

Abstract
BACKGROUND AND PURPOSE
Selective and potent antagonists for the β2-adrenoceptor are potentially interesting as experimental and clinical tools, and we sought to identify novel ligands with this pharmacology.
EXPERIMENTAL APPROACH
A range of pharmacological assays was used to assess potency, affinity, selectivity (β2-adrenoceptor vs. β1-adrenoceptor) and efficacy.
KEY RESULTS
Ten novel compounds were identified but none had as high affinity as the prototypical β2-adrenoceptor blocker ICI-118,551, although one of the novel compounds was more selective for β2-adrenoceptors. Most of the ligands were inverse agonists for β2-adrenoceptor-cAMP signalling, although one (5217377) was a partial agonist and another a neutral antagonist (7929193). None of the ligands were efficacious with regard to β2-adrenoceptor-β-arrestin signalling. The (2S,3S) enantiomers were identified as the most active, although unusually the racemates were the most selective for the β2-adrenoceptors. This was taken as evidence for some unusual enantiospecific behaviour.
CONCLUSIONS AND IMPLICATIONS
In terms of improving on the pharmacology of the ligand ICI-118,551, one of the compounds was more selective (racemic JB-175), while one was a neutral antagonist (7929193), although none had as high an affinity. The results substantiate the notion that β-blockers do more than simply inhibit receptor activation, and differences between the ligands could provide useful tools to investigate receptor biology.
Keywords: adrenoceptor, antagonist, cell signalling
Introduction
The β2-adrenoceptor is one of the prototypical members of a superfamily of transmembrane proteins known as G-protein coupled receptors (GPCR). The β2-adrenoceptor mediates the effects of catecholamines by predominantly coupling to the stimulating G-protein (Gs) and then subsequent activation of adenylate cyclase, increases in cAMP and activation of protein kinase A. More recently, signalling through the inhibitory G-protein (Gi) (Daaka et al., 1997; Zamah et al. 2002) and G-protein-independent signalling have also been demonstrated (Azzi et al., 2003; Baker et al. 2003b). There is substantial evidence for the potential benefits of inhibiting β2-adrenoceptor signalling in heart failure (CIBIS Investigators and Committees, 1994; Packer et al., 1996; MERIT-HF Study Group, 1999). There is also evidence from both basic and clinical studies that action at β2-adrenoceptors may be an important component of this beneficial effect (Poole-Wilson et al. 2003; Harding and Gong, 2004).
The recent description of crystal structures of the β2- and β1-adrenoceptors has given new insights into enduring issues in GPCR biology (Cherezov et al., 2007; Rasmussen et al., 2007; Rosenbaum et al., 2007; Warne et al., 2008). This work is the potential springboard to understanding in molecular detail the basis of agonism, antagonism and inverse agonism and how ligand selectivity might be achieved between two similar GPCR. A series of ligands that are selective and have well-defined pharmacologies is clearly important for the successful implementation of such work.
There is a relative paucity of suitably selective β2-adrenoceptor ligands for use as experimental tools to probe the receptor and to investigate the role of β2-adrenoceptor-blockade in experimental and clinical settings. The experimental compound ICI-118,551 is the only β2-adrenoceptor antagonist that is both potent and selective for the receptor (Bilski et al., 1983; Baker, 2005). However, it has a complex pharmacological profile that includes inverse agonism for β2-adrenoceptor-Gs signalling (Azzi et al., 2003; Baker et al. 2003b), agonism for β2-adrenoceptor-Gi (Gong et al., 2002), activation of MAPK signalling (Azzi et al., 2003), ion channel blockade (Teixeira et al., 2004) and a direct antiapoptotic action (Mikami et al., 2008). Therefore, experimental or clinical effects of ICI-118,551 may be obscured by events independent of simple blockade of β2-adrenoceptor. Other problems with ICI-118,551 that make it ill-suited to in vivo or clinical studies are that it is extensively metabolized by the liver into less selective derivatives and is potentially carcinogenic (Fitzgerald, 1991).
We sought to identify novel ligands that are potent and selective β2-adrenoceptor agents in order to generate pharmacological tools to elucidate the biological function of the receptor, while avoiding problems with the pharmacologies of existing ligands. Additionally, the role of β2-adrenoceptors in diseases such as heart failure could be more effectively investigated with such ligands, which could potentially lead to novel pharmacotherapies. To identify novel ligands, compounds were first chosen for potential β2-adrenoceptor antagonism using virtual field screening, a computational approach that predicts pharmacological similarities (Cheeseright et al., 2006; 2008;). These were then pharmacologically characterized for a number of β2-adrenoceptor-associated parameters, such as potency, affinity and efficacy. Their selectivity for β2-adrenoceptors over β1-adrenoceptors was investigated by comparing binding affinity at each receptor. A second approach for selecting potential ligands was to synthesize structural derivatives of ICI-118,551 using information gained from the active compounds in the virtual field screen. Additionally, the enantiospecific binding properties of some of the ligands were investigated, as this was predicted to provide further information regarding β-adrenoceptor function.
Methods
Materials
(−)-Adrenaline (+)-bitartate, histamine dihydrochloride (±)-ICI-118,551 hydrochloride, papaverine hydrochloride (±)-CGP-12177, cAMP, forskolin, 3-isobutyl-1-methylxanthine, Whatman® GF/B filter papers, [3H]-adenine, Dowex 50W × 4 hydrogen form 100–200 mesh and alumina oxide type WN-3 neutral were from Sigma-Aldrich (UK). cDNA clones encoding the genes for human β1-adrenoceptor and β2-adrenoceptor were from The Missouri S&T cDNA Resource Centre (USA). GFP-βarr2(1-380) DNA was a kind gift from Julie Pitcher (UCL). 25 mm diameter glass coverslips were from Menzel-Glaser. Cell culture materials were from Invitrogen (UK). (−)-[3H]-CGP-12177 was from GE Healthcare (UK). Ultima Gold™ liquid scintillation cocktail was from Perkin Elmer (UK). Poly-prep chromatography columns were from BIO-RAD (UK). T0502-1048 was purchased from Enamine (Ukraine). The 5731869, 5217377 and 7929193 were purchased from Chembridge (UK).
Virtual field screening
Drugs with very different chemical structures can bind at the same biological site with the same pharmacological outcome. Biological binding sites recognize molecular fields and not structural detail (Cheeseright et al., 2006). Virtual field screening was used to find new chemotypes by comparing the fields of a known active field template derived from ICI-118,551 to search through the fields of 3.4 M commercially available compounds (Cheeseright et al., 2008). Forty compounds with the most similar field patterns to the template were purchased and tested for β2-adrenoceptor activity and of these four were identified as active pharmacophores.
Synthesis
The details of the synthesis of analogues of ICI-118,551 are given in Appendix S1.
Organ bath studies
Methods were largely based on those described previously (Tanaka et al., 2005). Male and female guinea pigs of 300–600 g weight were killed by CO2 asphyxiation. The trachea was dissected and immersed in Krebs buffer (25 mM NaHCO3, 118 mM NaCl, 4.7 mM KCl, 1.2 mM KH2PO4, 1.2 mM MgSO4, 2 mM CaCl, 11.1 mM D(+)-glucose). Excess connective and adipose tissue was removed and it was cut into eight separate rings of approximately 2 mm length. These were epithelium-denuded by passing rolled-up tissue paper through the lumen of each ring. Two wire hooks, one connected to a stable foot and the other to a Grass FT03 force-displacement transducer, were passed through the rings, and they were then suspended in 25 mL water-jacketed organ baths containing carbogen bubbled Krebs buffer at 37°C. Applied tension of 2.5 ± 0.1 g was established and the tissue equilibrated by incubation for 60 min, with replacement of buffer every 20 min. After this period, the rings were contracted with 100 µM histamine for 15 min, followed by replacement of buffer. The 60 min equilibration period was then repeated (including replacement of buffer every 20 min), before the rings were contracted once more with 100 µM histamine. Test compounds being investigated for β2-adrenoceptor antagonism were added at this point. More than 30 min later, when the tissue had reached a steady state of contraction, the rings were relaxed by addition of cumulative concentrations of adrenaline from 1 nM to a concentration that elicited no further contraction, in half log unit increments. An adrenaline concentration was only applied when the previous concentration had caused a complete steady-state relaxation; 100 µM papaverine was then applied to induce maximal relaxation. The magnitude of each relaxant response from the histamine-induced contractile state was expressed as a percentage of the papaverine response, and concentration–response curves generated by fitting data to sigmoidal concentration–response (variable slope) curve function in GraphPad Prism 4. The adrenaline concentration–response curves in the presence of the range of antagonist concentrations used and control curves were compared globally using the Gaddum–Schild equation with GraphPad Prism 4 in order to determine pA2 values (a measure of antagonist potency). Owing to the low Schild-plot slope values of the compounds in this analysis, the data were also re-analysed by plotting the log (dose ratio – 1) of only the lowest concentration of antagonist applied and constraining the slope to unity, as described previously (Kenakin, 2006).
Competition binding assay
Human embryonic kidney cells (HEK293) lines stably expressing the human β1- or β2-adrenoceptor at 1.64 and 6.64 fmol·µg−1 receptor protein, respectively, were developed using previously described methods (Giblin et al., 1999). Once clonal isolates were established, cells were grown to confluency in 175 cm2 flasks. Culture medium was aspirated and the cells washed twice with ice-cold binding buffer (50 mM TRIS-base, pH 7.4). Cells were harvested into the same buffer, then an equal volume of a hypotonic buffer (10 mM TRIS-base, 10 mM EDTA, pH 7.4) was added and the mixture left on ice for 10 min to hypotonically shock the cells. Tonicity was restored by further addition of an equal volume of a sucrose buffer (10 mM TRIS-base, 500 mM sucrose, pH 7.4). Cells were homogenized with >60 strokes of a glass-on-glass Dounce homogenizer. The resulting homogenate was centrifuged (600×g, 15 min, 4°C) to pellet large cell debris and nuclei. The supernatant was centrifuged (100 000×g, 60 min, 4°C) and the pellet suspended in binding buffer to give a membrane stock, which was stored at −20°C.
Membranes were then thawed and diluted to 7 and 2 µg·mL−1 for β1- and β2-adrenoceptor expressing cells respectively. These were incubated with 0.3 nM [3H]-CGP-12177 (a roughly Kd concentration for this radioligand) in the presence of a range of concentrations of each test compound for 2 h at room temperature. This period of time is more than sufficient to allow equilibrium binding of the radioligand (Affolter et al., 1985). Additionally, non-specific binding was determined with incubation of a saturating concentration of cold CGP-12177 (3 µM, which is approximately a thousand times more concentrated than its Kd at the receptors). The total volume of each binding reaction was 250 µL. Experiments for β1- and β2-adrenoceptors were done in parallel and each treatment was performed in triplicate. Receptor-bound and unbound radioligand were separated by vacuum filtration through Whatman® grade GF/B filter papers, followed by 4 × 2 mL washes with binding buffer. Filter papers were then immersed in 5 mL scintillation cocktail and at least 15 h later disintegrations min−1 (dpm; binding signal) was measured for 2 min per sample with a Packard Tri-Carb 2100TR liquid scintillation counter. Specific binding was determined in each instance by subtraction of the average non-specific binding signal, and then expressed as a percentage of maximal binding. This was then plotted against the corresponding concentration of inhibitor compound. The Hill coefficients were not significantly different from one (not shown) and thus the data were fitted to one-site competition using nonlinear regression analysis with GraphPad Prism 4. The Cheng–Prusoff equation was used to calculate Ki values from these curves, and the binding affinity of the compounds for each β-adrenoceptor subtype was expressed as an average pKi ± SD for the number of experiments. Saturation binding analyses of the two cell lines was conducted (see Appendix S1, n = 3 in triplicate) to provide Kd values for [3H]-CGP-12177 at the β1-adrenoceptor (0.28 ± 0.16 nM) and β2-adrenoceptor (0.15 ± 0.02 nM) expressing cell lines for use in the Cheng–Prusoff equation. The β2-/β1-adrenoceptor selectivity ratio was calculated by dividing the Ki at β1-adrenoceptor by that at β2-adrenoceptor.
cAMP assay
Methods were largely based on those described previously (Salomon, 1991). β2-Adrenoceptor expressing HEK293 cells (as described above) were grown to 80–90% confluency in 12-well cell culture plates and incubated in minimum essential media (MEM) containing 5 µCi [3H]-adenine for 2 h at 37°C, 5% CO2. Cells were then washed with warm PBS and stimulated with reduced serum medium containing 0.1 mg·mL−1 bovine serum albumin, 0.1 mM 3-isobutyl-1-methylxanthine, 30 µM forskolin and the desired concentration of test compound(s) for 20 min at 37°C, 5% CO2. Basal cAMP levels were also determined for each experiment by incubating in the same media without forskolin or test compound. Each treatment was performed in triplicate. This media was then replaced by (1 mL per well) of a chilled 2.5% perchloric acid solution in dH2O containing 1 mM cAMP. The plates were then kept at 4°C for 30 min with periodic agitation; 900 µL of the resulting extract was transferred to a test tube and 90 µL of 4.2 M potassium hydroxide and 400 µL dH2O were added. In total, 100 µL of each sample was then added to a scintillation vial containing 5 mL scintillation cocktail to measure total [3H]-adenine uptake. The remaining volume was transferred to a chromatography column containing 1.2 mL Dowex 50. Once eluted, this was washed with 3 mL dH2O. The column was then directly suspended over a separate column containing 0.6 g alumina and 8 mL dH2O added. Once this had eluted from the alumina column, the column was suspended directly over a scintillation vial containing 5 mL scintillation cocktail. [3H]-cAMP was then eluted into these by addition of 5 mL elution buffer (100 mM TRIS-base, pH 7.5) to the column. At least 15 h later a dpm binding signal was measured for 2 min per sample with a Packard Tri-Carb 2100TR liquid scintillation counter, to determine the amount of [3H]-cAMP. cAMP responses were calculated as % conversion of the [3H]-cAMP extracted from total [3H]-adenine uptake. For each treatment the basal cAMP signal was subtracted and expressed as a % of the response to forskolin alone.
β-Arrestin recruitment assay
The HEK293 cells were seeded onto six-well cell culture plates containing 25 mm diameter glass coverslips and grown to ∼40% confluency. They were then simultaneously transiently transfected with DNA for human β2-adrenoceptor (2 µg per well) and GFP-βarr2(1–380) (a kind gift from Dr Julie Pitcher) using the Lipofectamine™ method. Approximately 24 h later the cells were washed with 3 × 2 mL HEPES-buffered reduced serum media (opti-MEM without phenol red) and then incubated for 3 h at 37°C, 5% CO2 in the same media. Each coverslip, with cells adhered to its surface, was then removed from the plate placed in a sealed confocal imaging chamber with 500 µL of the media. Images were taken with ×60 oil immersion objective with a Nikon Eclipse TE300 confocal microscope connected to a computer running Zeiss LaserSharp 2000™ software. A single laser line excitation at a wavelength of 488 nm was used, and images were acquired with a LP500 filter. Test ligand was added to a final concentration of 100 µM and the coverslip (remaining in the chamber and covered) was incubated at 37°C, 5% CO2 for 15 min before further images were taken of the cells. High quality images of a number of cells both before and after ligand treatment were acquired to investigate the effect of ligand exposure on GPF-βarr2(1–380) localization. At least 10 ligand-treated cells were examined thoroughly for a positive response. Each experiment was repeated at least twice.
Molecular modelling
All molecular mechanics modelling used the eXtended Electron Distribution (XED) force field (Vinter, 1994; Chessari et al., 2002; Cejas et al., 2008). Structures were generated using the in-house package ‘XEDRAW’ and the Discovery Studio Visualiser from Accelrys. Commercially available molecular dynamics and docking software produce less optimal results due to the use of monopolar electrostatics that mishandle aromatic interactions and hydrogen bond directions amongst others. The XED force field used electrostatic multipoles that avoid such approximations
Nomenclature
All drug and receptor nomenclature conforms to the British Journal of Pharmacology's Guide to Receptors and Channels (Alexander et al., 2009).
Results
Pharmacological characterization of compounds identified using virtual field screening
Four novel β2-adrenoceptor antagonists, T0502-1048, 5731869, 5217377 and 7929193 (Figure 1) were identified following a virtual field screen (see Methods). These were investigated using Gaddum–Schild analysis, whereby concentration-dependent increases in antagonism are used to quantify antagonist affinity (pA2 values). Adrenaline-mediated relaxation of guinea-pig tracheal smooth muscle was used for these experiments as this is predominantly a β2-adrenoceptor-driven response (Tanaka et al., 2005). ICI-118,551 was analysed for comparison, and was found to have a pA2 value of 9.49 (Table 1). The novel compounds had pA2 values ranging from 6.63 to 7.78, with 5731869 having the highest affinity (Table 1). The Schild-plot slopes were below unity in all instances (Table 1), which has been proposed to invalidate the analysis (O'Donnell and Wanstall, 1979;Kenakin, 1997). Therefore, an adapted method for pA2 derivation (Kenakin, 2006) was also used as an alternative to obtain the antagonist affinities of these ligands. This involved plotting a linear slope of unity through the log (dose ratio – 1) of only the lowest concentration of test compound applied. The re-analysis yielded a decrease in the pA2 values of the ligands and had an effect on their rank order of affinity (Table 1). Compared with control adrenaline concentration–response curves, none of the novel compounds (at the highest concentration applied) had an effect on the maximal relaxation response to adrenaline (anova Dunnett's test, P > 0.05, n = 3–4). However, ICI-118,551 caused a small but significant drop in the ability of adrenaline to elicit a maximal response (anova Dunnett's test, P < 0.05, n = 4).
Figure 1.
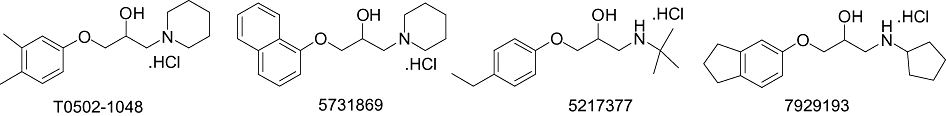
The chemical structures of active compounds isolated using a virtual field screen.
Table 1.
Gaddum–Schild analysis of ICI-118,551 and the compounds in Figure 1 in the guinea pig trachea
| Compound | n | pA2 | Schild-plot slope | pA2 (adapted analysis) |
|---|---|---|---|---|
| ICI-118,551 | 4 | 9.49 ± 0.14 | 0.61 ± 0.04 | 8.75 |
| T0502-1408 | 3 | 6.63 ± 0.20 | 0.68 ± 0.09 | 6.38 |
| 5731869 | 4 | 7.78 ± 0.17 | 0.73 ± 0.07 | 8.06 |
| 5217377 | 4 | 7.52 ± 0.13 | 0.88 ± 0.07 | 7.75 |
| 7929193 | 4 | 7.01 ± 0.08 | 0.72 ± 0.03 | 6.97 |
The interactions of these compounds with β2-adrenoceptors were confirmed by their ability to displace the radioligand [3H]-CGP-12177 to membranes isolated from HEK293 cells stably expressing the receptor. The Cheng–Prusoff correction (Cheng and Prusoff, 1973) was used to calculate the binding affinity of these compounds by the derivation of pKi values from the concentration-dependent displacement of [3H]-CGP-12177 binding to these membranes. The pKi values ranged from 5.99 to 7.68, with 5731869 having the highest affinity (Table 2). In comparison, ICI-118,551 had a higher affinity than these compounds (pKi = 9.47; Table 2).
Table 2.
[3H]-CGP-12177 competition binding data for ICI-118,551 and the compounds in Figure 1
| pKi | β2-Adrenoceptor selectivity (ratio) | |||
|---|---|---|---|---|
| Compound | n | β1-Adrenoceptor | β2-Adrenoceptor | |
| ICI-118,551 | 3 | 6.61 ± 0.09 | 9.47 ± 0.05 | 730.87 |
| T0502-1048 | 3 | 4.96 ± 0.20 | 5.99 ± 009 | 10.79 |
| 5731869 | 3 | 6.83 ± 0.08 | 7.68 ± 0.04 | 7.14 |
| 5217377 | 3 | 6.04 ± 0.12 | 7.06 ± 0.07 | 10.48 |
| 7929193 | 3 | 6.12 ± 0.11 | 6.63 ± 0.07 | 3.24 |
In order to establish β2-adrenoceptor selectivity, the binding affinity for β1-adrenoceptors was also determined for comparison, using HEK293 cells stably expressing this receptor. For all four compounds, the average pKi value at β2-adrenoceptors was significantly higher compared with β1-adrenoceptors (unpaired t-test, P≤ 0.05, n = 3–5; Table 2), which identified them as β2-selective ligands. The β2-adrenoceptor selectivity ratios ranged from <4 to >10, with T0502-1048 being the most selective (Table 2). ICI-118,551 was much more selective, with a >700-fold binding preference for β2-adrenoceptors (Table 2). Sample competition binding curves are shown in Figure 2.
Figure 2.
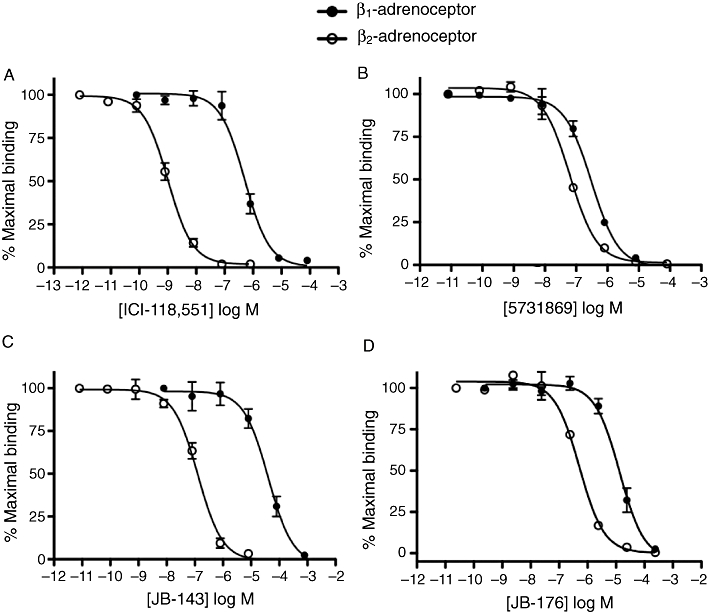
[3H]-CGP-12177 competition binding curves. These curves show the ability of ICI-118,551 (A), 5731869 (B), JB-143 (C) and JB-176 (D) to concentration-dependently displace [3H]-CGP-12177 binding to β1- and β2-adrenoceptors. The curves are an average ± SEM of three experiments performed in triplicate.
The ability of ICI-118,551 and the new compounds to influence intracellular cAMP levels was investigated to determine whether they were inverse agonists, partial agonists or neutral antagonists through β2-adrenoceptors. HEK293 cells stably expressing β2-adrenoceptors were stimulated with 30 µM as it activates adenylate cyclase in a manner that is synergistic with Gs and therefore increases the sensitivity of the assay to changes in Gs-coupling and constitutive receptor activity (Sutkowski et al., 1994; Alewijnse et al., 1997). This made the detection of inverse and partial agonist responses easier. Co-incubation with ICI-118,551, T0502-1048 or 5731869 caused a decrease in the ability of forskolin to elicit a cAMP response (anova Dunnett's test, P < 0.01, n = 3; Figure 3), which identified them as inverse agonists. In contrast, 5217377 significantly increased the forskolin response (anova Dunnett's test, P < 0.01, n = 3; Figure 3) and therefore acted as a partial agonist. The ligand 7929193 acted as a neutral antagonist as it had no effect on forskolin-stimulated cAMP response (anova Dunnett's test, P > 0.05, n = 3; Figure 3); 100 µM of each ligand was used in these experiments as this was predicted to achieve saturated receptor binding and maximal cAMP responses. Importantly, the respective inverse and partial agonist responses of ICI-118,551 and 5217377 were both sensitive to antagonism by 7929193 (Figure 4), confirming that they are mediated by β2-adrenoceptors, conform to the predictions of receptor activation models and that 7929193 is a true antagonist. Both produced significantly different cAMP responses when in the presence of the antagonist (unpaired t-test, P < 0.05, n = 3; Figure 4). In these experiments 100 µM 792913 and 1 µM of the efficacious ligands were used to optimize the detection of antagonism.
Figure 3.
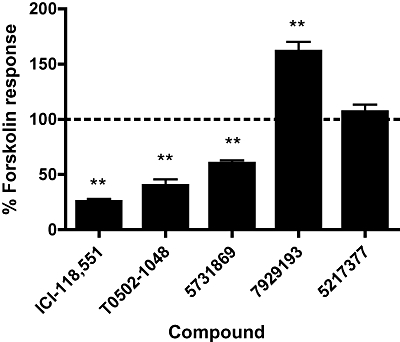
Efficacy of ICI-118,551 and the Cresset compounds for β2-adrenoceptor-mediated cAMP signalling. HEK239 cells stably expressing β2-adrenoceptors were co-incubated with 30 µM forskolin and 100 µM of each compound, and the resulting cAMP response is expressed as a percentage of the response to forskolin alone. Responses above and below 100% (dotted line) signified partial and inverse agonism respectively. Antagonism was signified by no deviation from this response. **P < 0.01 compared with forskolin alone. Data are an average ± SEM of three experiments performed in triplicate.
Figure 4.

Antagonism of inverse and partial agonists for the β2-adrenoceptor-mediated cAMP pathway. β2-adrenoceptor-expressing HEK293were incubated with 30 µM forskolin and stimulated with 1 µM ICI-118,551, JB-143 or 5217377 with or without 100 µM 7929193. The resulting cAMP responses from the treatments identified 7929193-mediated antagonism of both inverse and partial agonism. *P < 0.05 comparing the effect of antagonist on each ligand response. Data are an average ± SEM of three experiments performed in triplicate.
The compound's ability to evoke a β-arrestin recruitment response at β2-adrenoceptors was also investigated by imaging the subcellular translocation of a fluorescently tagged β-arrestin protein [GFP-βarr2(1–380)]. HEK293 cells were co-transfected with β2-adrenoceptors and the GFP-βarr2(1–380) reporter, and imaged with a confocal microscope. In transfected cells, GFP-βarr2(1–380) was predominantly located in the nucleus with a lesser, but still robust, cytoplasmic signal (Figure 5). After a 15 min incubation (at 37°C) with 100 µM ICI-118,551 or any of the compounds, no change in the subcellular location of GFP-βarr2(1–380) was identified (Figure 5). As a positive control, the ability of 100 µM adrenaline to elicit a response was tested. This treatment produced a noticeable subcellular translocation of GFP-βarr2(1–380) from the cytoplasm to the plasma membrane (Figure 5) in the majority of transfected cells (>75%), which was taken as evidence for β2-adrenoceptor-mediated β-arrestin recruitment. ICI-118,551 inhibited the ability of adrenaline to evoke this response (Figure 5), which suggests it acts as an antagonist.
Figure 5.
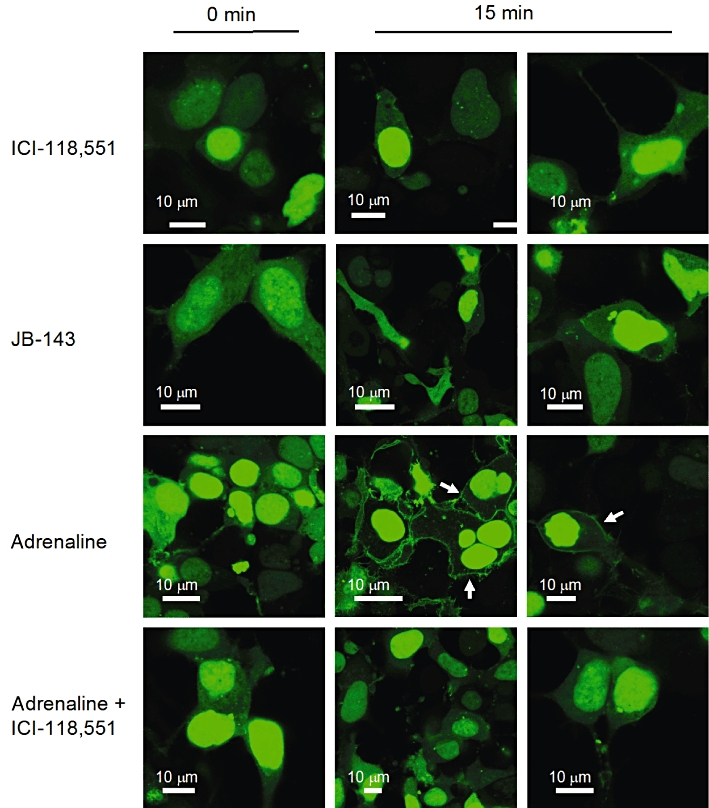
Ligand-mediated regulation of β2-adrenoceptor-β-arrestin interactions. HEK293 cells were transiently transfected with β2-adrenoceptors and GFP-βarr2(1–380), and subcellular localization of fluorescence was imaged by confocal microscopy. One image before (0 min) and two after (15 min) incubation with ligand are shown. Positive responses (membrane translocation) are highlighted with arrows. Images are a representation of at least 10 groups of cells from at least two separate experiments.
Pharmacological characterization of some structural derivatives of ICI-118,551
The compounds identified in the virtual field screen lacked desirable levels of potency, affinity and selectivity for β2-adrenoceptors. However, they displayed some notable structural features, including a piperidine ring in the case of T0502-1048 and 5731869 that are tolerated by β-adrenoceptors. We used this finding to design novel analogues of ICI-118,551 which would incorporate the piperidine or related cyclic amines, while retaining the core structure of ICI-118,551. Thus, six structural derivatives of ICI-118,551 were synthesized (JB-141, JB-142, JB-143, JB-163, JB-175, JB-176; Figure 6).
Figure 6.
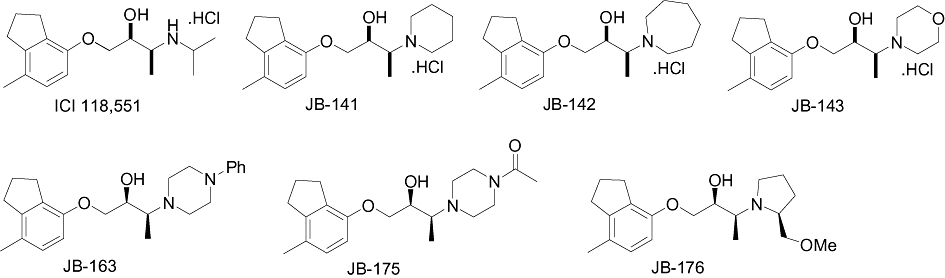
The chemical structures of ICI-118,551 and its derivatives. The compounds JB-163, JB-175 and JB-176 were synthesized and pharmacologically analysed as 2S,3S enantiopure preparations. The remaining compounds are racemates.
Gaddum–Schild analysis suggested that these compounds had higher pA2 values compared with the compounds identified by virtual field screening, although none were as potent as ICI-118,551. The pA2 values ranged from 7.23 to 9.03, with JB-175 displaying the highest affinity (Table 3). All the compounds produced Schild-plot slopes of below unity, with particularly low values of <0.5 for JB-141, JB-142 and JB-163 (Table 3). Re-analysis using only low concentrations of antagonist produced lower pA2 values and changed the rank order of potency of the compounds (Table 3). An example of the concentration-dependent antagonism that produces a low Schild-plot slope value is shown for JB-175 in Figure 7. In contrast to ICI-118,551, none of its structural derivatives had an effect on the ability of adrenaline to elicit a maximal relaxant response (anova Dunnett's test, P > 0.05, n = 3–4).
Table 3.
Gaddum–Schild analysis of structural derivatives of ICI-118,551 in the guinea pig trachea
| Compound | n | pA2 | Schild-plot slope | pA2 (adapted analysis) |
|---|---|---|---|---|
| JB-141 | 4 | 8.86 ± 0.12 | 0.49 ± 0.02 | 8.45 |
| JB-142 | 3 | 8.36 ± 0.24 | 0.49 ± 0.04 | 8.09 |
| JB-143 | 3 | 8.38 ± 0.10 | 0.57 ± 0.02 | 7.78 |
| JB-163 | 3 | 8.93 ± 0.41 | 0.26 ± 0.03 | 7.20 |
| JB-175 | 3 | 9.06 ± 0.16 | 0.50 ± 0.03 | 8.15 |
| JB-176 | 3 | 7.23 ± 0.11 | 0.57 ± 0.03 | 6.81 |
Figure 7.
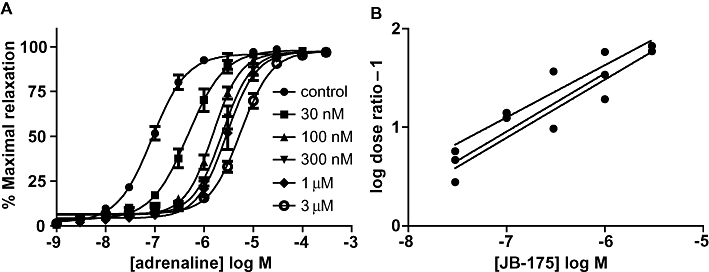
JB-175-mediated antagonism of adrenaline responses in the guinea-pig trachea. (A) Increasing concentrations of JB-175 produce increasing rightward shifts in the concentration–response curve to adrenaline. (B) The Schild-plot of these responses constructed by plotting log (dose ratio – 1) by concentration of JB-175. Results of each individual experiment are plotted together with their respective linear regression lines. This illustrates the low slope of these curves.
In [3H]-CGP-12177 competition binding experiments the structural derivatives of ICI-118,551 bound with a range of affinities to β2-adrenoceptors, although none had as high a binding affinity as ICI-118,551 Table 4). JB-175 had the highest affinity with a pKi of 7.84. These compounds all showed good β2-adrenoceptor selectivity with >30-fold to >480-fold binding preferences (Table 4). JB-141 was the most selective, although this did not reach the high levels of selectivity achieved with ICI-118,551.
Table 4.
[3H]-CGP-12177 competition binding data for structural derivatives of ICI-118,551
| pKi | β2-Adrenoceptor selectivity (ratio) | |||
|---|---|---|---|---|
| Compound | n | β1-Adrenoceptor | β2-Adrenoceptor | |
| JB-141 | 3 | 5.05 ± 0.14 | 7.74 ± 0.05 | 487.86 |
| JB-142 | 3 | 5.12 ± 0.18 | 7.09 ± 0.06 | 92.04 |
| JB-143 | 3 | 4.71 ± 0.12 | 7.37 ± 0.06 | 455.95 |
| JB-163 | 4 | 5.64 ± 0.10 | 7.20 ± 0.07 | 36.55 |
| JB-175 | 5 | 5.15 ± 0.07 | 7.84 ± 0.06 | 482.26 |
| JB-176 | 3 | 5.19 ± 0.09 | 6.76 ± 0.09 | 37.55 |
In the cAMP assay, 100 µM of all the structural derivatives of ICI-118,551 significantly inhibited the ability of forskolin to elicit a response (anova Dunnett's test, P < 0.01, n = 3; Figure 8), thus identifying them as inverse agonists. The sensitivity of the inverse agonism of JB-143 to receptor antagonism (with 7929193) was tested, and its negative responses were significantly less than without antagonism (unpaired t-test, P < 0.05, n = 3; Figure 4). This was taken as evidence that these are true inverse agonist effects. The pertussis toxin (PTX)-sensitivity of inverse agonism with ICI-118,551 and JB-143 was investigated to determine whether specific β2-adrenoceptor-Gi-coupling with these ligands plays a role in the negative responses. PTX-pretreatment had an effect on the ability of forskolin to elicit a cAMP response alone, so this difference was taken into account when comparing PTX-treated and untreated cells. The negative response to ICI-118,551 or JB-143 was unaffected by PTX (unpaired t-test, P > 0.05, n = 3; Figure 9). Equivalent inverse agonist, partial agonist and neutral antagonist effects of the novel ligands, were also identifiable without the use of forskolin stimulation (data not shown).
Figure 8.
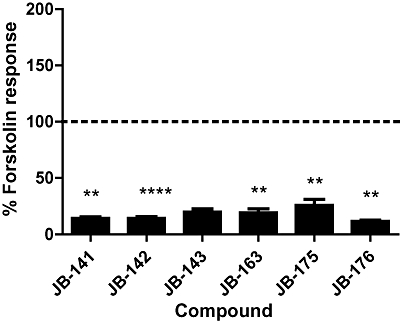
Efficacy of the structural derivatives of ICI-118,551 for β2-adrenoceptor-mediated cAMP signalling. HEK239 cells stably expressing β2-adrenoceptors were co-incubated with 30 µM forskolin and 100 µM of each compound, and the resulting cAMP response is expressed as a percentage of the response to forskolin alone. Responses above and below 100% (dotted line) signify partial and inverse agonism respectively. Antagonism was signified by no deviation from this response. **P < 0.01 compared with forskolin alone. Data are an average ± SEM of three experiments performed in triplicate.
Figure 9.
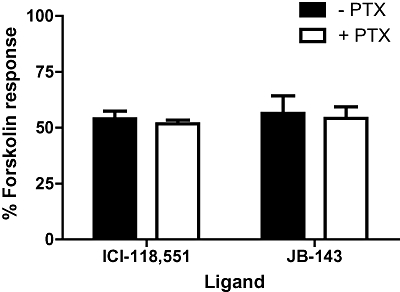
The effect of pertussis toxin (PTX) on the negative cAMP responses of ICI-118,551 and JB-143. HEK239 cells stably expressing β2-adrenoceptors were pretreated with or without 10 ng·mL−1 PTX for 15 h. They were then co-incubated with forskolin and ICI-118,551 or JB-143, and cAMP responses measured. As PTX had an effect on forskolin responses alone, each ligand-mediated response is expressed as a percentage of the respective forskolin response with or without PTX. Data are an average ± SEM of three experiments performed in triplicate.
None of the derivatives of ICI-118,551 had an effect on the subcellular localization of GFP-βarr2(1–380) (Figure 5). This suggested that they were not efficacious for this β2-adrenoceptor pathway, as was the case with the other β-blockers tested.
Stereoselective binding properties of JB-141, JB-143 and JB-175
The stereochemistry of the most potent and selective novel ligands JB-141, JB-143 and JB-175 were investigated with regards to their β-adrenoceptor binding affinities and selectivities, as determined previously for ICI-118,551 (Baker et al., 2010). Racemic (2S,3S)-enantiopure and (2R,3R)-enantiopure preparations of these ligands were subjected to [3H]-CGP-12177 competition binding analysis. The (2S,3S) enantiomers of these compounds had far higher affinity for both β1- and β2-adrenoceptors than the antipode (anova Bonferroni's test, P < 0.001, n≥ 3, Table 5). Interestingly, the relative enantiospecific differences in binding affinity appeared to be non-equivalent at the two receptors, which had an effect on the degree of β2-adrenoceptor selectivity with the enantiopure preparations compared with the racemates (Table 5). The (2S,3S) enantiomers were less selective than the racemates because the relative increases in β1-adrenoceptor binding were greater than that at β2-adrenoceptor. Conversely, the (2R,3R) enantiomers were also less selective than the racemates because the relative drops in β2-adrenoceptor binding were greater than at β1-adrenoceptors. Therefore, in all instances the racemate was the most selective for β2-adrenoceptors. In previous experiments, JB-175 was characterized as a (2S,3S) enantiomer, and upon investigation of the racemate this was found to be more selective for β2-adrenoceptors than ICI-118,551, having a >950-fold binding preference for this receptor.
Table 5.
[3H]-CGP-12177 competition binding analysis with racemic and enantiopure preparations of some β2-adrenoceptor selective β-blockers
| Compound (preparation) | pKi | β2-Adrenoceptor selectivity (ratio) | ||
|---|---|---|---|---|
| n | β1-Adrenoceptor | β2-Adrenoceptor | ||
| JB-141 | ||||
| Racemate | 3 | 5.05 ± 0.14 | 7.74 ± 0.05 | 488 |
| (2S,3S)-enantiomer | 4 | 5.70 ± 0.11 | 8.03 ± 0.08 | 213 |
| (2R,3R)-enantiomer | 3 | 4.02 ± 0.45 | 6.37 ± 0.12 | 222 |
| JB-143 | ||||
| Racemate | 3 | 4.71 ± 0.12 | 7.37 ± 0.06 | 456 |
| (2S,3S)-enantiomer | 3 | 4.98 ± 0.09 | 7.54 ± 0.05 | 364 |
| (2R,3R)-enantiomer | 3 | <3.94 | 5.92 ± 0.05 | >95 |
| JB-175 | ||||
| Racemate | 4 | 3.98 ± 0.44 | 6.97 ± 0.10 | 960 |
| (2S,3S)-enantiomer | 5 | 5.15 ± 0.07 | 7.84 ± 0.06 | 482 |
| (2R,3R)-enantiomer | 4 | 3.94 ± 0.47 | 6.42 ± 0.12 | 295 |
The β-adrenoceptor binding affinities (pKi) of racemic, (2S,3S)- and (2R,3R)-enantiopure preparations of JB-141, JB-143 and JB-175 were calculated. From these values, β2-adrenoceptor-selectivty was derived.
Discussion and conclusions
Summary
In an effort to develop novel β2-adrenoceptor-selective ligands we initially employed virtual field screening. The four novel ligands identified through this route had a variety of efficacies (inverse agonism, partial agonism and neutral antagonism) but showed only modest antagonism, affinity (low µM range) and selectivity (at most 10-fold), and were therefore not promising leads in their own right. However, analysis of the structure of T0502-1048 and 5731869 served to reveal to us that cyclic amines could be tolerated by the β2-adrenoceptor. We chose to combine this structural motif with the core of ICI-118,551 and we felt confident of accessing new β2-selective antagonists in this manner. We thus synthesized six novel ICI-118,551 analogues which varied just in the nature of cyclic amine (JB-141, JB-142, JB-143, JB-163, JB-175, JB-176; Figure 6). We found that they were all more pharmacologically similar to ICI-118,551, showing generally greater antagonism, higher affinity, moderate-to-good selectivity and inverse agonism. One of the compounds (racemic JB-175) had higher β2-adrenoceptor selectivity than ICI-118,551.
Pharmacology
The ligands inhibited adrenaline-mediated responses in the guinea-pig trachea and this response was attributed to β2-adrenoceptor antagonism because this receptor predominates in this tissue (Carswell and Nahorski, 1983; Tanaka et al., 2005). Gaddum–Schild analysis suggested that JB-175 had the highest affinity of the novel antagonists (pA2 = 9.06), although this was not as high that for ICI-118,551. These compounds had low Schild-plot slope values and the responses were re-analysed by an alternative method. This produced lower pA2 values and affected the rank order of antagonist affinity of these ligands. Low Schild-plot slope values in the guinea-pig trachea have been demonstrated to be influenced by the use of non-selective β-adrenoceptor agonists, a heterogeneous β-adrenoceptor population in the tissue, and an effect of tissue precontraction, while saturable agonist uptake sites and agonist degradation are also possible contributory factors (O'Donnell and Wanstall, 1979; 1980;Keith et al., 1986; Christopoulos and Kenakin, 2002). A likely explanation for the low slope values in this assay is that at higher concentrations adrenaline activates additional receptor subtypes to drive the functional responses and thus invalidate the assumption of the Schild equation that the agonist and antagonist are acting through a single receptor site. As it has been suggested that the β1-adrenoceptor is present in this tissue (O'Donnell and Wanstall, 1979; Tanaka et al., 2007), this is the most likely candidate. The observation that the slope values of the novel compounds are relatively variable between the ligands (0.26–0.88; Tables 1 and 3), suggests that pharmacological differences between them may account for at least part of this effect on the analysis. Allosteric receptor interactions, inverse or partial agonism, and off target effects have been identified as potential mechanisms for an unusual Schild-plot, as they can interfere with the assumption that the test compound acts as a neutral competitive antagonist (Ghosh et al., 1999; Christopoulos and Kenakin, 2002). Interestingly, ICI-118,551 acted as an insurmountable antagonist owing to its ability to decrease the maximal responses attainable by the agonist. This is in agreement with a number of previous reports (Hopkinson et al., 2000) and substantiates the notion that unusual pharmacological properties of β-blockers may influence their antagonist effects.
Radioligand competition binding experiments were used to determine the β-adrenoceptor binding affinities and the β2 selectivity of the ligands. All the ligands showed a binding preference for β2- over β1-adrenoceptors, although this varied greatly. ICI-118,551 had high affinity and selectivity for β2-adrenoceptors. None of the novel compounds were an improvement on ICI-118,551 in terms of these parameters, although JB-141, JB-143 and JB-175 demonstrated impressive selectivity levels (over 450-fold). Although β2-adrenoceptor-selective blockade is therefore attainable with these ligands, a far higher concentration compared with ICI-118,551 is needed to achieve this, owing to their lower affinities for β2-adrenoceptors.
The ligands were tested for efficacy at two β2-adrenoceptor-mediated pathways; Gs-cAMP and β-arrestin. Within the context of receptor activation models, efficacy is influenced by a ligand's ability to disturb the equilibrium between active (R*) and inactive (R) receptor conformations and therefore evoke a signalling response (Christopoulos and Kenakin, 2002). ICI-118,551 and most of the novel compounds were inverse agonists as their activity at β2-adrenoceptors caused a decrease in cellular cAMP levels suggesting a selective interaction with R. The 5217377 evoked a positive cAMP response, which suggests that this ligand selectively interacts with and enriches R* and therefore activates the receptor. The 7929193 had no effect on cellular cAMP, which was evidence that it has no binding preference for R or R*, does not influence the receptor equilibrium and is a neutral antagonist. This ligand was particularly interesting as receptor/ligand modelling systems and experimental work have led to the hypothesis that neutral antagonism is a rare occurrence (Kenakin, 2004).
Some β-blockers have been demonstrated to have positive effects (are agonists) for a number of β2-adrenoceptor-mediated signalling pathways including β-arrestin recruitment, activation of different MAPK cascades and/or Gi-coupling (Azzi et al., 2003; Baker et al., 2003b; Zheng et al., 2005; Wisler et al., 2007; Drake et al., 2008; Wenzel et al., 2009). That these occur in a ligand-specific manner is evidence that there are multiple active β2-adrenoceptor conformations that differentially interact with β-adrenoceptor ligands. None of these compounds affected the subcellular localization of β-arrestin or had a Gi dependence of action. Indeed, the ability of ICI-118,551 to inhibit adrenaline-mediated β-arrestin responses suggests it acts as an antagonist. These findings substantiate previous reports that ICI-118,551 is without effect alone and can block the effects of efficacious ligands (Wisler et al., 2007; Drake et al., 2008), although they contradict the findings of Azzi et al. (2003), who demonstrated a positive β-arrestin response with this ligand. It is probable that differences in cellular context and assay sensitivity underlie these different observations. For example, putative β-blocker-mediated β-arrestin responses may be smaller than those to agonists, and more sensitive experimental approaches, such as DiscoveRx (McGuinness et al., 2009) or fluorescence resonance energy transfer/bioluminescence resonance energy transfer (Azzi et al., 2003; Wisler et al., 2007), could demonstrate positive responses to these ligands. The efficacy of β-blockers for β2-adrenoceptor-β-arrestin signalling is important as it has been suggested that the improved clinical effects of carvedilol in heart failure pharmacotherapy are influenced by its ability to evoke a β-arrestin response (Wisler et al., 2007).
Stereochemistry
The stereoselectivity of β-blockers is well established for the β1-adrenoceptor with the (S) enantiomers usually showing greater activity than their antipodes (Morris and Kaumann, 1984; Wahlund et al., 1990). There are data showing that stereoisomers of fenoterol, a β2 selective agonist, may couple to different G-protein signalling pathways (Woo et al., 2009). It is unclear what the effect of varying the stereochemistry is on inverse agonists/antagonists at the β2-adrenoceptor. We therefore tested differences between racemates and individual enantiomers of the most promising novel compounds (JB-141, JB-143, JB-175). Radioligand binding experiments provided strong evidence that the (2S,3S) enantiomers of these ligands bound with the highest affinity. Unusually, however, racemic preparations were the most selective for the β2-adrenoceptor, and this is in line with our findings on ICI-118,551 itself (Baker et al., 2010). This can be explained by the fact that the relative enantiospecific differences in β1- and β2-adrenoceptor binding affinity are not equivalent: the gain in affinity of the (2S,3S) enantiomer is greater at β1-adrenoceptor while the drop in affinity of the (2R,3R) enantiomer is greater at β2-adrenoceptor. This occurrence is puzzling as the racemates comprise an equimolar combination of the two enantiomers, and their activity is therefore predicted to be an average of these (i.e. an intermediate selectivity). This is clearly not the case, which suggests that some form of functional interaction between the two enantiomers causes their respective binding affinities to be quantitatively different when they are in combination in the racemate. A proposed mechanism for this effect could involve enantiospecific allosteric interactions with either receptor that alters the orthosteric binding properties. An allosteric binding site on β1-adrenoceptor with affinity for β-blockers has been identified (Baker et al., 2003a; Joseph et al., 2004), while we know of no evidence that such a site exists on β2-adrenoceptor. This perhaps makes β1-adrenoceptors a more likely candidate for allosteric interactions. The unusual stereospecific β2-adrenoceptor selectivities of these ligands may provide novel insights into the activity of racemic preparations of β-blockers, and suggests that the highest receptor selectivity is attainable with the racemate. Indeed, JB-175, was found to be the most selective β2-adrenoceptor ligand when tested as a racemate, which suggests that it may have a better selectivity than ICI-118,551.
Molecular modelling
In a recent study, the available crystal structures of β2-adrenoceptors were used to screen chemical libraries using docking based approaches to identify novel ligands (Kolb et al., 2009). They succeeded in doing so although most were inverse agonists and they did not look at β1/β2-adrenoceptor selectivity. It is telling that the current crystal structures have been elucidated in the presence of an inverse agonist (Cherezov et al., 2007; Rasmussen et al., 2007; Rosenbaum et al., 2007; Warne et al., 2008). Thus we attempted to model the binding of JB-175 with the published X-ray structures [from protein data bank (PDB) entries 2RH1.PDB (β2-adrenoceptor/Carazolol) and 2VT4.PDB (β1-adrenoceptor/Cyanopindolol)]. Over a dozen ligands were incorporated into each receptor and computationally ‘relaxed’ to their lowest achievable energetic stable state. The act of computationally optimizing ligands in each receptor yields enthalpies that may indicate the strengths of ligand binding from which selectivity may be inferred. Because real binding strengths include entropy, solvation and other dynamic effects, the calculated enthalpies are approximate, but scrutiny of Table 6 suggests a correlation between major biological action and calculated binding strength. For example, propranolol, which has a small experimental β2 preference, is electrostatically bound by −18.2 kcal·mol−1 at β2 and by −17.8 kcal·mol−1 at β1. Atenolol shows the expected β1 preference and both tight binders; carazolol and cyanopindolol have strong binding enthalpies. Furthermore the selectivity of ICI-118,551 (and JB compounds) for β2 over β1 is also predicted from these calculations. Figure 10 shows the two β-receptors both containing JB-175 after molecular mechanics relaxation over the entire ensembles along with the superposition of the structures by least squares fit of the Cα backbone atoms showing little conformational variation of the backbone or the ligand position (Figure 10C). This insignificant change was observed for all ligands so far replaced into the receptors. This conclusion from molecular modelling is supported by the recent crystal structure of ICI-118,551 bound to the β2-receptor (Wacker et al., 2010). In addition, the predicted conformation of ICI-118,551 in the binding pocket agrees closely with that observed in the structure (not shown). Thus the small differences in the inverse agonist bound β1 and β2 crystal structures are enough to account for ligand selectivity with inverse agonists and antagonists. Perhaps more surprisingly agonist selectivity is also predicted with adrenaline showing no selectivity and salbutamol confirmed as strongly β2-selective. The enthalpies for the fenoterol enantiomers, although correctly showing β2 preference, suggest that the reported less active enantiomer has the higher binding enthalpy. There is much speculation as to the conformation of the agonist activated state (Audet and Bouvier, 2008). However, it is also possible that small variations in ligand structure may also lead to changes in electrostatic forces at specific points around the accessible surface of the receptor. Such subtleties could help explain the differences in biological action, such as ligand-selective agonism, seen with some pharmacophores.
Table 6.
Computed binding enthalpies of selected β1- and β2-adrenoceptor ligands
| β2-Adrenoceptor 2RH1 | β1-Adrenoceptor 2VT4 | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Ligand | Major action | Total | LB | coul LB | vdw LB | Total | LB | coul LB | vdw LB |
| None | 2760 | 2677 | |||||||
| Antagonists | |||||||||
| Cyanopindolol | β1/β2-adrenoceptor Ant | 2716 | −91.8 | −26.2 | −65.6 | 2633 | −92.2 | −27.9 | −64.8 |
| Carazolol | β1/β2-adrenoceptor In Ag | 2723 | −98.3 | −23.9 | −74.4 | 2641 | −90.6 | −20.7 | −69.9 |
| ICI118551 | β2-adrenoceptor In Ag | 2727 | −83.6 | −12.3 | −71.3 | 2620 | −75.2 | −8.5 | −66.7 |
| JB-175 | β2-adrenoceptor In Ag | 2743 | −86.7 | −12.2 | −74.5 | 2637 | −82.1 | −9.7 | −72.4 |
| JB-143 | β2-adrenoceptor In Ag | 2718 | −84.4 | −13.4 | −70.9 | 2597 | −84.7 | −7.9 | −76.8 |
| Atenolol | β1-adrenoceptor In Ag | 2708 | −78.7 | −22.6 | −56.1 | 2629 | −84.1 | −28.4 | −55.7 |
| Propranolol | β2-adrenoceptor In Ag | 2699 | −85.5 | −18.2 | −67.2 | 2622 | −81.7 | −17.8 | −63.8 |
| Agonists | |||||||||
| Salbutamol | β2-adrenoceptor Ag | 2702 | −72.8 | −19.1 | −53.7 | 2621 | −66.3 | −14.4 | −51.9 |
| Prenalterol | β1-adrenoceptor Ag | 2712 | −72.2 | −20.9 | −51.3 | 2628 | −67.7 | −16.6 | −51.1 |
| FenoterolRR | β2-adrenoceptor Ag | 2694 | −89.4 | −17.9 | −71.5 | 2629 | −77.4 | −15.9 | −61.6 |
| FenoterolSR | β2-adrenoceptor Ag | 2687 | −94.1 | −24.4 | −69.7 | 2628 | −77.9 | −13.4 | −64.5 |
| Adrenaline | β1/β2-adrenoceptor Ag | 2727 | −60.1 | −16.4 | −43.7 | 2632 | −55.6 | −16.6 | −39 |
Each of the 12 ligands in column 1 were placed into the two receptor X-ray structures in place of their native ligands and optimized to a minimum overall energy (columns 3 & 7) using the eXtended Electron Distribution molecular mechanics force field. Once optimized, the overall ligand binding enthalpy was calculated (LB columns 4 & 8) as two contributions; electrostatic (coulombic) enthalpy (coul LB columns 5 & 9) and van der Waals (hydrophobic) enthalpy (vdw LB columns 6 & 10). Antagonist, inverse agonist and agonist are abbreviated to Ant, In Ag and Ag respectively.
Figure 10.
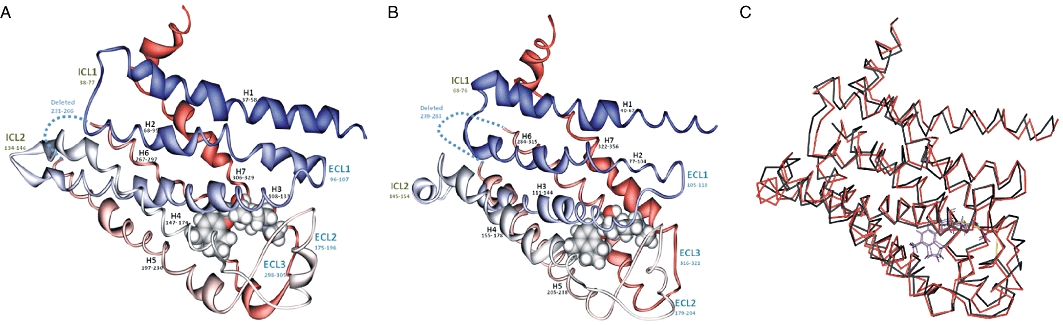
Conformation changes in the β-adrenoceptor on ligand binding. (A) Carazolol was replaced by JB-175 in the β2-adrenoceptor X-ray structure (2RH1.PDB) (B) Cyanopindolol was replaced by JB-175 in the β1-adrenoceptor X-ray structure (2VT4.PDB). In each case, the whole ensemble was fully optimized using the eXtended Electron Distribution molecular mechanics force field. The least squares Cα backbone root mean square difference between the fully optimized empty β2-receptor and the receptor containing JB-175 was 0.49 and that for the β1-adrenoceptor was 0.23 (C) Superimposition of the two optimized receptors indicates the relatively small change in ligand alignment (red; β1-adrenoceptor with magenta ligand, black; β2-adrenoceptor with grey ligand).
Conclusions
Ten novel β2-adrenoceptor antagonists were identified and found to have different pharmacological profiles. In terms of improving on the pharmacology of the ligand ICI-118,551, one of the compounds was more selective (racemic JB-175), while one was a neutral antagonist (7929193), although none were as potent. Further work may provide novel ligands that more successfully combine these pharmacologies. Nevertheless, the results substantiate the notion that β-blockers do more than simply inhibit receptor activation, and interesting variation between ligands could provide useful tools to investigate the receptor. Additionally, intriguing enantiospecific behaviour of some of the novel ligands provides insights into receptor binding.
Acknowledgments
This work was supported by the Dorset Foundation and the British Heart Foundation.
Glossary
Abbreviations
- HEK293
human embryonic kidney cells
Conflict of interest
The authors have no conflicts of interest to declare.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Appendix S1 Supplementary methods.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Affolter H, Hertel C, Jaeggi K, Portenier M, Staehelin M. (−)-S-[3H]CGP-12177 and its use to determine the rate constants of unlabeled beta-adrenergic antagonists. Proc Natl Acad Sci U S A. 1985;82:925–929. doi: 10.1073/pnas.82.3.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alewijnse AE, Smit MJ, Rodriguez Pena MS, Verzijl D, Timmerman H, Leurs R. Modulation of forskolin-mediated adenylyl cyclase activation by constitutively active G(S)-coupled receptors. FEBS Lett. 1997;419:171–174. doi: 10.1016/s0014-5793(97)01440-3. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 4th edition. Br J Pharmacol. 2009;158(Suppl. 1):S1–254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audet M, Bouvier M. Insights into signaling from the beta2-adrenergic receptor structure. Nat Chem Biol. 2008;4:397–403. doi: 10.1038/nchembio.97. [DOI] [PubMed] [Google Scholar]
- Azzi M, Charest PG, Angers S, Rousseau G, Kohout T, Bouvier M, et al. Beta-arrestin-mediated activation of MAPK by inverse agonists reveals distinct active conformations for G protein-coupled receptors. Proc Natl Acad Sci U S A. 2003;100:11406–11411. doi: 10.1073/pnas.1936664100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker JG. The selectivity of beta-adrenoceptor antagonists at the human beta1, beta2 and beta3 adrenoceptors. Br J Pharmacol. 2005;144:317–322. doi: 10.1038/sj.bjp.0706048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker JG, Hall IP, Hill SJ. Agonist actions of ‘beta-blockers’ provide evidence for two agonist activation sites or conformations of the human beta1-adrenoceptor. Mol Pharmacol. 2003a;63:1312–1321. doi: 10.1124/mol.63.6.1312. [DOI] [PubMed] [Google Scholar]
- Baker JG, Hall IP, Hill SJ. Agonist and inverse agonist actions of beta-blockers at the human beta 2-adrenoceptor provide evidence for agonist-directed signaling. Mol Pharmacol. 2003b;64:1357–1369. doi: 10.1124/mol.64.6.1357. [DOI] [PubMed] [Google Scholar]
- Baker JR, Hothersall JD, Fitzmaurice RJ, Tucknott M, Vinter A, Tinker A, et al. An efficient asymmetric synthesis of the potent beta-blocker ICI-118,551 allows the determination of enantiomer dependency on biological activity. Chem Commun. 2010;46:3953–3954. doi: 10.1039/c0cc00142b. [DOI] [PubMed] [Google Scholar]
- Bilski AJ, Halliday SE, Fitzgerald JD, Wale JL. The pharmacology of a beta 2-selective adrenoceptor antagonist (ICI 118,551) J Cardiovasc Pharmacol. 1983;5:430–437. doi: 10.1097/00005344-198305000-00013. [DOI] [PubMed] [Google Scholar]
- Carswell H, Nahorski SR. Beta-adrenoceptor heterogeneity in guinea-pig airways: comparison of functional and receptor labelling studies. Br J Pharmacol. 1983;79:965–971. doi: 10.1111/j.1476-5381.1983.tb10542.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cejas MA, Kinney WA, Chen C, Vinter JG, Almond HR, Jr, Balss KM, et al. Thrombogenic collagen-mimetic peptides: self-assembly of triple helix-based fibrils driven by hydrophobic interactions. Proc Natl Acad Sci U S A. 2008;105:8513–8518. doi: 10.1073/pnas.0800291105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheeseright T, Mackey M, Rose S, Vinter A. Molecular field extrema as descriptors of biological activity: definition and validation. J Chem Inf Model. 2006;46:665–676. doi: 10.1021/ci050357s. [DOI] [PubMed] [Google Scholar]
- Cheeseright TJ, Mackey MD, Melville JL, Vinter JG. FieldScreen: virtual screening using molecular fields. Application to the DUD data set. J Chem Inf Model. 2008;48:2108–2117. doi: 10.1021/ci800110p. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Prusoff WH. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, et al. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chessari G, Hunter CA, Low CM, Packer MJ, Vinter JG, Zonta C. An evaluation of force-field treatments of aromatic interactions. Chemistry. 2002;8:2860–2867. doi: 10.1002/1521-3765(20020703)8:13<2860::AID-CHEM2860>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Christopoulos A, Kenakin T. G protein-coupled receptor allosterism and complexing. Pharmacol Rev. 2002;54:323–374. doi: 10.1124/pr.54.2.323. [DOI] [PubMed] [Google Scholar]
- CIBIS Investigators and Committees. A randomized trial of beta-blockade in heart failure. The Cardiac Insufficiency Bisoprolol Study (CIBIS). CIBIS Investigators and Committees. Circulation. 1994;90:1765–1773. doi: 10.1161/01.cir.90.4.1765. [DOI] [PubMed] [Google Scholar]
- Daaka Y, Luttrell LM, Lefkowitz RJ. Switching of the coupling of the b2-adrenergic receptor to different G proteins by protein kinase A. 1997. pp. 88–91. [DOI] [PubMed]
- Drake MT, Violin JD, Whalen EJ, Wisler JW, Shenoy SK, Lefkowitz RJ. beta-arrestin-biased agonism at the beta2-adrenergic receptor. J Biol Chem. 2008;283:5669–5676. doi: 10.1074/jbc.M708118200. [DOI] [PubMed] [Google Scholar]
- Fitzgerald JD. The applied pharmacology of beta-adrenoceptor antagonists (beta blockers) in relation to clinical outcomes. Cardiovasc Drugs Ther. 1991;5:561–576. doi: 10.1007/BF03029726. [DOI] [PubMed] [Google Scholar]
- Ghosh K, Kowal D, Dawson LA, Tasse R. Design and models for estimating antagonist potency (pA2, Kd and IC50) following the detection of antagonism observed in the presence of intrinsic activity. Neuropharmacology. 1999;38:361–373. doi: 10.1016/s0028-3908(98)00185-3. [DOI] [PubMed] [Google Scholar]
- Giblin JP, Leaney JL, Tinker A. The molecular assembly of ATP-sensitive potassium channels: determinants on the pore forming subunit. J Biol Chem. 1999;274:22652–22659. doi: 10.1074/jbc.274.32.22652. [DOI] [PubMed] [Google Scholar]
- Gong H, Sun H, Koch WJ, Rau T, Eschenhagen T, Ravens U, et al. Specific beta(2)AR blocker ICI 118,551 actively decreases contraction through a G(i)-coupled form of the beta(2)AR in myocytes from failing human heart. Circulation. 2002;105:2497–2503. doi: 10.1161/01.cir.0000017187.61348.95. [DOI] [PubMed] [Google Scholar]
- Harding SE, Gong H. beta-adrenoceptor blockers as agonists: coupling of beta2-adrenoceptors to multiple G-proteins in the failing human heart. Congest Heart Fail. 2004;10:181–185. doi: 10.1111/j.1527-5299.2004.02052.x. [DOI] [PubMed] [Google Scholar]
- Hopkinson HE, Latif ML, Hill SJ. Non-competitive antagonism of beta(2)-agonist-mediated cyclic AMP accumulation by ICI 118551 in BC3H1 cells endogenously expressing constitutively active beta(2)-adrenoceptors. Br J Pharmacol. 2000;131:124–130. doi: 10.1038/sj.bjp.0703535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph SS, Lynham JA, Colledge WH, Kaumann AJ. Binding of (−)-[3H]-CGP12177 at two sites in recombinant human beta 1-adrenoceptors and interaction with beta-blockers. Naunyn Schmiedebergs Arch Pharmacol. 2004;369:525–532. doi: 10.1007/s00210-004-0884-y. [DOI] [PubMed] [Google Scholar]
- Keith RA, Donahue JY, Salama AI. The effect of varying carbachol concentration on the slope of Schild plots of selective beta-adrenoceptor antagonists in the carbachol-contracted guinea-pig trachea. J Pharm Pharmacol. 1986;38:107–112. doi: 10.1111/j.2042-7158.1986.tb04521.x. [DOI] [PubMed] [Google Scholar]
- Kenakin T. Differences between natural and recombinant G protein-coupled receptor systems with varying receptor/G protein stoichiometry. Trens Pharmacol Sci. 1997;18:456–464. doi: 10.1016/s0165-6147(97)01136-x. [DOI] [PubMed] [Google Scholar]
- Kenakin T. Efficacy as a vector: the relative prevalence and paucity of inverse agonism. Mol Pharmacol. 2004;65:2–11. doi: 10.1124/mol.65.1.2. [DOI] [PubMed] [Google Scholar]
- Kenakin TP. A Pharmacology Primer: Theory, Applications and Methods. 2nd edn. San Diego, CA: Academic Press; 2006. [Google Scholar]
- Kolb P, Rosenbaum DM, Irwin JJ, Fung JJ, Kobilka BK, Shoichet BK. Structure-based discovery of beta2-adrenergic receptor ligands. Proc Natl Acad Sci U S A. 2009;106:6843–6848. doi: 10.1073/pnas.0812657106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuinness D, Malikzay A, Visconti R, Lin K, Bayne M, Monsma F, et al. Characterizing cannabinoid CB2 receptor ligands using DiscoveRx PathHunter beta-arrestin assay. J Biomol Screen. 2009;14:49–58. doi: 10.1177/1087057108327329. [DOI] [PubMed] [Google Scholar]
- MERIT-HF Study Group. Effect of metoprolol CR/XL in chronic heart failure: metoprolol CR/XL Randomised Intervention Trial in Congestive Heart Failure (MERIT-HF) Lancet. 1999;353:2001–2007. [PubMed] [Google Scholar]
- Mikami M, Goubaeva F, Song JH, Lee HT, Yang J. beta-Adrenoceptor blockers protect against staurosporine-induced apoptosis in SH-SY5Y neuroblastoma cells. Eur J Pharmacol. 2008;589:14–21. doi: 10.1016/j.ejphar.2008.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris TH, Kaumann AJ. Different steric characteristics of beta 1- and beta 2-adrenoceptors. Naunyn Schmiedebergs Arch Pharmacol. 1984;327:176–179. doi: 10.1007/BF00500913. [DOI] [PubMed] [Google Scholar]
- O'Donnell SR, Wanstall JC. The importance of choice of agonist in studies designed to predict beta 2 : beta 1 adrenoceptor selectivity of antagonists from pA2 values on guinea-pig trachea and atria. Naunyn Schmiedebergs Arch Pharmacol. 1979;308:183–190. doi: 10.1007/BF00501381. [DOI] [PubMed] [Google Scholar]
- O'Donnell SR, Wanstall JC. Are the pA2 values of selective beta-adrenoceptor antagonists valid when obtained on guinea-pig tracheal preparations contracted with carbachol? J Pharm Pharmacol. 1980;32:413–416. doi: 10.1111/j.2042-7158.1980.tb12954.x. [DOI] [PubMed] [Google Scholar]
- Packer M, Bristow MR, Cohn JN, Colucci WS, Fowler MB, Gilbert EM, et al. The effect of carvedilol on morbidity and mortality in patients with chronic heart failure. U.S. Carvedilol Heart Failure Study Group. N Engl J Med. 1996;334:1349–1355. doi: 10.1056/NEJM199605233342101. [DOI] [PubMed] [Google Scholar]
- Poole-Wilson PA, Swedberg K, Cleland JG, Di Lenarda A, Hanrath P, Komajda M, et al. Comparison of carvedilol and metoprolol on clinical outcomes in patients with chronic heart failure in the Carvedilol Or Metoprolol European Trial (COMET): randomised controlled trial. Lancet. 2003;362:7–13. doi: 10.1016/S0140-6736(03)13800-7. [DOI] [PubMed] [Google Scholar]
- Rasmussen SG, Choi HJ, Rosenbaum DM, Kobilka TS, Thian FS, Edwards PC, et al. Crystal structure of the human beta2 adrenergic G-protein-coupled receptor. Nature. 2007;450:383–387. doi: 10.1038/nature06325. [DOI] [PubMed] [Google Scholar]
- Rosenbaum DM, Cherezov V, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, et al. GPCR engineering yields high-resolution structural insights into beta2-adrenergic receptor function. Science. 2007;318:1266–1273. doi: 10.1126/science.1150609. [DOI] [PubMed] [Google Scholar]
- Salomon Y. Cellular responsiveness to hormones and neurotransmitters: conversion of [3H]adenine to [3H]cAMP in cell monolayers, cell suspensions, and tissue slices. In: Johnson RA, Corbin JD, editors. Methods in Enzymology. New York: Academic Press; 1991. pp. 22–28. [DOI] [PubMed] [Google Scholar]
- Sutkowski EM, Tang WJ, Broome CW, Robbins JD, Seamon KB. Regulation of forskolin interactions with type I, II, V, and VI adenylyl cyclases by Gs alpha. Biochemistry. 1994;33:12852–12859. doi: 10.1021/bi00209a017. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Yamashita Y, Horinouchi T, Koike K. Adrenaline produces the relaxation of guinea-pig airway smooth muscle primarily through the mediation of beta(2)-adrenoceptors. J Smooth Muscle Res. 2005;41:153–161. doi: 10.1540/jsmr.41.153. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Yamashita Y, Michikawa H, Horinouchi T, Koike K. Pharmacological characterization of the beta-adrenoceptor that mediates the relaxant response to noradrenaline in guinea-pig tracheal smooth muscle. Naunyn Schmiedebergs Arch Pharmacol. 2007;375:51–64. doi: 10.1007/s00210-006-0130-x. [DOI] [PubMed] [Google Scholar]
- Teixeira CE, Baracat JS, Zanesco A, Antunes E, De NG. Atypical beta-adrenoceptor subtypes mediate relaxations of rabbit corpus cavernosum. J Pharmacol Exp Ther. 2004;309:587–593. doi: 10.1124/jpet.103.062026. [DOI] [PubMed] [Google Scholar]
- Vinter JG. Extended electron distributions applied to the molecular mechanics of some intermolecular interactions. J Comput Aided Mol Des. 1994;8:653–668. doi: 10.1007/BF00124013. [DOI] [PubMed] [Google Scholar]
- Wacker D, Fenalti G, Brown MA, Katritch V, Abagyan R, Cherezov V, et al. Conserved binding mode of human beta2 adrenergic receptor inverse agonists and antagonist revealed by X-ray crystallography. J Am Chem Soc. 2010;132:11443–11445. doi: 10.1021/ja105108q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahlund G, Nerme V, Abrahamsson T, Sjoquist PO. The beta 1- and beta 2-adrenoceptor affinity and beta 1-blocking potency of S- and R-metoprolol. Br J Pharmacol. 1990;99:592–596. doi: 10.1111/j.1476-5381.1990.tb12974.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warne T, Serrano-Vega MJ, Baker JG, Moukhametzianov R, Edwards PC, Henderson R, et al. Structure of a beta1-adrenergic G-protein-coupled receptor. Nature. 2008;454:486–491. doi: 10.1038/nature07101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzel D, Knies R, Matthey M, Klein AM, Welschoff J, Stolle V, et al. beta(2)-adrenoceptor antagonist ICI 118,551 decreases pulmonary vascular tone in mice via a G(i/o) protein/nitric oxide-coupled pathway. Hypertension. 2009;54:157–163. doi: 10.1161/HYPERTENSIONAHA.109.130468. [DOI] [PubMed] [Google Scholar]
- Wisler JW, DeWire SM, Whalen EJ, Violin JD, Drake MT, Ahn S, et al. A unique mechanism of beta-blocker action: carvedilol stimulates beta-arrestin signaling. Proc Natl Acad Sci U S A. 2007;104:16657–16662. doi: 10.1073/pnas.0707936104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo AY, Wang TB, Zeng X, Zhu W, Abernethy DR, Wainer IW, et al. Stereochemistry of an agonist determines coupling preference of 2-adrenoceptor to different g proteins in cardiomyocytes. Mol Pharmacol. 2009;75:158–165. doi: 10.1124/mol.108.051078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamah AM, Delahunty M, Luttrell LM, Lefkowitz RJ. Protein kinase A-mediated phosphorylation of the beta 2-adrenergic receptor regulates its coupling to Gs and Gi. Demonstration in a reconstituted system. J Biol Chem. 2002;277:31249–31256. doi: 10.1074/jbc.M202753200. [DOI] [PubMed] [Google Scholar]
- Zheng M, Zhu W, Han Q, Xiao RP. Emerging concepts and therapeutic implications of beta-adrenergic receptor subtype signaling. Pharmacol Ther. 2005;108:257–268. doi: 10.1016/j.pharmthera.2005.04.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.