

Abstract
BACKGROUND AND PURPOSE
The aim was to advance the understanding of Orai proteins and identify a specific inhibitor of the associated calcium entry mechanism in vascular smooth muscle cells (VSMCs).
EXPERIMENTAL APPROACH
Proliferating VSMCs were cultured from human saphenous veins. Intracellular calcium was measured using fura-2, whole-cell current was recorded using patch-clamp and cell migration quantified in modified Boyden chambers. Subcellular protein localization was determined by microscopy. Isometric tension was recorded from mouse aortic rings.
KEY RESULTS
Molecular disruption and rescue experiments indicated the importance of Orai1 in calcium entry caused by store depletion evoked passively or by platelet-derived growth factor (PDGF), suggesting the presence of Ca2+ release-activated Ca2+ (CRAC) channels like those of the immune system. The CRAC channel blocker, S66, was a potent inhibitor of the VSMC signals, IC50 26 nM, which was almost two orders of magnitude greater than with leucocytes. S66 had no effect on PDGF- and ATP-evoked calcium release, overexpressed transient receptor potential canonical (TRPC)5 channels, native TRPC1/5-containing channels, stromal interaction molecule 1 clustering, non-selective cationic current evoked by store depletion and phenylephrine-evoked aortic contraction. S66 reduced PDGF-evoked VSMC migration while having only modest effects on cell proliferation and no effect on cell viability.
CONCLUSIONS AND IMPLICATIONS
The data suggest that Orai1 has a role in human VSMC migration, and that a CRAC channel inhibitor has high potency and selectivity for the associated calcium entry, suggesting a distinct characteristic of vascular CRAC channels and the potential for selective chemical suppression of vascular remodelling.
Keywords: vascular smooth muscle, migration, calcium channel, store-operated calcium entry, platelet-derived growth factor, chemical blockade
Introduction
Contractile smooth muscle cells regulate the diameters of most blood vessels. Although they are relatively quiescent, throughout life they retain capacity for profound but controlled plasticity that enables them to change reversibly to non-contractile and mobile phenotypes that play critical roles in vascular development, tissue remodelling and responses to injury (Owens et al., 2004). This property of these cells contributes not only to physiology but to major disease phenomena and unwanted clinical responses, which include restenosis, atherosclerosis and neointimal hyperplasia of the saphenous vein (Angelini and Jeremy, 2002; Kumar et al., 2006). The molecular events controlling phenotypic switching in smooth muscle cells and the properties of the non-contractile cells, such as mobility, are starting to emerge (Owens et al., 2004).
Important events determining smooth muscle cell functions are plasma membrane ion fluxes such as Ca2+ entry (Beech, 2007). Voltage-gated Ca2+ entry channels (especially the dihydropyridine-sensitive L-type channels) have established roles in the regulation of blood pressure and the treatment of hypertension. However, other non-voltage-gated Ca2+ entry mechanisms exist and appear to make greater contributions in the non-contractile smooth muscle cells (Landsberg and Yuan, 2004; Beech, 2007). One of these mechanisms is store-operated Ca2+ entry, which was identified in vascular smooth muscle 30 years ago (Casteels and Droogmans, 1981). Various studies have suggested roles of transient receptor potential canonical (TRPC) channels (Alexander et al., 2008) in the store-operated mechanism and shown the channels to have an important role in non-contractile properties of smooth muscle cells (Sweeney et al., 2002; Kumar et al., 2006; Takahashi et al., 2007; Li et al., 2008). Nevertheless, experiments using TRPC channel blockers or molecular approaches for TRPC disruption suggest that TRPC channels explain only part of the store-operated Ca2+ entry signal of vascular smooth muscles (VSMCs) (Li et al., 2008) or even none of it according to some studies (Potier et al., 2009). Therefore, an additional mechanism is inferred. A candidate for the additional mechanism is the store-operated Ca2+ release-activated Ca2+ (CRAC) channel that has been associated primarily with immune cells (Hogan et al., 2010).
Determinants of CRAC channels are stromal interaction molecule 1 (STIM1) and Orai1 (Feske et al., 2006; Cahalan, 2009; Hogan et al., 2010). Orai1 is one of three related mammalian proteins encoded by distinct genes. The proteins are thought to contain four membrane-spanning segments and resemble, in some regards, the extensively-studied tetraspanin proteins, which have roles in mammalian cell morphology and motility. Tetraspanins had not been considered to be ion channel subunits, but various studies now suggest that Orai1 is the ion pore-forming subunit of CRAC channels (Hogan et al., 2010). Despite the focus on immune cells and absence of a vascular phenotype in patients with disrupted Orai1, reports have suggested that Orai proteins are expressed, and that Orai1 is functional in VSMCs (Berra-Romani et al., 2008; Baryshnikov et al., 2009; Potier et al., 2009; Bisaillon et al., 2010; Ng et al., 2010). One report has importantly described a whole-cell CRAC channel-like current in rat cultured VSMCs (Potier et al., 2009), but the very small size of the current and failure to detect the current in the presence of physiological Ca2+ have generated challenges for establishing its relevance to VSMC biology. Here we describe an investigation of the relevance of Orai proteins and CRAC channels to proliferating human saphenous vein VSMCs with particular focus on identification of a chemical agent that could facilitate studies of the mechanism in the cardiovascular field.
Methods
Cell culture
Freshly discarded human saphenous vein segments were obtained anonymously and with informed consent from patients undergoing open heart surgery in the General Infirmary at Leeds. Approval was granted by the Leeds Teaching Hospitals Local Research Ethics Committee. Proliferating VSMCs were prepared using an explant technique (Porter et al., 2001) and grown in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% fetal calf serum (FCS), penicillin/streptomycin and L-glutamine at 37°C in a 5% CO2 incubator. Experiments were performed on VSMCs passaged two to five times; all of the cells stained positively for smooth muscle α-actin and smooth muscle myosin heavy chain and were non-contractile. Tetracycline-induced expression of exogenous human TRPC5 in HEK 293 cells has been described previously (Zeng et al., 2004). HL-60 (human promyelocytic leukaemia) cells were from ATTC (Teddington, Middlesex, UK) and were cultured in RPMI 1640 with 10% FCS and penicillin/streptomycin.
VSMC transfections and knock-down validation
Cells (0.5–2 × 106) were centrifuged (100×g) for 10 min, resuspended in Basic Nucleofector solution (Amaxa GmbH, Lonza GmbH, Cologne, Germany), mixed with 1 µM short interfering (si) RNA (Ambion Europe Ltd, Applied Biosystems, Warrington, UK; Table S1) and transferred into a cuvette for electroporation (Amaxa). With this method, the siRNA concentration is higher than used with lipid transfection methods, but the exposure time to the siRNA is considerably shorter. The scrambled control siRNA was Silencer Negative Control #1, which is a 19 bp scrambled sequence with no significant homology to human gene sequences (Ambion). Cells were transferred from cuvettes to pre-warmed culture medium and incubated in a 5% CO2 incubator at 37°C. Culture medium was changed after 24 h, and measurements were made after a further 24 h. When two siRNA probes were used as a cocktail, each probe was used at one-half of its usual concentration. Full-length cDNA for Orai1 (BC013386) was purchased from Geneservice (Source Bioscience LifeSciences, UK) and subcloned into pcDNA6/V5-His (Invitrogen, Paisley, UK) at EcoRI/XhoI sites. cDNA encoding eYFP-STIM1 was a gift from T Meyer. Clones were sequenced to confirm identities. Transfections with 1–1.5 µg of DNA also occurred by electroporation.
RNA isolation and quantitative reverse transcription PCR
Total RNA was extracted from saphenous vein smooth muscle cells using a Tri-reagent protocol followed by DNase I (Ambion) treatment (Fountain et al., 2004). One microgram of total RNA was used for RT based on oligo-dT primers and AMV RT enzyme. The specificity of PCR was verified by reactions without RT (–RT) and by melt curve analysis of PCR products. Sequences of PCR primers are shown in Table S1. PCR products were electrophoresed on 2% agarose gels containing ethidium bromide. No PCR products occurred in the absence of RT. With RT, there was only a single product of the correct size, giving a single peak in the melt curve. All PCR products were sequenced to confirm identity (Lark Technologies,Takeley, Essex, UK). Real-time PCR was carried out using a Lightcycler (Roche, Welwyn Garden City, UK) largely as described previously (Fountain et al., 2004). Relative abundance of target RNA was normalized to β-actin RNA, which showed no difference between samples. PCR efficiency (E) was 10(−1/slope). Relative abundance of target RNA was calculated from (Eβ-actinCp) / (EtargetCp), where PCR cycle crossing points (CP) were determined by fit-points methodology. PCR reactions were at least in duplicate.
Western blotting
Cells were collected using 1 mL of ice-cold phosphate-buffered saline (PBS) and pelleted at 4°C and then lysed in Laemmli sample buffer supplemented with complete protease inhibitor cocktail (Roche). The lysate was centrifuged to remove particulate matter, and equal amounts of protein were separated by 8% sodium dodecyl sulphate polyacrylamide gel electrophoresis and transferred onto nitrocellulose membrane. Membranes were incubated with blocking buffer (containing 0.1% Tween 20 and 5% nonfat dry milk) for 2 h at room temperature, followed by incubation in dialysed rabbit anti-Orai1 antibody (Alomone Labs Ltd., Jerusalem, Israel, #ACC-060) overnight at 4°C. Anti-β-actin antibody (1:1000) was from Santa Cruz. Membranes (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA) were then washed and incubated with secondary antibody. Labelling was detected using SuperSignal West Pico chemiluminescent substrates (Pierce, Thermo FisherScientific Inc., Rockford, IL, USA).
Intracellular calcium measurements
Cells were pre-incubated with fura-2AM for 1 h at 37°C followed by a 0.5 h wash at room temperature. HL-60 cells were centrifuged in the plate at 2 × 105 cells per well. Measurements were made at room temperature on a 96-well plate reader (FlexStation, Molecular Devices Inc., Sunnyvale, CA, USA). The change (Δ) in intracellular calcium (Ca2+i) concentration is indicated as the ratio of fura-2 emission intensities for 340 and 380 nm excitation (F ratio). Wells within columns of the 96-well plate were loaded alternately for test and control conditions. The recording solution contained (mM): 130 NaCl, 5 KCl, 8 d-glucose, 10 HEPES and 1.2 MgCl2, titrated to pH 7.4 with NaOH. When indicated, 0.2 or 1.5 mM CaCl2 was added. EGTA was not included in the Ca2+-free recording solution because of the solution addition, rather than replacement, format of the FlexStation; therefore, contaminating Ca2+ was present (estimated to be 1–10 µM).
Deconvolution microscopy
VSMCs, 48 h post-transfection, were detached and transferred to glass coverslips with fresh culture medium and allowed to spread for 24 h. After treatments with reagents, the cells were fixed in methanol for 10 min at −20°C, washed and rehydrated in PBS, and mounted onto glass slides using ProLong Gold anti-fade (Molecular Probes®, Invitrogen, Paisley, UK). Cells were visualized on an Olympus IX-70 inverted microscope using a × 100 UPLAN objective (NA 1.35) supported by a DeltaVision deconvolution system (Applied Precision LLC Issaquah, WA, USA) with SoftWoRx image acquisition and analysis software. Images were captured on a Roper CoolSNAP HQ CCD camera, and epifluorescence was recorded using filter sets for FITC. Wide-field optical sections of 0.2 µm were acquired throughout the z plane of the cells and deconvolved using a constrained iterative algorithm assigned by Delta Vision.
Cell assays
Cell migration assays were performed in duplicate in modified Boyden chambers with polycarbonate membranes containing 8 µm pores (BD Biosciences, Oxford Science Park, Oxford, UK). Cells in suspension (1 × 105) were loaded in the upper chamber in DMEM supplemented with 0.4% FCS. The lower chamber contained DMEM (0.4% FCS) supplemented with the chemoattractant 10 ng mL−1 platelet-derived growth factor (PDGF)-BB (Invitrogen). After 8 h at 37°C in a 5% CO2 incubator, cells that had attached but not migrated were scraped from the upper surface, membranes were fixed in 70% ethanol at −20°C and the migrated cells were stained with haematoxylin and eosin and evaluated by counting cell nuclei in 10 randomly chosen fields under light microscopy. For Orai1 knock-down cell proliferation experiments, equal numbers of cells from the same patient were transfected and seeded in parallel into six-well tissue culture plates in DMEM culture medium plus 10% FCS. Medium was changed after 24 h, and cells were incubated for a further 24 h. Cells were collected after trypsinization, stained with trypan blue, and the cell number was determined in duplicate wells and counted at least twice with a haemocytometer. Trypsinized wells were observed microscopically to confirm that all cells had been released. For S66 experiments, cells at 40–50% confluence were seeded in 96-well plates for the indicated time periods in at least triplicate wells. At the end of the specified times, cells were fixed and stained with haematoxylin and eosin for counting in four random fields. The medium was replaced every 24 h to provide fresh S66.
Patch-clamp recordings
Recordings were made using the Patchliner planar patch-clamp system (Nanion Technologies, Munich, Germany) in whole-cell mode (Milligan et al., 2009). Prior to recordings, cells were detached from culture flasks with Detachin (Gelantis Inc., AMS Biotechnology Europe Ltd., Abingdon, UK, cat. no. T100110) or 0.05% Trypsin/EDTA and resuspended at a density of 1 × 106–5 × 107 mL−1 in extracellular solution that was the same as the recording solution for Ca2+ measurements except it contained 0.2 mM CaCl2 and 100 µM niflumic acid. The intracellular recording solution contained (mM): 40 EGTA, 17 CaCl2, 2 MgCl2, 8 NaCl, 1 Na2ATP, 10 HEPES, 66 L-glutamic acid, 66 CsOH, titrated to pH 7.2 with CsOH (calculated unbound Ca2+, 100 nM). Corrections were not made for liquid–liquid junction potentials. Voltage ramps were applied from −100 to +100 mV for 1 s every 10 s from a holding potential of 0 mV. Currents were filtered at 1 kHz and sampled at 3 kHz.
Isometric tension recording
For isometric tension recordings, 8 week old male C57/BL6 mice were killed by CO2 asphyxiation and cervical dissociation in accordance with Schedule 1 Code of Practice, UK Animals Scientific Procedures Act 1986. The thoracic aorta of the mouse was removed and placed in ice-cold Hank's solution. Fat was removed by dissection, and blood was flushed from the lumen with Hank's solution. Hanks solution contained (mM): NaCl, 137; KCl, 5.4; CaCl2, 0.01; NaH2PO4, 0.34; K2HPO4, 0.44; d-glucose, 8; HEPES, 5. Vessels were mounted on two 40 µm diameter wires for isometric tension recording in a 410A dual wire myograph system (Danish Myo Technology, Aarhus, Denmark). The wires were separated by 1 mm to provide basal tension, and the bath solution was at 37°C gassed continuously with 95% O2 and 5% CO2. The bath solution contained (mM): 125 NaCl, 3.8 KCl, 25 NaHCO3, 1.5 MgSO4, 1.2 KH2PO4, 8 d-glucose, 1.2 CaCl2, 0.02 EDTA. For Ca2+-free solution (0 Ca2+), Ca2+ was omitted.
Reagents
S66 [3-fluoropyridine-4-carboxylic acid (2′,5′'-dimethoxybiphenyl-4-yl)amide] was a gift from GSK (Oxbridge, Middlesex, UK) (GSK1349571A); its chemical identity was confirmed independently by liquid chromatography mass spectrometry analysis (Leeds). S66 is an abbreviation of ‘Synta 66’ from patent WO 2005/009954 (Ng et al., 2008; Di Sabatino et al., 2009). All other reagents were from Sigma (Sigma-Aldrich, Gillingham, Dorset, UK) unless indicated otherwise.
Data analysis
Mean data are presented as mean ± SEM, where n represents the number of independent experiments and N represents the number of wells of a 96-well plate used in a single experiment. For patch-clamp experiments, n was the number of recordings from individual cells. In all experiments, comparisons were made independently with cells from at least three patient samples. Experiments were all performed in test and control pairs, and so t-tests were used for comparisons. Each experiment had its own paired control (ctrl), even though only single control columns (white) are shown in the figures. *P-values <0.05, which were taken to indicate significant difference; no significant difference is indicated by NS. Data were analysed and presented, and curves were fitted using Origin software (OriginLab Corporation, Northampton, MA, USA).
Results
Orai1 function in Ca2+ entry evoked by store depletion in proliferating human saphenous VSMCs
Messenger RNAs encoded by the Orai genes were readily detected in proliferating VSMCs and had relatively high abundance (Figure S1). To investigate if the gene expression is functionally relevant, we measured the Ca2+ entry signal occurring when Ca2+ was added back to cells that had been store-depleted by the pharmacological agent thapsigargin (Figure S2). Specific knock down of expression was achieved using small interfering (si) RNAs (Figure S3 and S4, Table S1). Knock down of Orai1 by two different Orai1 siRNAs suppressed store-operated Ca2+ entry, while knock down of Orai2 and Orai3 had no effect (Figure 1A, Figure S5). A third Orai1 siRNA was investigated, but it failed to have any effect on the expression of Orai1 (Figure S3), and so it acted as a negative control (Orai1 si.negative). Because the two active Orai1 siRNAs may have had off-target effects, rescue experiments were performed using wild-type Orai1 cDNA, which had no effect on its own but recovered the Ca2+ entry that was suppressed by Orai1 siRNA (Figure 1B). Ca2+ entry was also suppressed by the R91W dominant-negative mutant form of Orai1 (Derler et al., 2009) (Figure 1A, Figure S5). The data support the hypothesis that Orai1 is functionally important for Ca2+ entry evoked by thapsigargin and suggest that CRAC channels are present.
Figure 1.
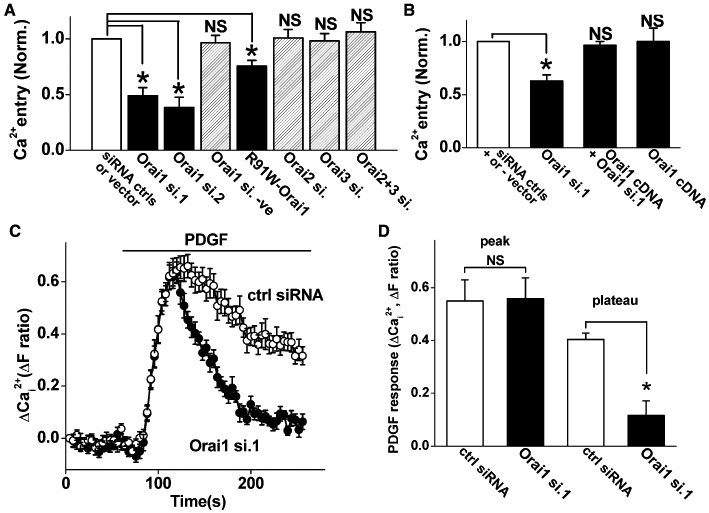
Effect of Orai1 knock down on Ca2+ entry of human saphenous vein vascular smooth muscle cells (VSMCs). (A, B) Prior to recording Ca2+ signals, VSMCs were divided into paired groups and transfected with control (siRNA ctrls, scrambled siRNA) or test siRNA (e.g. Orai1 si.1), or control DNA (vector) or vector expressing R91W-Orai1 or Orai1 (wild-type) cDNA. Although only one control column is shown in each graph, each test group had its own control, and the data were normalized (Norm.) to that control. NS indicates no significant difference compared with the matched control data. The ‘Orai1 si.negative’ was generated for the purpose of suppressing Orai1 expression but it failed to have any effect on the expression and so was used as a negative control. For Ca2+ recording, VSMCs were pretreated with 1 µM thapsigargin in Ca2+-free bath solution for 30 min and exposed to 0.2 mM extracellular Ca2+ as indicated. (A) Mean normalized (Norm.) data for experiments of the type illustrated in Figure S5 (n/N = 3–4/48–64). Raw mean control fura-2 ratio values for each of the seven groups were 0.55, 0.90, 0.54, 0.70, 0.56 and 0.57 (left to right). (B) Rescue of Ca2+ entry in Orai1 knock down cells by Orai1 cDNA (n/N = 3/48). Raw mean control fura-2 ratio values for each of the three groups were 0.50, 0.48 and 0.45 (left to right). (C) Typical Ca2+ responses to 100 ng·mL−1 PDGF in the presence of extracellular 1.5 mM Ca2+ after transfection with control (ctrl) or Orai1 siRNA 1 (N = 12 each). (D) Mean data for the type of experiment illustrated in (C) (n/N = 3/30).
Orai1 function in Ca2+ entry evoked by PDGF
PDGF is an important physiological stimulant of VSMCs that causes Ca2+ release. Direct application of PDGF in Ca2+-containing extracellular solution evoked a rise in intracellular Ca2+ that had rapid onset followed by decay towards a plateau phase of Ca2+ entry (Figure 1C). Knock down of Orai1 had no effect on the initial rise in Ca2+ but suppressed the Ca2+- entry (Figure 1C, D). The data suggest that PDGF-evoked Ca2+ entry depends substantially on Orai1.
Potent and specific chemical inhibition of Ca2+ entry
With studies of ion channels, it is helpful to have a potent and specific blocker that is easy to use and applicable to a range of assays. Therefore, we investigated if a CRAC channel blocker has effect on Ca2+ entry in VSMCs. The blocker tested was S66, which has previously been reported to specifically inhibit CRAC-related signals in immune cells with an IC50 of 1.4–3.0 µM (Ng et al., 2008; Di Sabatino et al., 2009) whether applied before or after store depletion (Ng et al., 2008). It was observed that the Ca2+ entry occurring upon returning Ca2+ to the medium (the add-back signal) in store-depleted VSMCs was strongly suppressed by S66 (Figure 2A). Unexpectedly, S66 had high potency, inhibiting the Ca2+ entry signal with an IC50 of 26 or 43 nM depending on whether the maximum or rate-of-rise Ca2+ signal was used (Figure 2B). We therefore made a direct comparison with human leucocytes using the same assay conditions. Consistent with previous reports, the leucocyte Ca2+ entry was much less sensitive, giving an IC50 of 1.76 µM (Figure 2B). The effect of S66 on VSMCs occurred relatively rapidly, with a time constant of ∼7 s (Figure 2C, D). S66 had no effect on the initial rise in Ca2+ evoked by PDGF but suppressed the plateau phase of Ca2+ entry (Figure 3A, B). Consistent with the lack of effect on Ca2+ release, in the absence of extracellular Ca2+, PDGF evoked a transient rise in intracellular Ca2+ that was unaffected by S66 (Figure 3C, D). Similar observations were made when ATP was the receptor agonist used to evoke Ca2+ release and Ca2+ entry (Figure 3E, F). The data suggest that S66 has high potency and specificity for store-operated Ca2+ entry in human VSMCs, and that the potency is almost two orders of magnitude greater compared with immune cells.
Figure 2.
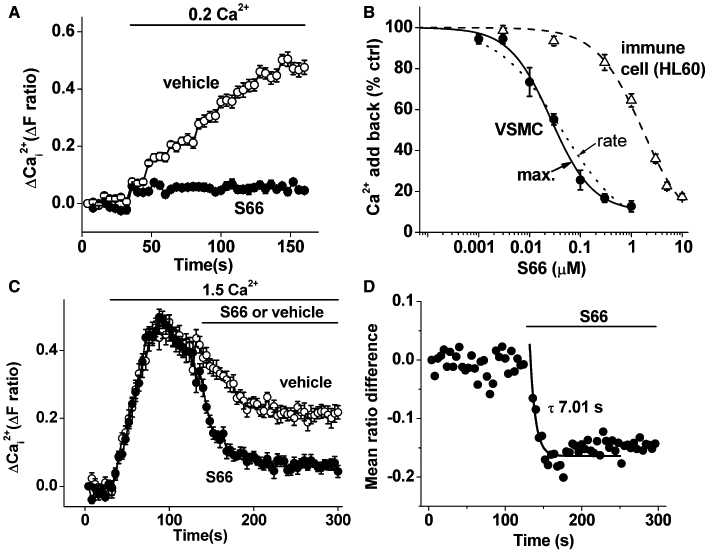
S66 inhibition of Ca2+ add-back responses in store-depleted VSMCs. (A) Example of VSMC paired experiment showing inhibition by 5 µM S66 of the Ca2+ entry signal observed when adding 0.2 mM Ca2+ to thapsigargin-treated cells (N = 10 each). (B) As for (A) but mean data for the maximum (max.) Ca2+ entry signal (n/N = 4/40) with a fitted Hill equation (IC50 26 nM, slope 1.05). The rate of rise of the same Ca2+ entry events (rate) was also analysed, and the mean data (not shown but see Figure S6) were fitted with a Hill equation (dotted curve, IC50 43 nM, slope 0.69). Comparative maximum Ca2+ entry data are shown for HL-60 cells, also with a fitted Hill equation (IC50 1.76 µM, slope 1.0). (C) As for (A) but 5 µM S66 or its vehicle were applied after add-back of 1.5 mM Ca2+ (N = 10 each). (D) As for (C) but the mean difference between S66 and vehicle data points (n = 3) with a fitted single exponential function (smooth curve, τ = 7.01 s).
Figure 3.
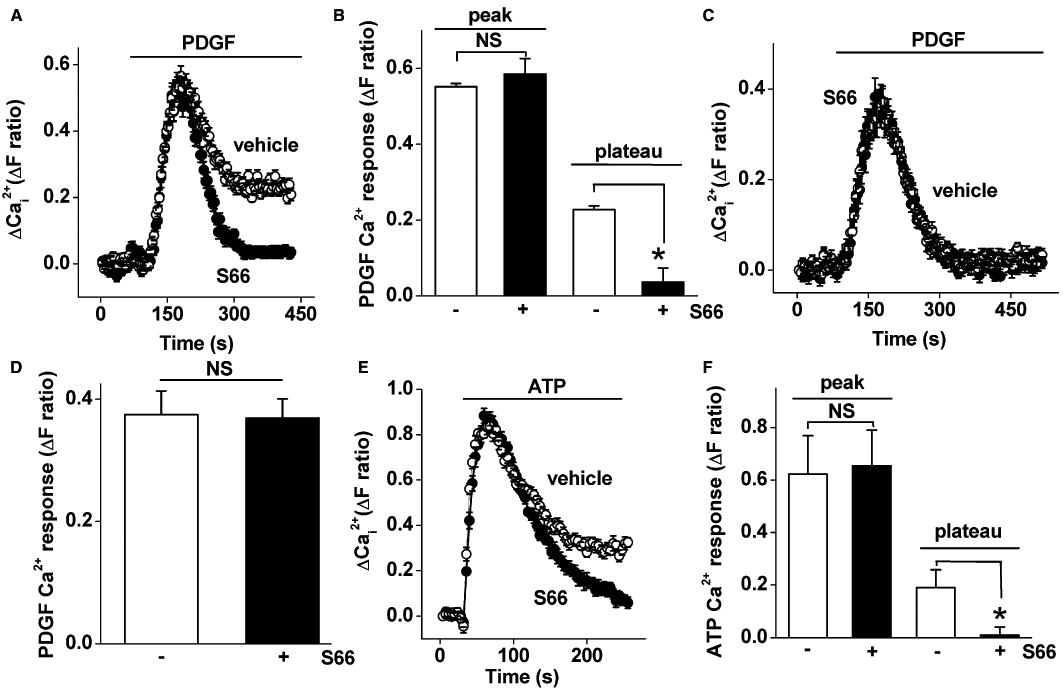
S66 inhibition of Ca2+ entry evoked by physiological agonists. Recordings were made from VSMCs. (A) Intracellular Ca2+ measurement showing responses to 100 ng·mL−1 PDGF in 1.5 mM extracellular Ca2+ and vehicle or 5 µM S66 (N = 32 each). (B) Mean data for the type of experiment illustrated in (A) comparing the vehicle control and S66 (n/N = 3/72). (C, D) As for (A, B) but in the absence of extracellular Ca2+ (n/N = 5/64) and therefore showing Ca2+ release only. (E, F) As for (A, B) but using 100 µM ATP instead of PDGF (n/N = 3/48).
Lack of effect of S66 on endogenous or overexpressed TRPC channels
The above data suggest that S66 has specificity because it lacked effect on the PDGF receptor or Ca2+ release mechanism. To further investigate the specificity, we tested S66 against the Ca2+ signal evoked by the endogenous oxidized phospholipid PGPC (1-palmitoyl-2-glutaroyl-phosphatidylcholine) (Al-Shawaf et al., 2010). Although PGPC evokes a transient rise in intracellular Ca2+, this effect is not accounted for by Ca2+ release but by Ca2+ entry mediated by TRPC1/5-containing channels (Al-Shawaf et al., 2010). The PGPC-evoked Ca2+ signal was completely unaffected by S66 at the high concentration of 5 µM (Figure 4A, B). We also investigated human TRPC5 conditionally overexpressed in HEK 293 cells and stimulated by the lanthanide ion, gadolinium (Zeng et al., 2004); this signal was also unaffected by S66 (Figure 4C). The data suggest that S66 does not inhibit endogenous TRPC1/5 or overexpressed TRPC5 channels.
Figure 4.
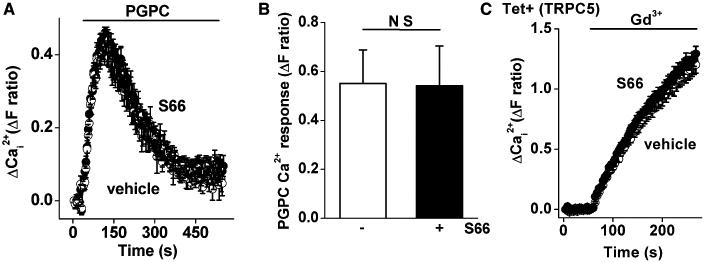
Resistance of TRPC1/5 channels to S66. Recordings were made from VSMCs (A, B) or HEK 293 cells (C). (A) Intracellular Ca2+ measurement showing responses to 5 µM 1-palmitoyl-2-glutaroyl-phosphatidylcholine (PGPC) in 1.5 mM extracellular Ca2+ and vehicle or 5 µM S66 (black circles) (N = 16 for each). (B) As for (A) but mean data for the vehicle control and S66 (n/N = 3/64). (C) As for (A) but using HEK 293 cells with overexpression of TRPC5 showing responses to 50 µM gadolinium (Gd3+) in the presence of vehicle or 5 µM S66 (N = 12, typical of n = 3).
Lack of effect of S66 on STIM1 clustering
A widely accepted mechanism for coupling of depleted Ca2+ stores to plasma membrane store-operated Ca2+ channels is the clustering of STIM1 proteins in sarcoendoplasmic reticulum membranes and the subsequent interaction with plasma membrane channels (Cahalan, 2009). Therefore, the effects of S66 on the clustering of STIM1 were investigated. YFP-tagged STIM1 was expressed in VSMCs and observed by fluorescence microscopy. In the absence of store depletion, STIM1 showed the expected microtubule-like localization pattern (Figure 5A), consistent with it binding a plus-end microtubule tracking protein (Cahalan, 2009). Also, as expected, application of thapsigargin caused striking re-arrangement of STIM1 into clusters (Figure 5B). S66, however, had no effect on the clustering process (Figure 5C). These data suggest that S66 does not act by inhibiting store depletion, its sensing by STIM1 or the subsequent re-arrangements of STIM1 that are considered to be necessary for activation of store-operated channels.
Figure 5.
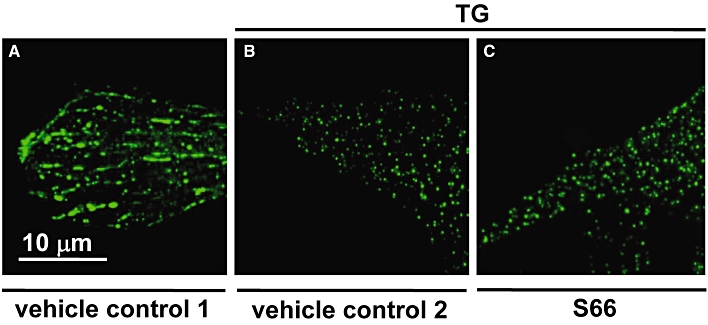
Resistance of STIM1 clustering to S66. The images all show VSMCs from the same batch transfected with plasmid expressing eYFP-STIM1 (green colour). VSMCs were then exposed for 5 min to: (A) the solvent for thapsigargin (vehicle control 1); (B) 1 µM thapsigargin (TG) and the solvent for S66 (vehicle control 2); or (C) 1 µM thapsigargin and 10 µM S66. The scale bar in (A) applies to all of the images. Data are representative of two independent experiments on VSMCs from different patients.
Lack of effect of S66 on store-operated non-selective cationic current
In human saphenous vein VSMCs, there is also a large non-selective cationic current that is evoked by thapsigargin and can be considered to be store-operated (Li et al., 2008). Other investigators have observed a similar ionic current in response to store depletion (Trepakova et al., 2001; Sweeney et al., 2002). This ionic current in human saphenous vein VSMCs was completely unaffected by S66 at 5 µM (Figure 6A–C). These data suggest that the action of S66 is not general for all types of store-operated channel.
Figure 6.
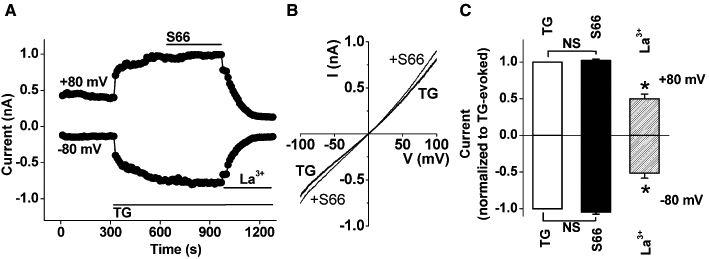
Resistance of non-selective store-operated cationic current to S66. Recordings were made from VSMCs. (A) Example of ionic current evoked by 2 µM thapsigargin (TG), showing effects of 5 µM S66 and 50 µM lanthanum (La3+). (B) As for (A) but example current–voltage relationships (I–V) for currents evoked by TG and then following the addition of S66. (C) As for (A) but mean data normalized to the current in the presence of TG alone, showing the effect of S66 and La3+ (n = 14 cells from 3 patients).
Lack of effect of S66 on phenylephrine-evoked contraction
The possible effect of S66 on the contractile function of VSMCs was investigated by making isometric tension recordings from freshly-isolated aortic rings of the mouse. Phenylephrine was used as an α1-adrenoceptor agonist to evoke contraction (Figure 7A). S66 (5 µM) had no effect on the evoked contraction whether applied before (Figure 7A, B) or acutely after phenylephrine (Figure 7B). Phenylephrine-evoked contraction did, nevertheless, depend substantially on Ca2+ entry (Figure 7B). These data further suggest the specificity of S66, as it had no effect on the α1-adrenoceptor-mediated signalling mechanisms, Ca2+ handling in contractile cells or the contractile machinery.
Figure 7.
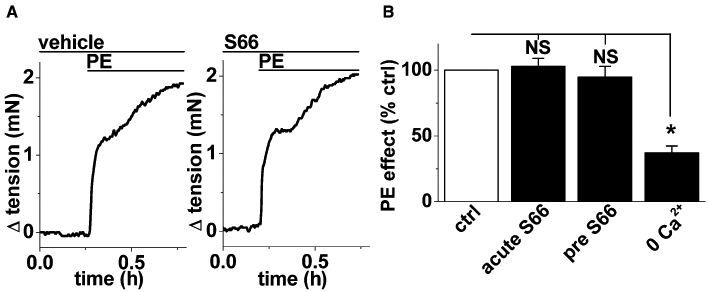
Resistance of aortic contraction to S66. Recordings were made from mouse aorta. (A) Sequential isometric tension recordings from a ring of mouse aorta showing responses to 1 µM phenylephrine (PE) in the presence of vehicle (DMSO, left panel) or 5 µM S66 (right panel). (B) Mean phenylephrine-evoked contractions normalized to the vehicle control data (ctrl) for the acute (15 min) effect of 5 µM S66 (acute S66) and effect of pretreatment and continuous application of 5 µM S66 (pre S66) (as in A). The effect of removing extracellular Ca2+ is shown for comparison (0 Ca2+).
Potent inhibition of VSMC migration by S66
Because of the high potency and selectivity of S66, we used it to investigate the relevance of the Ca2+ entry mechanism to VSMC migration and proliferation. Cell migration was equally suppressed by 0.1 and 5 µM S66 (Figure 8A, Figure S7), suggesting high potency against this cellular parameter that is similar to that observed against Ca2+ entry.
Figure 8.
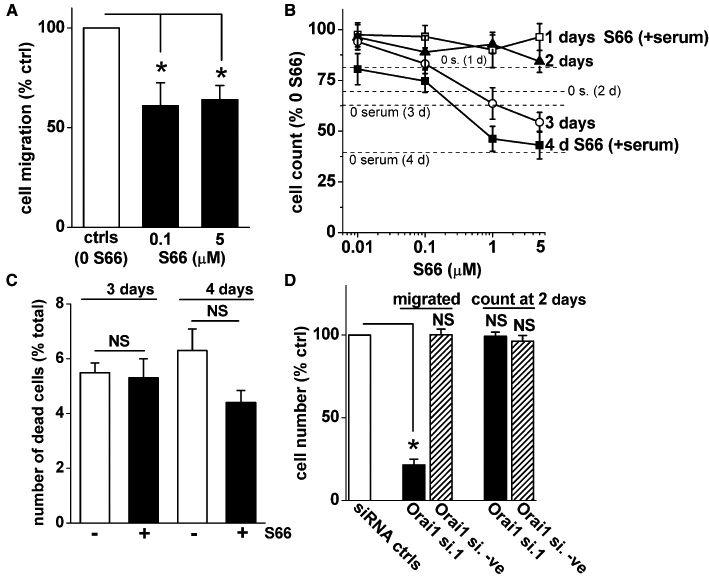
Effects of S66 on VSMC migration, proliferation and viability. (A, B) Mean data for S66 effects on cell migration (A) and cell count (B) as percentages of their own vehicle controls (ctrls). (B) Cell count was measured in the presence of serum and various concentrations of S66 at 1, 2, 3 and 4 day time points (1–4 days) (symbols with error bars and connecting lines). The dashed horizontal lines are comparative mean data for cell counts in serum-free conditions as percentages of their serum controls. (C) Mean numbers of cells that failed to exclude trypan blue (i.e. non-viable cells) as a percentage of the total number of cells for the vehicle control (white bar) and S66 (black bar) groups at 3 and 4 days. (D) Mean data for number of cells migrating in Boyden chambers (‘migrated’) and with trypan blue exclusion in the presence of serum over 2 days (‘count at 2 days’). Data obtained after transfection with Orai1 siRNA (Orai1 si.1) or Orai1 negative siRNA (Orai1 si.-ve) were normalized to their own scrambled siRNA controls (siRNA ctrls), even though only one control column is shown. (A, D) Raw mean migrated cell numbers in the controls (ctrls) were 28.9 and 32.5 (for the 0.1 and 5 S66 groups) and 30.4 and 31.8 (for the Orai1 si.1 and Orai1 si.-ve groups). (A–D) Data are from experiments performed at least twice with cells from at least four patients.
Modest effect of S66 on VSMC proliferation and lack of effect on cell viability
The saphenous vein VSMCs doubled in number over a period of ∼7 days in the presence of serum (data not shown), whereas in the absence of serum the cell number declined relative to the serum group (Figure 8B). The cell count in the presence of serum was quantified over 4 days in the presence of the S66 solvent (vehicle control) or different concentrations of S66. The cell counts were normalized to the vehicle controls at the specified time points (Figure 8B). At days 1 and 2, the cell count was unaffected by S66 and, similarly, concentrations of S66 below 1 µM lacked effects on the cell counts at days 3 and 4 (Figure 8B). There were, however, inhibitory effects of 1 and 5 µM S66 at days 3 and 4 (Figure 8B). Cell death, as indicated by trypan blue uptake, was small over these time periods in the presence of serum and was not affected by 5 µM S66 (Figure 8C). The data suggest that S66-sensitive Ca2+ entry has a positive but relatively modest effect on cell proliferation and no effect on cell viability.
Strong inhibition of VSMC migration by Orai1 siRNA
The data obtained with S66 suggested a relatively specific role of the Ca2+ entry channels in cell migration. It was therefore investigated if knock-down of Orai1 with siRNA had a similar effect (Figure 8D). Orai1 knock down had a strong inhibitory effect on VSMC migration and no effect on cell proliferation over 2 days (Figure 8D). The inhibition of cell migration was quantitatively greater than that observed with S66 (Figure 8D; cf. Figure 8A). The data suggest that CRAC channels and Orai1 impact positively on VSMC migration, and that Orai1 may not only contribute through the CRAC channel mechanism.
Discussion and conclusions
These data support the hypothesis that Orai1 has a role in Ca2+ influx evoked by store depletion in VSMCs and reveal a chemical inhibitor of the CRAC channel that is a promising tool for further investigation of the mechanism, showing ∼100-fold selectivity for VSMCs over leucocytes. The chemical showed excellent specificity, lacking effects on PDGF receptors, ATP receptors, Ca2+ release, TRPC1/5 channel-dependent Ca2+ entry, STIM1 clustering, non-selective cationic current evoked by store depletion and the entire mechanisms of α-adrenoceptor-mediated vascular contraction. In line with results from Orai1 knock-down experiments, S66 suppressed VSMC migration, although not as effectively as Orai1 knock down.
In addition to patent information (WO 2005/009954), effects of S66 on CRAC channel activity in rat basophilic leukaemia and Jurkat T cells have been reported in the scientific literature (Ng et al., 2008; Di Sabatino et al., 2009). S66 was found to have specificity because it did not affect plasma membrane Ca2+ ATPase, inward rectifier K+ current, hippocampal synaptic transmission or radioligand binding to a range of receptors and ion channels including the L-type Ca2+ channel (Ng et al., 2008; Di Sabatino et al., 2009). Our data not only expand the evidence for specificity but also show that S66 has two orders of magnitude greater potency at the CRAC mechanism in VSMCs. In confirmation of prior observations, and under identical conditions to those used for the VSMC recordings, we showed that the CRAC mechanism in leucocytes is less sensitive to S66. The data suggest pharmacological opportunity and a fundamental difference between the CRAC mechanisms of the vascular and immune cells, even though Orai1 and STIM1 are involved in both cases.
The ionic current of Figure 6 has been previously described as a non-selective cationic current evoked by thapsigargin in saphenous vein smooth muscle cells and inhibited partially by extracellular antibody targeted to STIM1, suggesting the importance of plasma membrane STIM1(Li et al., 2008). The lack of cationic selectivity of this current, its large amplitude and relatively linear current–voltage relationship suggest that it is not mediated by a CRAC channel. Other investigators have observed similar currents (Trepakova et al., 2001; Sweeney et al., 2002; Stiber et al., 2008). We show here that the current in saphenous vein smooth muscle cells is not sensitive to acute inhibition by S66, further distinguishing it from the CRAC mechanism. It might be expected that a CRAC channel current would have been evident in our electrophysiology experiments, but it was not observed, possibly because of the extremely small size of the current, which was not detectable in rat smooth muscle cells in the presence of extracellular calcium (Potier et al., 2009).
Conflicting conclusions are reported about the contributions of TRPC channels to store-operated Ca2+ entry in VSMCs. Nevertheless, specifically in relation to human saphenous vein VSMCs, our data consistently point to a role of TRPC channels in the Ca2+ add-back signal of thapsigargin-treated cells, based on anti-TRPC1 blocking antibody, TRPC1/4/5 RNA interference and dominant-negative mutant TRPC experiments (Kumar et al., 2006; Li et al., 2008). The effects have, nevertheless, been only partial (up to 40%). Therefore, at least 60% of the Ca2+ add-back signal would seem to have been mediated by other mechanisms (i.e. non-TRPC1/4/5 mechanisms). These findings should be considered in the context of the present study where we observed that S66 had no effect on TRPC activity evoked by non-store depletion mechanisms but inhibited store-operated Ca2+ entry by about 90%. It seems therefore that there is a quantitative discrepancy with the previous data based on disruption of TRPC subunits. A reason for the discrepancy (i.e. of ∼10% vs. ∼40% TRPC contribution) could be that the TRPC contribution to the store-operated Ca2+ add-back signal depends on CRAC channel activity. If this is the case, the TRPC channels would contribute to the Ca2+ add-back signal only if the CRAC channels were first activated by store depletion and then the CRAC channels stimulated the TRPC channels, perhaps via Ca2+ facilitation. This may be an over-simplification, however, because of compelling evidence that TRPC channels are activated directly by store depletion due to physical coupling with STIM1 (Zeng et al., 2008). Indeed, our immune-precipitation and immune-staining results have supported the conclusion that a pool of TRPC1 interacts with STIM1 in saphenous vein smooth muscle cells (Li et al., 2008). Although the relationship of TRPC channels to store depletion remains puzzling, it is clear that such channels do not require store depletion in order to be activated in VSMCs (Al-Shawaf et al., 2010).
The mechanism of action of S66 on Ca2+ entry is unknown. It acts relatively rapidly and so may function as an ion pore blocker of CRAC channels. Most simply, it would have such an effect by binding directly to Orai1. However, evidence to support such a hypothesis is lacking and the common role of Orai1 in Ca2+ entry of VSMCs and immune cells, and yet differential potencies of S66 on these cell types, argues against Orai1 as the target protein. However, if Orai1 is not the target protein, we do not have another clear candidate. Our data argue against S66 acting through disruption of the STIM1 mechanism for store-to-channel coupling (Figure 5) or as a general blocker of all types of store-operated channel (Figure 6). It may interfere with the physical coupling between STIM1 and Orai1, but it is unclear why the VSMC Ca2+ entry would be more potently affected.
One of the curious results of the study was that Orai1 knock down inhibited VSMC migration by about 80% whereas S66 inhibited it by only 40%. If Orai1 acted only as a subunit of CRAC channels, the percentage inhibition would be predicted to be similar or the effect of S66 may even be anticipated to be greater because it is such a strong inhibitor of the channels and siRNA knocks down the gene expression rather than deleting it. One explanation could be that Orai1 has a role in cell migration in addition to those that occur via the CRAC channel. However, additional evidence for such a hypothesis is needed because the possibility that S66 becomes less effective progressively in culture, either because of chemical modification or desensitization at its receptor, was not excluded.
In summary, the results of this study suggest the importance of Orai1 and CRAC channels as mediators of thapsigargin-, PDGF- and ATP-evoked Ca2+ entry in human saphenous vein VSMCs and that this mechanism potentiates migration of the cells but is not essential for migration. These findings support the hypothesis that CRAC channels are important in VSMCs, even though it has not, so far, been possible to detect an associated ionic current (i.e. I-CRAC) in physiological Ca2+ conditions. The study also reveals an easy-to-use small molecule inhibitor of the Ca2+ entry mechanism that has promising specificity and should be useful in further studies of the mechanism in a wide range of experiments. Perhaps most striking is the nanomolar potency of the inhibitor and approximate 100-fold selectivity for VSMCs over immune cells. This suggests that the CRAC channels of VSMCs are fundamentally different from those of immune cells, even though Orai1 and STIM1 are involved in both. From a pharmacological perspective, it would seem worthwhile to consider chemicals such as S66 as potential therapeutic inhibitors of vascular remodelling without negative implications for vascular tone or immune response.
Acknowledgments
The study was supported by the British Heart Foundation and the Wellcome Trust. A. Sedo provided technical assistance.
Glossary
Abbreviations
- CRAC
channel, Ca2+ release activated Ca2+ channel
- PDGF
platelet-derived growth factor
- PGPC
1-palmitoyl-2-glutaroyl-phosphatidylcholine
- siRNA
short interfering RNA
- STIM1
stromal interaction molecule 1
- S66
3-fluoropyridine-4-carboxylic acid (2′,5′–dimethoxybiphenyl-4-yl)amide
- TRPC
transient receptor potential canonical
- VSMC
vascular smooth muscle cell
Conflict of interest
The authors state no conflict of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 Relevant gene expressions in human saphenous vein VSMCs. (a) Typical gel showing PCR products for Orai1, 2 and 3 mRNAs with (+) but not without (−) reverse transcriptase (RT) reaction. A DNA marker ladder is on the left. (b) Using real-time RT-PCR quantification, relative abundances of the mRNAs encoding the indicated proteins (n = 3–4).
Figure S2 Example Ca2+ add-back signals in human saphenous vein VSMCs with and without store depletion and showing comparison of effects of two chemically different store depletion agents, thapsigargin (TG) and cyclopiazonic acid (CPA). Intracellular Ca2+ was measured in cells pretreated (as indicated below) in Ca2+-free bath solution for 30 min and then exposed to 0.2 mM extracellular Ca2+ as indicated by the horizontal bar labelled ‘add-back’, except for one group of cells where the Ca2+ was not added back (0 Ca2+). The pretreatments in addition to the Ca2+-free solution were 1 µM TG, 10 µM CPA, 1 µM TG plus 10 mM CPA (TG + CPA), 0.2% dimethylsulphoxide (DMSO) as the vehicle control for TG and CPA, or no TG, CPA or DMSO (none).
Figure S3 Validation and specificity of siRNA knock down of Orai expression. See Table S1 for annotations. (a) Validation of knock down, showing relative abundances of Orai1, 2 and 3 mRNAs after transfection of vascular smooth muscle cells with Orai1, 2 and 3 siRNAs as indicated (n/N = 3/6). The control was scrambled siRNA. (b) Validation of specificity, showing lack of effect on the non-target mRNAs (as indicated) after transfection of vascular smooth muscle cells with the indicated siRNAs (n/N = 2–3/4–6).
Figure S4 Endogenous Orai1 protein detection and knock down in human saphenous vein VSMCs. Western blot for lysates from cells transfected with control (scrambled) siRNA (siRNA ctrl) or Orai1 siRNA 1 (Orai1 si.1). The blot was probed with anti-Orai1 antibody (upper panel) and anti-β-actin antibody (lower panel). The predicted protein mass of Orai1 is 33 kDa (arrow).
Figure S5 Typical original traces for the mean data shown in Figure 1A. For N = 16 per individual experiment, intracellular Ca2+ measurements from VSMCs pretreated with 1 µM thapsigargin in Ca2+-free bath solution for 30 min and then exposed to 0.2 mM extracellular Ca2+ as indicated by the horizontal bars: comparing control (scrambled) siRNA with Orai1 siRNA 1 (a), Orai2 siRNA (b), Orai3 siRNA (c), Orai2 and Orai3 siRNAs (d) and Orai1 siRNA 2 (e); and comparing DNA vector with Orai1 R91W mutant (dominant negative) (f).
Figure S6 In support of the dotted curve in Figure 2B, the graph shows the mean rate of rise data points without normalization and with the fitted Hill equation. The y-axis is the linear slope of the initial rate of rise of the fura-2 fluorescence after extracellular Ca2+ was added to the medium.
Figure S7 Original data for VSMC migration assays. Typical bright-field images of human saphenous vein VSMCs that had moved through pores in the polycarbonate membrane for vehicle control (ctrl) and 1 µM S66. A cell is indicated by a white arrow and a pore by a black arrow.
Table SI PCR primer pairs (F, forward direction; R, reverse direction), PCR amplicon sizes, and siRNA probes sequences. The ‘Orai1 si.negative’ was generated for the purpose of suppressing Orai1 expression, but it failed to have any effect on the expression and so was used as a negative control
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Alexander SP, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 3rd edition. Br J Pharmacol. 2008;153(Suppl 2):S1–209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Shawaf E, Naylor J, Taylor H, Riches K, Milligan CJ, O'Regan D, et al. Short-term stimulation of calcium-permeable transient receptor potential canonical 5-containing channels by oxidized phospholipids. Arterioscler Thromb Vasc Biol. 2010;30:1453–1459. doi: 10.1161/ATVBAHA.110.205666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelini GD, Jeremy JY. Towards the treatment of saphenous vein bypass graft failure–a perspective of the Bristol Heart Institute. Biorheology. 2002;39:491–499. [PubMed] [Google Scholar]
- Baryshnikov SG, Pulina MV, Zulian A, Linde CI, Golovina VA. Orai1, a critical component of store-operated Ca2+ entry, is functionally associated with Na+/Ca2+ exchanger and plasma membrane Ca2+ pump in proliferating human arterial myocytes. Am J Physiol Cell Physiol. 2009;297:C1103–C1112. doi: 10.1152/ajpcell.00283.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beech DJ. Ion channel switching and activation in smooth-muscle cells of occlusive vascular diseases. Biochem Soc Trans. 2007;35:890–894. doi: 10.1042/BST0350890. [DOI] [PubMed] [Google Scholar]
- Berra-Romani R, Mazzocco-Spezzia A, Pulina MV, Golovina VA. Ca2+ handling is altered when arterial myocytes progress from a contractile to a proliferative phenotype in culture. Am J Physiol Cell Physiol. 2008;295:C779–C790. doi: 10.1152/ajpcell.00173.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisaillon JM, Motiani RK, Gonzalez-Cobos JC, Potier M, Halligan KE, Alzawahra WF, et al. Essential role for STIM1/Orai1-mediated calcium influx in PDGF-induced smooth muscle migration. Am J Physiol Cell Physiol. 2010;298:C993–1005. doi: 10.1152/ajpcell.00325.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahalan MD. STIMulating store-operated Ca2+ entry. Nat Cell Biol. 2009;11:669–677. doi: 10.1038/ncb0609-669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casteels R, Droogmans G. Exchange characteristics of the noradrenaline-sensitive calcium store in vascular smooth muscle cells or rabbit ear artery. J Physiol. 1981;317:263–279. doi: 10.1113/jphysiol.1981.sp013824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derler I, Fahrner M, Carugo O, Muik M, Bergsmann J, Schindl R, et al. Increased hydrophobicity at the N terminus/membrane interface impairs gating of the severe combined immunodeficiency-related ORAI1 mutant. J Biol Chem. 2009;284:15903–15915. doi: 10.1074/jbc.M808312200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Sabatino A, Rovedatti L, Kaur R, Spencer JP, Brown JT, Morisset VD, et al. Targeting gut T cell Ca2+ release-activated Ca2+ channels inhibits T cell cytokine production and T-box transcription factor T-bet in inflammatory bowel disease. J Immunol. 2009;183:3454–3462. doi: 10.4049/jimmunol.0802887. [DOI] [PubMed] [Google Scholar]
- Feske S, Gwack Y, Prakriya M, Srikanth S, Puppel SH, Tanasa B, et al. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature. 2006;441:179–185. doi: 10.1038/nature04702. [DOI] [PubMed] [Google Scholar]
- Fountain SJ, Cheong A, Flemming R, Mair L, Sivaprasadarao A, Beech DJ. Functional up-regulation of KCNA gene family expression in murine mesenteric resistance artery smooth muscle. J Physiol. 2004;556:29–42. doi: 10.1113/jphysiol.2003.058594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan PG, Lewis RS, Rao A. Molecular basis of calcium signaling in lymphocytes: STIM and ORAI. Annu Rev Immunol. 2010;28:491–533. doi: 10.1146/annurev.immunol.021908.132550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar B, Dreja K, Shah SS, Cheong A, Xu SZ, Sukumar P, et al. Upregulated TRPC1 channel in vascular injury in vivo and its role in human neointimal hyperplasia. Circ Res. 2006;98:557–563. doi: 10.1161/01.RES.0000204724.29685.db. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landsberg JW, Yuan JX. Calcium and TRP channels in pulmonary vascular smooth muscle cell proliferation. News Physiol Sci. 2004;19:44–50. doi: 10.1152/nips.01457.2003. [DOI] [PubMed] [Google Scholar]
- Li J, Sukumar P, Milligan CJ, Kumar B, Ma ZY, Munsch CM, et al. Interactions, functions, and independence of plasma membrane STIM1 and TRPC1 in vascular smooth muscle cells. Circ Res. 2008;103:e97–104. doi: 10.1161/CIRCRESAHA.108.182931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan CJ, Li J, Sukumar P, Majeed Y, Dallas ML, English A, et al. Robotic multiwell planar patch-clamp for native and primary mammalian cells. Nat Protoc. 2009;4:244–255. doi: 10.1038/nprot.2008.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng LC, Ramduny D, Airey JA, Singer CA, Keller PS, Shen XM, et al. Orai1 interacts with STIM1 and mediates capacitative Ca2+ entry in mouse pulmonary arterial smooth muscle cells. Am J Physiol Cell Physiol. 2010;299:C1079–C1090. doi: 10.1152/ajpcell.00548.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng SW, di Capite J, Singaravelu K, Parekh AB. Sustained activation of the tyrosine kinase Syk by antigen in mast cells requires local Ca2+ influx through Ca2+ release-activated Ca2+ channels. J Biol Chem. 2008;283:31348–31355. doi: 10.1074/jbc.M804942200. [DOI] [PubMed] [Google Scholar]
- Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767–801. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- Porter KE, Dickinson T, London NJ. Inhibition of neointima formation in an organ culture of human saphenous vein: a comparison of dual endothelin-converting enzyme/neutral endopeptidase and selective neutral endopeptidase inhibition. J Vasc Surg. 2001;34:548–554. doi: 10.1067/mva.2001.115960. [DOI] [PubMed] [Google Scholar]
- Potier M, Gonzalez JC, Motiani RK, Abdullaev IF, Bisaillon JM, Singer HA, et al. Evidence for STIM1- and Orai1-dependent store-operated calcium influx through ICRAC in vascular smooth muscle cells: role in proliferation and migration. FASEB J. 2009;23:2425–2437. doi: 10.1096/fj.09-131128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiber J, Hawkins A, Zhang ZS, Wang S, Burch J, Graham V, et al. STIM1 signalling controls store-operated calcium entry required for development and contractile function in skeletal muscle. Nat Cell Biol. 2008;10:688–697. doi: 10.1038/ncb1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeney M, Yu Y, Platoshyn O, Zhang S, McDaniel SS, Yuan JX. Inhibition of endogenous TRP1 decreases capacitative Ca2+ entry and attenuates pulmonary artery smooth muscle cell proliferation. Am J Physiol Lung Cell Mol Physiol. 2002;283:L144–L155. doi: 10.1152/ajplung.00412.2001. [DOI] [PubMed] [Google Scholar]
- Takahashi Y, Watanabe H, Murakami M, Ohba T, Radovanovic M, Ono K, et al. Involvement of transient receptor potential canonical 1 (TRPC1) in angiotensin II-induced vascular smooth muscle cell hypertrophy. Atherosclerosis. 2007;195:287–296. doi: 10.1016/j.atherosclerosis.2006.12.033. [DOI] [PubMed] [Google Scholar]
- Trepakova ES, Gericke M, Hirakawa Y, Weisbrod RM, Cohen RA, Bolotina VM. Properties of a native cation channel activated by Ca2+ store depletion in vascular smooth muscle cells. J Biol Chem. 2001;276:7782–7790. doi: 10.1074/jbc.M010104200. [DOI] [PubMed] [Google Scholar]
- Zeng F, Xu SZ, Jackson PK, McHugh D, Kumar B, Fountain SJ, et al. Human TRPC5 channel activated by a multiplicity of signals in a single cell. J Physiol. 2004;559:739–750. doi: 10.1113/jphysiol.2004.065391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng W, Yuan JP, Kim MS, Choi YJ, Huang GN, Worley PF, et al. STIM1 gates TRPC channels, but not Orai1, by electrostatic interaction. Mol Cell. 2008;32:439–448. doi: 10.1016/j.molcel.2008.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.