

Abstract
BACKGROUND AND PURPOSE
The angiotensin II type 1 receptor (AT1R) is a key regulator of blood pressure and cardiac contractility and is profoundly involved in development of cardiac disease. Since several microRNAs (miRNAs) have been implicated in cardiac disease, we determined whether miRNAs might be regulated by AT1R signals in a Gαq/11-dependent or -independent manner.
EXPERIMENTAL APPROACH
We performed a global miRNA array analysis of angiotensin II (Ang II)-mediated miRNA regulation in HEK293N cells overexpressing the AT1R and focused on separating the role of Gαq/11-dependent and -independent pathways. MiRNA regulation was verified with quantitative PCR in both HEK293N cells and primary cardiac myocytes and fibroblasts.
KEY RESULTS
Our studies revealed five miRNAs (miR-29b, -129-3p, -132, -132* and -212) that were up-regulated by Ang II in HEK293N cells. In contrast, the biased Ang II analogue, [Sar1, Ile4, Ile8] Ang II (SII Ang II), which selectively activates Gαq/11-independent signalling, failed to regulate miRNAs in HEK293N cells. Furthermore, Ang II-induced miRNA regulation was blocked following Gαq/11 and Mek1 inhibition. The observed Ang II regulation of miRNA was confirmed in primary cultures of adult cardiac fibroblasts. Interestingly, Ang II did not regulate miRNA expression in cardiac myocytes, but SII Ang II significantly down-regulated miR-129-3p.
CONCLUSIONS AND IMPLICATIONS
Five miRNAs were regulated by Ang II through mechanisms depending on Gαq/11 and Erk1/2 activation. These miRNAs may be involved in Ang II-mediated cardiac biology and disease, as several of these miRNAs have previously been associated with cardiovascular disease and were found to be regulated in cardiac cells.
Keywords: angiotensin II, AT1R, microRNA, cell signalling, Erk1/2, Gαq, biased agonist
Introduction
Angiotensin II (Ang II) is a key hormone in cardiovascular homeostasis and is involved in the development of multiple cardiac diseases (Mehta and Griendling, 2007). Hence, the angiotensin II type 1 receptor (AT1R) is a prominent drug target in cardiovascular medicine, as reflected by the use of AT1R blockers or inhibitors of Ang II synthesis such as losartan and ramipril in various cardiovascular diseases. The AT1R belongs to the family of seven transmembrane or G-protein coupled receptors (7TMR/GPCR). The classical description of AT1R signalling depicts heterotrimeric G-protein activation and downstream signalling. We and others have shown that the AT1R can also confer cellular signals independently of Gαq/11 activation (Ahn et al., 2004; Aplin et al., 2007a; Christensen et al., 2010a). The involvement of Gαq/11 protein-independent signalling (including β-arrestin-dependent signalling) in the regulation of various cellular processes is evolving and includes regulation of proliferation, protection against apoptosis, protein synthesis and migration (Revankar et al., 2004; Hunton et al., 2005; Aplin et al., 2007b; DeWire et al., 2008; Ahn et al., 2009). These observations are pharmacologically intriguing. Selective activation of some of these cellular processes, in combination with inhibition of Gαq/11-dependent signals, may prove pharmacologically superior to existing treatment of cardiac hypertrophy and heart failure (Aplin et al., 2008). Several recent findings indicate that Gαq/11-independent signalling can lead to transcriptional regulation (Morinelli et al., 2009; Szekeres et al., 2009; Christensen et al., 2010b). However, we and others have recently shown that AT1R-mediated gene regulation is largely G protein-dependent, and that the ability of G protein-independent/β-arrestin-dependent signalling to induce gene expression is reliant on crosstalk with G protein-induced pathways (Lymperopoulos et al., 2009; Christensen et al., 2010b).
Ang II activates the canonical MAP kinases Erk1/2, Jnk and p38 that in turn regulate gene transcription. MAP kinases are activated by both Gαq/11- and β-arrestin-dependent AT1R signalling (Figure 1A). Importantly, the MAP kinases Erk1/2 activated by Gαq/11-dependent signalling translocate to the nucleus, whereas Erk1/2 proteins activated by the β-arrestin-dependent pathway are sequestered in the cytosol (Ahn et al., 2004; Aplin et al., 2007a). It is therefore uncertain if MAP kinases activated by Gαq-independent signalling can activate transcription. Despite sequestered in the cytosol, MAP kinases may regulate transcription indirectly by phosphorylating cytosolic proteins that are free to translocate to the nucleus.
Figure 1.

The AT1R regulates miRNA expression profiles by Gαq-dependent mechanisms. (A) Schematic illustration of AT1R signal transduction. Erk1/2, Jnk and p38 can be activated by both Gαq-dependent and -independent mechanisms. Erk1/2 activated by Gαq-independent signalling is sequestered in the cytosol and it is therefore questionable whether MAP kinases activated by this pathway can regulate miRNA expression. (B) Representative Western blots comparing p-Erk1/2, p-p38 and p-Jnk activation in AT1R-HEK cells treated with angiotensin II (Ang II) or [Sar1, Ile4, Ile8] Ang II (SII Ang II) for 3 min. (C) Heatmap showing differential miRNA expression in AT1R-HEK cells treated with Ang II or SII Ang II for 24 h (n = 3). (D)q-PCR validation of regulated miRNAs. The expression is normalized against the stably expressed miRNAs miR-17 and miR-191. Note the difference in the normalized expression at the y-axis. *P < 0.05, tested with a paired two-tailed t-test.
The transcriptional apparatus regulates the expression of both protein coding and non-coding RNA. MiRNAs are small non-coding RNAs that by sequence homology regulate stability and translation of mRNA targets (Bartel, 2004). Several miRNAs are aberrantly expressed in cardiovascular diseases (Olson, 2006; Care et al., 2007; van Rooij et al., 2007) in which Ang II also holds a key role (Mehta and Griendling, 2007). Although AT1R-mediated mRNA regulation is mostly dependent on Gαq/11 activation, the mechanisms underlying miRNA regulation have not previously been studied. Therefore, we evaluated the ability of Ang II to directly regulate miRNA expression, as Ang II-regulated miRNAs may serve as suitable drug targets for manipulating Ang II signalling during disease progression. Moreover, as biased agonism of the AT1R is a potential future treatment for specific cardiovascular diseases (Aplin et al., 2008), we determined the involvement of Gαq/11-dependent and -independent signalling in miRNA regulation. For this purpose, we compared the effect on miRNA expression of both Ang II and the biased AT1R analogue [Sar1, Ile4, Ile8] Ang II (SII Ang II), which inhibits Gαq/11 protein activation while still activating Gαq/11-independent signalling (Holloway et al., 2002). In addition, we used Gαq/11, Mek1, p38 and Jnk inhibitors to distinguish the contribution of different signalling pathways to Ang II-mediated regulation of miRNA expression.
We found that a specific set of miRNAs is up-regulated by AT1R activation. These data further enhance the understanding of Ang II signalling mechanisms and the miRNAs identified could potentially play a role in cardiovascular disease.
Methods
Molecular target nomenclature conforms to BJP's Guide to Receptors and Channels (Alexander et al., 2009).
Cell culturing
AT1R-HEK cells
AT1R stably transfected HEK293N (AT1R-HEK) cells were a generous gift from Dr Robert J. Lefkowitz, Duke University Medical Center, Durham, North Carolina (Hunton et al., 2005). Cells were grown on plasticware coated with poly-l-lysine and maintained in Dulbecco's modified Eagle's medium (DMEM) (Lonza) with Glutamax supplemented with 10% fetal bovine serum (FBS) (Gibco), 50 U·mL−1 penicillin and 50 U·mL−1 streptomycin (Lonza) and 300 µg·mL−1 zeocin (Invitrogen). For miRNA microarray analysis, AT1R-HEK cells were kept overnight in serum-free DMEM and stimulated with 10 nM Ang II (Sigma) or 1.87 µM SII Ang II (Cleveland Clinic, USA). For all inhibitor assays and in fibroblasts and myocytes, we increased the concentrations by a factor 10. The different concentrations of Ang II and SII Ang II were chosen to obtain equal receptor occupancy (Miura and Karnik, 1999; Thomas et al., 2000). Inhibitors were added 30 min prior to stimulation. YM254890 was kindly provided by Astellas Pharma Inc. (Tokyo, Japan). U0126, SB203580 and SP600125 were from Sigma-Aldrich (Denmark).
Cardiac fibroblast cell culture
Adult fibroblasts were isolated by the principle of selective plating as previously described (Villarreal et al., 1993; Dubey et al., 1997; Andersen et al., 2009), with some minor modifications. In brief, ventricles from 8 week-old male Sprague-Dawley rats (Taconic, Denmark) were minced and digested twice for 10 min with 0.08% Trypsin (BD Difco), followed by 9500 U Collagenase Type 2 (Worthington) for 70 min. Red blood cells were lysed with a standard ammonium chloride/EDTA buffer and the resulting suspension was plated for 1 h in cell culture dishes in DMEM/10% FBS/1% penicillin-streptomycin. The cultures were washed thoroughly with PBS and the remaining adherent cells were cultivated for 3 days without signs of myofibroblast differentiation. Fibroblasts were deprived of serum for 4 h and then incubated for 48 h with the agonist and antagonist.
Neonatal cardiac myocytes
Neonatal cardiac myocytes were isolated by trypsin digestion of heart tissue combined with a buffer system and purification methods recommended for obtaining high-quality myocyte cultures from adult animals, as previously described (Simpson, 1983; Busk et al., 2005). In brief, neonatal ventricular cardiac myocytes were prepared from 1 to 3 days old Wistar rats (Charles River, Germany), minced in myocyte isolation buffer and digested in 0.08% trypsinbuffer. Our cardiac myocyte cultures have previously been reported to be more than 98% positive for the cardiac myocyte markers sarcomeric-α-actin and sarcomeric tropomyosin (Busk et al., 2005). Cells were cultivated in DMEM supplemented with 100 nM insulin, 656 mg·L−1 creatine, 396 mg·L−1 carnitine, 626 mg·L−1 taurine, 0.5 g·L−1 BSA, 0.1% FBS, 1% penicillin-streptomycin, 0.5 mM CaCl2, and 1% l-glutamine for 48 h. After 4 h in serum-free medium, the cells were treated for 48 h with 100 nM Ang II, 18.7 µM SII Ang II or vehicle in DMEM.
RNA purification
RNA was purified with TriReagent (Molecular Research Center) according to manufacturer's protocol. The quantity of the extracted total RNA was assessed on a nanodrop in TE-buffer (10 mM Tris-HCl, 1 mM EDTA, pH 7.5) and RNA quality was validated by NanoDrop measurements and agarose gel electrophoresis.
miRNA array
RNA samples were analysed by Exiqon A/S (Vedbaek, Denmark). Briefly 1 µg RNA was labelled with the miRCURY Hy3/Hy5 power labelling kit. The Hy3™-labelled samples (n = 3 in each treatment) and a Hy5™-labelled reference RNA sample were mixed pair-wise and hybridized to the miRCURY™ LNA array version 11.0 (Exiqon, Denmark), which contains capture probes targeting all miRNAs for human, mouse or rat registered in the miRBase version 11.0 at the Sanger Institute. The quantified signals were background corrected and normalized using the global Lowess (LOcally WEighted Scatterplot Smoothing) regression algorithm. To determine significantly regulated miRNAs, statistical analysis was performed by the significance analysis for microarrays function of the MeV v4.5 software, using two-class unpaired tests. MiRNAs regulated less than twofold were excluded, and the false discovery rate was set to 0 (corresponded to a tuning parameter Delta = 3.729).
Quantitative polymerase chain reaction (q-PCR)
A real-time analytical technique for q-PCR, TaqMan® MiRNA assay (commercially available primers from Applied Biosystems), was used to detect and quantify miRNA of interest according to the manufacturer's protocol on an Applied Biosystems 7900HT. q-PCR values for miRNA analyses were normalized against multiple experimentally-verified stably expressed miRNAs (let-7f, miR-17 and miR-191) by use of qBase software as previously described (Vandesompele et al., 2002; Hellemans et al., 2007; Andersen, 2010). SYBR green-based real-time q-PCR was used to detect and quantify mRNA expression according to the manufacturer's protocol on a 7900HT (Applied Biosystems). Primers were designed with the following sequences (DNA technology, Riskov, Denmark):
AT1aR: forward CAGCTCTGCCACATTCCCTGAGTT, reverse CTGGTGATCACTTTCTGGGAGGG. AT1bR: forward GCCACCAGGCTTGAAAGAAGCCC, reverse CAGCCTTGGGGCAGTCATCTTGGA. ATR2: forward CCCGTGACCAAGTCTTGAAGAT, reverse ATACCCATCCAGGTCAGAGCAT.
Specificity of primers was validated by sequencing of amplicons (DNA technology, Riskov, Denmark). q-PCR values for mRNA analyses were normalized against two experimentally verified stably expressed mRNAs (Rpl13a and β-actin) by use of qBasePlus software as previously described (Andersen et al., 2009).
Cell number and volume
Cell number and cell diameter were measured using a Beckman Coulter Multisizer Z2 (Ramcon, Denmark). Cells were seeded in six-well plates, stimulated for 24 h and trypsinized. Cells were resuspended and diluted in isotonic fluid and counted in the range of 8–24 µm. Cell diameter was used to estimate cell volume.
Western blotting
Cells were seeded in six-well plates, cultured for 2 days, deprived of serum overnight, stimulated with 100 nM Ang II, 18.7 µM SII Ang II or vehicle for the indicated time periods and lysed in 0.5% Triton X-100, 150 nM NaCl and 50 mM Tris-HCl, supplemented with protease and phosphatase inhibitors.
Equal amounts of lysates were loaded on SDS gels (Pagegel inc.) and Western blotting performed as wet blotting. Briefly, PVDF membranes (Amersham) were soaked in ethanol and placed in cold transfer buffer (25 mM Tris-HCl, 195 mM glycine, 20% ethanol, 0.1% SDS). Proteins were transferred in a Criterion blotter (Bio-Rad). Membranes were blocked using Enhanced ECL blocking reagent and probed against phosphorylated Erk1/2 and total Erk1/2, phosphorylated Jnk, or phosphorylated p38 (all antibodies were from Cell signalling). Total Erk1/2 was used as a control for equal loading. Protein bands were visualized using enhanced ECL (Amersham) and scanned in an Intelligent Dark Box II (Fuji).
Statistics
MiRNA expression values were log-transformed before further statistical analysis. Differential miRNA expression was tested with one- or two-way anova or paired t-test when relevant. P < 0.05 was considered statistically significant. Statistics were performed using GraphPad 5. All error bars indicate ±SEM. The number of experiments (n) for AT1R-HEK cells reflects different passages of cells treated independently. Primary cells were not passaged and thus ‘n’ reflects the number of cell batches treated as independent experiments.
Results
Differential miRNA expression profiles in Ang II and SII Ang II treated AT1R-HEK cells
To investigate how Ang II-induced signalling networks affect miRNA expression patterns and elucidate the role of Gαq-dependent and -independent signalling in this regulation, we compared the global miRNA expression in AT1R-HEK cells induced by Ang II and SII Ang II using the miRCURY™ miRNA array technology (Exiqon A/S, Denmark).
Both Ang II and SII Ang II, which does not activate Gαq/11 protein signalling, resulted in phosphorylation of Erk1/2 and Jnk, but only Ang II led to phosphorylation of p38, indicating that p38 is only regulated by Gαq-dependent mechanisms (Figure 1B). The global miRNA differential expression analysis revealed seven up-regulated and four down-regulated miRNAs with the applied statistical cut-offs (Figure 1C).
To validate these results, we performed q-PCR of these miRNAs using normalization against stably expressed miRNAs (miR-17 and miR-191; Figure 1D). This analysis confirmed six of the seven up-regulated miRNAs: miR-7, -29b, -129-3p, -132, -132* and -212. We also profiled down-regulated miRNAs with q-PCR but were unable to validate any miRNA as down-regulated (data not shown). Although SII Ang II readily regulated Erk1/2 and Jnk kinase signalling and mediated a moderate increase in cell volume (Figures 1B and 2B), this agonist failed to significantly regulate miRNA expression in AT1R-HEK cells (Figure 1C,D).
Figure 2.
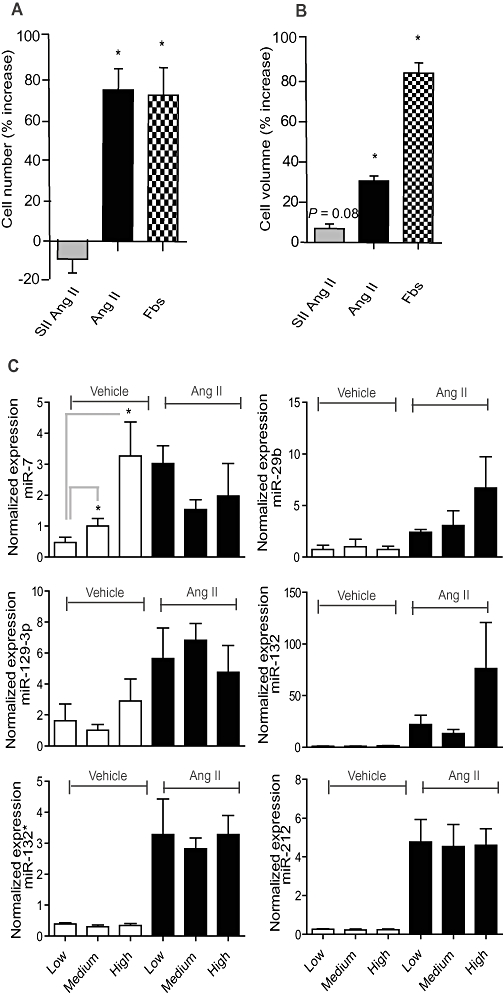
The effect of angiotensin II (Ang II) on cell number and volume of AT1R-HEK cells. (A,B) Cells were treated with 100 nM Ang II, 18.7 µM [Sar1, Ile4, Ile8] Ang II (SII Ang II), or 5% FBS for 24 h and cell number and volume were measured on a Coulter counter. *P < 0.05 tested with a paired t-test (n = 4). (C)The effect of confluence on miRNA expression in AT1R-HEK cells was investigated by treatment of cells seeded at low (10 000 cm−2), medium (20 000 cm−2) and high densities (60 000 cm−2) and with vehicle or Ang II for 24 h. The expression of miR-7 is significantly regulated by plating densities alone. The expression is normalized against the stably expressed miRNAs miR-17 and miR-191. The effect of confluence was tested for significance by two-way anova analysis (n = 3). Note the difference in expression at the y-axis.
Does cell confluence affect the miRNA expression profile?
Cell confluence has been reported to affect the levels of miRNA expression (Hwang et al., 2009). As Ang II increases cell number and volume (Figure 2A,B), it was important to rule out the possibility that the observed regulation of miRNAs was due to changes in confluence. We therefore tested expression changes in the Ang II-induced miRNAs at different degrees of cell confluence. Cells were grown in densities ranging from low (10 000 cells·cm−2) to medium (20 000 cells·cm−2) and high (60 000 cells·cm−2) and stimulated with Ang II or vehicle. Ang II treatment for 24 h increased cell numbers by 74% and cell volume by 31% compared with vehicle, while SII Ang II increased cell volume by 7% compared with vehicle at 20 000 cells·cm−2 (Figure 2A,B). The q-PCR analysis showed that miR-29b, -129-3p, -132, -132* and -212 were not affected by confluence alone, indicating that these miRNAs are directly regulated by Ang II signalling. In contrast, miR-7 expression was confluence-dependent (Figure 2C) and was therefore excluded from the following analyses. Importantly, expression of our reference miRNAs miR-17 and -191 were not dependent on confluence (data not shown).
Ang II-induced miRNA expression is Gαq/11-dependent
SII Ang II treatment, which does not activate Gαq/11, did not result in changes in miRNA expression, indicating that Gαq/11 activation is essential for Ang II-induced miRNA regulation. To further establish the Gαq-dependence, we used the Gαq/11-specific inhibitor YM254890 (Takasaki et al., 2004). MiR-29b, -129-3p -132, -132* and -212 were all sensitive to Gαq/11 inhibition (Figure 3), confirming that Gαq/11 protein activation is important for AT1R-induced miRNA regulation. Notably, not all the miRNAs were suppressed to the same degree by Gαq/11 inhibition (YM254890). We further found that Ang II induced up-regulation of miR-29b, -132, -132* and -212 expression was significantly reduced by Mek1 inhibition (U0126) but not by Jnk (SP600125) or p38 (SB203580) inhibition. As for miR-129-3p, U0126 only caused an insignificant reduction. Standard deviations for miR-129-3p expression were generally a little higher than for the other miRNAs, which possibly reflects their very low expression level.
Figure 3.

Angiotensin II (Ang II)-mediated expression of miR-29b, miR-129-3p, miR-132, miR-132* and miR-212 in AT1R-HEK cells is dependent on Gαq and Erk1/2. Cells were treated with 100 nM Ang II in combination with inhibitors for 24 h. (A) MicroRNA (miRNA) expression was revealed with q-PCR. Control (DMSO), Gαq-i (Gαq inhibitor/YM254890, 10 nM), Mek-i (Mek1 inhibitor/UO126, 10 µM), p38-i (p38 inhibitor/SB203580, 10 µM) and Jnk-i (Jnk inhibitor/ SP600125, 10 µM). The expression is normalized against the stably expressed miR-17 and miR-191. q-PCR values are illustrated with values for cells treated with Ang II or vehicle. (B) q-PCR values of Ang II treatments were divided by their respective vehicle expression value to achieve fold change values. Note the difference in fold change at the y-axis. Treatments were tested for significance by one-way anova, *P < 0.05 (n = 3).
Overall, Ang II-induced miRNA expression in HEK293 cells appears to be regulated by Gαq/11-dependent signalling in a Mek1/Erk1/2-dependent manner.
AT1R regulation of miRNA expression in primary cardiac fibroblasts and myocytes
In the miRNA array experiments we used HEK293N cells overexpressing the AT1R to identify as many Ang II-regulated miRNAs as possible. To determine whether the observed miRNA regulation is also relevant in endogenous Ang II target cells, we tested how the profiled miRNAs were affected by Ang II and SII Ang II treatment in primary cultures of adult cardiac fibroblasts and neonatal cardiac myocytes (Figure 4A). MiR-132, -132* and -212 were present in both cell types, miR-29b was expressed preferentially in fibroblasts, in agreement with previous observations (van Rooij et al., 2008) and miR-129-3p was highly expressed in cardiac myocytes but only at very low levels in cardiac fibroblasts (Figure 4A). MiR-132, -132* and -212 were significantly up-regulated by Ang II but not SII Ang II in cardiac fibroblasts. Interestingly, whereas no other miRNAs were regulated by Ang II or SII Ang II in neonatal cardiac myocytes, miR-129-3p was sixfold down-regulated by SII Ang II treatment (Figure 4A). We have previously shown that both Ang II and SII Ang II treatment of cardiac myocytes results in activation of Erk1/2 (Aplin et al., 2008). Contradictory, Erk1/2 was indeed activated by Ang II but not by SII Ang II in cardiac fibroblasts (Figure 4B). The miRNA regulation was significantly reduced by Mek1 inhibition in cardiac fibroblasts whereas the Gαq/11 inhibitor YM254890 appeared less efficient in this cell type (Figure 5). These data indicate that Erk1/2 and Gαq activation are overall important for miRNA regulation in primary cultures of cardiac fibroblasts but also that G protein-independent signalling may have an important role in miRNA regulation in cardiac myocytes, underlining significant differences between these two cardiac cell types.
Figure 4.
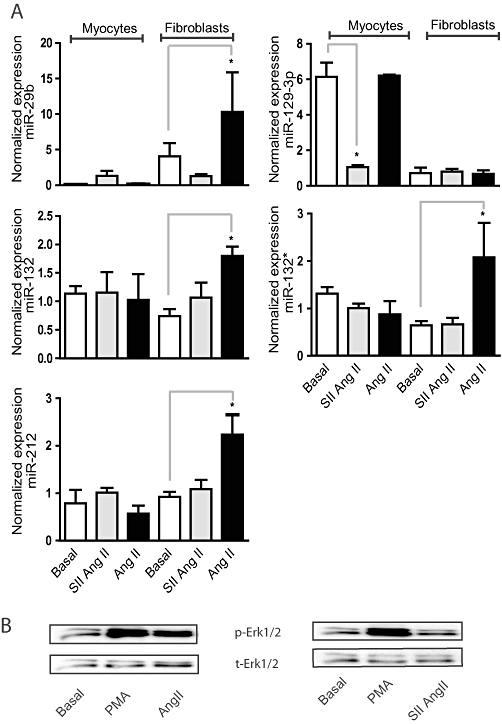
Angiotensin II (Ang II) and [Sar1, Ile4, Ile8] Ang II (SII Ang II)-regulated expression of miRNAs in primary neonatal cardiomyocytes and adult cardiac fibroblasts. (A) Rat cardiac fibroblasts and myocytes were treated with Ang II (100 nM) or SII Ang II (18.7 µM) for 48 h and miRNA expression was evaluated by q-PCR. The expression is normalized against the stably expressed miRNAs miR-17 and miR-191. Effect of treatment was tested for significance by paired two-tailed t-tests, *P < 0.05 (n = 3). (B) Representative Western blots of Erk1/2 phosphorylation (p-Erk1/2) in adult rat cardiac fibroblasts treated with 100 nM Ang II, 18.5 µM SII Ang II or 0.15 nM phorbol myristate acetate (PMA; positive control). Total Erk1/2 (t-Erk1/2) is shown as a loading control.
Figure 5.
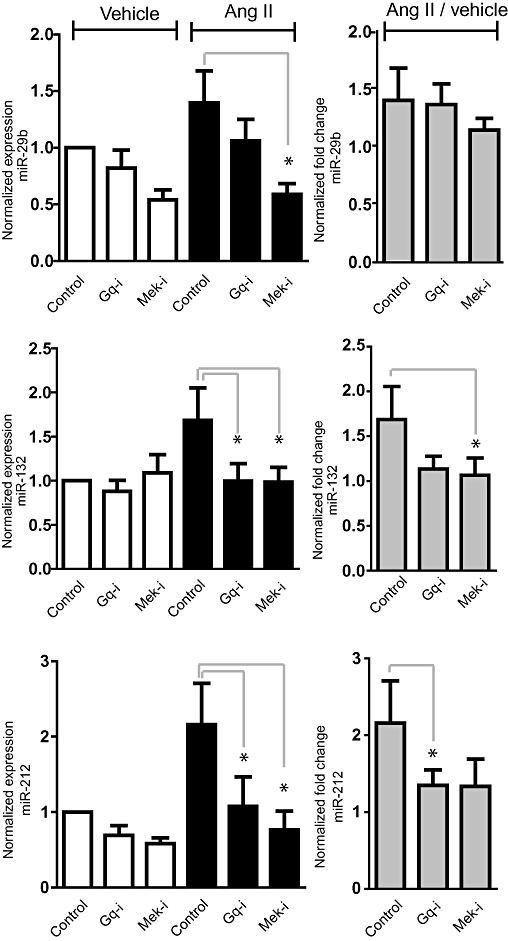
Angiotensin II (Ang II)-mediated microRNA (miRNA) expression in primary cardiac fibroblasts is dependent on Erk1/2. Cells were treated with Ang II (100 nM) in combination with inhibitors for 48 h. Control (DMSO), Gαq-i (Gαq inhibitor/YM254890, 10 nM), Mek-i (Mek1 inhibitor/UO126, 10 µM), p38-i (p38 inhibitor/SB203580, 10 µM) and Jnk-i (Jnk inhibitor/ SP600125, 10 µM). (Left-hand coumns) The expression is normalized against the stably expressed let-7f. q-PCR values are illustrated with values for cells treated with Ang II or vehicle. (Right-hand columns) q-PCR values of Ang II treatments were divided with the respective vehicle expression value to achieve fold change values. *P < 0.05 tested with Student's two-tailed t-test (n = 5).
The Ang II-induced regulation of miRNA in cardiac myocytes and fibroblasts was observed to be significantly different. The AT2R has been proposed to be able to counteract some AT1R-mediated effects in cardiac myocytes (Booz and Baker, 1996; van Kesteren et al., 1997), which has been questioned by other studies (Akishita et al., 2000; D'Amore et al., 2005). Thus, we wanted to determine the role of AT1R versus AT2R in the observed regulation. We therefore determined the expression of AT1aR, AT1bR and AT2R in our cultures of cardiac myocytes and fibroblasts by q-PCR (Figure 6). As a positive control we used embryonic rat hearts and expression levels were normalized against the experimentally verified stably expressed β-actin and Rpl13a mRNAs. TheAT1bR and AT2R are both expressed at very low levels in neonatal cardiac myocytes and adult cardiac fibroblasts. The AT1aR was weakly expressed in cardiac myocytes but significantly higher (26-fold) in cardiac fibroblasts. Thus, since miRNA regulation by Ang II was only observed in cardiac fibroblasts it is most likely that the AT1aR have a significant role in this regulation. To further explore this we stimulated cardiac fibroblasts with Ang II in the presence and absence of the specific AT1R inhibitor losartan (Figure 7). We observed that Ang II-induced expression of miRNA was consistently inhibited by losartan indicating an AT1R-mediated effect.
Figure 6.
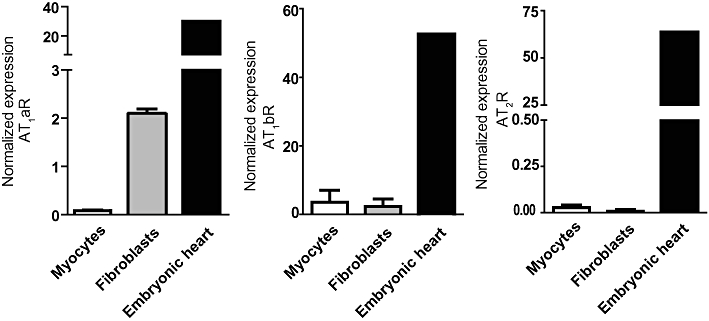
Expression profile of AT1aR, AT1bR and AT2R in cardiac myocytes and fibroblasts. SYBR green based real-time q-PCR was used to detect and quantify mRNA expression of the three ATR subtypes. As a positive control we used pooled mRNA extracted from three embryonic rat hearts. The expression was normalized against the stably expressed mRNAs for Rpl13a and β-actin (n = 3).
Figure 7.

Losartan inhibits the up-regulation of angiotensin II (Ang II)-induced miRNAs in cardiac fibroblasts. Rat cardiac fibroblasts were treated with 1 µM losartan 30 min prior to Ang II stimulation for 48 h and miRNA expression was evaluated by q-PCR. The expression is normalized against the stably expressed let-7f. Effect of treatment was tested for significance by a paired one-tailed t-test, *P < 0.05 (n = 5).
Discussion
This study presents a comprehensive investigation of miRNA regulation by AT1R signalling. We showed that Ang II-mediated regulation of miRNA expression in AT1R-HEK cells depends on Gαq/11 activation. Previous studies have established that the Gαq/11 activated pathway is the major player in Ang II-mediated gene regulation (Lee et al., 2008), but recent reports indicate that Gαq/11-independent signal transduction can also be involved in the regulation of gene expression (Morinelli et al., 2009; Szekeres et al., 2009; Christensen et al., 2010b). We therefore found it of importance to determine whether miRNAs regulated by Ang II are regulated by Gαq-dependent or -independent mechanisms. With the global array analysis of Ang II-induced miRNA expression, we initially identified seven up-regulated and four down-regulated miRNAs. Six of the up-regulated miRNAs were confirmed by q-PCR, while it was generally not possible to confirm the down-regulated miRNAs. The down-regulation observed on the arrays may reflect variations in very low expressed miRNAs, which could not be reproduced with q-PCR and can therefore be assumed to be of minor importance. MiR-7 was regulated by confluence alone, which shows that confluence is a major parameter in studies of growth factor-induced miRNA regulation, in accordance with a previous report (Hwang et al., 2009). The remaining five miRNAs were not affected by cell density and are considered to be direct Ang II-regulated targets. All five miRNAs were up-regulated after 24 h of Ang II treatment and all showed Gαq/11-dependent regulation as they were not regulated with SII Ang II and their Ang II-induced up-regulation could be repressed with the Gαq/11 inhibitor YM254890. We did not find any miRNA to be regulated by SII Ang II treatment in AT1R-HEK cells. Moreover, four of the five Ang II regulated miRNAs were highly sensitive to Mek1 inhibition (although the fifth, miR-129-3p, appeared to be inhibited by U0126, the effect was not significant), placing the Erk1/2 signalling cascade as a central hub in Ang II-mediated regulation of miRNA expression.
SII Ang II stimulation did not regulate any of the miRNAs annotated to miRBase 11.0. Although we cannot exclude the possibility that recently annotated miRNAs may be regulated, the overall conclusion is that SII Ang II does not regulate miRNA expression in AT1R-HEK cells. In a similar study considering mRNA regulation we observed that SII Ang II regulated more mRNA coding genes after 24 h than after 3 h treatment (Christensen et al., 2010a), which supports the choice of time-point. The sensitivity of all five regulated miRNAs to Gαq/11 inhibition supports the conclusion that AT1R-induced miRNA regulation is dependent on Gαq/11 protein activation. As depicted in Figure 3, the inhibition by the Gαq/11 inhibitor YM254890 is not complete and the level of inhibition also varies between miRNAs. This could indicate that other pathways are involved in the regulation, but might also reflect incomplete inhibition due to degradation of the inhibitor at these long-term stimulations. In relation to this, YM254890 inhibition of Ang II-induced miRNA expression in cardiac fibroblasts was not as efficient as in AT1R-HEK cells. This could indicate that alternative pathways leading to Ang II-induced miRNA regulation are more pronounced in cardiac fibroblasts or less likely it might reflect that the inhibitor is not as efficiently taken up in primary cells. SII Ang II did not regulate expression of miRNAs in either AT1R-HEK cells or cardiac fibroblasts and thus it appears that Gαq/11 activation is required for regulation of the miRNAs investigated in both cell types.
As Erk1/2 activated by Gαq/11-independent signalling is sequestered in the cytosol by tight binding to β-arrestin 2 (Ahn et al., 2004; Aplin et al., 2007a), these findings also suggest that Erk1/2 translocation to the nucleus is necessary for Ang II-induced miRNA regulation. All of the Ang II-induced miRNAs were regulated by Ang II, but not SII Ang II, in adult cardiac fibroblasts, with the exception of miR-129-3p, which was expressed in very low amounts. All the miRNAs investigated were expressed in cardiac myocytes, except miR-29b, which was expressed in very few copies. This difference between cardiac myocytes and cardiac fibroblasts with regards to miRNA expression is striking but in concurrence with previous reports (Thum et al., 2007; van Rooij et al., 2008). We did not observe SII Ang II-induced regulation of the miRNAs found to be regulated by Ang II in either AT1R-HEK cells or cardiac fibroblasts. In contrast, miR-129-3p was down-regulated sixfold in cardiac myocytes by SII Ang II treatment. This is a very interesting observation indicating that SII Ang II may have cell type-specific effects. Of note, cardiac myocytes appear to induce Erk1/2 phosphorylation by a β-arrestin-mediated pathway when stimulated by SII Ang II (Aplin et al., 2007a), whereas cardiac fibroblasts do not (Figure 4B). These data indicate that, in addition to the low receptor expression, the reason that cardiac myocytes show a different regulation pattern from the miRNAs investigated could be due to other differences between the cell types, including a different stoichiometry of other signalling molecules.
As the AT2R has been proposed to have a role in Ang II signalling in cardiac myocytes, we determined the expression of the AT1a, AT1b and AT2 receptor types in our cardiac myocytes and fibroblasts. Our profiling points to a major role of AT1aR in this regulation, and the much higher expression of AT1aR might be the reason why we observed a much more prominent miRNA regulation in fibroblasts than in myocytes. We can however not rule out the possibility that the weakly expressed AT2R can also affect the regulation. On speculation, the difference in AT1R-mediated miRNA expression changes and the very different AT1aR expression levels between the tested cell types might indicate that the effects of prolonged Ang II stimulation on the heart are primarily related to fibroblasts.
Although it is not the scope of this study to identify targets and functions of the Ang II regulated miRNAs, it is noteworthy that several of the miRNAs found have previously been identified in cardiovascular disease. MiR-29b is a key regulator of fibrotic genes and is known to inhibit collagen expression after myocardial infarction (van Rooij et al., 2008) as well as being up-regulated in dilated cardiomyopathy (Naga Prasad et al., 2009). MiR-212 and miR-129 are up-regulated in end-stage heart failure due to dilated cardiomyopathy (Thum et al., 2007) and have previously been reported to be induced by 7TMR activation (Yuen et al., 2009). MiR-212 and -132/132* are clustered closely in the genome and are transcribed together as an intergenic contig under the regulation of cAMP response element binding protein (Vo et al., 2005) and possibly also other redox sensitive transcription factors (Lee et al., 2007). This group of redox sensitive transcription factors is scaffolded by the transcription factor early growth response 1 (Egr-1) which is a known Ang II- and Erk1/2-regulated gene (Neyses et al., 1993; Khomenko et al., 2003; Christensen et al., 2010a). MiR-132 and miR-132* have not previously been shown to be regulated by Ang II or been associated with cardiovascular disease. Recently, miR-132 has been shown to act as an angiogenic switch and a growth promoter in the endothelium by targeting the Ras inhibitor p120RasGAP and thereby activating Ras signalling (Anand et al., 2010). Since Ang II can also induce both angiogenesis and growth of endothelial cells (Herr et al., 2008), it will be interesting to see whether this involves miR-132 regulation. Moreover, an investigation of the involvement of miR-132 in Ang II-mediated growth of fibroblasts is warranted.
In summary, we have identified five miRNAs which are induced by Ang II treatment in both AT1R-HEK293 cells and primary adult cardiac fibroblasts, several of which have previously been linked to cardiovascular diseases such as hypertrophy, myocardial infarction and fibrosis. Importantly, we showed that regulation of these miRNAs relies on Gαq/11-dependent signalling, which is also profoundly involved in the progression of maladaptive hypertrophy and causative of fibrosis. Moreover, the Ang II-induced Gαq/11-dependent regulation of miRNAs is also largely Erk1/2-dependent, placing Erk1/2 as a central protein in Ang II-induced miRNA regulation. Gαq/11-independent signalling induced by the biased agonist SII Ang II was not able to regulate miRNA expression in HEK293 cells and cardiac fibroblasts, but surprisingly regulated one miRNA in cardiac myocytes. This indicates an interesting bias in cell type-specific Ang II-induced regulation of miRNA. Future experiments should reveal the targets and functional roles of the Ang II-induced miRNAs identified in the cardiovascular system and address their potential as drug targets.
Acknowledgments
We thank Marlene Louise Christensen and Birte Kofoed for excellent technical assistance and Dr Robert J. Lefkowitz for kindly providing us with the stable transfected AT1R-HEK cells.
Glossary
Abbreviations
- Ang II
angiotensin II
- AT1R
angiotensin II type 1 receptor
- Erk1/2
extracellular regulated kinase1/2
- Jnk
c-jun terminal kinase
- Mek1
mitogen activated kinase 1
- miRNA
microRNA
- p38
p38 mitogen-activated kinase
- SII Ang II
[Sar1, Ile4, Ile8] Ang II
Sources of funding
S. P. S. is funded by the John and Birthe Meyer Foundation. J. L. H. is funded by the Danish National Research Foundation, The Danish Council for Independent Research | Medical Sciences, The Købmand i Odense Johan og Hanne Weimann f. Seedorffs legat and The Novo Nordisk Foundation. P. L. J. is funded by The Danish Cardiovascular Research Academy and the Tømrermester Alfred Andersen og Hustrus Foundation. G. L. C. is funded by The Danish Heart Foundation.
Conflict of interest
All authors declare no conflict of interest.
References
- Ahn S, Shenoy SK, Wei H, Lefkowitz RJ. Differential kinetic and spatial patterns of beta-arrestin and G protein-mediated ERK activation by the angiotensin II receptor. J Biol Chem. 2004;279:35518–35525. doi: 10.1074/jbc.M405878200. [DOI] [PubMed] [Google Scholar]
- Ahn S, Kim J, Hara MR, Ren XR, Lefkowitz RJ. {beta}-Arrestin-2 mediates anti-apoptotic signaling through regulation of bad phosphorylation. J Biol Chem. 2009;284:8855–8865. doi: 10.1074/jbc.M808463200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akishita M, Iwai M, Wu L, Zhang L, Ouchi Y, Dzau VJ, et al. Inhibitory effect of angiotensin II type 2 receptor on coronary arterial remodeling after aortic banding in mice. Circulation. 2000;102:1684–1689. doi: 10.1161/01.cir.102.14.1684. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 4th edition. Br J Pharmacol. 2009;158(Suppl 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand S, Majeti BK, Acevedo LM, Murphy EA, Mukthavaram R, Scheppke L, et al. MicroRNA-132-mediated loss of p120RasGAP activates the endothelium to facilitate pathological angiogenesis. Nat Med. 2010;16:909–914. doi: 10.1038/nm.2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen DC, Andersen P, Schneider M, Jensen HB, Sheikh SP. Murine ‘cardiospheres’ are not a source of stem cells with cardiomyogenic potential. Stem Cells. 2009;27:1571–1581. doi: 10.1002/stem.72. [DOI] [PubMed] [Google Scholar]
- Andersen DAJC, Schneider M, Nossent AY, Eskildsen TE, Hansen JL, Teisner B, et al. MicroRNA-15a finetunes the level of Delta-like 1 homologue (DLK 1) in proliferating 3T3-L1 Preadipocytes. Exp Cell Res. 2010;316:1681–1691. doi: 10.1016/j.yexcr.2010.04.002. [DOI] [PubMed] [Google Scholar]
- Aplin M, Christensen GL, Schneider M, Heydorn A, Gammeltoft S, Kjolbye AL, et al. The angiotensin type 1 receptor activates extracellular signal-regulated kinases 1 and 2 by G protein-dependent and -independent pathways in cardiac myocytes and langendorff-perfused hearts. Basic Clin Pharmacol Toxicol. 2007a;100:289–295. doi: 10.1111/j.1742-7843.2007.00063.x. [DOI] [PubMed] [Google Scholar]
- Aplin M, Christensen GL, Schneider M, Heydorn A, Gammeltoft S, Kjolbye AL, et al. Differential extracellular signal-regulated kinases 1 and 2 activation by the angiotensin type 1 receptor supports distinct phenotypes of cardiac myocytes. Basic Clin Pharmacol Toxicol. 2007b;100:296–301. doi: 10.1111/j.1742-7843.2007.00064.x. [DOI] [PubMed] [Google Scholar]
- Aplin M, Christensen GL, Hansen JL. Pharmacologic perspectives of functional selectivity by the angiotensin II type 1 receptor. Trends Cardiovasc Med. 2008;18:305–312. doi: 10.1016/j.tcm.2009.01.003. [DOI] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- Booz GW, Baker KM. Role of type 1 and type 2 angiotensin receptors in angiotensin II-induced cardiomyocyte hypertrophy. Hypertension. 1996;28:635–640. doi: 10.1161/01.hyp.28.4.635. [DOI] [PubMed] [Google Scholar]
- Busk PK, Hinrichsen R, Bartkova J, Hansen AH, Christoffersen TE, Bartek J, et al. Cyclin D2 induces proliferation of cardiac myocytes and represses hypertrophy. Exp Cell Res. 2005;304:149–161. doi: 10.1016/j.yexcr.2004.10.022. [DOI] [PubMed] [Google Scholar]
- Care A, Catalucci D, Felicetti F, Bonci D, Addario A, Gallo P, et al. MicroRNA-133 controls cardiac hypertrophy. Nat Med. 2007;13:613–618. doi: 10.1038/nm1582. [DOI] [PubMed] [Google Scholar]
- Christensen GL, Kelstrup CD, Lyngso C, Sarwar U, Bogebo R, Sheikh SP, et al. Quantitative phosphoproteomics dissection of seven-transmembrane receptor signaling using full and biased agonists. Mol Cell Proteomics. 2010a;9:1540–1553. doi: 10.1074/mcp.M900550-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen GL, Knudsen S, Schneider M, Aplin M, Gammeltoft S, Sheikh SP, et al. AT(1) receptor Galphaq protein-independent signalling transcriptionally activates only a few genes directly, but robustly potentiates gene regulation from the beta2-adrenergic receptor. Mol Cell Endocrinol. 2010b;331:49–56. doi: 10.1016/j.mce.2010.08.004. [DOI] [PubMed] [Google Scholar]
- D'Amore A, Black MJ, Thomas WG. The angiotensin II type 2 receptor causes constitutive growth of cardiomyocytes and does not antagonize angiotensin II type 1 receptor-mediated hypertrophy. Hypertension. 2005;46:1347–1354. doi: 10.1161/01.HYP.0000193504.51489.cf. [DOI] [PubMed] [Google Scholar]
- DeWire SM, Kim J, Whalen EJ, Ahn S, Chen M, Lefkowitz RJ. Beta-arrestin-mediated signaling regulates protein synthesis. J Biol Chem. 2008;283:10611–10620. doi: 10.1074/jbc.M710515200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubey RK, Gillespie DG, Mi Z, Jackson EK. Exogenous and endogenous adenosine inhibits fetal calf serum-induced growth of rat cardiac fibroblasts: role of A2B receptors. Circulation. 1997;96:2656–2666. doi: 10.1161/01.cir.96.8.2656. [DOI] [PubMed] [Google Scholar]
- Hellemans J, Mortier G, De Paepe A, Speleman F, Vandesompele J. qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol. 2007;8:R19. doi: 10.1186/gb-2007-8-2-r19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herr D, Rodewald M, Fraser HM, Hack G, Konrad R, Kreienberg R, et al. Regulation of endothelial proliferation by the renin-angiotensin system in human umbilical vein endothelial cells. Reproduction. 2008;136:125–130. doi: 10.1530/REP-07-0374. [DOI] [PubMed] [Google Scholar]
- Holloway AC, Qian H, Pipolo L, Ziogas J, Miura S, Karnik S, et al. Side-chain substitutions within angiotensin II reveal different requirements for signaling, internalization, and phosphorylation of type 1A angiotensin receptors. Mol Pharmacol. 2002;61:768–777. doi: 10.1124/mol.61.4.768. [DOI] [PubMed] [Google Scholar]
- Hunton DL, Barnes WG, Kim J, Ren XR, Violin JD, Reiter E, et al. Beta-arrestin 2-dependent angiotensin II type 1A receptor-mediated pathway of chemotaxis. Mol Pharmacol. 2005;67:1229–1236. doi: 10.1124/mol.104.006270. [DOI] [PubMed] [Google Scholar]
- Hwang HW, Wentzel EA, Mendell JT. Cell-cell contact globally activates microRNA biogenesis. Proc Natl Acad Sci U S A. 2009;106:7016–7021. doi: 10.1073/pnas.0811523106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Kesteren CA, van Heugten HA, Lamers JM, Saxena PR, Schalekamp MA, Danser AH. Angiotensin II-mediated growth and antigrowth effects in cultured neonatal rat cardiac myocytes and fibroblasts. J Mol Cell Cardiol. 1997;29:2147–2157. doi: 10.1006/jmcc.1997.0448. [DOI] [PubMed] [Google Scholar]
- Khomenko T, Deng X, Jadus MR, Szabo S. Effect of cysteamine on redox-sensitive thiol-containing proteins in the duodenal mucosa. Biochem Biophys Res Commun. 2003;309:910–916. doi: 10.1016/j.bbrc.2003.08.092. [DOI] [PubMed] [Google Scholar]
- Lee J, Li Z, Brower-Sinning R, John B. Regulatory circuit of human microRNA biogenesis. PLoS Comput Biol. 2007;3:e67. doi: 10.1371/journal.pcbi.0030067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MH, El-Shewy HM, Luttrell DK, Luttrell LM. Role of beta-arrestin-mediated desensitization and signaling in the control of angiotensin AT1a receptor-stimulated transcription. J Biol Chem. 2008;283:2088–2097. doi: 10.1074/jbc.M706892200. [DOI] [PubMed] [Google Scholar]
- Lymperopoulos A, Rengo G, Zincarelli C, Kim J, Soltys S, Koch WJ. An adrenal beta-arrestin 1-mediated signaling pathway underlies angiotensin II-induced aldosterone production in vitro and in vivo. Proc Natl Acad Sci U S A. 2009;106:5825–5830. doi: 10.1073/pnas.0811706106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta PK, Griendling KK. Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Physiol. 2007;292:C82–C97. doi: 10.1152/ajpcell.00287.2006. [DOI] [PubMed] [Google Scholar]
- Miura S, Karnik SS. Angiotensin II type 1 and type 2 receptors bind angiotensin II through different types of epitope recognition. J Hypertens. 1999;17:397–404. doi: 10.1097/00004872-199917030-00013. [DOI] [PubMed] [Google Scholar]
- Morinelli TA, Kendall RT, Luttrell LM, Walker LP, Ullian ME. Angiotensin II-induced cyclooxygenase 2 expression in rat aorta vascular smooth muscle cells does not require heterotrimeric G protein activation. J Pharmacol Exp Ther. 2009;330:118–124. doi: 10.1124/jpet.109.151829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naga Prasad SV, Duan ZH, Gupta MK, Surampudi VS, Volinia S, Calin GA, et al. Unique microRNA profile in end-stage heart failure indicates alterations in specific cardiovascular signaling networks. J Biol Chem. 2009;284:27487–27499. doi: 10.1074/jbc.M109.036541. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Neyses L, Nouskas J, Luyken J, Fronhoffs S, Oberdorf S, Pfeifer U, et al. Induction of immediate-early genes by angiotensin II and endothelin-1 in adult rat cardiomyocytes. J Hypertens. 1993;11:927–934. doi: 10.1097/00004872-199309000-00006. [DOI] [PubMed] [Google Scholar]
- Olson EN. Gene regulatory networks in the evolution and development of the heart. Science. 2006;313:1922–1927. doi: 10.1126/science.1132292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revankar CM, Vines CM, Cimino DF, Prossnitz ER. Arrestins block G protein-coupled receptor-mediated apoptosis. J Biol Chem. 2004;279:24578–24584. doi: 10.1074/jbc.M402121200. [DOI] [PubMed] [Google Scholar]
- van Rooij E, Sutherland LB, Qi X, Richardson JA, Hill J, Olson EN. Control of stress-dependent cardiac growth and gene expression by a microRNA. Science. 2007;316:575–579. doi: 10.1126/science.1139089. [DOI] [PubMed] [Google Scholar]
- van Rooij E, Sutherland LB, Thatcher JE, DiMaio JM, Naseem RH, Marshall WS, et al. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc Natl Acad Sci USA. 2008;105:13027–13032. doi: 10.1073/pnas.0805038105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson P. Norepinephrine-stimulated hypertrophy of cultured rat myocardial cells is an alpha 1 adrenergic response. J Clin Invest. 1983;72:732–738. doi: 10.1172/JCI111023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szekeres M, Turu G, Orient A, Szalai B, Supeki K, Cserzo M, et al. Mechanisms of angiotensin II-mediated regulation of aldosterone synthase expression in H295R human adrenocortical and rat adrenal glomerulosa cells. Mol Cell Endocrinol. 2009;302:244–253. doi: 10.1016/j.mce.2008.12.015. [DOI] [PubMed] [Google Scholar]
- Takasaki J, Saito T, Taniguchi M, Kawasaki T, Moritani Y, Hayashi K, et al. A novel Galphaq/11-selective inhibitor. J Biol Chem. 2004;279:47438–47445. doi: 10.1074/jbc.M408846200. [DOI] [PubMed] [Google Scholar]
- Thomas WG, Qian H, Chang C-S KS. Agonist-induced phosphorylation of the angiotensin II (AT1A) receptor requires generation of a conformation that is distinct from the inositol phosphate-signaling state. J Biol Chem. 2000;275:2893–2900. doi: 10.1074/jbc.275.4.2893. 1074/jbc.275.4.2893. [DOI] [PubMed] [Google Scholar]
- Thum T, Galuppo P, Wolf C, Fiedler J, Kneitz S, van Laake LW, et al. MicroRNAs in the human heart: a clue to fetal gene reprogramming in heart failure. Circulation. 2007;116:258–267. doi: 10.1161/CIRCULATIONAHA.107.687947. [DOI] [PubMed] [Google Scholar]
- Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3:RESEARCH0034. doi: 10.1186/gb-2002-3-7-research0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villarreal FJ, Kim NN, Ungab GD, Printz MP, Dillmann WH. Identification of functional angiotensin II receptors on rat cardiac fibroblasts. Circulation. 1993;88:2849–2861. doi: 10.1161/01.cir.88.6.2849. [DOI] [PubMed] [Google Scholar]
- Vo N, Klein ME, Varlamova O, Keller DM, Yamamoto T, Goodman RH, et al. A cAMP-response element binding protein-induced microRNA regulates neuronal morphogenesis. Proc Natl Acad Sci USA. 2005;102:16426–16431. doi: 10.1073/pnas.0508448102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen T, Ruf F, Chu T, Sealfon SC. Microtranscriptome regulation by gonadotropin-releasing hormone. Mol Cell Endocrinol. 2009;302:12–17. doi: 10.1016/j.mce.2008.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]