

Abstract
BACKGROUND AND PURPOSE
Ligustilide, the main lipophilic component of Danggui, has been reported to protect the brain against ischaemic injury. However, the mechanisms are unknown. Here, we investigated the roles of erythropoietin (EPO) and the stress-induced protein RTP801 in neuroprotection provided by ligustilide against ischaemia-reperfusion (I/R) damage to the brain.
EXPERIMENTAL APPROACH
The efficacy of ligustilide against I/R damage was assessed by neurological deficit, infarct volume and cell viability, using the middle cerebral artery occlusion model in rats in vivo and rat cultured neurons in vitro. EPO and RTP801 were analysed by Western blot. Over-expression of RTP801 was achieved by transfection of an expression plasmid.
KEY RESULTS
Ligustilide decreased the neurological deficit score, infarct volume and RTP801 expression and increased EPO transcription in I/R rats, and increased cell viability and EPO and decreased LDH release and RTP801 in I/R neurons. Also, ligustilide increased ERK phosphorylation (p-ERK). The positive effects of ligustilide on p-ERK, cell viability and EPO were blocked by PD98059, but not LY294002 and SB203580. In addition, transfection of SH-SY5Y cells with RTP801 plasmid increased RTP801 and LDH release, while ligustilide inhibited the effects of transfection on RTP801 expression and also increased cell viability.
CONCLUSION AND IMPLICATIONS
Ligustilide exerts neuroprotective effects against I/R injury by promoting EPO transcription via an ERK signalling pathway and inhibiting RTP801 expression, This compound could be developed into a therapeutic agent to prevent and treat ischaemic disorders.
Keywords: ligustilide, neuroprotection, pharmacological mechanisms, ischaemia-reperfusion in vivo and in vitro, erythropoietin, RTP801, neurological deficit score, infarct volume, cell viability, ERK phosphorylation, PD98059, transfection
Introduction
Radix Angelicae Sinensis, known as Danggui in Chinese, is the root of Angelica Sinensis (Oliv.) Diels (Umbelliferae). It is a popular traditional Chinese medicinal herb which has long been used as a medicinal plant and is included in a number of traditional Sino-Japanese herbal prescriptions (Hou et al., 2004; Rhyu et al., 2005). The chemical constituents of the Danggui extract are classified into essential oil and water soluble components (Huang et al., 2004)and the essential oil is believed to contain its main pharmacologically active compounds (Huang and Song, 2001)and the major active agent is ligustilide (Gijbels et al., 1982; Naito et al., 1992).
Ligustilide protected PC12 cells against injury-induced by hydrogen peroxide (Yu et al., 2008), and also the brain from damage induced by transient forebrain cerebral ischaemia in male ICR mice (Swiss Hauschka strain) (Kuang et al., 2006) and by permanent focal ischaemia in rats (Peng et al., 2007). It has been suggested that the antioxidant and anti-apoptotic properties of ligustilide might contribute to its neuroprotective role in cerebral ischaemic damage (Kuang et al., 2006) and hydrogen peroxide-induced cell injury (Peng et al., 2007). However, the precise molecular mechanisms underlying the neuroprotective effect of ligustilide have not been fully understood.
In the present study, we first tested whether ligustilide has a protective effect against the damage induced by ischaemia-reperfusion (I/R) in the cerebral circulation. Then we investigated the effects of ligustilide on the expression of erythropoietin (EPO) and the stress-induced protein RTP801 (also known as REDD1, regulated in development and DNA damage responses 1) in I/R rats in vivo and I/R neurons in vitro. EPO was chosen in this study as a potential target of ligustilide because the increased transcription of EPO, promoted by hypoxia-inducible factor-1α (HIF-1α), plays a dominant role in neuroprotection in ischaemic stroke and intracerebral haemorrhage (Rabie and Marti, 2008; van der Kooij et al., 2008; Fan et al., 2009). RTP801 was also measured because this novel protein, the product of the Ddit4 gene, was strongly up-regulated by hypoxia in vitro and in vivo (Shoshani et al., 2002) and an increase in RTP801 appeared to be toxic to non-dividing neuron-like PC12 cells and increased their sensitivity to ischaemic injury (Shoshani et al., 2002).
We proposed that ligustilide might increase transcription of EPO, an endogenous protective factor, and might inhibit expression of RTP801, a detrimental factor. We found that the protection by ligustilide of the brain in vivo and neurons in vitro, from damage induced by I/R was mediated by up-regulation of EPO and down-regulation of RTP801, supporting our original hypothesis. In addition, we demonstrated that the increased EPO transcription induced by ligustilide was mediated by activation of the ERK pathway.
Methods
Animals
All animal care and experimental procedures complied with the guidelines developed by the Health Department of Local Government and Experimentation Ethics Committee of the Universities and approved by the Department of Health of the Hong Kong Government and the Animal Ethics Committee of The Chinese University of Hong Kong. Male Sprague-Dawley rats (3-month-old, 250 to 270 g, n = 51) were provided by the Animal Centers of The Chinese University of Hong Kong and Nantong University. The animals were housed in pairs in stainless steel cages at 21 ± 2°C and had free access to food and water. Rooms were in a cycle of 12 h of light (7:00 to 19:00) and 12 h of darkness (from 19:00 to 7:00).
Pharmacological treatments and experimental design
Danggui was purchased from the Danggui Cultivating Base of Good Agricultural Practice in Nin Xian County, Gansu Province, China and ligustilide was prepared by a well-established procedure as described previously (Qian et al., 2005). Its purity was over 98% based on the percentage of total peak area in the high performance liquid chromatography analysis.
To test whether ligustilide has a protective effect against the damage induced by I/R, this compound (suspended in 0.5% sodium carboxymethylcellulose) was administered by oral gavage at a dose of 20, 40 or 80 mg·kg−1, at 3 and 0.5 h before the middle cerebral artery occlusion (MCAO) procedure, according to Zhang et al. (2009). Rats in the sham group received volume-matched vehicle. Nimodipine was used as a positive control as it is known that nimodipine pretreatment attenuated infarct volumes after MCAO, by reducing Ca2+ entry (Mcculloch, 1992). Nimodipine in 0.5% sodium carboxymethylcellulose solution was given by oral gavage once at a dose of 12 mg·kg−1 at 1 h prior to onset of the ischaemia.
To investigate the effect of ligustilide on cell viability and LDH in cultured neurons, ligustilide was freshly prepared as a stock solution in dimethyl sulphoxide and diluted with neuronal culture medium before the experiments. Neurons were pretreated with ligustilide (0, 0.625, 1.25, 2.5, 5, 10 or 20 µmol·L−1) for 2 h before being exposed to oxygen-glucose deprivation (OGD) for 4 h (Peng et al., 2007). The concentration of dimethyl sulphoxide in the final culture medium was 0.1% (v/v) which had no effect on cell viability.
To study the effects of ligustilide on EPO and RTP801 expression, neurons were pretreated with ligustilide (0, 1.25, 5 and 10 µmol·L−1) before exposing to OGD. The effects of inhibitors of PI3K, ERK and p38 MAPK were investigated by pre-incubating neurons with 0 or 50 µmol·L−1 of LY294002, 0 or 25 µmol·L−1 of PD98059, or 0 or 50 µmol·L−1 of SB203580 for 1 h, respectively, before being treated with 5 µmol·L−1 of ligustilide for 2 h followed by OGD.
The rats were randomized to treatment or placebo group. The investigators who induced the stroke, analysed the infarct volumes and assessed the neurological deficits were unaware of the group identity of each animal.
Ischaemia-reperfusion model in vivo and determination of neurological deficit and infarct volume
Ischaemia-reperfusion was induced in rats by MCAO followed by reperfusion as described by Saleem et al. (2008). Three out of a total of 45 rats subjected to MCAO surgery died before the endpoint of reperfusion. Post-mortem examination revealed that these were caused by trauma, such as internal bleeding, caused during the surgical procedure and these animals were therefore excluded from any data analysis. The animals were fasted overnight but allowed free access to water. They were then anesthetized with chloral hydrate (400 mg·kg−1, i.p.). Throughout the surgery, rectal temperature was maintained at 37°C by a heating pad. A 4-0 silicon-coated monofilament nylon suture with a round tip was advanced from the right external carotid artery into the lumen of the internal carotid artery to occlude the origin of the middle cerebral artery and maintained for 2 h. Reperfusion was introduced by withdrawing the monofilament after occlusion. As a control, sham-operated rats underwent identical surgery but did not have the suture inserted. After recovery from anaesthesia, the rats were returned to their cage with free access to water and food. Twenty-four hours after reperfusion, rats were scored for neurological function based on a modified Longa EZ test, which employs a six-point scale (Longa et al., 1989) with some modifications: 0 = no apparent deficits; 1 = failure to extend left forepaw fully; 2 = circling to the left; 3 = falling to the left; 4 = did not walk spontaneously and had a depressed level of consciousness; 5 = death (excluding causation in operation).
The brains of rats were then removed, sliced coronally into five 2 mm thick sections starting at 1 mm from the frontal pole, and incubated with 0.5% 2,3,5-triphenyltetrazolium chloride for 30 min at 37°C followed by 4% formaldehyde solution overnight. Infarct volume was then measured as described previously (Kuang et al., 2006). The normal area of brain was stained dark red based on intact mitochondrial function whereas infarct tissue remained unstained (white). The infarct area from each section was measured using the Pro-plus 6.0 image analysis software (Image J, Bethesda, MD, USA). The total volume of infarction was calculated as the sum of the infarct areas (all sections) × thickness (2 mm). The infarct volume was expressed as a percentage of contralateral hemisphere.
Neuronal culture and a model of oxygen-glucose deprivation (OGD) in vitro
The primary cortical neuronal culture was prepared using a method described by Ho et al. (2003) and Wu et al. (2009). In brief, the cortex was aseptically removed from 15–16 day-old mouse embryos, minced with sterile surgical blades, incubated in 0.125% trypsin and dissociated by trituration in DNase/trypsin inhibitor solution. The cortical neurons were suspended in complete DMEM containing 2.5 mmol·L−1 glucose and 10% fetal bovine serum, and plated in poly-L-lysine-coated six-well or 96-well plates (Corning) at a density of 2.0 × 105 cells·cm−2. Cultures were kept in an incubator at 5% CO2 (NAPCO 5400) at 37°C. The medium was replaced by Neurobasal medium with supplemental B27 (Invitrogen) 4 h after cells were seeded. After 5 days in culture, purity of the neurons was assessed by staining with neuron-specific antibody against microtubule associated protein 2 (Millipore). In our case, over 98% of cells were positively stained. The cultures were therefore used for the following experiments.
To investigate the effect of ligustilide, cultured neurons were pretreated with this compound for 2 h before being exposed to OGD. OGD was achieved by placing cells in DMEM without glucose and in a dedicated chamber (NAPCO 7101FC-1) with 1% O2, 94% N2 and 5% CO2 for 4 h at 37°C as previously described (Wu et al., 2009). After OGD, neurons were incubated with the original medium in a normoxic incubator for 24 h.
Assessment of cell viability
The cell viabilities were measured using a MTT (3-(4,5-dimethylthazol-2-yl)-2,5-diphenyltetrazolinum bromide) assay as described previously (Du et al., 2008; He et al., 2008). Briefly, a total of 25 µL MTT (1 g·L−1 in PBS) was added to each well before the conduction of incubation at 37°C for 4 h. The reaction was stopped by the addition of a 100 µL lysis buffer (20% SDS in 50% N'Ndimethylformamide, pH 4.7). Optical density was measured at the 570 nm wavelength by the use of an ELX-800 microplate assay reader (Bio-tek, USA) and the results were expressed as a percentage of absorbance measured in the control cells.
LDH release
The intracellular enzyme LDH leaks into the culture medium when cell membranes are damaged (Zhu et al., 2007) and the quantity of LDH (unit·mL−1·min−1) released into the medium was determined by the decrease in absorbance at 340 nm for NADH disappearance within 3 min (Zhu et al., 2008). Briefly, the cells were treated, 500 µL of supernatant was then collected from each well and mixed with 1.3 mL of NADH (0.217 mmol·L−1) and 1.3 mL of sodium pyruvate (1.77 mmol·L−1) in the modified Krebs-Henseleit buffer (118 mmol·L−1 NaCl, 4.8 mmol·L−1 KCl, 1 mmol·L−1 KH2PO4, 24 mmol·L−1 NaHCO3, 3 mmol·L−1 CaCl2, 0.8 mmol·L−1 MgPO4, pH 7.4) for 30 s at 37°C. The activity was spectrophotometrically measured (Mod-756) at optical density of 340 nm.
Immunofluorescence
After being deeply anesthetized, the rats were transcardially perfused with normal saline solution, followed by 4% paraformaldehyde in 0.1 M PBS 24 h after I/R. Brains were removed and post-fixed in 4% paraformaldehyde for 4 h, then transferred into 30% sucrose solution until the brains sank to the bottom of the container. Coronal sections (25 µm) were made using a Leica CM3050S cryostat (Leica Microsystems, Germany). Sections were blocked with 3% normal goat serum (diluted in PBS containing 0.3% Triton X-100) for 1 h and incubated with primary antibodies (anti-EPO and anti-RTP801, 1:200) overnight at 4°C. After rinsing with PBS, sections were incubated with FITC-conjugated donkey anti-goat IgG (for RTP801) or Alexa 586-conjugated goat anti-rabbit IgG (for EPO) as secondary antibodies (1:200) for 2 h at room temperature. The specificity of antibody was tested using a negative control which was performed by adding only second antibody without primary antibody pre-incubation in brain slices. The immunofluorescence images from at least four fields each from three rats were examined using the same brightness and exposure settings. Image-Pro Plus software (Media Cybernetics, Silver Spring, MD, USA) was used to analyse the intensity of immunofluorescence for EPO or RTP801 in each photograph. All the sections were stained within the same immunohistochemistry run and the results were shown as the percentage of the control.
Transfection
The pcDNA3.1-RTP801 construct was prepared by subcloning the open reading frame of human RTP801 cDNA (Genebank, NM_019058) into the HidIII/XhoI sites of expression plasmid pcDNA3.1 (Invitrogen Co., Carlsbad, CA, USA). The cDNA fragment was obtained by PCR amplification using RTP801-specific primer 5′-CCCAAGCTTATGCCTAGCCTTTGGGAC-3′ and 5′-CCGCTCGAGTCAACACTCCTCAATGAG-3′ with the added HindIII and XhoI restriction sites (indicated by underlines respectively). SH-SY5Y cells grown to 80% confluence in 24-well plates were transfected with 0.8 µg of pcDNA3.1-RTP801 plasmid DNA using Lipofectamine 2000 reagent. The empty vector was used as a control.
Western blot analysis
Neurons receiving different treatments were washed with ice-cold PBS, homogenized in ice-cold lysis buffer [50 mM Tris-HCl (pH 7.4), 1 mM EDTA, 1% SDS, 1% Nonide P-40, 1 mM Na3VO4, 1 mM NaF, 5% β-mercaptoethanol, 400 uM phenylmethysulphonyl fluoride, 2 µg·mL−1 each of pepstatin, aprotinin and leupeptin]. After centrifugation at 12 000×g for 10 min at 4°C, the supernatant was collected, and protein concentration in the extracts was determined by Bradford assay kit (Bio-Rad). Aliquots of the cell extract containing equal amounts of protein was boiled in a protein loading buffer for 5 min, separated on a 10% SDS-polyacrylamide gels, and transferred to PVDF membranes. Transfer membranes were blocked with 5% non-fat milk in Tris-buffered saline for 1 h and incubated in primary antibodies (anti-EPO and anti-RTP801, 1:500; anti-ERK42/44 and phospho-ERK42/44, 1:5000) overnight at 4°C (He et al., 2008; Li et al., 2008). Then, the membranes were washed three times and incubated with appropriate secondary antibodies (1:2000) for 2 h followed by three washes. Signal detection was performed with an enhanced chemiluminescence kit (Pierce Biotechnology). β-Actin monoclonal antibody (1:20 000) was used as internal protein controls.
Statistical analysis
Data are presented as mean ± SEM. The differences among neurological deficit scores were determined by Kruskall–Wallis test followed by Mann–Whitney test for multiple comparisons. Analysis of other parametric assay was performed using one-way or two-way anova in appropriate experiments followed by Newman–Keuls post hoc test. A probability value of P < 0.05 was taken to be statistically significant.
Materials
Unless otherwise stated, all chemicals were obtained from Sigma Chemical Company, St. Louis, MO, USA. Neurobasal medium with supplemental B27, Dulbecco's modified Eagle's medium (DMEM), Trizol and Lipofectamine 2000 were purchased from Invitrogen Co., Carlsbad, CA, USA. Fetal bovine serum was obtained from Hyclone, Logan, UT, USA. Primary polyclonal rabbit anti-ERK42/44 and phospho-ERK42/44 were bought from Cell Signalling Technology, Beverly, MA, USA. Primary polyclonal goat anti-RTP801, rabbit anti-EPO antibody and FITC-labelled secondary donkey anti-goat IgG were bought from Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA. Alexa 586-labelled goat anti-rabbit IgG was purchased from Molecular Probes, Leiden, the Netherlands. Nimodipine was provided by Zhenzhou Rikang Pharmaceutical Company Limited, China. Drug and molecular target nomenclature follow Alexander et al., (2009).
Results
In vivo study
Ligustilide protected brain from injury induced by I/R
We first investigated whether ligustilide could protect the brain against damage induced by I/R. Our results based on the modified Longa EZ test clearly showed that I/R induced a significant increase in neurological deficit score, compared with rats treated with sham procedures only. Pretreatment with 20, 40 or 80 mg·kg−1 of ligustilide significantly reduced neurological deficit score in a dose-dependent manner in I/R rats. The scores in all ligustilide groups were lower than those in the vehicle group (all P < 0.01, Figure 1A). Nimodipine pretreatment, as a positive control, at a dose of 12 mg·kg−1, also decreased the neurological deficit score (Figure 1A).
Figure 1.
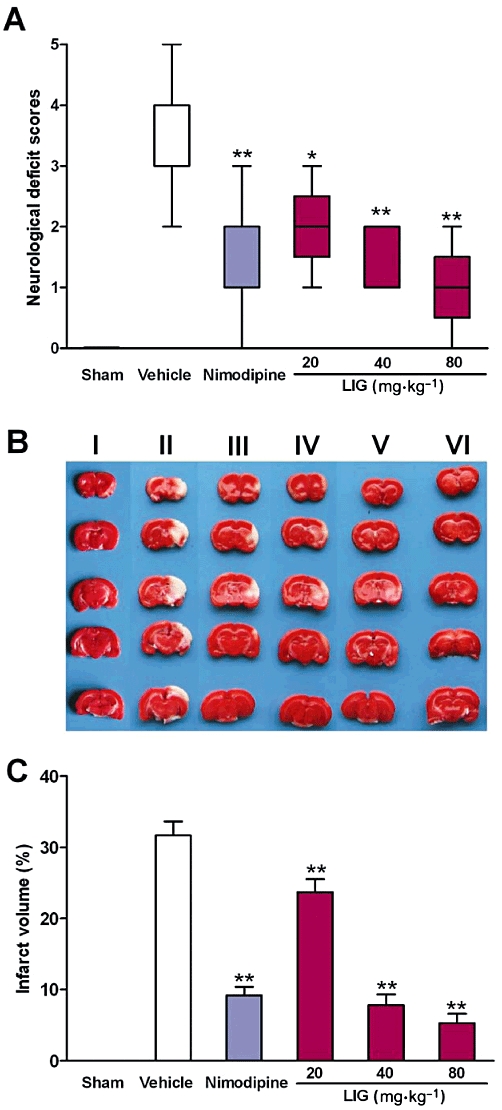
Ligustilide (LIG) protected brain from injury induced by ischaemia-reperfusion in rat. (A) Neurological deficit scores. (B) Representative photographs of brain slices stained with 2,3,5-triphenyltetrazolium chloride. (C) Infarct volume expressed as the percentage of brain volume. Animals were subjected to sham operation (I), administration of vehicle only (II), nimodipine at a dose of 12 mg·kg−1 (III), Ligustilide at a dose of 20 (IV), 40 (V) or 80 mg·kg−1 (VI) at 3 h and 0.5 h before undergoing MCAO for 2 h followed by 24 h reperfusion. Neurological deficit scores were analysed using Kruskall–Wallis test followed by Mann–Whitney test for multiple comparisons to identify which was different from the ‘vehicle’ group. The infarct volume was analysed by one-way anova followed by Newman–Keuls post hoc test. Parametric data are presented as mean ± SEM (n = 6) and non-parametric data as box and whisker plots with the minimum and maximum values (n = 9). *P < 0.05, **P < 0.01 significantly different from ‘vehicle’.
We also assessed the effects of ligustilide pretreatment on infarction induced by I/R and found that this compound (20, 40 or 80 mg·kg−1) dose-dependently reduced infarct volume. Also, infarct volume in the rats pretreated with nimodipine (12 mg·kg−1) was significantly lower than that in the vehicle-treated rats (Figure 1B,C). These findings demonstrated that ligustilide protected the brain against I/R-induced damage.
Ligustilide increased EPO and decreased RTP801 expression in rats treated with I/R
To find out whether the protective effects of ligustilide were associated with changes in EPO and RTP801, we then investigated EPO and RTP801 expression in the brain of I/R rats. Figure 2A shows representative photographs of EPO and RTP801 expression by immunohistochemistry in the rats received different treatments (I. sham; II. 0; III. 20; IV. 40; and V. 80 mg·kg−1 ligustilide). Immunofluorescence analysis (Figure 2B,C) shows that EPO expression within the infarct region of cortex in the vehicle-treated group was slightly increased compared with that of the sham-operated group. Prior administration of ligustilide enhanced EPO expression dose-dependently. The EPO contents in the rats of all ligustilide groups were significantly higher than those in the rats treated without ligustilide (P < 0.05 or 0.01) (Figure 2A,B). In the case of RTP801, I/R induced a significant increase, while pretreatment with ligustilide resulted in a significant decrease in expression of the protein in the infarct region of cortex, also in a dose-dependent manner. The levels of RTP801 in the rats of all ligustilide groups were significantly lower than those in the rats without ligustilide treatment (P < 0.01) (Figure 2A,C).
Figure 2.
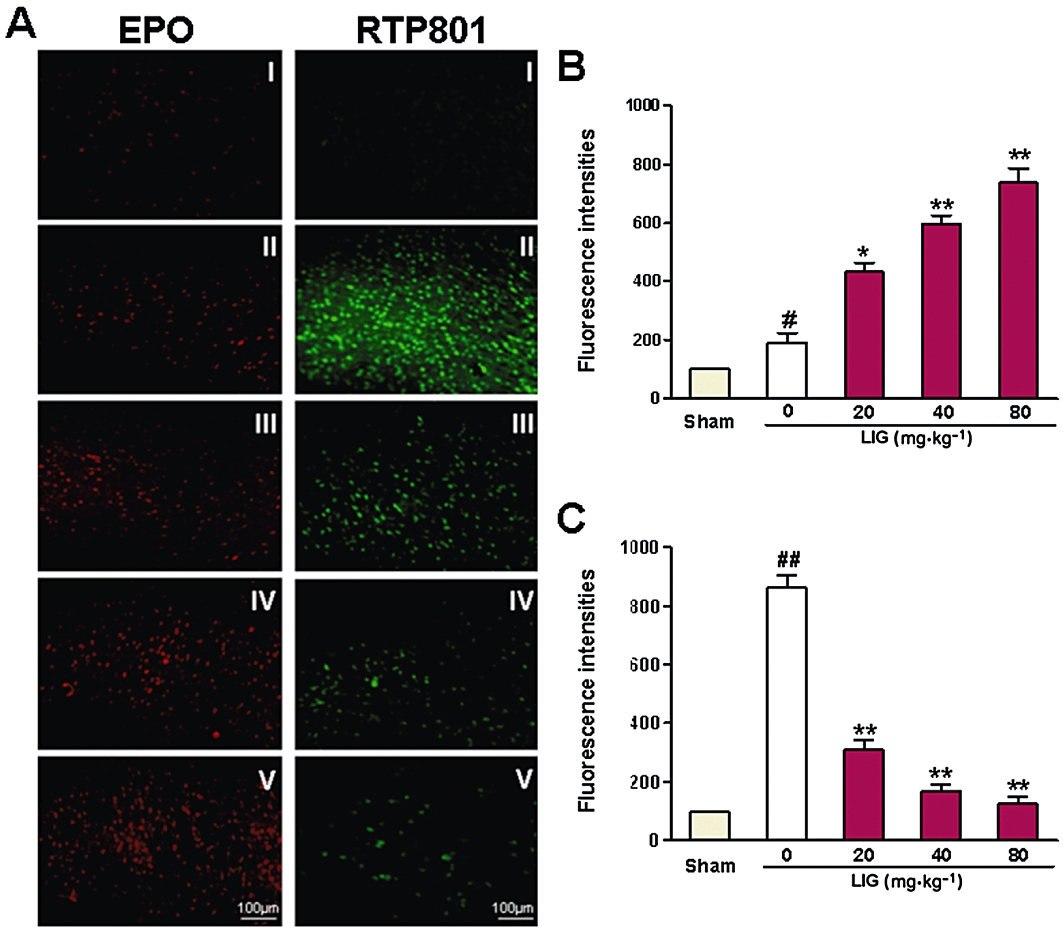
Effects of ligustilide (LIG) on erythropoietin (EPO) and RTP801 expression in cerebral cortex after ischaemia-reperfusion in rats. Rats were subjected to ligustilide treatment (0 (vehicle), 20, 40 or 80 mg·kg−1) given at 3 h and 0.5 h before undergoing MCAO for 2 h followed by 24 h reperfusion. (A) Representative photographs of EPO and RTP801 expression, assayed immunohistochemically. (B,C) Data were normalized to the sham group, which was taken as 100% and presented as mean ± SEM. Statistical significance was assessed by two-way anova followed by a Newman–Keuls post hoc test. #P < 0.05, ##P < 0.01 significantly different from sham (control) group; *P < 0.05, **P < 0.01 significantly different from vehicle group. Three independent experiments were carried out in each group.
In vitro study
The above in vivo studies suggested that the protective effects of ligustilide on brain I/R damage were associated with up-regulation of EPO and down-regulation of RTP801 expression. To provide more direct evidence of the involvement of EPO and RTP801 and to elucidate the underlying molecular mechanisms, we performed a series of in vitro studies.
Ligustilide increased cell viability and decreased LDH release in neurons exposed to OGD
We first investigated the effects of ligustilide on cell viability and LDH release in cultured neurons exposed to OGD. The neurons were pre-incubated with ligustilide (0, 0.625, 1.25, 2.5, 5, 10 or 20 µmol·L−1) for 2 h before undergoing OGD for 4 h followed by normal conditions for 24 h. As expected, OGD induced a significant decrease in cell viability (Figure 3A) and a marked increase in LDH release (Figure 3B) in neuron cultures. In OGD neurons pretreated with ligustilide, the cell viabilities increased and LDH decreased progressively with the increase of ligustilide concentrations, attaining the highest (cell viability) and lowest (LDH release) at 5 µmol·L−1. Above this concentration, cell viability decreased again and LDH release increased (10 and 20 µmol·L−1 ligustilide; Figure 3). These data suggested that ligustilide had protective effects on neurons against the injury induced by OGD in vitro, at the lower concentrations but at concentrations greater than 5 µM may cause cytotoxicity in cultured neurons. The findings also confirmed the results of our in vivo studies and provided the basis for using the in vitro model to further study the mechanisms underlying the effect of ligustilide.
Figure 3.
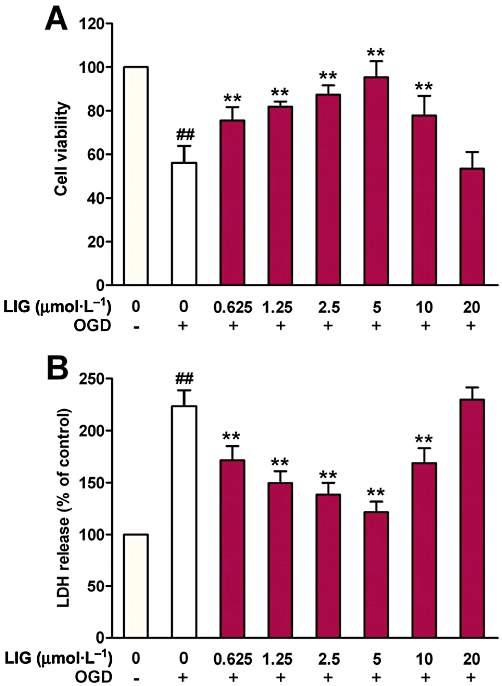
Effect of ligustilide (LIG) on cell viability and LDH release in neurons treated with OGD. Neurons were pretreated with ligustilide (0, (OGD alone) 0.625, 1.25, 2.5, 5, 10 or 20 µmol·L−1) for 2 h before undergoing OGD for 4 h followed by a return to normal media and oxygenation for 24 h. Statistical significance was assessed by two-way anova followed by a Newman–Keuls post hoc test. (A) Cell viability by MTT assay. (B) LDH release. Data are presented as mean ± SEM (percentage of control) (n = 8), ##P < 0.01 significantly different from control; **P < 0.01 significantly different from OGD alone group.
Ligustilide up-regulated EPO and down-regulated RTP801 expression in neurons exposed to OGD
The effects of ligustilide at different concentrations on EPO and RTP801 expression were investigated by pre-incubating neurons with ligustilide (1.25, 5 and 10 µmol·L−1) before undergoing OGD. Western blot analysis showed that ligustilide at a dosage of 1.25 or 5 µmol·L−1 could induce a significant increase in EPO expression in neurons exposed to OGD, while 10 µmol·L−1 of ligustilide produced a much weaker effect (Figure 4A,B). The EPO content of neurons pretreated with 1.25 or 5 µmol·L−1 of ligustilide were significantly higher than those in the controls (0 µmol·L−1 ligustilide).
Figure 4.
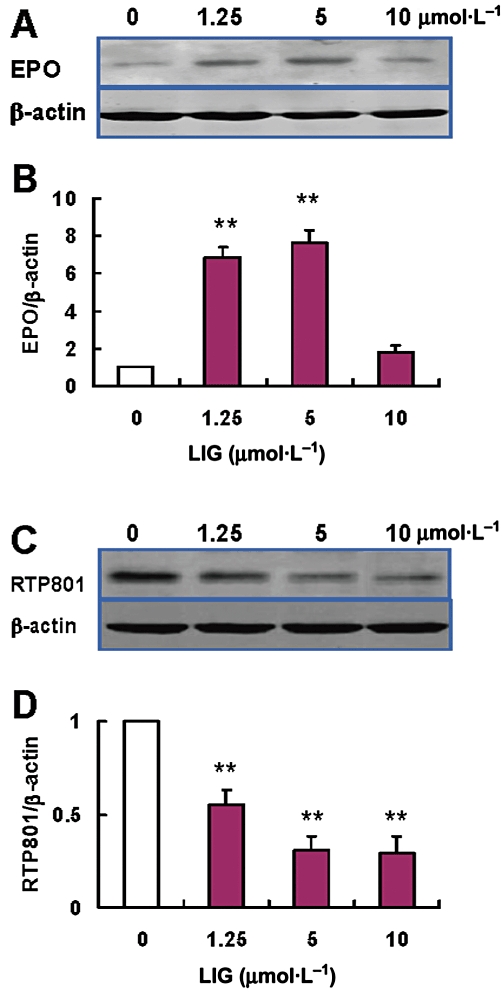
Effect of ligustilide (LIG) on erythropoietin (EPO) and RTP801 expression in neurons treated with OGD. Neurons were pretreated with ligustilide (0 (OGD alone), 1.25, 5 or 10 µmol·L−1 for 2 h before undergoing OGD for 4 h followed by a return to normal media and oxygenation for 24 h. (A) A representative Western blot of EPO. (B) Quantification of expression of EPO. (C) A representative Western blot of RTP801. (D) Quantification of expression of RTP801 (n = 3 in all cases). Data are presented as mean ± SEM. Statistical significance was assessed by one-way anova followed by a Newman–Keuls post hoc test. **P < 0.01 significantly different from OGD alone group.
Exposure to OGD also induced a significant increase (about three-fold control) in the expression of RTP801 in neurons, as was found in the cortex in vivo. The increased RTP801 induced by OGD could be significantly inhibited by pretreatment with ligustilide in a dose-dependent manner. (Figure 4C, D). The RTP801 content of neurons treated with 1.25, 5 and 10 µmol·L−1 of ligustilide were all significantly lower than those in the neurons without ligustilide treatment (all P < 0.01) (Figure 4C,D). The effects of ligustilide on EPO and RTP801 expression were consistent with the protection by ligustilide of neurons against I/R injury, suggesting correlation of the protective effect of ligustilide with the up-regulation of EPO and down-regulation of RTP801 expression.
Inhibition of ERK reduced significantly the effects of ligustilide on cell viability and EPO expression in neurons exposed to OGD
To understand the signalling pathway involved in the protective effect of ligustilide on neurons, we then investigated the effects of inhibitors of PI3K, ERK and p38 MAPK on cell viability and LDH release. The neurons were pre-incubated with 0 or 50 µmol·L−1 of LY294002 (a specific inhibitor of PI3K), 0 or 25 µmol·L−1 of PD98059 (a specific inhibitor of ERK), or 0 or 50 µmol·L−1 of SB203580 (a specific inhibitor of p38 MAPK) (Harada et al., 2001; Wu et al., 2009) for 1 h before being treated with 5 µmol·L−1 of ligustilide for 2 h and exposed to OGD. As shown above, treatment with ligustilide increased cell viability and decreased LDH release in neurons exposed to OGD. These effects of ligustilide, however, could be blocked by PD98059 but not LY294002 and SB203580 (Figure 5A,B). In control experiments, LY294002, PD98059 and SB203580 did not affect the cell viability of neurons, in the absence of ligustilide (data not shown).
Figure 5.
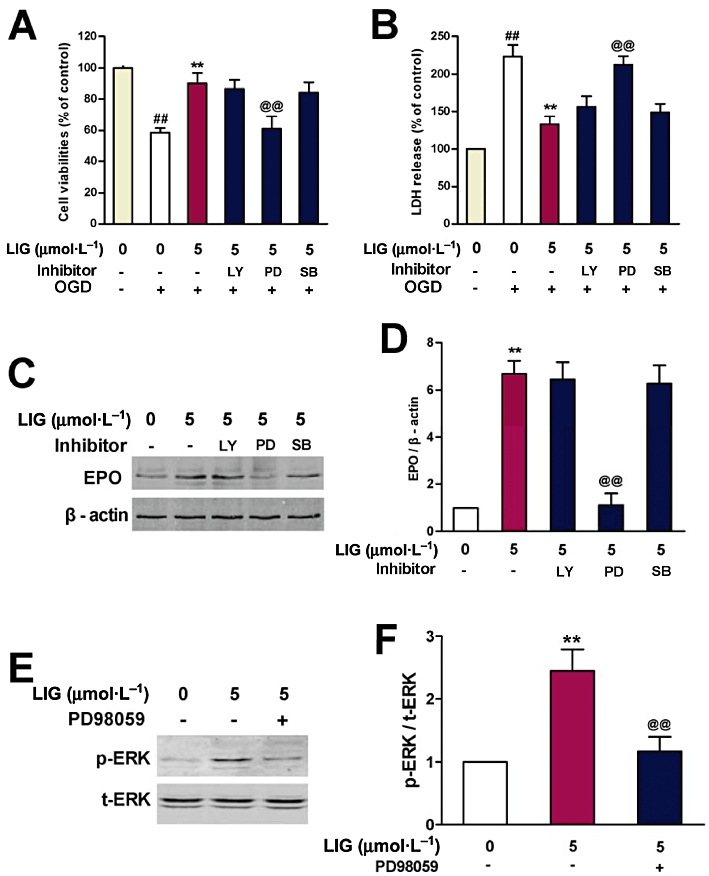
Effects of inhibitors of PI3K, ERK and p38 MAPK on cell viability, LDH release and erythropoietin (EPO) expression in neurons treated with ligustilide (LIG) and then exposed to OGD. Neurons were pre-incubated with or without LY294002 (LY, a specific inhibitor of PI3K, 50 µmol·L−1), PD98059 (PD, a specific inhibitor of ERK, 25 µmol·L−1) or SB203580 (SB, a specific inhibitor of p38 MAPK, 50 µmol·L−1) for 1 h before being treated with ligustilide (5 µmol·L−1) for 2 h and then exposed to OGD for 4 h followed by a return to normal media and oxygenation for 24 h. (A) Cell viability by MTT assay (n = 8). (B) LDH release (n = 8). (C) A representative Western blot of EPO. (D) Quantification of expression of EPO (n = 3). (E) A representative experiment of Western blot of p-ERK. (F) Quantification of expression of p-ERK (n = 3). Statistical significance was assessed by two-way anova followed by a Newman–Keuls post hoc test. Data (mean ± SEM) are expressed as a percentage of control or fold increase over control. ##P < 0.01 significantly different from control group; **P < 0.01 significantly different from OGD alone; @@P < 0.01 significantly different from 5 µmol·L−1 of ligustilide with OGD.
To further explore the relationship between ligustilide, EPO and ERK, we studied the effects of PD98059, LY294002 and SB203580 on EPO expression and ERK phosphorylation (p-ERK) in neurons exposed to OGD. The positive effects of ligustilide on EPO expression were inhibited by PD98059 but not LY294002 and SB203580 (Figure 5C,D). Furthermore, ligustilide increased the amount of p-ERK and this effect was also blocked by PD98059 (Figure 5E,F). In the absence of ligustilide, LY294002, PD98059 and SB203580 had no effect on EPO expression.
Ligustilide reduced RTP801 expression and LDH release in SH-SY5Y cells transfected with pcDNA3.1-RTP801 plasmid DNA
To provide further evidence for the involvement of RTP801 in ligustilide-induced protective effects on I/R neurons, we next investigated the effects of ligustilide on RTP801 expression and LDH release in SH-SY5Y cells, transfected with 0.8 µg of pcDNA3.1-RTP801 plasmid DNA. The transfection increased RTP801 expression and LDH release in the cell cultures (P < 0.01, Figure 6), implying that the increased expression of RTP801 is associated with increased cellular damage, as indicated by the increased LDH release. Treatment of cells with ligustilide inhibited the effects of the transfection on RTP801 expression, and also increased cell viability. In the plasmid-transfected cells, RTP801 expression and LDH release were both significantly lower in the ligustilide-treated group than the group without ligustilide treatment (Figure 6).
Figure 6.
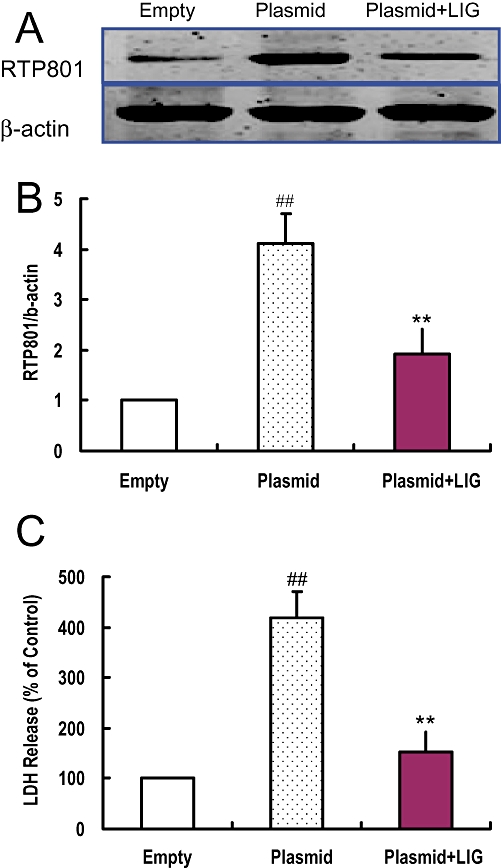
Ligustilide (LIG) reduced RTP801 expression and LDH release in SH-SY5Y cells transfected with pcDNA3.1-RTP801 plasmid. SH-SY5Y cells were transfected with empty pcDNA3.1 plasmid (Empty) or recombinant pcDNA3.1-RTP801 plasmid respectively. The cells that were transfected with recombinant pcDNA3.1-RTP801 plasmid were treated with 0 or 5 µmol·L−1 of ligustilide as described in Methods. The RTP801 expression and cell viability in the cells in different groups were measured by Western blot and LDH assay, respectively, after transfection for 48 h. (A) A representative Western blot of RTP801. (B) Quantification of expression of RTP801. (C) LDH release. Statistical significance was assessed by two-way anova followed by a Newman–Keuls post hoc test. Data are presented as mean ± SEM (n = 6). ##P < 0.01 significantly different from control group (transfected with empty pcDNA3.1 plasmid). **P < 0.01 significantly different from 5 µmol·L−1 ligustilide group transfected with pcDNA3.1-RTP801 plasmid.
Discussion
In spite of a long history of clinical application of Danggui in the treatment of ischaemic disorders of the cardiovascular and cerebrovascular systems in traditional Chinese medicine, very little is known about which of the compound(s) in Danggui is the active ingredient(s) that improves syndromes associated with the diseases. In previous studies, we demonstrated the neuroprotective effects of ligustilide, a major component of the Danggui extract, in different models including hydrogen peroxide-induced neuronal damage in PC12 cells as well as brain damage induced by transient forebrain cerebral ischaemia and permanent focal ischaemia in mice and rats respectively (Kuang et al., 2006; Peng et al., 2007; Yu et al., 2008). In the present study, we found that pretreatment with ligustilide reduced neurological deficit scores and infarct volume in a dose-dependent manner in I/R rats in vivo and increased cell viability with a corresponding decrease in LDH release, in neurons exposed to OGD in vitro. These findings provide further evidence that ligustilide is one of the active ingredients of Danggui and also suggest that ligustilide might have a protective effect against I/R-induced damage to the brain. However, it should be pointed out that because infarct volume was assessed at 24 h in the present study, the compound might slow down the evolution of damage, rather than actually reduce final magnitude of the infarct. To rule out that possibility, further observation until at least 48 h in this model is needed.
Unravelling the precise molecular mechanisms of ligustilide is important in order to design neuroprotective therapies for hypoxic-ischaemic brain injury based on this compound. Ligustilide induced a significant increase in the activities of glutathione peroxidase and superoxide dismutase, and Bcl-2 expression in the ischaemic brain tissues, implying that the antioxidant and anti-apoptotic properties of ligustilide may also contribute to its neuroprotective role in cerebral ischaemic damage (Kuang et al., 2006) and hydrogen peroxide-induced cell injury (Peng et al., 2007). The precise molecular mechanisms underlying the neuroprotective effect of ligustilide have not been fully elucidated. In the present study, we investigated whether ligustilide had any effects on the expression of EPO (an endogenous protective factor) and RTP801 (an endogenous detrimental factor) in I/R rats in vivo and OGD neurons in vitro and whether these effects induced by ligustilide had any connections with its neuroprotective role.
The protein EPO was originally recognized as a humoral mediator involved in the maturation and proliferation of erythroid progenitor cells (van der Kooij et al., 2008; Fan et al., 2009). Recent studies have found EPO mRNA and protein in the brain of a variety of mammals including humans (Fan et al., 2009) and the EPO receptor was widely expressed in most cerebral cell types, including astrocytes, neurons, endothelial cells and microglial cells (Nagai et al., 2001; Marti, 2004). The increased expression of EPO induced by HIF-1 has a dominant role in neuroprotection after ischaemic stroke and intracerebral haemorrhage (Yatsiv et al., 2005; Rabie and Marti, 2008; van der Kooij et al., 2008; Fan et al., 2009). Consistent with this, application of EPO increased survival and function of retinal ganglion cells in rats suffering from optic neuritis (Sattler et al., 2004). The presence of EPO and its receptor in the brain and the function of EPO in neuroprotection led us to propose that EPO might be one of pharmacological targets of ligustilide and that ligustilide may play a role in promoting EPO transcription. An increase in EPO might be one of the mechanisms for the neuroprotection by ligustilide. In the present study, a significant increase in EPO was found in I/R rats in vivo and also in cultured neurons pretreated with ligustilide and exposed to OGD. These findings were accompanied by a significant reduction in neurological deficit score and infarct volume in vivo as well as an increase in cell viability and a decrease in LDH release in vitro. These results suggested that EPO might be one of the essential mediators in ligustilide-induced neuroprotection. In a recent study, Zheng et al. (2010) demonstrated that Danggui Buxue Tang (DBT), a Chinese herbal decoction prepared from Radix Astragali and Radix Angelicae Sinensis, triggered EPO expression in cultured HEK293T cells. Based on the findings in the present study, it is reasonable to speculate that the DBT-induced increase in EPO expression might be partly associated with ligustilide and this compound could be one of the major active ingredients of DBT.
A previous study in vitro (Siren et al., 2001) demonstrated that inhibition of MAPK and PI3K blocked EPO-mediated protection of rat hippocampal neurons against hypoxia. Using ERK-1/-2 and Akt inhibitors, Kilic et al. (2005) showed that activation of these proteins was essential for EPO-mediated neuroprotection in an animal model of focal cerebral ischaemia. These findings implied that PI3K, ERK and p38 MAPK are all associated with EPO expression and EPO-mediated neuroprotection in hypoxia and ischaemia (Rabie and Marti, 2008). To understand how ligustilide increased EPO expression and EPO-mediated neuroprotection in our experiments, we investigated the effects of inhibitors of PI3K, ERK and p38 MAPK on cell viability, EPO expression and p-ERK in neurons pretreated with ligustilide and exposed to OGD. Our data showed that ligustilide increased the level of p-ERK, along with increased cell viability and EPO. EPO-mediated neuroprotection was significantly blocked by inhibition of ERK using PD98059, while the specific inhibitors of PI3K and p38 MAPK had no such roles. These data indicated that the ligustilide-induced increase in EPO might be mediated only by ERK and was not associated with PI3K and p38 MAPK as seen in other neuroprotective models (Asomugha et al., 2010). We would propose that ligustilide promotes the phosphorylation of ERK and this increased p-ERK then increased the phosphorylation of HIF-1. The latter change, in turn, increased the transactivating activity of HIF-1 and hence, EPO expression (Figure 7). This scheme is consistent with the findings of previous studies which had revealed that the transactivating activity of HIF-1 is increased by phosphorylation of HIF-1 (Berra et al., 2000; Minet et al., 2001) and that the PI3K/Akt and the ERK pathway have been proposed to be involved in HIF-1 phosphorylation (Jewell et al., 2001; Minet et al., 2001; Stolze et al., 2002).
Figure 7.
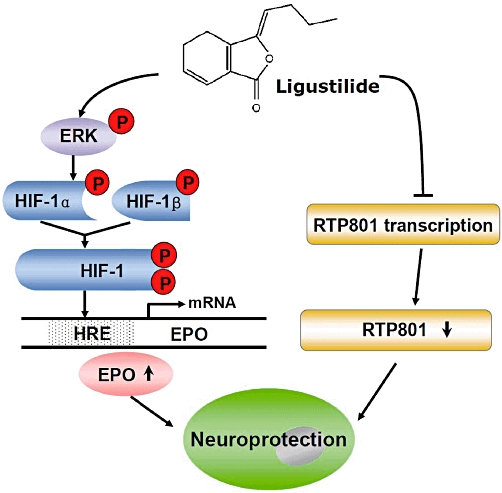
Proposed mechanisms for the neuroprotective role of ligustilide (LIG). The neuroprotective effect of ligustilide against ischemia-reperfusion injury is mediated by up-regulation of erythropoietin (EPO) and down-regulation of RTP801. Ligustilide might promote the phosphorylation of ERK (p-ERK). The increased p-ERK then induces an increase in the phosphorylation of hypoxia-inducible factor-1α (HIF-1α). The latter leads to an increase in the transactivating activity of HIF-1 and then EPO expression. In addition, ligustilide also protects the cells by inhibiting the expression of the pro-apoptotic protein RTP801.
RTP801 is the product of a recently cloned gene and is strongly up-regulated by hypoxia in vitro and in vivo (Shoshani et al., 2002). It appears to contribute to apoptotic cell death in several contexts (Shoshani et al., 2002; Bakker et al., 2007) although the molecular mechanisms are largely unknown. We therefore speculated that RTP801 might be another potential target of ligustilide such that this compound might inhibit the expression of RTP801 and thus protect neurons against I/R damage. Our findings showed that transfection of SH-SY5Y cells with pcDNA3.1-RTP801 plasmid DNA increased RTP801 expression as well as LDH release, implying that the increased expression of RTP801 was associated with the increased cellular damage as indicated by increased LDH release. The results are in line with the earlier studies of Shoshani et al. (2002) and Bakker et al. (2007) as mentioned above. We also demonstrated that pretreatment of the cells with ligustilide inhibited the effects of transfection on RTP801 expression and also increased cell viability. In addition, ligustilide reduced RTP801 expression in both rat brain and cultured neurons in I/R or OGD respectively. These results imply that neuroprotection of ligustilide might be partly mediated by its ability to inhibit RTP801 expression in brain following I/R in vivo and following OGD in neurons in vitro (Figure 7).
In conclusion, the present study demonstrated for the first time that ligustilide pretreatment protected brain against I/R injury and neurons in vitro against OGD injury by promoting EPO transcription via an ERK signalling pathway and inhibiting RTP801 expression. Our findings also provide further evidence that ligustilide is one of the active ingredients of Danggui. Thus ligustilide has the potential to be developed into an effective therapeutic agent in preventing and treating ischaemic disorders of the cardiovascular and cerebrovascular systems.
Acknowledgments
The studies in our laboratories were supported by Key Project Grant of Jiangsu Province (BG2007607), the Applied Science Foundation of Nantong City (K2007021), National 973 programme (2011CB510004), The Competitive Earmarked Grants of The Hong Kong Research Grants Council (PolyU562309), NSFC-RGC Joint Research Grant (N-CUHK433/08), Direct Grant of CUHK (2009.2.036), National Natural Science Foundation of China (30971197) and grants from South-west Hospital of The third Military Medical University and Shenzhen-Hong Kong Innovation Circle Programa (2008, 2009).
Glossary
Abbreviations
- EPO
erythropoietin
- I/R
ischaemia-reperfusion
- MCAO
middle cerebral artery occlusion
- OGD
oxygen-glucose deprivation
- p-ERK
phosphorylated ERK
- REDD1
(regulated in development and DNA damage responses 1)
Conflict of interest
The authors state no conflict of interest.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 4th edition. Br J Pharmacol. 2009;158(Suppl. 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asomugha CO, Linn DM, Linn CL. ACh receptors link two signaling pathways to neuroprotection against glutamate-induced excitotoxicity in isolated RGCs. J Neurochem. 2010;112:214–226. doi: 10.1111/j.1471-4159.2009.06447.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakker WJ, Harris IS, Mak TW. FOXO3a is activated in response to hypoxic stress and inhibits HIF1-induced apoptosis via regulation of CITED2. Mol Cell. 2007;28:941–953. doi: 10.1016/j.molcel.2007.10.035. [DOI] [PubMed] [Google Scholar]
- Berra E, Milanini J, Richard DE, Le Gall M, Viñals F, Gothié E, et al. Signaling angiogenesis via p42/44 MAP kinase and hypoxia. Biochem Pharmacol. 2000;60:1171–1178. doi: 10.1016/s0006-2952(00)00423-8. [DOI] [PubMed] [Google Scholar]
- Du F, Zhu L, Qian ZM, Wu X, Yung W, Xu W, et al. Expression of bystin in reactive astrocytes induced by ischemia/reperfusion and chemical hypoxia in vitro. Biochim Biophys Acta Mol Basis Dis. 2008;1782:658–663. doi: 10.1016/j.bbadis.2008.09.007. [DOI] [PubMed] [Google Scholar]
- Fan X, Heijnen CJ, van der Kooij MA, Groenendaal F, van Bel F. The role and regulation of hypoxia-inducible factor-1alpha expression in brain development and neonatal hypoxic-ischemic brain injury. Brain Res Rev. 2009;62:99–108. doi: 10.1016/j.brainresrev.2009.09.006. [DOI] [PubMed] [Google Scholar]
- Gijbels MJ, Scheffer JJ, Svendsen AB. Phthalides in the essential oil from roots of LeVisticumofficinale. Planta Med. 1982;44:207–211. doi: 10.1055/s-2007-971448. [DOI] [PubMed] [Google Scholar]
- Harada T, Morooka T, Ogawa S, Nishida E. ERK induces p35, a neuron-specific activator of Cdk5, through induction of Egr1. Nat Cell Biol. 2001;3:453–459. doi: 10.1038/35074516. [DOI] [PubMed] [Google Scholar]
- He W, Qian Zhong M, Zhu L, Christopher Q, Du F, Yung WH, et al. Ginkgolides mimic the effects of hypoxic preconditioning to protect C6 cells against ischemic injury by up-regulation of hypoxia-inducible factor-1 alpha and erythropoietin. Int J Biochem Cell Biol. 2008;40:651–662. doi: 10.1016/j.biocel.2007.10.013. [DOI] [PubMed] [Google Scholar]
- Ho KP, Li L, Zhao L, Qian ZM. Genistein protects primary cortical neurons from iron-induced lipid peroxidation. Mol Cell Biochem. 2003;247:219–222. doi: 10.1023/a:1024142004575. [DOI] [PubMed] [Google Scholar]
- Hou YZ, Zhao GR, Yang J, Yuan YJ, Zhu GG, Hiltunen R. Protective effect of Ligusticum chuanxiong and Angelica sinensis on endothelial cell damage induced by hydrogen peroxide. Life Sci. 2004;75:1775–1786. doi: 10.1016/j.lfs.2004.05.005. [DOI] [PubMed] [Google Scholar]
- Huang LF, Li BY, Liang YZ, Guo FQ, Wang YL. Application of combined approach to analyze the constituents of essential oil from Dong Quai. Anal Bioanal Chem. 2004;378:510–517. doi: 10.1007/s00216-003-2309-z. [DOI] [PubMed] [Google Scholar]
- Huang WH, Song CQ. Research progresses in the chemistry and pharmacology of Angelica sinensis (Oliv.) Diel. Zhongguo Zhong Yao Za Zhi. 2001;26:147–151. [PubMed] [Google Scholar]
- Jewell UR, Kvietikova I, Scheid A, Bauer C, Wenger RH, Gassmann M. Induction of HIF-1α in response to hypoxia is instantaneous. FASEB J. 2001;15:1312–1324. [PubMed] [Google Scholar]
- Kilic E, Kilic U, Soliz J, Bassetti CL, Gassmann M, Hermann DM. Brain-derived erythropoietin protects from focal cerebral ischemia by dual activation of ERK-1/-2 and Akt pathways. FASEB J. 2005;19:2026–2028. doi: 10.1096/fj.05-3941fje. [DOI] [PubMed] [Google Scholar]
- van der Kooij MA, Groenendaal F, Kavelaars A, Heijnen CJ, van Bel F. Neuroprotective properties and mechanisms of erythropoietin in in vitro and in vivo experimental models for hypoxia/ischemia. Brain Res Rev. 2008;59:22–33. doi: 10.1016/j.brainresrev.2008.04.007. [DOI] [PubMed] [Google Scholar]
- Kuang X, Yao Y, Du JR, Liu YX, Wang CY, Qian ZM. Neuroprotective role of Z-ligustilide against forebrain ischemic injury in ICR mice. Brain Res. 2006;1102:145–153. doi: 10.1016/j.brainres.2006.04.110. [DOI] [PubMed] [Google Scholar]
- Li Z, Ya K, Xiao-Mei W, Lei Y, Yang L, Ming QZ. Ginkgolides protect PC12 cells against hypoxia-induced injury by p42/p44 MAPK pathway-dependent upregulation of HIF-1alpha expression and HIF-1DNA-binding activity. J Cell Biochem. 2008;103:564–575. doi: 10.1002/jcb.21427. [DOI] [PubMed] [Google Scholar]
- Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20:84–91. doi: 10.1161/01.str.20.1.84. [DOI] [PubMed] [Google Scholar]
- McCulloch J. Excitatory amino acid antagonists and their potential for the treatment of ischaemic brain damage in man. Br J Clin Pharmacol. 1992;34:106–114. doi: 10.1111/j.1365-2125.1992.tb04118.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marti HH. Erythropoietin and the hypoxic brain. J Exp Biol. 2004;207(Pt 18):3233–3242. doi: 10.1242/jeb.01049. [DOI] [PubMed] [Google Scholar]
- Minet E, Michel G, Mottet D, Raes M, Michiels C. Transduction pathways involved in hypoxia-inducible factor-1 phosphorylation and activation. Free Radic Biol Med. 2001;31:847–855. doi: 10.1016/s0891-5849(01)00657-8. [DOI] [PubMed] [Google Scholar]
- Nagai A, Nakagawa E, Choi HB, Hatori K, Kobayashi S, Kim SU. Erythropoietin and erythropoietin receptors in human CNS neurons, astrocytes, microglia, and oligodendrocytes grown in culture. J Neuropathol Exp Neurol. 2001;60:386–392. doi: 10.1093/jnen/60.4.386. [DOI] [PubMed] [Google Scholar]
- Naito T, Katsuhara T, Niitsu K, Ikeya Y, Okada M, Mitsuhashi H. Two phthalides from Ligusticum Chuangxiong. Phytochemistry. 1992;31:639–642. [Google Scholar]
- Peng HY, Du JR, Zhang GY, Kuang X, Liu YX, Qian ZM, et al. Neuroprotective effect of Z-ligustilide against permanent focal ischemic damage in rats. Biol Pharm Bull. 2007;30:309–312. doi: 10.1248/bpb.30.309. [DOI] [PubMed] [Google Scholar]
- Qian ZM, Wang CY, Du JR. A method for the preparation of high purity Ligustilide. 2005. Patent no. ZL 2005 1002 1261.2.
- Rabie T, Marti HH. Brain protection by erythropoietin: a manifold task. Physiology (Bethesda) 2008;23:263–274. doi: 10.1152/physiol.00016.2008. [DOI] [PubMed] [Google Scholar]
- Rhyu MR, Kim JH, Kim EY. Radix angelica elicits both nitric oxide-dependent and calcium influx-mediated relaxation in rat aorta. J Cardiovasc Pharmacol. 2005;46:99–104. doi: 10.1097/01.fjc.0000164092.88821.49. [DOI] [PubMed] [Google Scholar]
- Saleem S, Zhuang H, Biswal S, Christen Y, Dore S. Ginkgo biloba extract neuroprotective action is dependent on heme oxygenase 1 in ischemic reperfusion brain injury. Stroke. 2008;39:3389–3396. doi: 10.1161/STROKEAHA.108.523480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattler MB, Merkler D, Maier K, Stadelmann C, Ehrenreich H, Bahr M, et al. Neuroprotective effects and intracellular signaling pathways of erythropoietin in a rat model of multiple sclerosis. Cell Death Differ. 2004;11(Suppl. 2):S181–S192. doi: 10.1038/sj.cdd.4401504. [DOI] [PubMed] [Google Scholar]
- Shoshani T, Faerman A, Mett I, Zelin E, Tenne T, Gorodin S, et al. Identification of a novel hypoxia-inducible factor 1-responsive gene, RTP801, involved in apoptosis. Mol Cell Biol. 2002;22:2283–2293. doi: 10.1128/MCB.22.7.2283-2293.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siren AL, Fratelli M, Brines M, Goemans C, Casagrande S, Lewczuk P, et al. Erythropoietin prevents neuronal apoptosis after cerebral ischemia and metabolic stress. Proc Natl Acad Sci U S A. 2001;98:4044–4049. doi: 10.1073/pnas.051606598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolze I, Berchner-Pfannschmidt U, Freitag P, Wotzlaw C, Rössler J, Frede S, et al. Hypoxia-inducible erythropoietin gene expression in human neuroblastoma cells. Blood. 2002;100:2623–2628. doi: 10.1182/blood-2001-12-0169. [DOI] [PubMed] [Google Scholar]
- Wu X, Qian ZM, Ke Y, Du F, Zhu L. Ginkgolide B preconditioning protects neurons against ischaemia-induced apoptosis. J Cell Mol Med. 2009;13:4474–4483. doi: 10.1111/j.1582-4934.2008.00551.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yatsiv I, Grigoriadis N, Simeonidou C, Stahel PF, Schmidt OI, Alexandrovitch AG, et al. Erythropoietin is neuroprotective, improves functional recovery, and reduces neuronal apoptosis and inflammation in a rodent model of experimental closed head injury. FASEB J. 2005;19:1701–1703. doi: 10.1096/fj.05-3907fje. [DOI] [PubMed] [Google Scholar]
- Yu Y, Du JR, Wang CY, Qian ZM. Protection against hydrogen peroxide-induced injury by Z-ligustilide in PC12 cells. Exp Brain Res. 2008;184:307–312. doi: 10.1007/s00221-007-1100-3. [DOI] [PubMed] [Google Scholar]
- Zhang L, Du JR, Wang J, Yu DK, Chen YS, He Y, et al. Z-ligustilide extracted from Radix Angelica Sinensis decreased platelet aggregation induced by ADP ex vivo and arterio-venous shunt thrombosis in vivo in rats. Yakugaku Zasshi. 2009;129:855–859. doi: 10.1248/yakushi.129.855. [DOI] [PubMed] [Google Scholar]
- Zheng KY, Choi RC, Xie HQ, Cheung AW, Guo AJ, Leung KW, et al. The expression of erythropoietin triggered by Danggui Buxue Tang, a Chinese herbal decoction prepared from Radix Astragali and Radix Angelicae Sinensis, is mediated by the hypoxia-inducible factor in cultured HEK293T cells. J Ethnopharmacol. 2010;132:259–267. doi: 10.1016/j.jep.2010.08.029. [DOI] [PubMed] [Google Scholar]
- Zhu L, Xu YJ, Du F, Qian ZM. Ginkgolides protect primary cortical neurons from potassium cyanide-induced hypoxic injury. Exp Brain Res. 2007;179:665–671. doi: 10.1007/s00221-006-0823-x. [DOI] [PubMed] [Google Scholar]
- Zhu L, Du F, Yang L, Wu XM, Qian ZM. Nerve growth factor protects the cortical neurons from chemical hypoxia-induced injury. Neurochem Res. 2008;33:784–789. doi: 10.1007/s11064-007-9495-6. [DOI] [PubMed] [Google Scholar]