrated data would give an assessment of the risk of a candidate drug causing an ECG change in humans. However, such data are not designed to assess the pro-arrhythmic risk per se. Ultimately, the only way to determine if a drug has pro-arrhythmic potential is to conduct a large and expensive longitudinal trial in patients to assess cardiovascular risk. Therefore, screening out any liability to interact with cardiac ion channels early in the drug discovery process is the simplest route to reduce potential cardiac safety issues.
Safety margins for compounds with activity at hNav1.5 channels
Given the importance of preclinical screening on hNav1.5 channels, the interpretation of such data should also be given serious consideration – that is, how does a drug discovery programme place the IC50 at hNav1.5 channels in context? To address this question, we set out to define what should be considered a safe margin between a hNav1.5 IC50 and the free drug concentration that is achieved in man. A similar approach has been taken by several authors for the hERG K+ channel (Webster et al., 2002; Redfern et al., 2003).
The data presented here indicate that a ratio of 30–100 between hNav1.5 IC50 and fCmax would be sufficient to ensure a suitable degree of safety in terms of drug-induced QRS complex prolongation. This 30–100 ratio represents the optimal range over which false positives and false negatives are at a minimum (see Figure 3B). A number of investigators have come to a similar conclusion regarding safety margins applied to blockade of hERG channels (Webster et al., 2002; Redfern et al., 2003).
We would recommend this safety margin to drug discovery projects that have liability to block hNav1.5 channels and a reliable estimate of the fCmax in human. However, this ratio should only be regarded as an initial guide – that is, the IC50 at hNav1.5 channels within a 30- to 100-fold margin should act as a safety flag. Once in vivo ECG data are available, this should also be taken into account when drawing up an integrated risk assessment (Valentin and Hammond, 2008). In addition to this, the indication for which the drug is intended should be considered. For example, a 10-fold safety margin may be warranted for life-threatening indications (e.g. late-stage cancers or patients with severe hospitalized infections) whereas a 100-fold safety margin would be more appropriate for indications such as eczema or seasonal rhinitis. The safety profile of existing therapies is also important – a drug that does not affect the QRS complex might be favoured over one that does have this liability. Other factors that also influence the risk assessment include the duration of treatment and the likely patient population (for instance, do patients have underlying cardiac disease?).
Relationship between IC50 at hNav1.5 channels and plasma concentrations evoking a QRS change
We wanted to understand how hNav1.5 IC50 data compared with the free plasma concentration that evokes a QRS change in man ([QRS]free), which we have called the therapeutic window). For the 37 drugs for which we could obtain human pharmacodynamic and pharmacokinetic data, there was a wide range in this therapeutic window. Nonetheless, these data suggest that, on average, a QRS change can be evoked by a free plasma concentration 15-fold less than the corresponding IC50 for hNav1.5 channels – that is, QRS complex prolongation can occur to the left of the hNav1.5 IC50. A preliminary analysis, based on a limited number of compounds, by Cordes et al. (2009) has come to a similar conclusion. These analyses suggest that <10% inhibition of the hNav1.5 channel could be associated with prolongation of the QRS complex in man.
The reasons why there is such a wide range in the IC50 : [QRS] therapeutic window are likely to be diverse. One possible explanation could include the varying quality of the clinical data. For some drugs, the QRS data come from patients who have taken a drug overdose, or from patients with underlying cardiac disease, whereas other drug data come from controlled clinical trials. Secondly, in some cases, measurement of blood plasma levels and QRS intervals may not be optimal. For example, plasma samples may only have been collected as a single time point during dosing, while manual measurement of the QRS complex duration can also be prone to error. Finally, the kinetics with which drugs bind to the hNav1.5 channel may also influence the therapeutic window. Drugs with rapid on/off kinetics may require high plasma concentrations to affect the duration of the QRS complex duration – and this may only occur at high heart rates (Vaughan Williams, 1991; Takanaka et al., 1994).
Outlying drugs
Analysis of the calculated ratios has identified a number of outliers, that is, drugs where the IC50 : fCmax ratio is not what is expected for that category. For example, outliers in the Class I antiarrhythmic group are disopyramide and procainamide, with an IC50 : fCmax ratio of 47 and 57, respectively. Other drugs in the same category tend to have ratios below 30. The reason for this discrepancy is not clear. A plausible explanation is that the hNav1.5 assay employed in this study underestimated the potency of these compounds (see Limitations of this study). Yatani and Akaike (1985) obtained an IC50 for disopyramide of 28 µM, determined using the Na+ current measured in isolated cardiac myocytes. This value is 10-fold less than the IC50 value determined using the IonWorks hNav1.5 assay (302 µM) and would lead to an IC50 : fCmax ratio of 4.3, which is in line with the ratios for other Class I anti-arrhythmics.
Amodiaquine is an outlier in category 2, with a margin of 166. However, its main metabolite, desethylamodiaquine, has a margin of only 19. Both parent and metabolite molecules are approximately equipotent at hNav1.5 channels, although the metabolite has a sixfold higher free plasma concentration. It is plausible that other drugs have metabolites with either higher plasma concentrations or higher potencies at hNav1.5 channels. Screening these metabolites in the hNav1.5 channel assay would test this hypothesis.
Some of the outliers in category 3, such as chloramphenicol and cinnarizine, have lower ratios compared with the category as a whole. Given these low ratios, one would expect these drugs to cause QRS prolongation in a clinical setting. It is possible that the fCmax data for these drugs are not reflective of plasma concentrations seen in normal clinical practice (e.g. chloramphenicol is mainly used as a topical eye treatment in developed countries). Alternatively, measurements of QRS complex prolongation may have been under-reported for patients taking these drugs.
A plausible explanation is not obvious for many of the other outlying drugs. However, one possibility is differential accumulation of the compound in the myocardium – a phenomenon recognized for a number of drugs (Jensen and Nielsen-Kudsk, 1988; Yoshida and Furuta, 1999; Titier et al., 2004). In addition, impulse conduction in the heart is determined by three factors: (i) cellular excitability (e.g. hNav1.5 channel activity); (ii) electrical coupling (e.g. connexin43 in the ventricle); and (iii) cellular/tissue architecture (e.g. fibrosis, myocyte size and shape). If a compound affects any of these parameters, then it could also potentially affect QRS duration.
It should be noted that it was not possible to obtain IC50 values for 34 drugs as these were either inactive or only partially active at hNav1.5 channels. Of these 34 drugs, three are associated with QRS complex prolongation in man (amantadine, atenolol and sotalol), placing them into category 2 (Freedman et al., 1992; Schwartz et al., 2008). These drugs caused 25% to 30% inhibition in the hNav1.5 channel assay. The remaining 31 drugs are not associated with QRS complex prolongation in man, which places them in category 3.
Limitations of this study
There are a number of considerations that need to be taken into account that limit the interpretation of this study. The primary limitation is the lack of clinical data where drug-induced QRS changes have been accurately measured, and pharmacokinetic data have been simultaneously determined. This makes it difficult to estimate the therapeutic window between IC50 at hNav1.5 channels and the concentration causing a QRS change. Much of the data relating to drug-induced QRS effects are derived from isolated case studies (including drug overdose) and from clinical trials conducted on patients with underlying cardiac disease. Clearly, use of such data is sub-optimal, but until more comprehensive clinical data are available, no other analysis is possible. It is our hope that more comprehensive pharmacokinetic/pharmacodynamic data relating to drug-induced QRS changes in man will emerge as the importance of this cardiotoxicity receives more attention. When more comprehensive clinical data are available, we may also be able to determine if specific patient populations are more susceptible to QRS prolonging drugs (e.g. patients with ischaemic heart disease or those with impaired cardiac conduction).
It should also be noted that we could not take into account the magnitude of any drug-induced QRS complex prolongation, as these data were either unavailable or quite variable. For example, we have not differentiated between marked increases in QRS complex duration seen with some Class I anti-arrhythmics (e.g. the ∼30% increase seen with encainide) and small QRS complex changes seen with some other drugs (e.g. amodiaquine). The magnitude of QRS complex prolongation is likely to be important, as evidence from patients with right bundle branch block indicates that mortality increases in line with prolongation of the QRS complex (Adesanya et al., 2008). Until more detailed clinical data are available, it remains difficult to quantify what constitutes a ‘safe’ degree of QRS complex prolongation.
Another limitation is the reliance upon Cmax and human plasma protein binding data. During the compilation of these data, it was apparent that some drugs have a wide range of reported therapeutic plasma concentration values (e.g. 65 to 17 103 nM for ajmaline). Furthermore, it was also obvious that the fCmax calculation was dependent upon the plasma protein binding data. Clearly, for the ratio used here, an inaccurate fCmax value will return an equally inaccurate ratio. However, it is our contention that although these variables do add a certain amount of ‘noise’ to the data, they do not affect the overall conclusions reached.
We have chosen to use the IC50 at hNav1.5 channels, as this is the most accurate measurement that can be made from a sigmoidal log10 concentration–effect curve. An alternative approach would have been to use IC10 or IC20 values. The latter method was excluded on the grounds that it relies upon setting a smaller margin based on an intercept derived from an unreliable part of the sigmoidal log10 concentration–effect curve, barely above the background noise. However, for compounds that inhibit hNav1.5 channels by less that 50% (and thus do not have an IC50 value), the use of IC10 or IC20 may be of some value.
The basis of this paper was to describe the IC50 : fCmax ratios for a range of drugs. We addressed this challenge by separating the drugs into three categories (Class I anti-arrhythmics, other known QRS prolonging drugs, drugs not known to prolong QRS). However, a better approach would have been to stratify these drugs into groups of varying incidence of causing QRS prolongation (i.e. high, low or no QRS prolongation). Unfortunately, these data are not currently available in the literature.
In this study we employed a single assay to determine blocking potencies at hNav1.5 channels. This has the advantage of a single data set generated using a robust and reliable method, with minimal experimental variation. This contrasts with previous studies, which examined the relationship between hERG IC50 and fCmax– these employed hERG IC50 values quoted in the literature (Webster et al., 2002; Redfern et al., 2003). Nonetheless, the IonWorks™ hNav1.5 channel assay does have the potential to generate variable data in the following respects. Firstly, the assay has a defined experimental design (i.e. hNav1.5 currents are generated at 3 Hz, and compounds are incubated for a fixed 3 min period). It is therefore possible that some compounds may not have reached steady-state inhibition of the hNav1.5 channel, resulting in underestimates of the IC50. Secondly, the hNav1.5 assay has a false negative rate of 8% when compared to assay of the effects on the upstroke of the canine Purkinje fibres action potential (Harmer et al., 2008). This raises the possibility that some compounds may cause more pronounced block of the cardiac Na+ channel when measured using native systems. Finally, although planar patch clamp recordings are considered fit for purpose within a screening context, the data generated using this method do not always correlate with that generated using conventional electrophysiology. For example, lipophilic compounds can adsorb non-specifically to plastic surfaces in the automated systems, potentially causing a rightward shift in compound IC50 values (Dunlop et al., 2008).
A subject that is often raised when considering safety margins is the use of total versus free plasma Cmax values. Drug that is bound to protein is not able to interact with ion channels on the cell surface. Therefore, it seems intuitive to use free drug concentrations. Indeed, several other studies have successfully adopted the free-drug approach when comparing in vitro and clinical data for the purposes of cardiac risk assessment (Kang et al., 2001; Webster et al., 2002; Redfern et al., 2003). The data presented here indicate that the use of total plasma concentrations is no better than free concentrations (see Figure 3B). We have therefore focused on setting hNav1.5 IC50 safety margins based on free plasma concentrations given the clear scientific rationale for using this approach.
Despite the clear limitations of this study, we feel that the data are of sufficient value to support the conclusion reached in this paper.
Conclusions
The principal aim of this study was to generate data that would provide evidence for setting provisional safety margins, and aid in the interpretation of potency data for blockade of hNav1.5 channels, obtained during preclinical safety screening. Based on the data presented here, we have concluded that a safety margin of 30- to 100-fold should be adopted early in the drug discovery process, where no other supporting data are available. However, inhibition of hNav1.5 channels should only be considered as a safety flag – an integrated preclinical assessment of all in vitro and in vivo cardiovascular data is essential in order to fully understand the risk/benefit of a compound, prior to progression into human trials.
Acknowledgments
The authors would like to thank Wendy Dong and Clare Sefton for technical assistance in running the hNav1.5 assay. We would also like to thank Will Redfern for reviewing the manuscript and for helpful discussions throughout the course of this study.
Glossary
Abbreviations
- fCmax
unbound (free) plasma concentration attained during clinical use
- PPB %
human plasma protein binding
- [QRS]free
the free drug concentration that is associated with prolongation of the QRS complex in humans
- TdP
Torsade de Pointes
Conflict of interest
None declared.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Table S1 Class I anti-arrhythmics
Table S2 Drugs associated with QRS complex prolongation
Table S3 Drugs not associated with QRS complex prolongation
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Adesanya CO, Yousuf KA, Co C, Gaur S, Ahmed S, Pothoulakis A, et al. Is wider worse? QRS duration predicts cardiac mortality in patients with right bundle branch block. Ann Noninvasive Electrocardiol. 2008;13:165–170. doi: 10.1111/j.1542-474X.2008.00216.x. DOI: 10.1111/j.1542-474X.2008.00216.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 4th edn. Br J Pharmacol. 2009;158(Suppl 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anonymous. ICH S7A safety pharmacology studies for human pharmaceuticals. 2000. London, 16 November 2000. CPMP/ICH/539/00. ICH S7B. CPMP/ICH/539/00.
- Anonymous. The clinical evaluation of QT/QTc interval prolongation and proarrhythmic potential for non-antiarrhythmic drugs. 2005a. ICH E14. CHMP/ICH/2/04. [PubMed]
- Anonymous. The nonclinical evaluation of the potential for delayed ventricular repolarisation (QT interval prolongation) by human pharmaceuticals. 2005b. ICH S7B. CHMP/ICH/423/02. [PubMed]
- Breidthardt T, Christ M, Matti M, Schrafl D, Laule K, Noveanu M, et al. QRS and QTc interval prolongation in the prediction of long-term mortality of patients with acute destabilised heart failure. Heart. 2007;93:1093–1097. doi: 10.1136/hrt.2006.102319. DOI: 10.1136/hrt.2006.102319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen MX, Helliwell RM, Clare JJ. In vitro profiling against ion channels beyond hERG as an early indicator of cardiac risk. Curr Opin Mol Ther. 2009;11:269–281. [PubMed] [Google Scholar]
- Cordes J, Lib C, Dugasa J, Austin-LaFrancea R, Lightbownc I, Engwalla M, et al. Translation between in vitro inhibition of the cardiac Nav1.5 channel and pre-clinical and clinical QRS widening. J Pharmacol Toxicol Methods. 2009;60:221. [Google Scholar]
- Darpo B. The thorough QT/QTc study 4 years after the implementation of the ICH E14 guidance. Br J Pharmacol. 2010;159:49–57. doi: 10.1111/j.1476-5381.2009.00487.x. DOI: 10.1111/j.1476-5381.2009.00487.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delk C, Holstege CP, Brady WJ. Electrocardiographic abnormalities associated with poisoning. Am J Emerg Med. 2007;25:672–687. doi: 10.1016/j.ajem.2006.11.038. [DOI] [PubMed] [Google Scholar]
- Dunlop J, Bowlby M, Peri R, Vasilyev D, Arias R. High-throughput electrophysiology: an emerging paradigm for ion-channel screening and physiology. Nat Rev Drug Discov. 2008;7:358–368. doi: 10.1038/nrd2552. DOI: 10.1038/nrd2552. [DOI] [PubMed] [Google Scholar]
- Echt DS, Liebson PR, Mitchell LB, Peters RW, Obias-Manno D, Barker AH, et al. Mortality and morbidity in patients receiving encainide, flecainide, or placebo. the cardiac arrhythmia suppression trial. N Engl J Med. 1991;324:781–788. doi: 10.1056/NEJM199103213241201. [DOI] [PubMed] [Google Scholar]
- Freedman RA, Karagounis LA, Steinberg JS. Effects of sotalol on the signal-averaged electrocardiogram in patients with sustained ventricular tachycardia: relation to suppression of inducibility and changes in tachycardia cycle length. J Am Coll Cardiol. 1992;20:1213–1219. doi: 10.1016/0735-1097(92)90380-6. [DOI] [PubMed] [Google Scholar]
- Harmer AR, Abi-Gerges N, Easter A, Woods A, Lawrence CL, Small BG, et al. Optimisation and validation of a medium-throughput electrophysiology-based hNav1.5 assay using IonWorks. J Pharmacol Toxicol Methods. 2008;57:30–41. doi: 10.1016/j.vascn.2007.09.002. DOI: 10.1016/j.vascn.2007.09.002. [DOI] [PubMed] [Google Scholar]
- Horwich T, Lee SJ, Saxon L. Usefulness of QRS prolongation in predicting risk of inducible monomorphic ventricular tachycardia in patients referred for electrophysiologic studies. Am J Cardiol. 2003;92:804–809. doi: 10.1016/s0002-9149(03)00887-7. [DOI] [PubMed] [Google Scholar]
- Jensen HK, Nielsen-Kudsk F. Pharmacokinetics and dynamic effects of diltiazem in the isolated guinea-pig heart. Pharmacol Toxicol. 1988;62:166–171. doi: 10.1111/j.1600-0773.1988.tb01866.x. [DOI] [PubMed] [Google Scholar]
- Kalahasti V, Nambi V, Martin DO, Lam CT, Yamada D, Wilkoff BL, et al. QRS duration and prediction of mortality in patients undergoing risk stratification for ventricular arrhythmias. Am J Cardiol. 2003;92:798–803. doi: 10.1016/s0002-9149(03)00886-5. [DOI] [PubMed] [Google Scholar]
- Kang J, Wang L, Chen XL, Triggle DJ, Rampe D. Interactions of a series of fluoroquinolone antibacterial drugs with the human cardiac K+ channel HERG. Mol Pharmacol. 2001;59:122–126. doi: 10.1124/mol.59.1.122. [DOI] [PubMed] [Google Scholar]
- Krejsa CM, Horvath D, Rogalski SL, Penzotti JE, Mao B, Barbosa F, et al. Predicting ADME properties and side effects: the BioPrint approach. Curr Opin Drug Discov Devel. 2003;6:470–480. [PubMed] [Google Scholar]
- Madias JE. Drug-induced QRS morphology and duration changes. Cardiol J. 2008;15:505–509. [PubMed] [Google Scholar]
- Pollard CE, Valentin JP, Hammond TG. Strategies to reduce the risk of drug-induced QT interval prolongation: a pharmaceutical company perspective. Br J Pharmacol. 2008;154:1538–1543. doi: 10.1038/bjp.2008.203. DOI: 10.1038/bjp.2008.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard CE, Abi Gerges N, Bridgland-Taylor MH, Easter A, Hammond TG, Valentin JP. An introduction to QT interval prolongation and non-clinical approaches to assessing and reducing risk. Br J Pharmacol. 2010;159:12–21. doi: 10.1111/j.1476-5381.2009.00207.x. DOI: 10.1111/j.1476-5381.2009.00207.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redfern WS, Carlsson L, Davis AS, Lynch WG, MacKenzie I, Palethorpe S, et al. Relationships between preclinical cardiac electrophysiology, clinical QT interval prolongation and torsade de pointes for a broad range of drugs: evidence for a provisional safety margin in drug development. Cardiovasc Res. 2003;58:32–45. doi: 10.1016/s0008-6363(02)00846-5. [DOI] [PubMed] [Google Scholar]
- Schulz M, Schmoldt A. Therapeutic and toxic blood concentrations of more than 800 drugs and other xenobiotics. Pharmazie. 2003;58:447–474. [PubMed] [Google Scholar]
- Schwartz M, Patel M, Kazzi Z, Morgan B. Cardiotoxicity after massive amantadine overdose. J Med Toxicol. 2008;4:173–179. doi: 10.1007/BF03161197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seger DL. A critical reconsideration of the clinical effects and treatment recommendations for sodium channel blocking drug cardiotoxicity. Toxicol Rev. 2006;25:283–296. doi: 10.2165/00139709-200625040-00008. [DOI] [PubMed] [Google Scholar]
- Takanaka C, Lee JK, Nonokawa M, Sugiyama T, Yame S. Frequency dependent effects of class I antiarrhythmic agents studied in patients with implanted pacemakers. Pacing Clin Electrophysiol. 1994;17:2100–2105. doi: 10.1111/j.1540-8159.1994.tb03808.x. [DOI] [PubMed] [Google Scholar]
- Tan HL, Bezzina CR, Smits JP, Verkerk AO, Wilde AA. Genetic control of sodium channel function. Cardiovasc Res. 2003;57:961–973. doi: 10.1016/s0008-6363(02)00714-9. [DOI] [PubMed] [Google Scholar]
- Thanacoody HK, Thomas SH. Tricyclic antidepressant poisoning: cardiovascular toxicity. Toxicol Rev. 2005;24:205–214. doi: 10.2165/00139709-200524030-00013. [DOI] [PubMed] [Google Scholar]
- Titier K, Canal M, Deridet E, Abouelfath A, Gromb S, Molimard M, et al. Determination of myocardium to plasma concentration ratios of five antipsychotic drugs: comparison with their ability to induce arrhythmia and sudden death in clinical practice. Toxicol Appl Pharmacol. 2004;199:52–60. doi: 10.1016/j.taap.2004.03.016. [DOI] [PubMed] [Google Scholar]
- Valentin JP. Reducing QT liability and proarrhythmic risk in drug discovery and development. Br J Pharmacol. 2010;159:5–11. doi: 10.1111/j.1476-5381.2009.00547.x. DOI: 10.1111/j.1476-5381.2009.00547.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentin JP, Hammond T. Safety and secondary pharmacology: successes, threats, challenges and opportunities. J Pharmacol Toxicol Methods. 2008;58:77–87. doi: 10.1016/j.vascn.2008.05.007. DOI: 10.1016/j.vascn.2008.05.007. [DOI] [PubMed] [Google Scholar]
- Valentin JP, Pollard C, Lainee P, Hammond T. Value of non-clinical cardiac repolarization assays in supporting the discovery and development of safer medicines. Br J Pharmacol. 2010;159:25–33. doi: 10.1111/j.1476-5381.2009.00530.x. DOI: 10.1111/j.1476-5381.2009.00530.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan Williams EM. Significance of classifying antiarrhythmic actions since the cardiac arrhythmia suppression trial. J Clin Pharmacol. 1991;31:123–135. doi: 10.1002/j.1552-4604.1991.tb03695.x. [DOI] [PubMed] [Google Scholar]
- Webster R, Leishman D, Walker D. Towards a drug concentration effect relationship for QT prolongation and torsades de pointes. Curr Opin Drug Discov Devel. 2002;5:116–126. [PubMed] [Google Scholar]
- Wible BA, Kuryshev YA, Smith SS, Liu Z, Brown AM. An ion channel library for drug discovery and safety screening on automated platforms. Assay Drug Dev Technol. 2008;6:765–780. doi: 10.1089/adt.2008.171. DOI: 10.1089/adt.2008.0171. [DOI] [PubMed] [Google Scholar]
- Yatani A, Akaike N. Blockage of the sodium current in isolated single cells from rat ventricle with mexiletine and disopyramide. J Mol Cell Cardiol. 1985;17:467–476. doi: 10.1016/s0022-2828(85)80051-1. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Furuta T. Tissue penetration properties of macrolide antibiotics–comparative tissue distribution of erythromycin-stearate, clarithromycin, roxithromycin and azithromycin in rats. Jpn J Antibiot. 1999;52:497–503. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.