

Abstract
BACKGROUND AND PURPOSE
Resolution of inflammation is mediated by endogenous molecules with anti-inflammatory and pro-resolving activities and they have generated new possibilities for the treatment of inflammatory diseases. Here, we have investigated the possible anti-hyperalgesic effects of two lipids, aspirin-triggered resolvin D1 (AT-RvD1) and its precursor, 17(R)-hydroxy-4Z,7Z,10Z,13Z,15E,17R,19Z-docosahexaenoic acid (17(R)HDoHE).
EXPERIMENTAL APPROACH
The anti-hyperalgesic effects of both lipid mediators were evaluated, using mechanical and thermal stimuli, at different time-points in adjuvant-induced arthritis in rats. Cytokine levels were measured, and immunohistochemistry and real-time PCR for pro-inflammatory mediators were also performed.
KEY RESULTS
The precursor of resolvin D series, 17(R)HDoHE, given systemically, inhibited the development and the maintenance of mechanical hyperalgesia in acute inflammation. Such effects were likely to be associated with modulation of both NF-κB and COX-2 in dorsal root ganglia and spinal cord. 17(R)HDoHE was also effective against sub-chronic pain. Unexpectedly, repeated treatment with 17(R)HDoHE did not modify paw and joint oedema in the sub-chronic model, while joint stiffness was prevented. Notably, AT-RvD1 exhibited marked anti-hyperalgesic effects in acute inflammation when given systemically. The efficacy of long-term treatment with either 17(R)HDoHE or AT-RvD1 was partly related to decreased production of TNF-α and IL-1β in rat hind paw.
CONCLUSIONS AND IMPLICATIONS
Our findings provide fresh evidence for the anti-hyperalgesic properties of 17(R)HDoHE and its pro-resolution metabolite AT-RvD1. Such lipid mediators might be useful for treating pain associated with acute or chronic inflammation.
LINKED ARTICLE
This article is commented on by Xu and Ji, pp. 274–277 of this issue. To view this commentary visit http://dx.doi.org/10.1111/j.1476-5381.2011.01348.x
Keywords: RvD series precursor, 17(R)HDoHE, AT-RvD1, inflammatory pain, arthritis
Introduction
Rheumatoid arthritis (RA) is a progressive and destructive chronic autoimmune disease of the joints, characterized by inflammation of the synovial membranes, joint stiffness and pain. Synovial inflammation is elicited by the infiltration of cells of the immune system into the joint and the release of pro-inflammatory and pro-nociceptive mediators (Firestein, 2003; Müller-Ladner et al., 2005; Brennan and McInnes, 2008). Joint hyperalgesia is expressed in response to the inflammatory mediators, which may sensitize primary nociceptive fibres innervating the joints (Schaible and Grubb, 1993; Schaible et al., 2002; Pinto et al., 2010). During the last two decades, great efforts have been made to determine the mechanisms underlying the physiopathology of RA and also to develop new therapies to treat the devastating outcomes of this disease (Quan et al., 2008; van Vollenhoven, 2009). Current therapy includes the disease-modifying anti-rheumatic drugs, such as methotrexate, and the biological agents, such as anti-TNF-α inhibitors and the newly approved anti-IL-6 receptor agent, tocilizumab. Moreover, non-steroidal anti-inflammatory drugs and the selective inhibitors of the COX-2 enzyme are also used for the treatment of joint pain. However, all these drugs have limited efficacy in most patients, and induce several side effects, such as gastrointestinal bleeding and/or perforation (Quan et al., 2008; González et al., 2010). Accordingly, new therapies with increased efficacy and without or with fewer side effects are urgently required for the treatment of joint pain.
Interesting potential candidates for such analgesics are the endogenous lipid mediators, such as lipoxins, resolvins, protectins and the newly described group of maresins, derived from ω-3-fatty acids (Sommer and Birklein, 2010), the latter demonstrating beneficial effects in pathological states, including Crohn's disease (Belluzzi et al., 1996), coronary heart disease (GISSI-Prevenzione Investigators, 1999) and sudden cardiac death (Albert et al., 2002). In addition, it was shown that i.v. administration of ω-3 fatty acids leads to clinical improvement of patients with RA (Leeb et al., 2006). The two main ω-3 fatty acids present in fish oil are eicosapentaenoic acid (EPA, C20:5) and docosahexaenoic acid (DHA, C22:6).
The resolvins (resolution phase interaction products) are a family of lipid mediators derived from both EPA and DHA (Serhan et al., 2000; 2002; Hong et al., 2003), denoted the E series (RvE) and the D series (RvD) resolvins (Serhan et al., 2002). DHA is the precursor for two groups of resolvins, referred to as the 17S- and 17R-resolvin D series, which are produced by different biosynthetic routes during the resolution phase of inflammation (Serhan et al., 2002; Hong et al., 2003). Endogenous DHA is converted in vivo via lipoxygenase (LOX)-catalysed mechanisms to the 17S-hydroxy-containing series of four resolvins, known as resolvin D1 to resolvin D4 (Serhan et al., 2002; Hong et al., 2003). Specifically, RvD1 biosynthesis involves sequential oxygenations by 15-LOX and 5-LOX. However, in the case of aspirin treatment, aspirin-acetylated COX-2 generates 17R-hydroxy-DHA, which on subsequent oxygenation by 5-LOX results in production of 17-epi-RvD1, also known as aspirin-triggered resolvin D1, (AT-RvD1) (Serhan et al., 2002; Serhan and Clish, 2003; Serhan, 2004). Recently, the structural elucidation of RvD1 and AT-RvD1 was confirmed by total organic synthesis and their complete stereochemistry were established as 7S,8R,17S-trihydroxy-4Z,9E,11E,13Z,15E,19Z-docosahexaenoic acid and 7S,8R,17R-trihydroxy-4Z,9E,11E,13Z,15E,19Z-docosahexaenoic acid respectively (Serhan et al., 2002; Sun et al., 2007).
The resolvins are not only anti-inflammatory, but they also promote resolution of the inflammation, back to the non-inflamed state (Gilroy et al., 2004; Bannenberg et al., 2005; Serhan, 2007; Serhan, 2009; Serhan et al., 2009). Initially, Levy et al. (2001) have demonstrated a lipid mediator class switching during the spontaneous resolution of acute inflammatory response. Furthermore, RvDs were demonstrated to display anti-inflammatory actions in an animal model of kidney ischaemia/reperfusion (Duffield et al., 2006). Specifically, both RvD1 and AT-RvD1 mediators limit the transendothelial migration (Serhan et al., 2002; Sun et al., 2007) and infiltration (Kasuga et al., 2008) of polymorphonuclear (PMN) leukocytes in vivo and regulate leukocyte trafficking as well as clearance of neutrophils from mucosal surfaces (Campbell et al., 2007). More recently, RvD1 was shown to diminish leukocyte and prostanoid generation in a murine model of oxidative stress, a common feature of the inflammatory process (Filep, 2009; Spite et al., 2009).
Most of the work addressing the potential effects of pro-resolving lipid mediators has been concerned with inflammatory processes. Recent reports suggest that the resolvins RvE1 and RvD1 could modulate the nociceptive state that usually accompanies inflammatory disease (Svensson et al., 2007; Bang et al., 2010; Xu et al., 2010). Therefore, considering the release of inflammatory mediators, such as cytokines and chemokines, which contribute to hyperalgesia, the major goal of this study was to evaluate the potential effect of the precursor of the resolvin D series, 17(R)HDoHE, as well as AT-RvD1, in modulating inflammatory pain and disease parameters in a rat model of arthritis. Efforts have also been made to investigate some of the mechanisms through which these mediators exert their anti-hyperalgesic actions.
Methods
Animals
All animal care and experimental procedures used in the present study followed the National Institute of Health Animal Care Guidelines (NIH publications) and were approved by the Ethics Committee of the Universidade Federal de Santa Catarina (Protocol Number 043/CEUA/PRPe/2008). The number of animals and intensity of noxious stimuli used were the minimum necessary to demonstrate consistent effects of the drug treatment. Male Hannover rats (180–250 g) kept under controlled temperature (22 ± 2°C) and humidity (60–80%) under a 12:12 h light–dark cycle were housed. Food and water were provided ad libitum, except during the experiments. The animals were acclimatized to the laboratory for at least 1 h prior to experimental procedures.
Complete Freund's Adjuvant (CFA)-induced inflammatory pain and arthritis
The protocol was conducted as previously described (Stein et al., 1988). Briefly, animals were anesthetized with a mixture of isoflurane-oxygen (2.5%–2.5%) and received an intraplantar (i.pl.) injection of CFA emulsified in phosphate-buffered saline (PBS). CFA (1 mg·mL–1 heat-killed and dried M. tuberculosis, each mL of vehicle containing 0.85 mL paraffin oil plus 0.15 mL mannide monooleate) and PBS were emulsified in a 1:1 ratio by the syringe-extrusion method with two syringes connected by a three-way stopcock until a stable emulsion was obtained (∼5 min). Each animal received a total of 200 µL in the right hind paw (ipsilateral). This model of adjuvant-induced arthritis (AIA) allows us to initially evaluate the acute inflammatory pain, resulting in a progressive diffuse inflammatory reaction characteristic of arthritis (Bendele, 2001; Nagakura et al., 2003).
Assessment of pain behaviour
Hind paw withdrawal response induced by von Frey hairs
Mechanical hyperalgesia induced by CFA was evaluated according to the method previously described (Nagakura et al., 2003), with some modifications. Rats were placed individually in clear Plexiglas boxes (13.8 cm × 18.0 cm × 68.2 cm) on elevated wire mesh platforms (23.0 cm × 39.8 cm × 72.7 cm) to allow access to the plantar surface of both hind paws. The animals were acclimatized for at least 1 h prior to behavioural testing. The paw withdrawal response frequency (%), was quantified following 10 applications of 8.0 g von Frey Hairs (Stoelting, USA) with a duration of ∼3 s each. Stimuli were applied on the plantar surface of each hind paw separately. The nociceptive responses were evaluated at different times (1 to 24 h, 72 h, 14 and 30 days), following CFA injection. All groups were evaluated before CFA injection in order to determine the baseline mechanical thresholds.
Thermal threshold evaluation
Thermal hyperalgesia of the hind paw was assessed using the plantar test (Ugo Basile, Italy), according to the methodology described already (Hargreaves et al., 1988). Briefly, the rats were acclimatized to an apparatus consisting of individual Perspex boxes on an elevated glass table. An infrared radiant heat (40 W) source was directed to the plantar surface of the hind paw, and the time spent to remove the hind paw was defined as the paw withdrawal latency. The cut-off point was set at 20 s in order to prevent tissue damage. The apparatus was calibrated to give a paw withdrawal latency of approximately 15 s in naïve rats.
Evaluation of disease parameters: paw oedema, joint oedema and joint stiffness
Paw oedema and joint diameter
Oedema of both right (ipsilateral) and left (contralateral) hind paws were measured by means of a plethysmometer (Ugo Basile, Italy) before and at several times after the i.pl. injection of CFA during the period of study (30 days). Moreover, the tibio-tarsal joint oedema was measured by means of a manual caliper oriented in a mediolateral plane across the joint line (McDougall et al., 2009), with minimal compression of the joint. This parameter was evaluated in triplicate, and the mean value was used as an index of joint oedema.
Joint stiffness
The joint stiffness score was evaluated according to the method previously described (Nagakura et al., 2003). The animals were restrained by the experimenter with one hand, and the bending and extension movement of the ankle joint (once in each direction) was conducted with the other hand. A score was attributed as the following: score 0, no restriction of movement; score 1, a restriction of movement in bending or extension; score 2, restrictions of movement in bending and extension.
Immunohistochemical studies
Rats were deeply anesthetized with 7% chloral hydrate (8 mL·kg–1; i.p.) and perfused with fresh 4% paraformaldehyde in 0.2 M sodium phosphate, pH 7.4. Immunohistochemical analysis was performed on paraffin-embedded spinal cord (L4–L5) and in the dorsal root ganglion (DRG; L4–L5) (5 µm) sections using polyclonal rabbit anti-COX-2 (1:500) and monoclonal mouse anti-phospho-p65 NF-κB (, 1:50) antibodies, according to the method described previously with some modifications (Vitor et al., 2009). After quenching of endogenous peroxidase with 1.5% hydrogen peroxide in methanol (v/v) for 20 min, high-temperature antigen retrieval was performed by immersion of the slides in a water bath at 95 to 98°C in 10 mM–1 trisodium citrate buffer, pH 6.0, for 45 min. The slides were then processed using the VECTASTAIN Elite ABC reagent, according to the manufacturer's instructions. After the appropriate biotinylated secondary antibody, immune complexes were visualized with 0.05% 3,3'-diaminobenzidine tetrahydrochloride + 0.03% H2O2 in PBS (for exactly 1 min), the reaction was stopped by means of washing the slides in water and counterstained with Harris's haematoxylin. Besides staining untreated animals as negative controls, sections were incubated with isotype-matched irrelevant primary antibodies, or the primary antibody was omitted. Despite antigen retrieval, these controls resulted in little or no staining. Images were acquired using a Sight DS-5 M-L1 digital camera connected to an Eclipse 50i light microscope (both from Nikon, USA). Settings for image acquisition were identical for control and experimental tissues. Five ocular fields per section (five mice per group) at 400 × magnification, of the DRG and superficial (lamina I and II) and neck region (lamina V and VI) of the dorsal horn of spinal cord regions associated primarily with the termination of nociceptive primary afferents were captured and a threshold optical density was obtained using the NIH ImageJ 1.36b imaging software (NIH, USA). The nucleus proprius (lamina III and IV) and ventral horn of spinal cord were not evaluated. For all analysis, total pixels intensity was determined, and data was expressed as optical density.
RNA extraction and real-time PCR
Total RNA from spinal cord (L4–L5) and DRG (L4–L5) was extracted using the TRizol® protocol, and its concentration was determined using a NanoDrop 1100 (NanoDrop Technologies, USA). A reverse transcription assay was performed as described in the M-MLV Reverse Transcriptase protocol, according to the manufacturer's instructions. cDNA (300 ng) was amplified in triplicate using the TaqMan® Universal PCR Master Mix Kit with specific TaqMan Gene Expression target genes, the 3' quencher MGB and FAM-labelled probes for rat COX-2 (Rn00568225_m1), NF-κB (Rn01399583) and β-actin (Rn00667869_m1) that was used as an endogenous control for normalization. The PCR reactions were performed in a 96-well Optical Reaction Plate (Applied Biosystems, USA). The thermocycler parameters were as follows: 50°C for 2 min, 95°C for 10 min, 50 cycles of 95°C for 15 s and 60°C for 1 min. Expression of the target genes was calibrated against conditions found in control animals (i.e. animals that had received vehicle).
Determination of cytokine levels
Levels of the pro-inflammatory cytokines TNF-α and IL-1β were determined in spinal cord samples (lumbar segments L4–L5) and in the hind paw tissue as previously described (Manjavachi et al., 2010). Briefly, samples were homogenized in phosphate buffer containing 0.05% Tween 20, 0.1 mM phenylmethylsulphonyl fluoride, 0.1 mM benzethonium chloride, 10 mM ethylene diamine tetracetic acid and 20 UI aprotinin A. The homogenate was centrifuged at 7000×g for 10 min and supernatants were stored at −70°C until further analysis. Cytokine levels were evaluated using an elisa kit according to the manufacturer's recommendations (R&D Systems, USA).
General protocols of treatment
The following series of experiments were designed to evaluate the potential effect of 17(R)HDoHE and AT-RvD1 on inflammatory pain and disease parameters, as well as the possible mechanisms of action in the AIA model in rats. (i) To determine the effect of the lipid precursor on the genesis of inflammatory hyperalgesia, the animals were pre-treated with 17(R)HDoHE (300 ng/i.p.), 30 min before the induction of AIA. (ii) In order to evaluate the effect of 17(R)HDoHE or AT-RvD1, after hyperalgesia had been already established, the animals were treated at different periods of the experimental model – development of hyperalgesia (the third day after AIA) – 17(R)HDoHE (300 ng/i.p.) and AT-RvD1 (100 or 300 ng/i.p.); sub-chronic inflammatory state (14th day after AIA) – 17(R)HDoHE (300, 600 or 900 ng/i.p.) and chronic inflammatory state (30th day after AIA) – 17(R)HDoHE (300 or 600 ng/i.p.). (iii) To assess the effects of repeated treatment with 17(R)HDoHE and AT-RvD1 to modulate the development of inflammatory hyperalgesia and pro-inflammatory mediators production, the animals were treated with 17(R)HDoHE (300 ng/i.p., given for 5 days, once a day) and AT-RvD1 (100 ng/i.p., given for 4 days, twice a day), starting 3 days after AIA induction. (iv) In order to further evaluate the possible mechanisms involved in the analgesic actions of 17(R)HDoHE, we next assessed whether systemic treatment with 17(R)HDoHE (300 ng/i.p., 3 days after AIA induction) was able to decrease the activation of NF-κB and COX-2 in the DRG and the spinal cord. For this reason, 4 h after the last treatment, the DRG and lumbar spinal cord were collected and immunohistochemically evaluated as described above. (v) We also evaluate the possible effect of 17(R)HDoHE on mRNA levels of both NF-κB and COX-2 in the spinal cord and DRG. For this purpose, 3 days after AIA, animals were treated with 17(R)HDoHE (300 ng/i.p.) and 4 h after treatment, the DRG and lumbar spinal cord were collected and processed for real-time PCR assay. (vi) In another set of experiments, we compared the analgesic efficacy of 17(R)HDoHE with standard, clinically used analgesics: indomethacin (5 mg·kg–1, i.p.), morphine (0.5 mg·kg–1, s.c.), gabapentin (70 mg·kg–1, p.o.) and dexamethasone (5 mg·kg–1, s.c.) in AIA induced hyperalgesia. All experiments were conducted in a double-blinded manner.
Statistical analysis
The results are expressed as mean ± SEM from 5–6 animals. Behavioural data were analysed by two-way analysis of variance (anova) with repeated measures, followed by Bonferroni's post hoc test. The non-parametric Kruskal–Wallis test was employed for analysing ordinal parameters. For elisa, immunohistochemistry and real-time PCR data, one-way anova followed by Bonferroni's post hoc test was used to determine the differences among groups. The accepted level of significance for the tests was *P < 0.05. All tests were performed using the GraphPad® 4 Software (USA).
Materials
17(R)HDoHE and AT-RvD1 were purchased from Cayman Chemicals (Ann Arbor, USA). CFA, dexamethasone, indomethacin, gabapentin, haematoxylin, eosin, hydrogen peroxide, tetramethylbenzidine, Tween-20, Tween-80, paraffin, PBS, sodium citrate, phenylmethylsulphonyl fluoride, paraformaldehyde, leupeptin, aprotinin, ethylene diamine tetracetic acid and benzethonium chloride were all purchased from Sigma Chemical Company (USA). Rabbit polyclonal anti-COX-2 was obtained from Cell Signaling Technology (USA). Rabbit polyclonal anti-phospho-p65 NF-κB was obtained from Santa Cruz Biotech. Inc. (USA). Rat TNF-α and IL-1β DuoSet kits were obtained from R&D Systems (USA). Morphine, sodium chloride and sodium phosphate were obtained from Merck (Germany). Acetone, xylol, ethyl alcohol and methyl alcohol were purchased from LabSynth (Brazil). Citric acid and sodium citrate were purchased from Merck (Brazil). Chloral hydrate was purchased from Vetec (Brazil). Harris's haematoxylin solution was purchased from Merck (Germany). 3,3'-Diaminobenzidine tetrahydrochloride and streptavidin-biotin peroxidase were obtained from Dako Cytomation (USA). Primers and probes for rat COX-2, NF-κB and β-actin were purchased from Applied Biosystems (UK). All receptors and channel nomenclature follow Alexander et al.(2009).
Results
Effect of pretreatment with 17(R)HDoHE on the genesis of mechanical and thermal hyperalgesia in AIA in rats
Our model of AIA is one of the most commonly used animal models to assess the preclinical efficacy of potential new agents to treat arthritis (Hegen et al., 2008). In order to evaluate the potential of 17(R)HDoHE in preventing the induction of mechanical and thermal hyperalgesia evoked by the acute inflammatory pain in response to AIA, rats were pretreated with 17(R)HDoHE (300 ng/i.p.), 30 min before the induction of AIA. The choice of the dose for each drug was based on pilot experiments (data not shown) and on previous data described in the literature (Dornelles et al., 2009). In the present study, i.pl. administration of CFA into the right hind paw markedly increased the mechanical (Figure 1A) and thermal hyperalgesia (Figure 1D) as early as 1 h after AIA induction (P < 0.05). Nonetheless, 1 h after pretreatment with 17(R)HDoHE, the animals displayed a significant reduction of the mechanical hyperalgesia, in the ipsilateral paw, compared with the control group. Moreover, 17(R)HDoHE exhibited its maximum effect 4 h after treatment and lasted for up to 6 h (Figure 1A,B). However, the pre-treatment with 17(R)HDoHE produced a moderate and transient reduction of the thermal hypersensitivity only at 2 h (Figure 1D,E). The mechanical and thermal threshold of the contralateral paw (Figure 1C,E,F) and the baseline threshold (data not shown) were not altered by pretreatment with 17(R)HDoHE.
Figure 1.
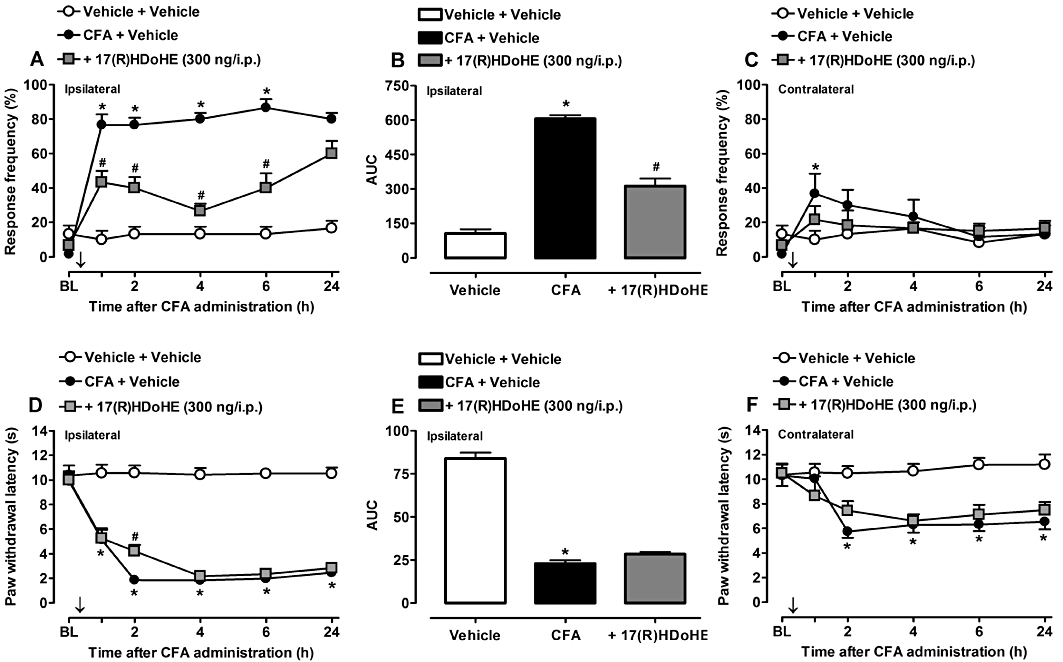
Pretreatment with the resolvin D series precursor, 17(R)HDoHE (300 ng/i.p., 30 min before), prevented the induction of mechanical (A,B) and thermal (D,E) hyperalgesia in ipsilateral hind paws of rats with adjuvant-induced arthritis (AIA). (C) and (F) represent mechanical and thermal hyperalgesia, respectively, in the contralateral hind paws of rats with AIA. Data are presented as the mean ± SEM of five to six animals per group. *P < 0.05 and #P < 0.05 are significantly different from the vehicle and complete Freund's Adjuvant (CFA) groups respectively. The arrow (↓) above X-axis represents the treatment with 17(R)HDoHE. BL, baseline.
Effect of post-treatment with 17(R)HDoHE on the development of mechanical and thermal hyperalgesia in AIA rats
As the resolvin D series precursor 17(R)HDoHE clearly prevented the genesis of mechanical, but only slightly affected that of the thermal hyperalgesia induced by AIA, we next attempted to evaluate whether 17(R)HDoHE could also inhibit the same parameters in established inflammatory pain. Accordingly, 3 days after AIA induction, rats were treated with a single dose of 17(R)HDoHE (300 ng/i.p.), and mechanical and thermal hyperalgesia were evaluated as described previously. Interestingly, this therapeutic treatment with 17(R)HDoHE significantly inhibited the mechanical hyperalgesia in the ipsilateral paw from 1 h to 12 h, with an inhibition of about 59%, compared with AIA group (P < 0.05, P < 0.001) (Figure 2A,B), demonstrating the clinical relevance of treatment with 17(R)HDoHE after hyperalgesia is already established. Conversely, treatment with 17(R)HDoHE did not inhibit thermal hyperalgesia in ipsilateral hind paw (Figure 2D,E). Furthermore, the treatment with 17(R)HDoHE also failed to significantly modify the mechanical and thermal hyperalgesia in contralateral paws (Figure 2C,F).
Figure 2.
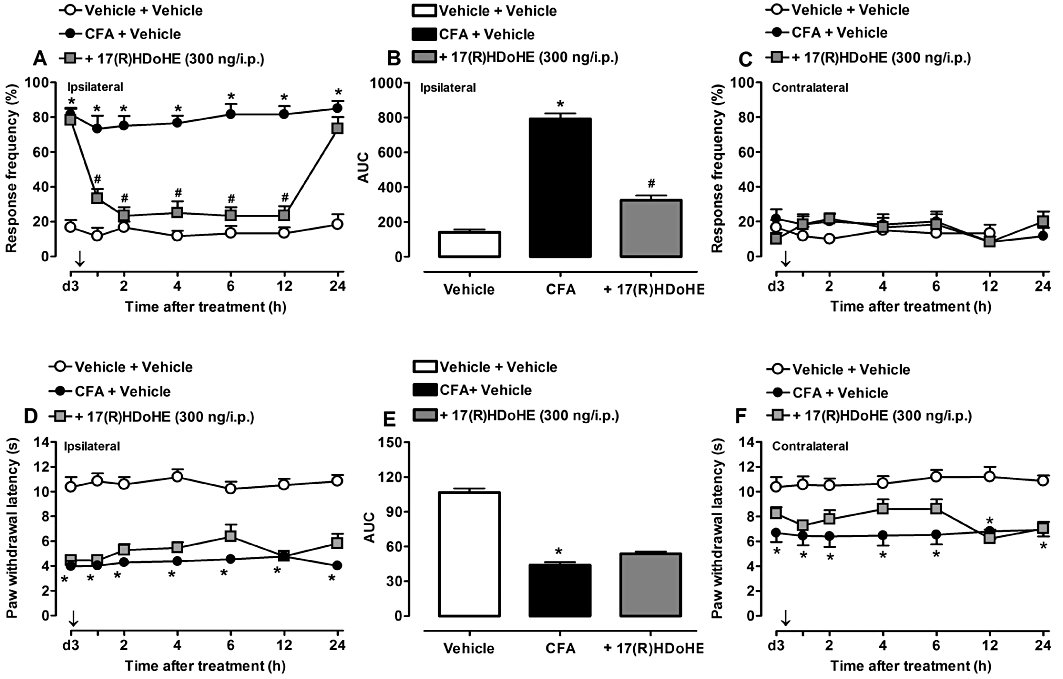
Post-treatment with 17(R)HDoHE (300 ng/i.p., 3 days after), almost abolished the mechanical (A,B) but not thermal (D,E) hyperalgesia in ipsilateral hind paws of rats with adjuvant-induced arthritis (AIA). (C) and (F) represent mechanical and thermal hyperalgesia, respectively, in the contralateral hind paws of rats with AIA. Data are presented as the mean ± SEM of five to six animals per group. *P < 0.05 and #P < 0.05 are significantly different from the vehicle and complete Freund's Adjuvant (CFA) groups respectively. The arrow (↓) above X-axis represents the treatment with 17(R)HDoHE.; d3, day 3 after AIA induction.
Effect of classic clinically used drugs on the development of mechanical hyperalgesia in AIA rats
In order to compare the effect of the post-treatment with the precursor 17(R)HDoHE with classic drugs used in clinic for the treatment of painful states, 3 days after AIA induction, the rats were treated with a single dose of 17(R)HDoHE (300 ng/i.p.), or the non-selective COX inhibitor indomethacin (5 mg·kg–1, i.p.), morphine (0.5 mg·kg–1, s.c.), an opioid analgesic drug, gabapentin (70 mg·kg–1, p.o.), an anti-epileptic drug, or dexamethasone (5 mg·kg–1, s.c.), a glucocorticoid agent. Injection of CFA into the rat hind paw induced a significant reduction in nociceptive threshold, which was strongly inhibited by post-treatment with the precursor 17(R)HDoHE, with an inhibition of 72 ± 3% (Table 1). However, post-treatment with indomethacin, morphine and gabapentin resulted in less inhibition of mechanical hyperalgesia, inhibitions of 35 ± 5%, 34 ± 4% and 42 ± 4% respectively (Table 1). Conversely, the treatment with dexamethasone (5 mg·kg–1, s.c.) caused a mild but statistically not significant (12 ± 3%) reduction in the mechanical hyperalgesia induced by adjuvant arthritis (Table 1). All drugs were administered 1 h before the measurement of pain parameters, with exception of dexamethasone, which was given 4 h before evaluation.
Table 1.
Effect of 7(R)HDoHE and different classes of drugs in a rat model of AIA
| Drug | Mechanism of action | Dose | Inhibition (%)a |
|---|---|---|---|
| 17(R)HDoHE | Unknown | 300 ng/i.p. | 72 ± 3* |
| Indomethacin | Cyclooxygenase inhibitor | 5 mg·kg–1 (i.p.) | 35 ± 5* |
| Morphine | Opioid analgesic | 0.5 mg·kg–1 (s.c.) | 34 ± 4* |
| Gabapentin | Anti-epileptic | 70 mg·kg–1 (p.o.) | 42 ± 4* |
| Dexamethasone | Glucocorticoid | 5 mg·kg–1 (s.c.) | 12 ± 3 |
Asterisk denote significant differences from CFA-treated group (*P < 0.05).
Mean ± SEM (n = 5–6 rats/group). The inhibition is given as the difference (in percentage) between the mean area under the time-response curve (1–6 h values) of the responses in the drug-treated group and in relation to the CFA-treated group. All drugs were administered on the third day after induction of adjuvant arthritis. 17(R)HDoHE, indomethacin, morphine and gabapentin were administered 1 h before and dexamethasone 4 h before the evaluation of pain response.
Effect of post-treatment with 17(R)HDoHE on the mechanical and thermal hyperalgesia in a sub-chronic and chronic inflammatory state in AIA rats
Here, we addressed the possible effects of 17(R)HDoHE in the mechanical and thermal hyperalgesia in a sub-chronic inflammatory pain state. To achieve this, we decided to treat the animals 14 days after AIA induction, when the animals started to present signs of systemic inflammation of the joints (i.e. joint stiffness and paw oedema in both ipsi- and contralateral hind paws), that is, parameters related to development of the arthritis. Fourteen days after AIA, the animals were treated with a single dose of 17(R)HDoHE (300, 600 or 900 ng/i.p.) and both mechanical and thermal hyperalgesia were evaluated. As shown in Figure 3A, the low dose of 17(R)HDoHE (300 ng/i.p.) was not able to inhibit the mechanical hyperalgesia induced by AIA. On the other hand, when rats were treated with higher doses (600 and 900 ng/i.p.) a significant reduction of mechanical hyperalgesia was observed for up to 6 h after treatment (P < 0.05) (Figure 3A,B). None of the tested doses of 17(R)HDoHE (300, 600 and 900 ng/i.p.) was effective in inhibiting the mechanical hyperalgesia in the contralateral hind paw (Figure 3C). Also, thermal hyperalgesia evaluated 4 h after 17(R)HDoHE administration was not significantly inhibited by any of the tested doses of 17(R)HDoHE (Figure 3D). Finally, when evaluated in a established chronic inflammatory state (i.e. 30 days after AIA induction), 17(R)HDoHE (300 and 600 ng/i.p.) was not effective in inhibiting either mechanical or thermal hyperalgesic responses (Figure 3E,F).
Figure 3.
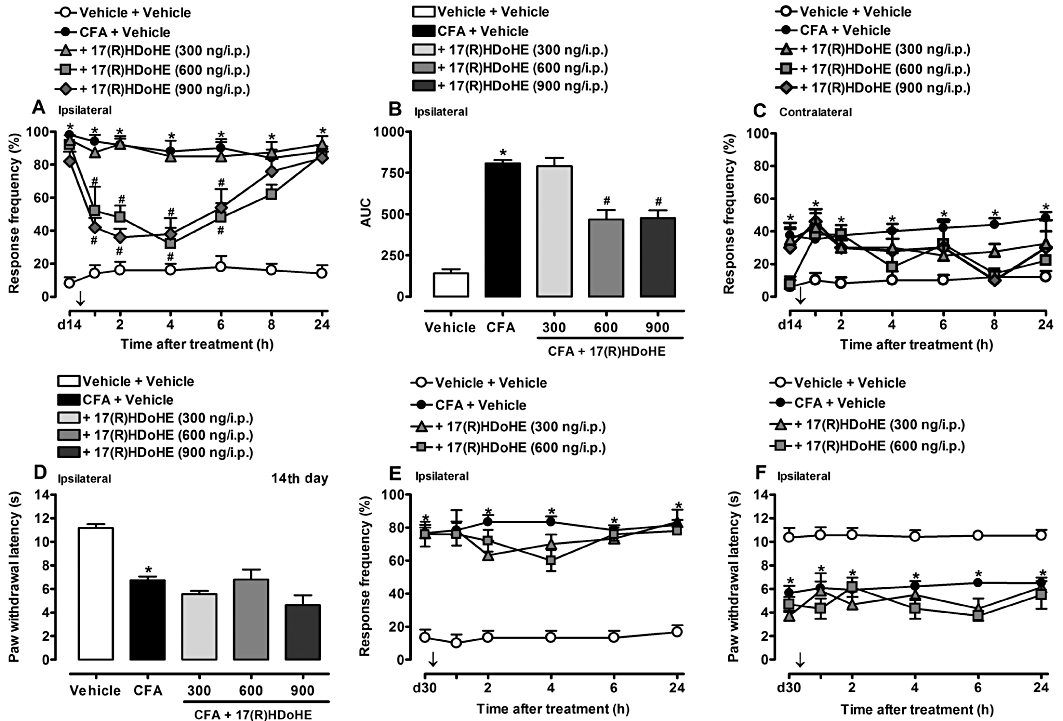
Post-treatment with 17(R)HDoHE (300, 600 or 900 ng/i.p., 14 days after), diminished mechanical (A,B) but not thermal (D) hyperalgesia in ipsilateral hind paws of rats with adjuvant-induced arthritis (AIA). (C) Represents the mechanical hyperalgesia in the contralateral hind paws of rats with AIA. Post-treatment with 17(R)HDoHE (300 or 600 ng/i.p., 30 days after) did not alter mechanical (E) nor thermal (F) hyperalgesia in ipsilateral hind paws of rats with AIA. Data are presented as the mean ± SEM of five to six animals per group. *P < 0.05 and #P < 0.05 are significantly different from the vehicle and complete Freund's Adjuvant (CFA) groups respectively. The arrow (↓) above X-axis represent the treatment with 17(R)HDoHE.; d14, day 14 after AIA induction; d30, day 30 after AIA induction.
Effect of 17(R)HDoHE on clinical signs of arthritis
Animals treated with a single dose of 17(R)HDoHE (300 ng/i.p.) 30 min before, 3 days and 30 days after AIA induction did not display any significant alterations in paw oedema, joint oedema and joint stiffness of both ipsi- and contralateral hind paws (data not shown). However, when animals received 17(R)HDoHE (300 ng/i.p.) for 6 consecutive days beginning on day 14 after AIA induction, the development of joint stiffness (Figure 4A), but not paw and joint oedema (Figure 4C,E), was significantly attenuated in comparison with AIA group (P < 0.05). No significant alterations were detected in the contralateral paws (Figure 4B,D,E).
Figure 4.
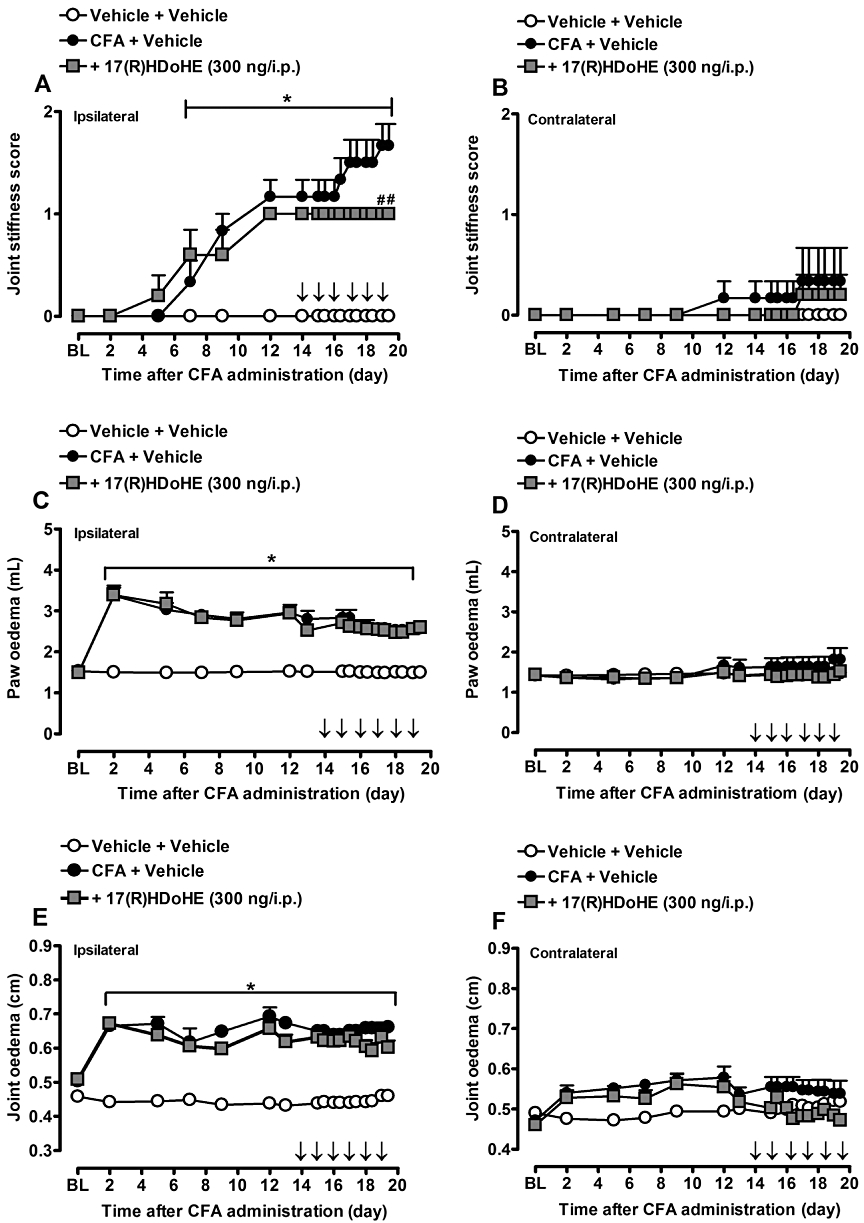
Repeated post-treatment with 17(R)HDoHE (300 ng/i.p., 14 days after AIA induction, for 6 days, once a day), prevented joint stiffness score (A) but not paw (C) and joint oedema (E) increase in ipsilateral hind paws of rats with adjuvant-induced arthritis (AIA). (B), (D) and (F) represent joint stiffness score, paw and joint oedema, respectively, of contralateral hind paw of rats with AIA. Data are presented as the mean ± SEM of five to six animals per group. *P < 0.05 and #P < 0.05 are significantly different from the vehicle and complete Freund's Adjuvant (CFA) groups respectively. The arrow (↓) above X-axis represents the treatment with 17(R)HDoHE. BL, baseline.
Effect of post-treatment with AT-RvD1 on the mechanical hyperalgesia in AIA rats
AT-RvD1 is one of the resolvins of the D series that can be produced by the metabolism of the precursor 17(R)HDoHE (Serhan et al., 2002). Thus, we addressed the potential of AT-RvD1 to inhibit the development of mechanical hyperalgesia in the established acute inflammatory state (the third day after AIA induction). As shown in Figure 5A,B, a single dose of AT-RvD1 (100 or 300 ng/i.p.) significantly inhibited the hyperalgesia caused by i.pl. injection of CFA for up to 6 h (P < 0.05). The treatment with AT-RvD1 did not significantly alter the baseline threshold of mechanical nociception (data not shown).
Figure 5.
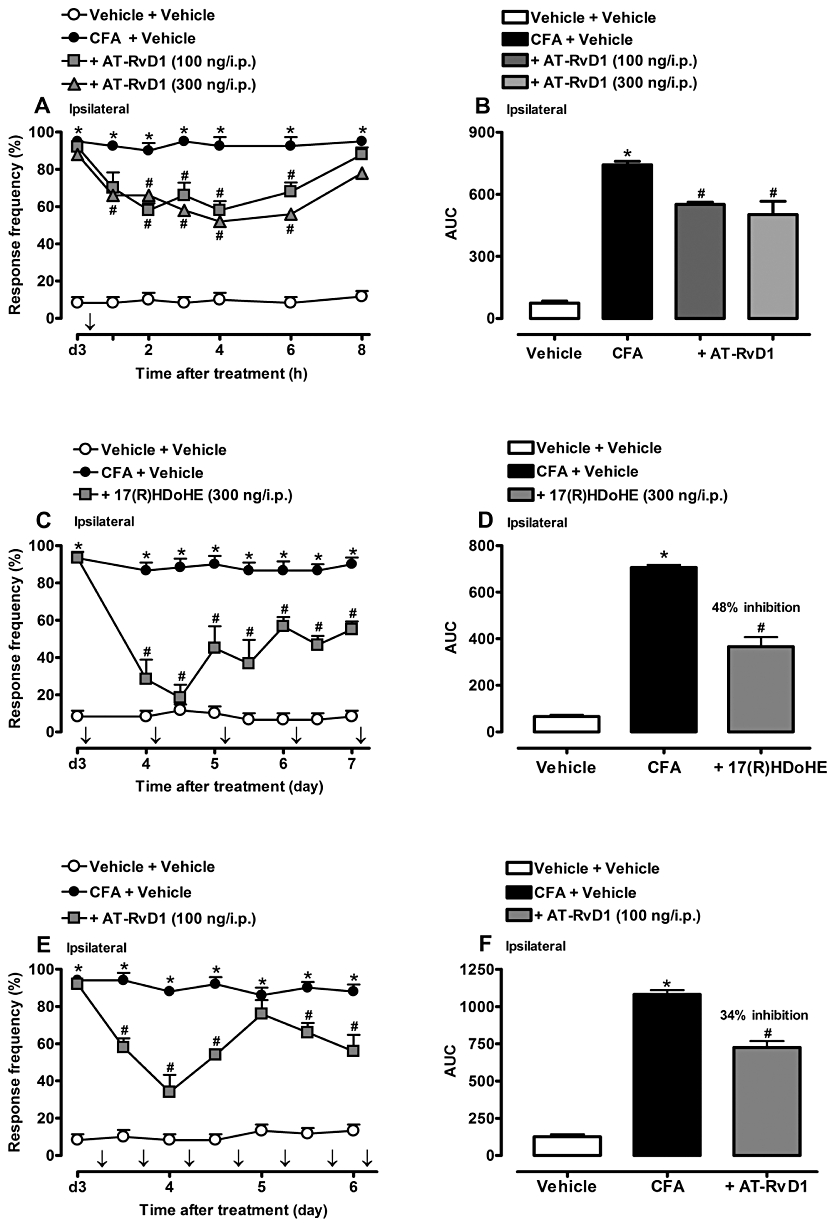
Post-treatment with AT-RvD1) (100 or 300 ng/i.p., 3 days after) (A,B), with the resolvins D series precursor 17(R)HDoHE (300 ng/i.p., 3 days after, for 5 days) (C,D) or with AT-RvD1 (100 ng/i.p., 3 days after, for 4 days) (E,F) partially inhibited mechanical hyperalgesia (ipsilateral hind paw) induced by adjuvant-induced arthritis (AIA). Data are presented as the mean ± SEM of four to six animals per group. *P < 0.05 and #P < 0.05 are significantly different from the vehicle and complete Freund's Adjuvant (CFA) groups respectively. The arrow (↓) above X-axis represent the treatment with AT-RvD1 or 17(R)HDoHE.; d3, day 3 after AIA induction.
Effect of repeated treatment with 17(R)HDoHE or AT-RvD1 on the mechanical hyperalgesia in AIA rats
In the next series of experiments, we assessed the ability of repeated treatment with 17(R)HDoHE and AT-RvD1 to inhibit the development of inflammatory hyperalgesia, as assessed 3 days after AIA induction. As shown in Figure 5C, systemic treatment with 17(R)HDoHE (300 ng/i.p.; given once a day, for 5 days) inhibited the mechanical hyperalgesia induced by i.pl. CFA, an effect that lasted for up to 6 h (Figure 5C) with a reduction of 48% (P < 0.05) (Figure 5D). In addition, repeated treatment with AT-RvD1 (100 ng/i.p.; given for 4 days, twice a day) markedly inhibited the development of inflammatory hyperalgesia induced by i.pl CFA-injection, as assessed 3 days after AIA induction, an effect that lasted for up to 6 h (Figure 5E) with an inhibition of 34% (P < 0.05) (Figure 5F).
Effect of repeated treatment with 17(R)HDoHE or AT-RvD1 on the levels of the pro-inflammatory cytokines in spinal cord and hind paw tissue of AIA rats
By the end of the repeated treatment with 17(R)HDoHE (300 ng/i.p.; for 5 days; once a day) or AT-RvD1 (100 ng/i.p.; for 4 days; twice a day), as described above, the spinal cord and paw tissue of both hind paws (ipsi- and contralateral) were collected and TNF-α and IL-1β levels were determined by elisa assay. As expected, AIA resulted in a significant enhancement in the levels of both TNF-α and IL-1β in the ipsilateral hind paw tissue (P < 0.05) (Figure 6A–D). Repeated treatment with 17(R)HDoHE resulted in a significant decrease of both TNF-α (Figure 6A) and IL-1β (Figure 6B) in the ipsilateral hind paw tissue (P < 0.05), but not in the spinal cord (Figure 6E,F). Moreover, treatment with AT-RvD1 was more effective in reducing the levels of TNF-α (Figure 6C) than IL-1β (Figure 6D) in the hind paw tissue (P < 0.05). Similar to 17(R)HDoHE treatment, AT-RvD1 did not significantly reduce the levels of both cytokines in the spinal cord (Figure 6G,H).
Figure 6.
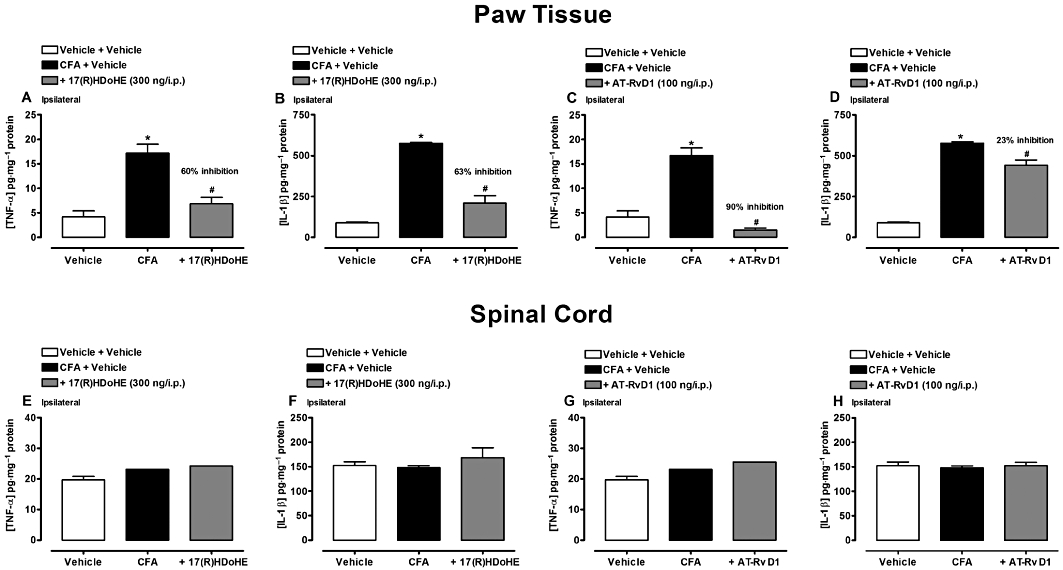
Repeated post-treatment with 17(R)HDoHE, or AT-RvD1 attenuated TNF-α and IL-1β levels in rat hind paw tissue, but not in the spinal cord after induction of adjuvant arthritis (AIA). TNF-α and IL-1β levels in the paw tissue (A–D) and in the spinal cord (E–H) after 17(R)HDoHE (300 ng/i.p., 3 days after, for 5 days, once a day) and AT-RvD1(100 ng/i.p., 3 days after, for 4 days, twice a day) treatment respectively. Data are presented as the mean ± SEM of four to six animals per group. *P < 0.05 and #P < 0.05 are significantly different from the vehicle and complete Freund's Adjuvant (CFA) groups respectively.
Effect of 17(R)HDoHE on NF-κB and COX-2 immunostaining in the spinal cord and DRG of AIA rats
To further define some of the signalling pathways that could mediate the analgesic effects of 17(R)HDoHE, we assessed, using immunohistochemical assays, the actions of 17(R)HDoHE (300 ng/i.p.), 4 h after treatment, on the activation of the transcription factor NF-κB or COX-2 expression in (L4–L5) and superficial (lamina I and II) and neck region (lamina V and VI) of the dorsal horn of spinal cord. As expected, AIA led to a pronounced phosphorylation of the p65 subunit of NF-κB (Figure 7) and increased COX-2 expression (Figure 7) (P < 0.05), either in the superficial and neck region of the dorsal horn of spinal cord, or in the DRG neurons (L4-L6), which correlates with persistent inflammatory hypersensitivity. Relevantly, treatment with 17(R)HDoHE (300 ng/i.p.) significantly reduced the activation of NF-κB (P < 0.05) and inhibited the increase of COX-2 expression (P < 0.05) in the dorsal horn of lumbar spinal cord (Figure 7A,C,E) and in DRG neurons (Figure 7B,D,F).
Figure 7.
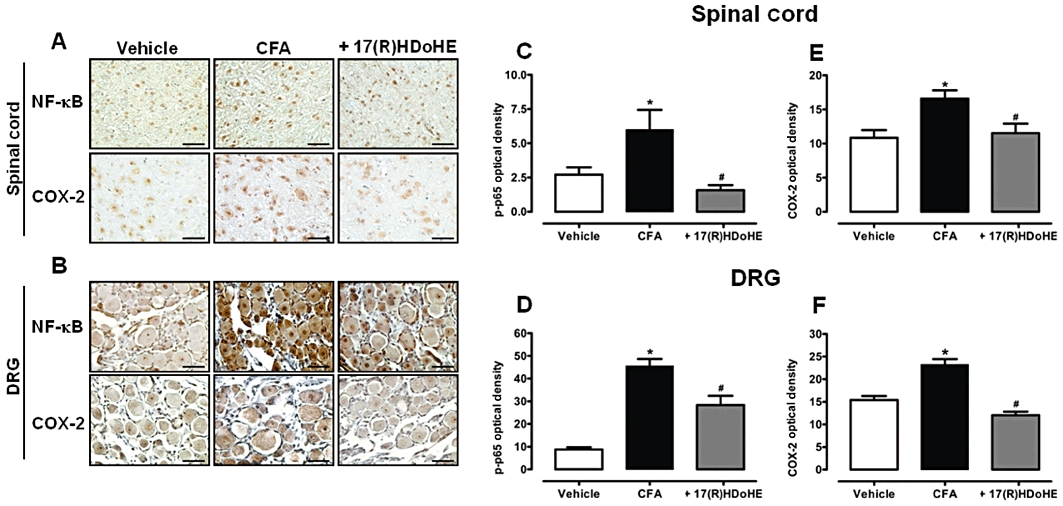
A single treatment with 17(R)HDoHE (300 ng/i.p., 3 days after), inhibited the increase of NF-κB and COX-2 in the spinal cord and dorsal root ganglion (DRG) of rats with adjuvant-induced arthritis (AIA). Representative images of NF-κB (upper panel) and COX-2 (lower panel) immunoreactivity in the spinal cord (A) and DRG (B) (scale bar = 100 µM). Graphic representation of the NF-κB (C,D) and COX-2 (E,F) immunostaining in the spinal cord and DRG respectively. Data are presented as the mean ± SEM of five to six animals per group. *P < 0.05 and #P < 0.05 are significantly different from the vehicle and complete Freund's Adjuvant (CFA) group respectively.
Effect of 17(R)HDoHE on NF-κB and COX-2 mRNA levels in the spinal cord and DRG of AIA rats
Another set of experiments was conducted to evaluate the effect of 17(R)HDoHE on NF-κB and COX-2 mRNA levels in both the lumbar spinal cord and DRG of rats subjected to AIA. As can be seen in Figure 8, the mRNA levels of the transcription factor NF-κB were not modified either in the spinal cord (Figure 8A) or in the DRG (Figure 8C), 3 days after AIA induction. However, in the same period of evaluation, COX-2 mRNA levels were increased in the spinal cord (Figure 8B), but not in the DRG (Figure 8D). Furthermore, treatment with 17(R)HDoHE decreased COX-2 mRNA levels in the spinal cord (Figure 8B).
Figure 8.
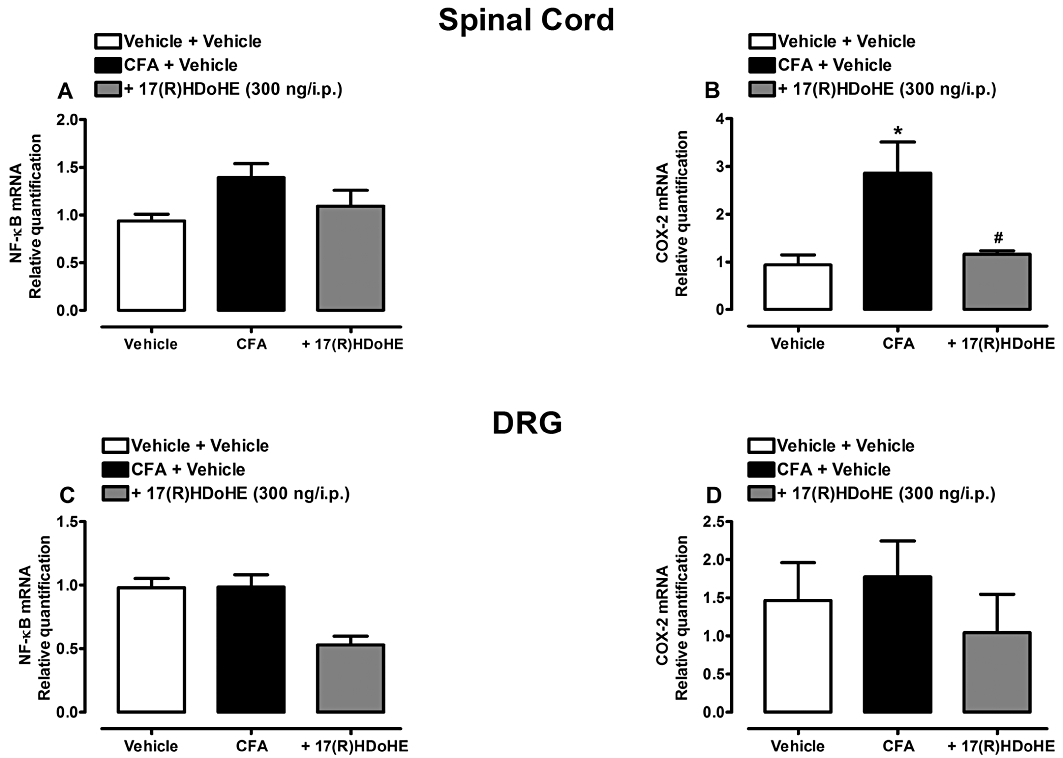
A single treatment with 17(R)HDoHE (300 ng/i.p., 3 days after), modulates(COX-2 mRNA levels in the spinal cord but not in dorsal root ganglion (DRG) nor (NF-κB mRNA levels in both tissues of rats with AIA. (A–D) Represent mRNA levels of NF-κB and COX-2 in the spinal cord and DRG respectively. Real-time PCR assay was performed in duplicate and β-actin mRNA was used to normalize the relative amount of mRNA. Data are presented as the mean ± SEM of three to five animals per group. *P < 0.05 and #P < 0.05 are significantly different from the vehicle and complete Freund's Adjuvant (CFA) groups respectively.
Discussion and conclusions
The main results emerging from the present study are, to the best of our knowledge, the first evidence showing that systemic administration of the resolvin D series precursor [17(R)HDoHE] or the aspirin-triggered resolvin D1 epimer (AT-RvD1) caused pronounced and long-lasting anti-hyperalgesic effects when assessed in a model of AIA in rats. Our data also show that mechanisms underlying the anti-hyperalgesic actions of 17(R)HDoHE and AT-RvD1 were primarily associated with the inhibition of pro-inflammatory and pro-nociceptive mediators, namely TNF-α and IL-1β, and through the blockade of COX-2 expression and NF-κB activation.
The AIA is a well-characterized model of persistent hyperalgesia, which progresses to chronic inflammation of the joints (Hegen et al., 2008). It is well known that the release of several pro-inflammatory mediators, for example, prostaglandin E2 and pro-inflammatory cytokines such as TNF-α and IL-1β, occurs after an inflammatory insult in the periphery, which in turn promotes nociceptor sensitization and a decrease in nociceptive threshold (Kassuya et al., 2007; Coderre, 2009; Schaible et al., 2009; Uçeyler et al., 2009). Furthermore, IL-1β also produces inflammation and induces synthesis of several nociceptor sensitizers and directly activates nociceptors to induce pain hypersensitivity (Binshtok et al., 2008). Thus, the inhibition of mechanical hyperalgesia observed soon after 17(R)HDoHE administration might be a result of an indirect modulation of nociceptor sensitization, because the precursor could attenuate the onset of inflammation and consequently the release of pro-inflammatory mediators, diminishing sensitization of afferent fibres.
In the present study, we observed that 17(R)HDoHE has suppressed the mechanical but not the thermal hyperalgesia induced by CFA. Earlier studies have demonstrated differences between tactile allodynia and thermal hyperalgesia, that is, the former being mediated through large diameter Aβ afferent fibres, and the latter being mediated through small diameter unmyelinated C-fibres (Xu et al., 1997; Ossipov et al., 1999). In addition, rats that received capsaicin neonatally (significantly reducing the number of C-fibres) failed to develop thermal hyperalgesia, but displayed heightened responses to mechanical nociceptive stimuli after chronic constriction injury (Shir and Seltzer, 1990). Therefore, during CFA-induced inflammation, the mechanism responsible for thermal hyperalgesia seems likely to be different from that of the mechanical hyperalgesia (Leem et al., 2001). Also, it has been suggested that thermal hyperalgesia involves both spinal and supraspinal circuits, while mechanical allodynia depends on a supraspinal loop (Wegert et al., 1997). This difference might reflect that afferent inputs might be associated with different types of fibre. It is tempting to suggest that 17(R)HDoHE might modulate inflammatory pain via sensitization of central and/or peripheral Aβ afferent fibres, mainly in the supraspinal circuits. However, additional experiments are needed to verify this hypothesis.
Concerning the parameters related to the disease model evaluated here (i.e. paw oedema, joint oedema and stiffness score), the marked prevention of joint stiffness by the treatment with 17(R)HDoHE is of great relevance, because one of the most common complaints of patients with arthritis is stiff joints, which together with joint pain results in great disability (Croci and Zarini, 2007; Montecucco et al., 2009).
AT-RvD1 can be generated from 17(R)HDoHE in the presence of aspirin by a series of reactions that include enzymatic epoxidation and hydrolysis (Sun et al., 2007; Serhan et al., 2008). In murine peritonitis, RvD1 was found to be equipotent to AT-RvD1, limiting PMN infiltration in a dose-dependent fashion (Sun et al., 2007). Our present results demonstrated that AT-RvD1, when administered at relatively low doses (100–300 ng), 3 days after AIA induction raised the threshold of mechanical hyperalgesia, demonstrating that not only the precursor 17(R)HDoHE, but also one of its end products, AT-RvD1, was very effective in modulating the acute inflammatory pain observed in the AIA model. However, we cannot rule out the possible conversion of the precursor 17(R)HDoHE to other 17R-RvD series, because previous studies have suggested that this precursor could generate different aspirin-triggered resolvins (Serhan et al., 2000; 2002; Hong et al., 2003). Furthermore, the possible conversion of 17(R)HDoHE to AT-RvD1 requires cells that possess 5-LOX, which is present or may be induced in several types of cells during inflammation, for example, in the paw, peripheral nerve fibre, infiltrated neutrophils, neurons in the spinal cord and DRG, and therefore might be a site of aspirin-triggered resolvin biosynthesis (Cortes-Burgos et al., 2009). Thus, because 17(R)HDoHE may be endogenously converted to other members of the AT-RvD series, that is, AT-RvD1 or AT-RvD2, we cannot be sure that the anti-hyperalgesic effect of 17(R)HDoHE observed in this study could be attributed to the molecule itself or the formation of other pro-resolution metabolites at the inflammatory site.Further studies would be required to confirm this hypothesis.
Extensive data already published has highlighted the involvement of pro-inflammatory cytokines as mediators of inflammatory pain (Firestein, 2003; Schaible et al., 2006; 2009;). Here, the reduction of mechanical hyperalgesia observed following repeated treatment with 17(R)HDoHE or AT-RvD1 might be, at least in part, due to their ability to decrease TNF-α and IL-1β in the rat paw. Recently, Xu et al., (2010) have reported a marked analgesic effect of resolvin E1 (RvE1) in several models of acute and persistent pain by regulating production of pro-inflammatory cytokines. In addition, RvD1 was shown to possess acute antinociceptive effect in several inflammatory conditions, probably due to inhibition of thermoTRP channels, including TRPA1, TRPV3 and TRPV4 (Bang et al., 2010). Thus, our results reinforce and extend the recently discovered analgesic properties of AT-RvD1 and its precursor, by showing that these lipids regulate the levels of important pro-inflammatory cytokines.
We have confirmed earlier data by showing that peripheral inflammation resulting from AIA increases the activation of NF-κB in both spinal cord and DRG (Chan et al., 2000; Lee et al., 2004). Noteworthy, we have demonstrated that 17(R)HDoHE (300 ng/i.p.) significantly decreased the phosphorylation of p65 subunit of NF-κB in the spinal cord and DRG, 3 days after AIA induction. NF-κB is known to have a pivotal role in immune and inflammatory responses through the regulation of several genes that encode pro-inflammatory molecules, including cytokines and COX-2 (Lawrence et al., 2002). Although COX-2 drives the onset of inflammation through the production of pro-inflammatory prostaglandin E2, it also drives the resolution of inflammation through the synthesis of the anti-inflammatory PG, 15deoxy-Δ12,14-PGJ2 (15dPGJ2). In macrophage and lymphocyte cell lines, 15dPGJ2 inhibited the activation of IKKβ, which regulates the activation of NF-κB in response to pro-inflammatory stimuli (Rossi et al., 2000; Straus et al., 2000). Our results confirm and also extend these findings by demonstrating that the resolvin D series precursor, 17(R)HDoHE, inhibited NF-κB activation in the DRG and dorsal horn of spinal cord. However, the exact mechanisms by which these lipid mediators inhibit the NF-κB expression should be further examined.
Considering mRNA levels of the transcription factor NF-κB, we did not detect a significant increase in the spinal cord and DRG in the acute inflammatory state. This data is in part supported by the study of Chan et al. (2000) who have demonstrated that the p50/p65 heterodimer of NF-κB increased as early as 30 min in the spinal cord, and started to decrease 2 h following peripheral inflammation, demonstrating that NF-κB expression/activity is dependent, at least in part, on the period of evaluation. Thus, we did not detect differences in the NF-κB mRNA levels probably because of the time at which we evaluated this variable in this study.
Moreover, marked increases in COX-2 expression occur locally at the sites of inflammation or in different cell types in the spinal cord and brain (Samad et al., 2001). It has been shown that COX-2 is induced after peripheral inflammation (Yacoubian and Serhan, 2007), which indirectly sensitizes peripheral terminals of sensory fibres (Serhan et al., 2008). Of note, 17(R)HDoHE (300 ng/i.p.) treatment elicited a significant decrease in the immunostaining for COX-2 in the spinal cord and DRG in acute inflammation. We have also observed an increase in the levels of COX-2 mRNA 3 days after AIA induction. Some authors have shown that COX-2 mRNA levels are increased at least for up to 24 h after peripheral inflammation (Samad et al., 2001) and others have observed a decrease in COX-2 mRNA levels after 3 days (Hay et al., 1997). The increase in COX-2 mRNA in spinal cord 6 h and 3 days after chronic inflammation in mice (Narita et al., 2008) would be in accordance with our results,. Also, we detected a significant reduction of COX-2 mRNA in the spinal cord of rats treated with 17(R)HDoHE. When evaluating COX-2 mRNA expression in DRG, no significant differences were observed. The DRG from animals with CFA-induced inflammation showed a minimal increase in COX-2 mRNA contrasting to a significant induction of COX-2 mRNA in the spinal cord (Amaya et al., 2009).
To summarize, our present data show that 17(R)HDoHE, a precursor of the resolvin D series, modulated both the genesis and the maintenance of mechanical hyperalgesia in the AIA model of arthritis in rats. Additionally, when comparing the effect of both 17(R)HDoHE and AT-RvD1 with other currently used analgesics (Table 1), we can observe that pro-resolution lipid mediators induced a greater percentage of inhibition of mechanical hyperalgesia. Moreover, the anti-hyperalgesic effect of 17(R)HDoHE in acute inflammation seems likely to be mediated by inhibition of both NF-κB and COX-2 in spinal cord and DRG levels. Furthermore, the anti-hyperalgesic effects reported here for AT-RvD1 are likely to rely on the modulation of TNF-α and IL-1β production in the hind paw tissue. Given the anti-hyperalgesic efficacy of resolvins and the safety associated with endogenous mediators, the present findings bring new evidence that lipid mediators, such as 17(R)HDoHE and AT-RvD1, may represent a new family of analgesics useful in treating inflammation-associated pain states such as arthritic pain.
Acknowledgments
This work was supported by grants from the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) and the Fundação de Apoio à Pesquisa do Estado de Santa Catarina (FAPESC). J. F. L. G., R. C. D. and K. A. B. S. S. are PhD students funded by CNPq. E. M. M. is a recipient of a postdoctoral grant from CNPq.
Glossary
Abbreviations
- 17(R)HDoHE
17(R)-hydroxy-4Z,7Z,10Z,13Z,15E,17R,19Z-docosahexaenoic acid
- AIA
adjuvant-induced arthritis
- AT-RvD1
aspirin-triggered resolvin D1, 7S,8R,17R-trihydroxy-4Z,9E,11E,13Z,15E,19Z-docosahexaenoic acid
- RA
rheumatoid arthritis
Conflict of interest
The authors declare that there are no conflicts of interest.
References
- Albert CM, Campos H, Stampfer MJ, Ridker PM, Manson JE, Willett WC, et al. Blood levels of long-chain n-3 fatty acids and the risk of sudden death. N Engl J Med. 2002;346:1113–1118. doi: 10.1056/NEJMoa012918. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 4th edition. Br J Pharmacol. 2009;150(Suppl 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaya F, Samad TA, Barrett L, Broom DC, Woolf CJ. Periganglionic inflammation elicits a distally radiating pain hypersensitivity by promoting COX-2 induction in the dorsal root ganglion. Pain. 2009;142:59–67. doi: 10.1016/j.pain.2008.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bang S, Yoo S, Yang TJ, Cho H, Kim YG, Hwang SW. Resolvin D1 attenuates activation of sensory transient receptor potential channels leading to multiple anti-nociception. Br J Pharmacol. 2010;161:707–720. doi: 10.1111/j.1476-5381.2010.00909.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannenberg GL, Chiang N, Ariel A, Arita M, Tjonahen E, Gotlinger KH, et al. Molecular circuits of resolution: formation and actions of resolvins and protectins. J Immunol. 2005;174:4345–4355. doi: 10.4049/jimmunol.174.7.4345. [DOI] [PubMed] [Google Scholar]
- Belluzzi A, Brignola C, Campieri M, Pera A, Boschi S, Miglioli M. Effect of an enteric-coated fish-oil preparation on relapses in Crohn's disease. N Engl J Med. 1996;334:1557–1560. doi: 10.1056/NEJM199606133342401. [DOI] [PubMed] [Google Scholar]
- Bendele AM. Animal models of rheumatoid arthritis. J Musculoskelet Neuronal Interact. 2001;1:377–385. [PubMed] [Google Scholar]
- Binshtok AM, Wang H, Zimmermann K, Amaya F, Vardeh D, Shi L, et al. Nociceptors are interleukin-1β sensors. J Neurosci. 2008;28:14062–14073. doi: 10.1523/JNEUROSCI.3795-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan FM, McInnes IB. Evidence that cytokines play a role in rheumatoid arthritis. J Clin Invest. 2008;118:3537–3545. doi: 10.1172/JCI36389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell EL, Louis NA, Tomassetti SE, Canny GO, Arita M, Serhan CN, et al. Resolvin E1 promotes mucosal surface clearance of neutrophils: a new paradigm for inflammatory resolution. FASEB J. 2007;21:3162–3170. doi: 10.1096/fj.07-8473com. [DOI] [PubMed] [Google Scholar]
- Chan C-F, Sun W-Z, Lin J-K, Lin-Shiau S-Y. Activation of transcription factors of nuclear factor kappa B, activator protein-1 and octamer factors in hyperalgesia. Eur J Pharmacol. 2000;402:61–68. doi: 10.1016/s0014-2999(00)00431-3. [DOI] [PubMed] [Google Scholar]
- Coderre TJ. Spinal cord mechanisms of hyperalgesia and allodynia. In: Basbaum AI, Bushnell MC, editors. Science of Pain. San Diego: Academic Press; 2009. pp. 339–380. [Google Scholar]
- Cortes-Burgos LA, Zweifel BS, Settle SL, Pufahl RA, Anderson GD, Hardy MM, et al. CJ-13610, an orally active inhibitor of 5-lipoxygenase is efficacious in preclinical models of pain. Eur J Pharmacol. 2009;617:59–67. doi: 10.1016/j.ejphar.2009.06.058. [DOI] [PubMed] [Google Scholar]
- Croci T, Zarini E. Effect of the cannabinoid CB1 receptor antagonist rimonabant on nociceptive responses and adjuvant-induced arthritis in obese and lean rats. Br J Pharmacol. 2007;150:559–566. doi: 10.1038/sj.bjp.0707138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dornelles FN, Santos DS, Van Dyke TE, Calixto JB, Batista EL, Jr, Campos MM. In vivo up-regulation of kinin B1 receptors after treatment with Porphyromonas gingivalis lipopolysaccharide in rat paw. J Pharmacol Exp Ther. 2009;330:756–763. doi: 10.1124/jpet.109.155762. [DOI] [PubMed] [Google Scholar]
- Duffield JS, Hong S, Vaidya VS, Lu Y, Fredman G, Serhan CN, et al. Resolvin D series and protectin D1 mitigate acute kidney injury. J Immunol. 2006;177:5902–5911. doi: 10.4049/jimmunol.177.9.5902. [DOI] [PubMed] [Google Scholar]
- Filep JG. Lipid mediator interplay: resolvin D1 attenuates inflammation evoked by glutathione-conjugated lipid peroxidation products. Br J Pharmacol. 2009;158:1059–1061. doi: 10.1111/j.1476-5381.2009.00235.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firestein GS. Evolving concepts of rheumatoid arthritis. Nature. 2003;423:356–361. doi: 10.1038/nature01661. [DOI] [PubMed] [Google Scholar]
- Gilroy DW, Lawrence T, Perretti M, Rossi AG. Inflammatory resolution: new opportunities for drug discovery. Nat Rev Drug Discov. 2004;3:401–415. doi: 10.1038/nrd1383. [DOI] [PubMed] [Google Scholar]
- GISSI-Prevenzione Investigators. Dietary supplementation with n-3 polyunsaturated fatty acids and vitamin E after myocardial infarction: results of the GISSI Prevenzione trial. Gruppo Italiano per lo Studio della Sopravvivenza nell'Infarto miocardico. Lancet. 1999;354:447–455. [PubMed] [Google Scholar]
- González ELM, Patrignani P, Tacconelli S, Rodríguez LAG. Variability among nonsteroidal antiinflammatory drugs in risk of upper gastrointestinal bleeding. Arthritis Rheum. 2010;62:1592–1601. doi: 10.1002/art.27412. [DOI] [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- Hay CH, Trevethick MA, Wheeldon A, Bowers JS, De Belleroche JS. The potential role of spinal cord cyclooxygenase-2 in the development of Freund's complete adjuvant-induced changes in hyperalgesia and allodynia. Neurosci. 1997;78:843–850. doi: 10.1016/s0306-4522(96)00598-2. [DOI] [PubMed] [Google Scholar]
- Hegen M, Keith JC, Jr, Collins M, Nickerson-Nutter CL. Utility of animal models for identification of potential therapeutics for rheumatoid arthritis. Ann Rheum Dis. 2008;67:1505–1515. doi: 10.1136/ard.2007.076430. [DOI] [PubMed] [Google Scholar]
- Hong S, Gronert K, Devchand PR, Moussignac R-L, Serhan CN. Novel docosatrienes and 17S-Resolvins generated from docosahexaenoic acid in murine brain, human blood, and glial cells. Autacoids in anti-inflammation. J Biol Chem. 2003;278:14677–14687. doi: 10.1074/jbc.M300218200. [DOI] [PubMed] [Google Scholar]
- Kassuya CAL, Ferreira J, Claudino RF, Calixto JB. Intraplantar PGE2 causes nociceptive behaviour and mechanical allodynia: the role of prostanoid E receptors and protein kinases. Br J Pharmacol. 2007;150:727–737. doi: 10.1038/sj.bjp.0707149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasuga K, Yang R, Porter TF, Agrawal N, Petasis NA, Irimia D, et al. Rapid appearance of resolvin precursors in inflammatory exudates: novel mechanisms in resolution. J Immunol. 2008;181:8677–8687. doi: 10.4049/jimmunol.181.12.8677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence T, Willoughby DA, Gilroy DW. Anti-inflammatory lipid mediators and insights into the resolution of inflammation. Nat Rev Immunol. 2002;2:787–795. doi: 10.1038/nri915. [DOI] [PubMed] [Google Scholar]
- Lee K-M, Kang B-S, Lee H-L, Son S-J, Hwang S-H, Kim D-S, et al. Spinal NF-κB activation induces COX-2 upregulation and contributes to inflammatory pain hypersensitivity. Eur J Neurosci. 2004;19:3375–3381. doi: 10.1111/j.0953-816X.2004.03441.x. [DOI] [PubMed] [Google Scholar]
- Leeb BF, Sautner J, Andel I, Rintelen B. Intravenous application of omega-3 fatty acids in patients with active rheumatoid arthritis. The ORA-1 Trial. An open pilot study. Lipids. 2006;41:29–34. doi: 10.1007/11745-006-5066-x. [DOI] [PubMed] [Google Scholar]
- Leem JW, Hwang JH, Hwang SJ, Park H, Kimb MK, Choib Y. The role of peripheral N-methyl-d-aspartate receptors in Freund's complete adjuvant induced mechanical hyperalgesia in rats. Neurosci Lett. 2001;297:155–158. doi: 10.1016/s0304-3940(00)01662-1. [DOI] [PubMed] [Google Scholar]
- Levy BD, Clish CB, Schmidt B, Gronert K, Serhan CN. Lipid mediator class switching during acute inflammation: signals in resolution. Nat Immunol. 2001;2:612–619. doi: 10.1038/89759. [DOI] [PubMed] [Google Scholar]
- McDougall JJ, Zhang C, Cellars L, Joubert E, Dixon CM, Vergnolle N. Triggering of proteinase-activated receptor 4 leads to joint pain and inflammation in mice. Arthritis Rheum. 2009;60:728–737. doi: 10.1002/art.24300. [DOI] [PubMed] [Google Scholar]
- Manjavachi MN, Quintão NL, Campos MM, Deschamps IK, Yunes RA, Nunes RJ, et al. The effects of the selective and non-peptide CXCR2 receptor antagonist SB225002 on acute and long-lasting models of nociception in mice. Eur J Pain. 2010;14:23–31. doi: 10.1016/j.ejpain.2009.01.007. [DOI] [PubMed] [Google Scholar]
- Montecucco C, Cavagna L, Caporali R. Pain and rheumatology: an overview of the problem. Eur J Pain. 2009;3(Suppl):105–109. [Google Scholar]
- Müller-Ladner U, Pap T, Gay RE, Neidhart M, Gay S. Mechanisms of disease: the molecular and cellular basis of joint destruction in rheumatoid arthritis. Nat Clin Pract Rheumatol. 2005;1:102–110. doi: 10.1038/ncprheum0047. [DOI] [PubMed] [Google Scholar]
- Nagakura Y, Okada M, Kohara A, Kiso T, Toya T, Iwai A, et al. Allodynia and hyperalgesia in adjuvant-induced arthritic rats: time course of progression and efficacy of analgesics. J Pharmacol Exp Ther. 2003;306:490–497. doi: 10.1124/jpet.103.050781. [DOI] [PubMed] [Google Scholar]
- Narita M, Shimamura M, Imai S, Kubota C, Yajima Y, Takagi T, et al. Role of interleukin-1β and tumor necrosis factor-α dependent expression of cyclooxygenase-2 mRNA in thermal hyperalgesia induced by chronic inflammation in mice. Neurosci. 2008;152:477–486. doi: 10.1016/j.neuroscience.2007.10.039. [DOI] [PubMed] [Google Scholar]
- Ossipov MH, Bian D, Malan TP, Jr, Lai J, Porreca F. Lack of involvement of capsaicin-sensitive primary afferents in nerve-ligation injury induced tactile allodynia in rats. Pain. 1999;79:127–133. doi: 10.1016/s0304-3959(98)00187-0. [DOI] [PubMed] [Google Scholar]
- Pinto LA, Cunha TM, Vieira SM, Lemos HP, Verri WA, Jr, Cunha FQ, et al. IL-17 mediates articular hypernociception in antigen-induced arthritis in mice. Pain. 2010;148:247–256. doi: 10.1016/j.pain.2009.11.006. [DOI] [PubMed] [Google Scholar]
- Quan LD, Thiele GM, Tian J, Wang D. The development of novel therapies for rheumatoid arthritis. Expert Opin Ther Pat. 2008;18:723–738. doi: 10.1517/13543776.18.7.723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi A, Kapahi P, Natoli G, Takahashi T, Chen Y, Karin M, et al. Anti-inflammatory cyclopentenone prostaglandins are direct inhibitors of IkB kinase. Nature. 2000;403:103–108. doi: 10.1038/47520. [DOI] [PubMed] [Google Scholar]
- Samad TA, Moore KA, Sapirstein A, Billet S, Allchorne A, Poole S, et al. Interleukin-1β-mediated induction of Cox-2 in the CNS contributes to inflammatory pain hypersensitivity. Nature. 2001;410:471–475. doi: 10.1038/35068566. [DOI] [PubMed] [Google Scholar]
- Schaible HG, Grubb BD. Afferent and spinal mechanisms of joint pain. Pain. 1993;55:5–54. doi: 10.1016/0304-3959(93)90183-P. [DOI] [PubMed] [Google Scholar]
- Schaible HG, Ebersberger A, Von Banchet GS. Mechanisms of pain in arthritis. Ann N Y Acad Sci. 2002;966:343–354. doi: 10.1111/j.1749-6632.2002.tb04234.x. [DOI] [PubMed] [Google Scholar]
- Schaible HG, Schmelz M, Tegeder I. Pathophysiology and treatment of pain in joint disease. Adv Drug Deliv Rev. 2006;58:323–342. doi: 10.1016/j.addr.2006.01.011. [DOI] [PubMed] [Google Scholar]
- Schaible HG, Richter F, Ebersberger A, Boettger MK, Vanegas H, Natura G, et al. Joint pain. Exp Brain Res. 2009;196:153–162. doi: 10.1007/s00221-009-1782-9. [DOI] [PubMed] [Google Scholar]
- Serhan CN. 2004. U.S. Patent No. 2004/0116408 A1.
- Serhan CN. Resolution phase of inflammation: novel endogenous anti-inflammatory and proresolving lipid mediators and pathways. Annu Rev Immunol. 2007;25:101–137. doi: 10.1146/annurev.immunol.25.022106.141647. [DOI] [PubMed] [Google Scholar]
- Serhan CN. Systems approach to inflammation resolution: identification of novel anti-inflammatory and pro-resolving mediators. J Thromb Haemost. 2009;7:44–48. doi: 10.1111/j.1538-7836.2009.03396.x. [DOI] [PubMed] [Google Scholar]
- Serhan CN, Clish CB. 2003. U.S. Patent No. 6,670,396 B2.
- Serhan CN, Clish CB, Brannon J, Colgan SP, Chiang N, Gronert K. Novel functional sets of lipid-derived mediators with antiinflammatory actions generated from omega-3 fatty acids via cyclooxygenase 2–nonsteroidal anti-inflammatory drugs and transcellular processing. J Exp Med. 2000;192:1197–1204. doi: 10.1084/jem.192.8.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serhan CN, Hong S, Gronert K, Colgan SP, Devchand PR, Mirick G, et al. Resolvins: a family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter proinflammation signals. J Exp Med. 2002;196:1025–1037. doi: 10.1084/jem.20020760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serhan CN, Yacoubian S, Yang R. Anti-inflammatory and proresolving lipid mediators. Annu Rev Pathol Mech Dis. 2008;3:279–312. doi: 10.1146/annurev.pathmechdis.3.121806.151409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serhan CN, Yang R, Martinoid K, Kassuga K, Pillai PM, Porter TF, et al. Maresins: novel macrophage mediators with potent anti-inflammatory and proresolving actions. J Exp Med. 2009;206:15–23. doi: 10.1084/jem.20081880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shir Y, Seltzer Z. A-fibers mediate mechanical hyperesthesia and allodynia and C-fibers mediate thermal hyperalgesia in a new model of causalgiform pain disorders in rats. Neurosci Lett. 1990;115:62–67. doi: 10.1016/0304-3940(90)90518-e. [DOI] [PubMed] [Google Scholar]
- Sommer C, Birklein F. Fighting off pain with resolvins. Nature Med. 2010;16:518–520. doi: 10.1038/nm0510-518. [DOI] [PubMed] [Google Scholar]
- Spite M, Summers L, Porter TF, Srivastava S, Bhatnagar A, Serhan CN. Resolvin D1 controls inflammation initiated by glutathione-lipid conjugates formed during oxidative stress. Br J Pharmacol. 2009;158:1062–1073. doi: 10.1111/j.1476-5381.2009.00234.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein C, Millan MJ, Herz A. Unilateral inflammation of the hindpaw in rats as a model of prolonged noxious stimulation: alterations in behavior and nociceptive thresholds. Pharmacol Biochem Behav. 1988;2:451–455. doi: 10.1016/0091-3057(88)90372-3. [DOI] [PubMed] [Google Scholar]
- Straus DS, Pascual G, Li M, Welch JS, Ricote M, Hsiang C-H, et al. 15-Deoxy-D12,14-prostaglandin J2 inhibits multiple steps in the NF-kB signaling pathway. Proc Natl Acad Sci U S A. 2000;97:4844–4849. doi: 10.1073/pnas.97.9.4844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun YP, Oh SF, Uddin J, Yang R, Gotlinger K, Campbell E, et al. Resolvin D1 and its aspirin-triggered 17R epimer: stereochemical assignments, anti-inflammatory properties, and enzymatic inactivation. J Biol Chem. 2007;282:9323–9334. doi: 10.1074/jbc.M609212200. [DOI] [PubMed] [Google Scholar]
- Svensson CI, Zattoni M, Serhan CN. Lipoxins and aspirin-triggered lipoxin inhibit inflammatory pain processing. J Exp Med. 2007;204:245–252. doi: 10.1084/jem.20061826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uçeyler N, Schäfers M, Sommer C. Mode of action of cytokines on nociceptive neurons. Exp Brain Res. 2009;196:67–78. doi: 10.1007/s00221-009-1755-z. [DOI] [PubMed] [Google Scholar]
- Vitor CE, Figueiredo CP, Hara DB, Bento AF, Mazzuco TL, Calixto JB. Therapeutic action and underlying mechanisms of a combination of two pentacyclic triterpenes, alpha- and beta-amyrin, in a mouse model of colitis. Br J Pharmacol. 2009;157:1034–1044. doi: 10.1111/j.1476-5381.2009.00271.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Vollenhoven RF. Treatment of rheumatoid arthritis: state of the art. Nat Rev Rheumatol. 2009;5:531–534. doi: 10.1038/nrrheum.2009.182. [DOI] [PubMed] [Google Scholar]
- Wegert S, Ossipov MH, Nichols ML, Bian D, Vanderah TW, Malan TP, et al. Differential activities of intrathecal MK-801 or morphine to alter responses to thermal and mechanical stimuli in normal or nerve-injured rats. Pain. 1997;71:57–64. doi: 10.1016/s0304-3959(97)03337-x. [DOI] [PubMed] [Google Scholar]
- Xu X-J, Farkas-Szallasi T, Lundberg JM, Hiikfelt T, Wiesenfeld-Hallin Z, Szallasi A. Effects of the capsaicin analogue resiniferatoxin on spinal notice mechanisms in the rat: behavioral, electrophysiological and in situ hybridization studies. Brain Res. 1997;752:52–60. doi: 10.1016/s0006-8993(96)01444-8. [DOI] [PubMed] [Google Scholar]
- Xu ZZ, Zhang L, Liu T, Park JY, Berta T, Yang R, et al. Resolvins RvE1 and RvD1 attenuate inflammatory pain via central and peripheral actions. Nature Med. 2010;16:592–597. doi: 10.1038/nm.2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yacoubian S, Serhan CN. New endogenous anti-inflammatory and proresolving lipid mediators: implication for rheumatic diseases. Nat Clin Pract Rheumatol. 2007;3:570–579. doi: 10.1038/ncprheum0616. [DOI] [PubMed] [Google Scholar]