

Abstract
Cryptophycins are a group of cyanobacterial depsipeptides with activity against drug-resistant tumors. Although shown to be promising, further efforts are required to return these highly potent compounds to the clinic through a new generation of analogs with improved medicinal properties. Herein, we report a chemosynthetic route relying on the multifunctional enzyme CrpD-M2 that incorporates a 2-hydroxy-acid moiety (unit D) into cryptophycin analogs. CrpD-M2 is a unique non-ribosomal peptide synthetase (NRPS) module comprised of condensation-adenylation-ketoreduction-thiolation (C-A-KR-T) domains. We interrogated A-domain 2-keto and 2-hydroxy acid activation and loading, and KR domain activity in the presence of NADPH and NADH. The resulting 2-hydroxy acid was elongated with three synthetic cryptophycin chain elongation intermediate analogs (SNAC-ABC) through ester bond formation catalyzed by CrpD-M2 C domain. Finally, the enzyme bound seco-cryptophycin products were macrolactonized by the Crp thioesterase (TE). The analysis of these sequential steps was enabled through liquid chromatography Fourier transform ion cyclotron resonance mass spectrometry (LC-FTICR-MS) analysis of enzyme bound intermediates and products. This novel chemoenzymatic synthesis of cryptophycin involves four sequential catalytic steps leading to the incorporation of a 2-hydroxy acid moiety in the final chain elongation intermediate. This is the first example where a NRPS-embedded KR domain is employed for assembly of a fully elaborated natural product, and serves as a proof-of-principle for chemoenzymatic synthesis of new cryptophycin analogs.
Natural products have been widely applied to fight disease and offer chemical scaffolds for development of new analogs with improved/modified functions, achieved through semi-, total-, or chemoenzymatic synthesis.1–3 Cryptophycins are potent anticancer agents at picomolar concentrations and exert their cytotoxic effects in both vinca alkaloid- and taxol-resistant cancer cells that contribute to the proliferation of drug-resistant tumors.4 Their clinical potential as well as synthetic challenges have stimulated the development of alternative strategies to provide suitable amounts of material and new analogs with improved physiochemical properties for clinical studies.5 The cryptophycin gene cluster was recently elucidated and offers unique opportunities for assembly of the drug and new analogs using chemoenzymatic approaches.6 The gene cluster is comprised of two type I poly-ketide synthase (PKS) genes, crpA and crpB, two NRPS genes, crpC and crpD, and four tailoring enzyme genes including a key P450 epoxidase gene (crpE). Previous studies from this laboratory have demonstrated the feasibility and efficiency of biocatalysts from this metabolic pathway to properly macrocyclize and regio- and stereo-specifically epoxidize cryptophycin intermediates to generate these natural products and novel analogs.6–8
CrpD is a bimodular NRPS involved in late-stage assembly of the cryptophycin chain elongation intermediate. Bioinformatic analysis and precursor incorporation studies revealed that the substrate of its first module is methyl-β-alanine converted from l-aspartic acid by CrpG, a β-methylaspartate-α-decarboxylase.6,9 These studies also suggested that 2-ketoisocaproic acid (2KIC, (1)) instead of l-2-hydroxyisocaproic acid (l-2HIC, (2)) was the substrate of CrpD module 2 (CrpD-M2), which was incorporated into cryptophycin as unit D (Figure 1).6 Several natural cryptophycin analogs contain unit D variations, including 3-methyl-2-hydroxyvalerate, 2-hydroxyvalerate, and 3-methyl-2-hydroxybutyrate (Figure S1).6 Altered bioactivity of these analogs suggest the importance of the unit D in anticancer activity, but limited efforts have been made to query analogs carrying unnatural unit D structures.10
Figure 1.

CrpD-M2 biosynthetic scheme. (A) Enzymes used in this study, CrpD-M2 and Crp TE. Domains within the CrpD-M2 polypeptide are noted with squares, and the phosphopantetheinyl arm is denoted by the linked SH group. (B) Reactions catalyzed by CrpD-M2 and Crp TE mediated cyclization. (B) SNAC-ABC analogs utilized (3–5).8 (D) Cyclic cryptophycins (6–8),11–13 and linear seco-cryptophycins (9–11) generated by CrpD-M2 and Crp TE8. 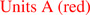 ,
, 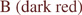 ,
,  , and
, and  .
.
Four distinct steps are catalyzed by CrpD-M2 in cryptophycin biosynthesis (Figure 1). In step I, the free-acid extender unit (e.g., 1) is activated by the CrpD-M2 A domain to form the corresponding acyl-AMP. This intermediate is then loaded onto the T domain active site bound phosphopantetheine arm through a transthioesterification reaction to form the acyl enzyme intermediate in step II. In the presence of reducing cofactors the 2KIC enzyme intermediate is converted stereoselectively to l-2HIC in step III by the unique 2-KR domain based upon analysis of known products. The l-2-hydroxy group in unit D is condensed with cryptophycin unit ABC biosynthetic intermediate transferred from CrpD module 1 T domain through formation of an atypical (in an NRPS module) ester linkage by the C domain (Figure 1, step IV).
In this report, four sequential steps were assessed to investigate the unique incorporation of a 2-hydroxy acid subunit into cryptophycin natural products, and to probe the intrinsic substrate flexibility and synthetic potential of CrpD-M2. Chemoenzymatic synthesis of cryptophycins 3,11 24,12 and 5113 (6–8) with CrpD-M2 and Crp TE (Figure 1, step V) serves as a proof-of-principle for efforts to generate cryptophycin analogs with unnatural structures of unit C and unit D through synthetic and biochemical methods.
A CrpD-M2 expression construct was generated by amplifying a DNA fragment consisting of C, A, KR, and T domains by PCR, and cloning into the BamHI and XhoI sites of pET28a. This construct was overexpressed in E. coli BAP1 strain for production of phosphopantetheinylated proteins.14 The N-terminal His-tagged protein was purified with Ni-NTA resin to ~ 80 % purity (Figure S2). The integrity of the purified protein was verified by peptide map fingerprinting and FTICR-MS (Figure S3A, Table S1). The CrpD-M2 T domain active site was also identified and proper post-translational modification of the T domain active site Ser was verified by MS/MS (Figure S3B, Table S2).15
Bioinformatic analysis can reliably predict NRPS A domain specificity based upon binding pocket residue motifs.16 The conserved Asp235 involved in ionic interaction with the amino group of the substrate amino acid is replaced by Val235 in CrpD-M2 A domain (Table S3). Similar to unit D of cryptophycin, a 2- hydroxy acid moiety is also involved in at least nine other natural products including bacillaene,17 barbamide,18 bassianolide,19 beauvericin,20 cereulide,21 enniatin,22 hectochlorin,23 kutzneride,24 and valinomycin.21 A similar replacement of Asp235 is conserved across all A domains responsible for incorporation of 2-hydroxy acid into these natural products (Table S3), predicting that the CrpD-M2 A domain prefers 2-hydroxy- and/or 2-keto-acids.
The well-established ATP-PPi exchange assay was employed to biochemically determine the substrate specificity of the CrpD-M2 A domain with ten acyl-acid substrates (Figure 2). CrpD-M2 activated 2-KIC (1) about 20 times more efficiently than its cognate amino acid l-leucine (14), consistent with bioinformatic prediction and previous feeding experiments.6 Interestingly, l-2HIC (2), which was not incorporated into the cryptophycin final structure during an in vivo precursor incorporation study6 was among the best substrates in the assay. A similar high level of selectivity for the natural substrate 2-oxovalerate (15) was observed. CrpD-M2 specificity to two other natural unit D fragments, 3-methyl-2-oxovalerate (13) and 3-methyl-2-oxobutyrate (16), was decreased approximately 50 % and 90 %, respectively. This result, along with the observed weak activation of unnatural substrates 2-oxobutyrate (17) and phenyl pyruvate (19), suggests that the size and bulk of substrate side chains are important in CrpD-M2 A domain recognition.
Figure 2.

Examination of CrpD-M2 A domain substrate specificity using the radio-PPi exchange assay. (A) Relative activity of the A-domain normalized to 2. (B) Extender units investigated in this assay.
In an effort to assess further the flexibility of the CrpD-M2 A domain, we tested 2-keto-γ-(methylthio) butyrate (AKGB, 12) as a substrate. Its effective activation (Figure 2) demonstrates the potential of native CrpD-M2 in producing novel cryptophycin analogs with an altered unit D moiety. Moreover, the weak activation of 4-methylvalerate (18) by CrpD-M2 A domain reveals the importance of the α-position functional group in enzyme recognition. Therefore, CrpD-M2 A domain has relatively relaxed substrate specificity and exhibits a similar selectivity toward 2-keto and 2-hydroxy acids (Figure 1, step I). ATP-PPi exchange assays have been applied to examine substrate preference of A domains in the biosynthesis of bacillaene,17 barbamide,18 cereulide,21 enniatin,22 hectochlorin,23 and kutzneride.24 Only HctEIVA from the hectochlorin pathway displayed a similar selectivity to 2-keto and 2-hydroxy acids compared to the CrpD-M2 A domain.23
We next monitored substrate loading directly on the T-domain of CrpD-M2 (Figure 1, step II).26–28 Enzyme reactions were terminated by proteolysis with trypsin, and the active-site peptide bound with extender units were separated and analyzed by LC-FTICR-MS and LC iontrap-MS/MS (LC-IT-MS/MS, Figure 3, Tables S4–S5, Figures S26–34). As shown in Figure 3A and 3D, T domain active site peptides bound with l-2HIC and 2-KIC showed masses of 4116.21 and 4114.15, respectively, at a charge state of 4+, matching theoretical values within ±30 ppm (Table S4). d-2HIC (20) was also loaded on CrpD-M2 T domain active site, as shown by the observed mass of 4116.18 (Figure 3B).
Figure 3.

CrpD-M2 T domain active-site bound with extender unit intermediates monitored by LC FTICR-MS. A zoomed mass spectrum averaged over the elution window is presented. The deconvoluted monoisotopic mass, and observed charge state are shown. Reactions of CrpD-M2 with ATP and: (A)  (2), (B)
(2), (B) 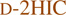 (20), (C)
(20), (C)  (1), (D)
(1), (D)  , (E)
, (E) 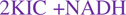 . Further data are provided such as LC IT-MS/MS25 (Table S4–5 and Figures S26–34).
. Further data are provided such as LC IT-MS/MS25 (Table S4–5 and Figures S26–34).
Since only l-2HIC containing cryptophycin analogs have been isolated and characterized from the cyanobacterium Nostoc sp. ATCC 53789, we suspect that factors other than A domain selectivity, such as substrate availability and/or downstream processing, determine the final outcome. It is well-known that 2-keto acids are indispensible intermediates in amino acid biosynthesis such as 2KIC (1), 3-methyl-2-oxovalerate (13) and 3-methyl-2-oxobutyrate (16) in the biosynthesis of leucine, isoleucine, and valine, respectively. The availability of free 2-hydroxy acid may be ascribed to a pathway-specific enzyme. For example, A domains for the biosynthesis of bassianolide,19 beauvericin,20 and enniatin,22 are specific to d-2-hydroxy isovalerate (d-2HIV), and a pathway-specific NADPH-dependent reductase is essential for stereospecific reduction of 2-keto isovalerate (2KIV).19,20,29 Since a corresponding reducing enzyme is not encoded in the cryptophycin gene cluster,6 it is possible that 2KIC is the native substrate of CrpD-M2 A domain.
The loaded 2KIC is proposed to be reduced into l-2HIC by the α-KR domain of CrpD-M2 (Figure 1 step III). This type of KR domain is also embedded in NRPS modules of cereulide,21 hectochlorin,23 kutzneride,24 and valinomycin21, and its α-keto reduction activity and stereospecificity were biochemically confirmed in the cereulide system.21 Bioinformatic analysis indicates that CrpD-M2 KR domain is grouped with these NRPS KR domains, and is phylogenetically distinct from the more thoroughly studied PKS β-KR domains (Figure S4A). The stereochemical outcome of PKS β-KR domains can be predicted based on conserved amino acid sequence motifs.30–32 A similar analysis was performed to predict 2-hydroxy chirality introduced by KR domains from CrpD-M2, CesA and CesB (Figure S4B). The corresponding KR domain in CesA and CesB produces l-2HIC and d-2HIV, respectively. However, none of these enzymes contain the conserved motifs expected for either type A or type B β-KR polypetides. Given the phylogenetic distance and positional difference of the keto group in their respective substrates between PKS β-KR domains and NRPS α-KR domains, this sequence divergence was not unexpected. The first KR domain in the hybrid PKS/NRPS PksJ involved in bacillaene biosynthesis catalyzes both β- and β-ketone reduction with 10-fold preference toward the former reaction.17 Based on structural analysis, the product of β-ketone reduction was consistent with a the KR A-type sequence motif. However, was with NRPS α-KR domains, the PksJ KR cannot be grouped as type A or B (Figure S4B). Therefore, it is not possible to classify NRPS α-KR domains using current PKS β-KR domain bioinformatic tools,30–32 and product chirality must be determined experimentally (Figure S5). In the CrpD-M2 KR reaction, the S-configuration (e.g., l-2HIC) is expected since all natural cryptophycins contain this stereochemistry.
After loading of 2KIC (Figure 3D), addition of reducing cofactors (e.g., NAD(P)H) to the CrpD-M2 reaction resulted in a mass shift observed by FTICR-MS (Figure 3E–F). The increase of 0.5 m/z units (a 2-Da shift in the deconvoluted mass) is consistent with 2KIC conversion to 2HIC as the product of the α-ketoreduction reaction. Both NADH and NADPH operated within a similar (1–2 fold) efficiency as hydride donors based on peak intensity (Figure 3E–F). Future structural analysis of the CrpD-M2 α-KR domain may contribute to understanding stereochemical control in this enzyme subclass.
Next, the ability of CrpD-M2 to form the seco-cryptophycin intermediate was investigated using synthetic SNAC-ABC chain elongation intermediates (3–5) as the starting point (Figure 1B). The synthetic scheme followed our previously established route33 and the SNAC-ABC intermediates were confirmed using NMR and high resolution mass spectrometry (Supporting Information). The intermediate with the monomethylated unit C (3-amino-2(R)-methylpropionyl, 3) was then combined with CrpD-M2 and l-2HIC (2). The C domain of CrpD-M2 is proposed to catalyze the formation of an ester bond with the 2-hydroxy group of unit D as the nucleophile (Figure 1, step IV). The formation of the ester bond was confirmed by detecting the reaction products released from CrpD-M2 T domain following addition of the excised Crp TE (Figure 1 step V, Figure 4A–B). Both cyclic cryptophycin 3 (6) (Figure 4A) and linear product (9) (Figure 4B) were observed in the extracted ion chromatograms (EIC). Previously, a didomain NRPS (T-C), Fum14p, was shown to form a C-O bond in the biosynthesis of the fungal mycotoxin fumonisin.34 The only other C domain shown to catalyze C-O bond formation is a discrete enzyme SgcC5 in C-1027 biosynthesis.35 In both cases, donor substrates are tethered to T domains while the nucleophile (-OH) is from a small molecule extender unit. This study represents the first example of a C domain in a complete NRPS module that catalyzes the incorporation of non-amino acid moieties as an ester synthase.
Figure 4.

FTICR-MS analysis of cryptophycin products from the reaction of unit C monomethyl chain elongation intermediate (3-amino-2(R)-methylpropionyl, 3) with l/d-2HIC, ATP, CrpD-M2, and Crp TE. EICs are presented at +/− 15 ppm as time versus absolute signal. Inset mass spectra are time averaged over the 1 minute elution window corresponding to the asterisk in the extracted ion chromatogram. Inset mass spectra are presented as m/z versus absolute signal. Monoisotopic mass MH+ and the experimental mass error in ppm are reported. Reactions with l-2HIC and d-2HIC monitor the formation of cyclic (A and C) and linear products (B and D). No enzyme control reactions for cyclic (E) and linear product formation (F) are provided.
Assuming both cyclic and linear products share similar ionization efficiency in the positive ion mode, we conclude that cyclic cryptophycin 3 (6) was formed as the predominant species over (linear) seco-cryptophycin 3 (9) (Figure 4A–B). This result demonstrates chemoenzymatic synthesis of cryptophycin 3 (6) through five catalytic steps (Figure 1, step I–V). Formation of (6) was confirmed by MS/MS (Figure S6A) and LC co-elution with an authentic standard (Figure S7). Observed isotope patterns of cyclic product and MS2 fragments were also consistent with the +2 37Cl shift from unit B.
Similarly, the synthetic chain elongation intermediate with desmethyl (3-amino-propionyl, 4) or gem-dimethyl (3-aminodimethylpropionyl, 5) unit C and l-2HIC (2) were used as substrates in the CrpD-M2 reaction along with Crp TE for offloading/cyclization. Both cryptophycin 24 (7) and cryptophycin 51 (8) were generated by this chemoenzymatic route, indicating the versatility of the CrpD-M2 C domain and Crp TE (Figure S8A, E). The corresponding hydrolyzed linear products (10, 11) were also observed (Figure S8B, F). Assuming similar ionization intensity between linear and cyclic products, seco-cryptophycin 24 (10) was the predominant species compared to cyclic cryptophycin 24 (7) (Figure S8A–B), and cyclic cryptophycin 51 (8) was present in similar abundance compared to its linear counterpart (11) (Figure S8E–F). Using SNAC-ABCD analogs as native substrate mimics, Beck et al. investigated the impact of unit C methylation on Crp TE mediated macrocylization.8 The analog bearing the 3-amino-dimethylpropionyl group was found to produce more cyclic product (6:1) than the one with a 3-aminopropionyl moiety (5:1) but less than the one with a 3-amino-2(R)-methylpropionyl group (10:1). We found a similar order of unit C reactivity when the native T domain bound substrates generated by CrpD-M2 were supplied to CrpD TE.
In an effort to further probe the flexibility of CrpD-M2 to substrate stereochemistry, d-2HIC (20) was substituted for l-2HIC (2) in the chemoenzymatic reaction. Following loading of chain elongation intermediate 3, no cyclic depsipeptide product was detected (Figure 4C), but a small amount of linear product was formed (Figure 4D). Suggesting that the C domain of CrpD-M2 was able to recognize the stereoisomer of its acceptor substrate and catalyze ester bond formation with its donor ABC chain elongation intermediate, albeit at a lower level. It also indicated that the natural outcome of the unit D KR reaction was l-2HIC, followed by esterification and macrocyclization. The failure to macrocyclize ABC-d-2HIC substrates suggests that the “gate-keepers” for unit D stereochemical selection are the CrpD-M2 α-KR domain and Crp TE rather than its C domain or A domain.
In summary, non-amino acid extender subunits selected and processed by NRPS enzymes have been found in a handful of natural products isolated from bacteria and fungi. However, a complete mechanism for understanding their incorporation has not been established. In this study, CrpD-M2 coupled with Crp TE was used to further explore unit D selection, loading, reduction, elongation through an ester bond, and final product formation. FTICR-MS was employed to assess these five sequential biochemical reactions that occurred through a complete NRPS module including A, C, KR, T, and TE domains. This is also the first study in which a NRPS bearing an embedded KR domain was used to directly generate bioactive compounds from fully elaborated natural and unnatural chain elongation intermediates to provide cyclic cryptophycins 3 (6), 24 (7), and 51 (8). Thus, CrpD-M2 as a chemoenzymatic reagent or as part of a microbial-derived production strategy has the potential to generate novel cryptophycin analogs. Future efforts will focus on new analogs with improved physicochemical properties that may be beneficial as clinical agents for treatment of malignant diseases.
Supplementary Material
ACKNOWLEDGMENTS
YD was supported by a Rackham Predoctoral Fellowship. This work was supported by NIH grant CA108874 and the Hans W. Vahlteich Professorship (to DHS). Work in KH’s laboratory is supported by an NSF Career Award (CHE-05-47699).
Footnotes
SUPPORTING INFORMATION AVAILABLE. Supporting information including methods, figures, and tables is available online.
REFERENCES
- 1.Nicolaou KC, Pfefferkorn JA, Roecker AJ, Cao GQ, Barluenga S, Mitchell HJ. J. Am. Chem. Soc. 2000;122:9939–9953. [Google Scholar]
- 2.Wohlleben W, Pelzer S. Chem. & Biol. 2002;9:1163–1164. doi: 10.1016/s1074-5521(02)00267-3. [DOI] [PubMed] [Google Scholar]
- 3.Ran N, Rui E, Liu J, Tao J. Curr. Pharm. Des. 2009;15:134–152. doi: 10.2174/138161209787002924. [DOI] [PubMed] [Google Scholar]
- 4.Borst P, Evers R, Kool M, Wijholds J. J. Nat. Cancer Inst. 2000;92:1295–1302. doi: 10.1093/jnci/92.16.1295. [DOI] [PubMed] [Google Scholar]
- 5.Kerksiek K, Mejilano MR, Schwartz RE, Georg GL, Himes RH. FEBS Lett. 1995;377:59–61. doi: 10.1016/0014-5793(95)01271-0. [DOI] [PubMed] [Google Scholar]
- 6.Magarvey NA, Beck ZQ, Golakoti T, Ding Y, Huber U, Hemscheidt TK, Abelson D, Moore RE, Sherman DH. ACS Chem. Biol. 2006;1:766–779. doi: 10.1021/cb6004307. [DOI] [PubMed] [Google Scholar]
- 7.Ding Y, Seufert WH, Beck ZQ, Sherman DH. J. Am. Chem. Soc. 2008;130:5492–5498. doi: 10.1021/ja710520q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beck ZQ, Aldrich CC, Magarvey NA, Georg GL, Sherman DH. Biochemistry. 2005;44:13457–13466. doi: 10.1021/bi051140u. [DOI] [PubMed] [Google Scholar]
- 9.Beck ZQ, Burr DA, Sherman DH. Chembiochem. 2007;8:1373–1375. doi: 10.1002/cbic.200700162. [DOI] [PubMed] [Google Scholar]
- 10.Eggen M, Georg GI. Med. Res. Rev. 2002;22:85–101. doi: 10.1002/med.10002. [DOI] [PubMed] [Google Scholar]
- 11.Trimurtulu G, Ohtani I, Patterson GML, Moore RE, Corbett TH, Valeriote FA, Demchik L. J. Am. Chem. Soc. 1994;116:4729–4737. [Google Scholar]
- 12.Kobayashi M, Aoki S, Ohyabu N, Kurosu M, Wang W, Kitagawa I. Tet. Lett. 1994;35:7969–7972. [Google Scholar]
- 13.Ghosh AK, Swanson L. J. Org. Chem. 2003;68:9823–9826. doi: 10.1021/jo035077v. [DOI] [PubMed] [Google Scholar]
- 14.Pfeifer BA, Admiraal SJ, Gramajo H, Cane DE, Khosla C. Science. 2001;291:1790–1792. doi: 10.1126/science.1058092. [DOI] [PubMed] [Google Scholar]
- 15.Dorrestein PC, Bumpus SB, Calderone CT, Garneau-Tsodikova S, Aron ZD, Straight PD, Kolter R, Walsh CT, Kelleher NL. Biochemistry. 2006;45:12756–12766. doi: 10.1021/bi061169d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Challis GL, Ravel J, Townsend CA. Chem. Biol. 2000;7:211–224. doi: 10.1016/s1074-5521(00)00091-0. [DOI] [PubMed] [Google Scholar]
- 17.Calderone CT, Bumpus SB, Kelleher NL, Walsh CT, Magarvey NA. Proc. Nat. Acad. Sci. USA. 2008;105:12809–12811. doi: 10.1073/pnas.0806305105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chang Z, Flatt P, Gerwick WH, Nguyen VA, Willis CL, Sherman DH. Gene. 2002;296:235–247. doi: 10.1016/s0378-1119(02)00860-0. [DOI] [PubMed] [Google Scholar]
- 19.Xu Y, Orozco R, Kithsiri Wijeratne EM, Espinosa-Artiles P, Leslie Gunatilaka AA, Patricia Stock S, Molnar I. Fungal Genet. Biol. 2009;46:353–364. doi: 10.1016/j.fgb.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 20.Xu Y, Orozco R, Wijeratne EM, Gunatilaka AA, Stock SP, Molnar I, Wijeratne EM, Gunatilaka AA, Stock SP, Molnar I. Chem. Biol. 2008;15:898–907. doi: 10.1016/j.chembiol.2008.07.011. [DOI] [PubMed] [Google Scholar]
- 21.Magarvey NA, Ehling-Schulz M, Walsh CT. J. Am. Chem. Soc. 2006;128:10698–10699. doi: 10.1021/ja0640187. [DOI] [PubMed] [Google Scholar]
- 22.Haese A, Schubert M, Herrmann M, Zocher R. Mol. Microbiol. 1993;7:905–914. doi: 10.1111/j.1365-2958.1993.tb01181.x. [DOI] [PubMed] [Google Scholar]
- 23.Ramaswamy AV, Sorrels CM, Gerwick WH. J. Nat. Prod. 2007;70:1977–1986. doi: 10.1021/np0704250. [DOI] [PubMed] [Google Scholar]
- 24.Fujimori DG, Hrvatin S, Neumann CS, Strieker M, Marahiel MA, Walsh CT. Proc. Natl. Acad. Sci. USA. 2007;104:16498–16503. doi: 10.1073/pnas.0708242104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meluzzi D, Zheng WH, Hensler M, Nizet V, Dorrestein PC. Bioorg.Med. Chem. Lett. 2008;18:3107–3111. doi: 10.1016/j.bmcl.2007.10.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dorrestein PC, Kelleher NL. Nat. Prod. Rep. 2006;23:893–918. doi: 10.1039/b511400b. [DOI] [PubMed] [Google Scholar]
- 27.Gu L, Geders TW, Wang B, Gerwick WH, Hakansson K, Smith JL, Sherman DH. Science. 2007;318:970–973. doi: 10.1126/science.1148790. [DOI] [PubMed] [Google Scholar]
- 28.Gu L, Wang B, Kulkarni A, Geders TW, Grindberg RV, Gerwick L, Håkansson K, Wipf P, Smith JL, Gerwick WH, Sherman DH. Nature. 2009;459:731–735. doi: 10.1038/nature07870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee C, Gorisch H, Kleinkauf H, Zocher R. J. Biol. Chem. 1992;267:11741–11744. [PubMed] [Google Scholar]
- 30.Caffrey P. Chem. Biol. 2005;12:1060–1062. doi: 10.1016/j.chembiol.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 31.Caffrey P. Chembiochem. 2003;4:654–657. doi: 10.1002/cbic.200300581. [DOI] [PubMed] [Google Scholar]
- 32.Siskos AP, Baerga-Ortiz A, Bali S, Stein V, Mamdani H, Spiteller D, Popovic B, Spencer JB, Staunton J, Weissman KJ, Leadlay PF. Chem. Biol. 2005;12:1145–1153. doi: 10.1016/j.chembiol.2005.08.017. [DOI] [PubMed] [Google Scholar]
- 33.Seufert W, Beck ZQ, Sherman DH. Angewandte Chemie Int Ed. 2007;46:9298–9300. doi: 10.1002/anie.200703665. [DOI] [PubMed] [Google Scholar]
- 34.Zaleta-Rivera K, Xu C, Yu F, Butchko RAE, Proctor RH, Hidalgo-Lara ME, Raza A, Dussault PH, Du L. Biochemistry. 2006;45:2561–2569. doi: 10.1021/bi052085s. [DOI] [PubMed] [Google Scholar]
- 35.Lin S, Van Lanen SG, Shen B. Proc. Nat. Acad. Sci. USA. 2009;106:4183–4188. doi: 10.1073/pnas.0808880106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.