

Abstract
Glutamate (Glu) is the primary excitatory neurotransmitter in the central nervous system and plays a critical role in the neuroplasticity of nociceptive networks. We aimed to examine the role of spinal astroglia in the modulation of glutamatergic neurotransmission in a model of chronic psychological stress-induced visceral hyperalgesia in male Wistar rats. We assessed the effect of chronic stress on different glial Glu control mechanisms in the spinal cord including N-methyl-d-aspartate receptors (NMDARs), glial Glu transporters (GLT1 and GLAST), the Glu conversion enzyme glutamine synthetase (GS), and glial fibrillary acidic protein (GFAP). We also tested the effect of pharmacological inhibition of NMDAR activation, of extracellular Glu reuptake, and of astrocyte function on visceral nociceptive response in naive and stressed rats. We observed stress-induced decreased expression of spinal GLT1, GFAP, and GS, whereas GLAST expression was upregulated. Although visceral hyperalgesia was blocked by pharmacological inhibition of spinal NMDARs, we observed no stress effects on NMDAR subunit expression or phosphorylation. The glial modulating agent propentofylline blocked stress-induced visceral hyperalgesia, and blockade of GLT1 function in control rats resulted in enhanced visceral nociceptive response. These findings provide evidence for stress-induced modulation of glia-controlled spinal Glu-ergic neurotransmission and its involvement in chronic stress-induced visceral hyperalgesia. The findings reported in this study demonstrate a unique pattern of stress-induced changes in spinal Glu signaling and metabolism associated with enhanced responses to visceral distension.
Keywords: chronic stress, glutamate, spinal cord, glia, pain
glutamate (Glu) is the primary excitatory neurotransmitter in the central nervous system (CNS) and plays a critical role in the neuroplasticity of nociceptive networks (2, 48). At the spinal level, Glu is released from the central terminals of primary afferents and acts on postsynaptic receptors, including ionotropic Glu receptors, the α-amino-3-hydroxy-5-methyl-4-izoxazolepropionic acid receptor, the kainate receptor, the N-methyl-d-aspartate receptor (NMDAR), and metabotropic Glu receptors, mediating rapid excitatory postsynaptic potentials in dorsal horn neurons (22). Upregulation of glutamatergic signaling plays an important role in central sensitization and associated hyperalgesia and may be involved in the pathophysiology of chronic pain (33). In experimental models of pathological pain induced by inflammation or injury, sustained activation of primary afferents leading to increased Glu release in the spinal dorsal horn is a well-established mechanism contributing to the generation and maintenance of hyperalgesia (23).
The homeostasis of extracellular Glu is tightly controlled by a system regulating the cellular uptake from the synaptic space and the intracellular Glu recycling cycle. Extracellular Glu clearance through uptake is dependent on the activity of high-affinity excitatory amino acid transporters (EAAT) present in the plasma membrane of glia as well as neurons (7). In the spinal cord, l-Glu-l-aspartate transporter (GLAST) and the Glu transporter 1 (GLT-1) are mostly expressed on mature and differentiated astrocytes. These Glu transporter proteins are concentrated in the superficial dorsal horn of the spinal cord and are responsible for over 80% of the total of Glu transport into the cells (21, 34, 41). The excitatory amino acid carrier 1 (EAAC1) is expressed on neurons. The intracellular Glu-glutamine metabolic cycle is another important component in the homeostasis of the Glu-ergic synapses (30, 31). In astrocytes, the activity of the glutamine synthetase (GS) catalyzes the condensation of Glu with ammonium ion to produce glutamine (6). Glutamine released from astrocytes is either transported into the bloodstream or taken up by neurons and hydrolyzed by neuronal glutaminase into Glu (9, 18). Several studies have demonstrated a link between protein expression and activity of astrocytic Glu transporters and GS activity, both being modulated by the level of extracellular and intracellular Glu (50).
Disturbance of this control mechanism such as change in Glu transporter expression or reuptake ability has been described in models of inflammation or nerve injury associated with hyperalgesia (28, 39). A pronociceptive effect of pharmacological inhibition of spinal Glu transporters in the spinal cord has been confirmed in several studies (46). More recently, the role of Glu transporters in the modulation of visceral pain has been demonstrated in mice overexpressing the glial Glu transporter EAAT2 (24) and in a rat model of visceral hyperalgesia induced by early life stress (16). The link between stress and altered Glu transporter function or expression is supported by reports of high extracellular Glu concentration in the CNS in stressful conditions (26, 32). Also a modulatory role of glucocorticoids on Glu transporter expression (20, 43) has been reported. In addition, there is evidence that stress exposure may induce changes in brain astrocytes population in patient populations affected with depression as well as in animal models (1).
In the present study, we used a model of chronic psychological stress in rats, which has previously been shown to be associated with stress-induced visceral hyperalgesia (3). Our overall goal was to assess the role of spinal astroglia in the modulation of glutamatergic neurotransmission in stress-induced visceral hyperalgesia. We examined the effect of chronic stress on different glial Glu control mechanisms in the spinal cord including NMDARs, glial Glu transporters (GLT1 and GLAST), the Glu conversion enzyme GS, and glial fibrillary acidic protein (GFAP), a key cytoskeletal protein of astrocytes. In addition, we tested the effect of pharmacological inhibition of NMDARs, Glu reuptake, and astrocytes function on visceral nociceptive response in naive and stressed rats. We observed a stress-induced alteration in the expression of GLT1, GLAST, GFAP, and GS. Although stress-induced visceral hyperalgesia was sensitive to pharmacological inhibition of NMDARs, we observed no stress effects on NMDAR subunit expression or phosphorylation. These findings suggest a key role of astrocytes and spinal Glu transmission in stress-induced visceral hyperalgesia.
MATERIAL AND METHODS
Water Avoidance Stress
Adult male Wistar rats (250–275 g) (Harlan, IN) were maintained on a normal light-dark cycle and provided with food and water ad libitum. All protocols were approved by the Institutional Animal Care and Use Committee at the Veterans Affairs Greater Los Angeles Healthcare System.
Rats were placed on a block (8 × 8 × 10 cm) affixed to the center of a Plexiglas cage (25 × 25 × 45 cm) filled with fresh room temperature water (25°C) to within 1 cm of the top of the block [water avoidance (WA)], or kept empty (sham WA) for 1 h daily, for 10 consecutive days (3).
Western Blotting
Rats were euthanized by decapitation following isoflurane overdose. Spinal cords were hydroextruded with iced saline, and lumbar L6-S1 segments were dissected and processed for Western blotting. Samples were transferred to extraction buffer [50 mM Tris buffer, pH 8.0, containing 3% SDS, 0.5% Triton X-100, 150 mM NaCl, 1 mM EDTA, protease inhibitor cocktail (1:100, Sigma), phosphatase inhibitor cocktail I and II (1:100, Sigma) and immediately homogenized by sonication]. Then 25 μg of protein was electrophoresed on 3–8% NuPage Tris-acetate gels and transferred to polyvinylidene fluoride (PVDF) membranes.
Blots were probed with primary antibodies anti-phospho-NR1 (Ser896) (P-NR1, 1:1,000, Millipore, Temecula, Ca), anti-GFAP (1:65,000, Millipore), anti-GLT1 (1:1,000, Cell Signaling), anti-GLAST (1:5,000, Millipore), and anti-GS (1:2,000, Abcam). Membranes were stripped and reblotted with anti-NR1 (1:200, Santa Cruz Biotechnology) or anti-actin (1:1,000, Cell Signaling). The intensity of immunoreactive bands was quantified by use of ImageQuant software (Molecular Dynamics, Sunnyvale, CA). P-NR1 was expressed relative to NR1, and GFAP, GLT1, GLAST, GS, and NR1 are expressed relative to actin.
Immunoprecipitation for NR2 and P-NR2
Spinal cord samples were collected and homogenized in ice cold RIPA-B buffer [50 mM Tris-Cl, pH 8.0, 10 mM NaF, 2 mM EDTA, 1 mM Na3VO4, 1% Nonidet P-40, 1% sodium deoxycholate, 0.05% SDS, and “complete” protease inhibitor cocktail (Roche Diagnostics, Mannheim, Germany)]. The homogenate was agitated for 2 h at 4°C before centrifugation at 14,000 rpm for 10 min. The supernatant was collected and the protein concentration was measured by the detergent-compatible BCA method (Thermo Scientific, Rockford, IL). A volume of extract equivalent to 500 μg of protein was transferred to a fresh tube and equal volume of RIPA-A buffer (RIPA-B buffer without deoxycholate) was added together with 5 μg rabbit anti-NR2B antibody (Millipore). The tube was incubated with rotation overnight at 4°C and immune complexes were isolated by use of protein A/G-Sepharose beads (Santa Cruz Biotechnology). The samples were boiled in SDS sample buffer before electrophoresis on 3–8% NuPage Tris-acetate gels (Invitrogen) and transfer to PVDF membranes. The membranes were blocked with 3% nonfat dry milk in Tris-buffered saline, incubated with anti-phosphotyrosine 4G-10 (1:2,000, Millipore) overnight at 4°C, washed, and incubated with anti-mouse IgG horseradish peroxidase (1:4,000, Millipore). Labeled bands were visualized with ECL reagent (GE Healthcare, Buckingham, UK) and a Fujifilm LAS-4,000 Image analyzer. To control for protein loading and transfer efficiency, the membranes were stripped (Restore Western Blotting Stripping Buffer; Thermo Scientific) and then reprobed with the antibody to NR2B (1:1,000, Millipore) followed by an anti-rabbit peroxidase-conjugated antibody (1:1,000, Amersham, GE Healthcare). The degree of tyrosine phosphorylation was obtained by normalizing the ∼180-kDa band on the anti-phosphotyrosine immunoblot with the corresponding band in the NR2B immunoblot from the same membrane.
Real-Time Quantitative PCR for NMDARs
Total RNA was isolated from L6-S1 samples and real-time PCR was performed by using the ABI Prism 7700 Sequence Detection System (Applied Biosystems, Foster City, CA) with SYBR green detection in a two-step reaction, as previously described in Ref. 29. Primer pairs for the NR2 subunits and the reference gene r36B4 (rat acidic ribosomal phosphoprotein P0) and PCR conditions have been previously described in detail (29). PCR primers for the NR1 subunit were NR1-forward: 5-GTTCTTCCGCTCAGGCTTTG, NR1-reverse: 5-AGGGAAACGTTCTGCTTCCA, product: 66 bp. Results of the RT-PCR analysis were expressed as threshold cycle (CT) values, which were used to determine the amount of target gene mRNA in relation to the amount of reference gene mRNA (ABI Prism 7700 Sequence Detection System User Bulletin 2). ΔCT indicates the difference between the number of cycles necessary to detect the PCR products for each NR subunit compared with the reference gene. Data were expressed as 2−ΔCT to give a linear estimate of the amount of target mRNA present in the tissue relative to the reference gene.
Immunohistochemistry
Animals were perfused with 4% paraformaldehyde and spinal cords were processed as previously described (5). Sections from both sham WA and WA rats were stained for GFAP (1:1,000, Millipore) followed by the secondary antibody (mouse green Alexa a21202, 1:2,000, Invitrogen). Images were captured by using a Zeiss Axioskop epifluorescent microscope with a Zeiss ×40 Plan Apo objective and ×20 Plan Apo (1.0 NA, Carl Zeiss, Jena, Germany) Images were acquired by using a SPOT charge-coupled device digital camera (Diagnostic Instruments, Sterling Heights, MI) and analyzed for GFAP immunofluorescence density and GFAP-positive cell count in 38 × 38 μm regions in the superficial laminae by use of the NIH Image J program.
Surgical Implantation of Chronic Intrathecal Catheter and EMG Electrodes and Assessment of Visceromotor Response to CRD
Rats were deeply anesthetized with Nembutal (Abbott Laboratories, North Chicago, IL; 50 mg/kg ip) and surgically equipped with a chronic intrathecal (it) catheter (8.5 cm polyethylene tubing-5, OD 0.14 in., ID 0.006 in., Spectranetics, Colorado Springs, CO), connected to a 4-cm PE10 (polyethylene tubing-10), inserted through the atlanto-occipital membrane of the cisterna magna. Following surgery, rats were housed singly, received injection of buprenorphine (0.03 mg/kg sc) and monitored for 3 days and allowed to recuperate for at least 5 days before colorectal distension (CRD) testing. Wounds were tested for tenderness to ensure complete recovery from surgery prior to testing. Rats exhibiting any sign of neurological or motor impairment, as evidenced by paralysis, abnormal gait, weight loss, or negligent grooming, were excluded from the study and euthanized (1 of 16). For chronic intrathecal treatment, the tip of the catheter was connected to an osmotic minipump (Alzet, model 2002, Cupertino, CA) positioned under the skin between the scapulas. Catheters were primed with vehicle or drugs before implantation. After completion of testing, the catheter position was verified in each animal by postmortem examination of the spinal cord.
EMG electrodes (Teflon-coated stainless steel wire, Cooner Wire, CA) were stitched into the external oblique musculature, for electromyographic (EMG) recordings as previously described (5). The visceral stimulus employed was distension of the descending colon and rectum by a well-established and validated method (5). Briefly, under light Isoflurane anesthesia, a flexible latex balloon (6 cm) was inserted intra-anally (after the distal part of the rectum was gently cleared by massage) such that its end was 1 cm proximal to the anus. Once recovered from anesthesia, animals equipped with the balloon were placed in a Plexiglas cylinder for 30 min before the CRD procedure was initiated. The CRD procedure consisted of two series of phasic CRDs to constant pressures of 10, 20, 40, and 60 mmHg (20-s duration; 4-min interstimulus interval). The visceromotor response (VMR) to CRD was quantified by measuring EMG activity in the external oblique musculature. EMG activity was recorded 20 s before (baseline), 20 s during, and 20 s after termination of CRD. The EMG activity was rectified, and the increase in the area under the curve of EMG amplitude during CRD over the baseline period before CRD was recorded as the response. In the following, we will use the term EMG referring to the VMR to CRD.
Drugs
Dihydrokainic acid (DHK), propentofylline, and methionine sulfoximine were purchased from Sigma-Aldrich and dissolved in sterile water or 0.9% saline. MK-801 [Tocris, (+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d] cyclohepten-5,10-imine maleate], was dissolved in sterile water.
Experimental Design
Effect of the NMDAR antagonist MK-801 on WA stress-induced visceral hypersensitivity.
We evaluated the effect of spinal administration of the selective NMDAR antagonist MK-801 on stress-induced visceral hyperalgesia at day 11. Animals were first equipped with intrathecal catheters and EMG electrodes, then 5 days later baseline measurements of the EMG response were recorded. Rats were divided into groups and exposed to either chronic WA stress or sham WA (n = 7–8 in each group). Animals received a single intrathecal injection with the NMDAR antagonist MK-801 (10 nmol, 5 μl it) or vehicle (saline) 10 min before measurement of EMG at day 11. The VMR to CRD recorded at day 11 was compared with baseline and the mean difference from baseline in treated and vehicle groups was compared in both sham WA and WA rats.
Effect of WA stress on expression and phosphorylation of the NMDAR subunits NR1 and NR2 and expression of spinal glial Glu transporters, glutamine synthetase, and GFAP.
Groups of rats without surgery for implantation of electrodes or catheters were exposed to either chronic WA or sham WA, and spinal cord samples were collected 24 h after the last session (day 11), divided, and processed for RNA, protein, and immunohistochemistry as described above. Western blotting for NR1 and P-NR1 and immunoprecipitation for NR2B and P-NR2B were used to measure protein expression and phosphorylation of the NMDAR subunits (n = 5 in each group). Western blots were also performed to measure levels of GFAP, GLT1, GLAST, and GS (n = 4–7 in each group). Quantitative RT-PCR was used to measure mRNA expression levels of the NMDAR subunits NR1, NR2A, and NR2B (n = 6 in each group). We also performed immunohistochemistry for GFAP (n = 4 in each group). All samples were collected from the lumbar L6-S1 region.
Effect of the glial modulating agent propentofylline on stress-induced visceral hyperalgesia.
We tested whether spinal infusion of the glia modulating agent propentofylline during the 10-day stress exposure affects the response to CRD at day 11. Animals were equipped with EMG electrodes, and baseline measurements of the EMG response were recorded. Then rats were implanted with a chronic intrathecal catheter connected to a subcutaneously implanted osmotic minipump delivering propentofylline (10 μg in 12 μl per day; 0.03 μM) or saline, and exposed to sham and WA. VMR to CRD was recorded again at day 11 and compared with baseline, and the mean difference from baseline in treated and vehicle groups was compared in both sham and WA groups (n = 5–7 in each group).
Effect of selective inhibition of GLT1 on basal visceral nociception.
Groups of naive rats equipped with EMG electrodes were treated with the selective GLT1 inhibitor DHK (20 mM in saline, 10 μl it) or vehicle (saline, 10 μl). The EMG response was started 15–30 min after injection of DHK (n = 11–15 in each group).
Data Presentation and Statistical Analysis
Western blot results are presented as the relative density to the appropriate reference protein (actin or nonphosphorylated form). EMG data are presented as the mean change from baseline in treated animals vs. vehicle (EMG day 11 or EMG posttreatment; EMG baseline in rats exposed to 10 days WA or sham WA or naive rats, respectively). The effect of stress and treatments on different markers expression and EMG responses were analyzed by a repeated-measure one-way ANOVA, Student's t-test, and two-way ANOVA followed by Bonferroni posttest comparisons, as indicated.
RESULTS
Effect of the NMDAR Antagonist MK-801 on Chronic WA Stress-Induced Visceral Hypersensitivity
Rats exposed to chronic WA stress and injected with vehicle at day 11 showed an increased EMG response to CRD compared with baseline (P < 0.05) consistent with stress-induced visceral hyperalgesia as has been previously described (4). Because spinal dorsal horn NMDARs are known to be involved in central sensitization leading to hyperalgesia following inflammation or injury (8), we first investigated the effect of an NMDAR antagonist on stress-induced visceral hyperalgesia. Visceral hyperalgesia was inhibited by intrathecal application of the NMDAR antagonist, MK-801 (10 nmol, 5 μl). The mean change from baseline in stressed rats treated with MK-801 vs. vehicle was significantly reduced at distension pressures of 40 and 60 mmHg (+3.3 ± 13.20 and −6 ± 19 compared with +9.9 ± 19.6 and +52 ± 30.7, P < 0.05; Fig. 1). In contrast, MK-801 treatment of control rats did not affect the EMG response to CRD as previously reported by another group (14).
Fig. 1.
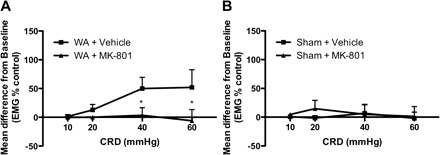
Inhibition of visceral hyperalgesia by MK-801. The N-methyl-d-aspartate receptor (NMDAR) antagonist MK-801 [10 nmol, 5 μl it, 10 min before colorectal distension (CRD)] completely blocked stress-induced visceral hyperalgesia at day 11 in water avoidance (WA)-stressed rats (A) but had no effect in sham WA rats (B) (n = 7–8). EMG, electromyography. Data are means ± SE. *P < 0.05, significantly different between vehicle- and MK-801-treated rats (2-way ANOVA followed by Bonferroni posttest).
Expression of NMDARs in the Spinal Cord After Chronic WA Stress
Since phosphorylation of NMDAR subunits by either by tyrosine or serine-threonine kinases leads to their upregulation in several models of chronic pain (8), we next investigated whether the NR1 or NR2B subunits were phosphorylated after chronic WA stress. The protein levels for the phosphorylated forms of NMDA-NR1 (P-NR1) (77.66 ± 12.20 vs. 100 ± 26.38%) and P-NR2B (94.92 ± 34.10 vs. 100 ± 14.38%) relative to the nonphosphorylated forms (NR1 and NR2B) were unchanged in WA compared with sham WA. (Fig. 2, A and B). When normalized to the level of actin, protein levels for NR1 and NR2B were unchanged in WA compared with control. Furthermore, there was no difference in the mRNA levels of the different subunits (NR1, NR2A, NR2B) in WA compared with controls, suggesting that neither gene or protein expression nor subunit phosphorylation was changed in the spinal cord after chronic WA stress (Fig. 2C).
Fig. 2.
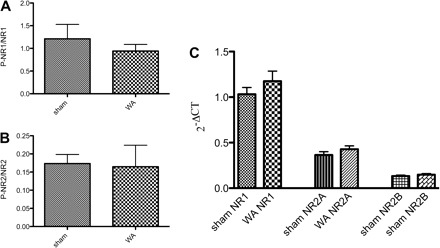
Expression and phosphorylation of the NMDAR subunits NR1 and NR2 in WA and sham WA. Phosphorylation of NR1 (A) and NR2B (B) was unchanged in WA-stressed rats compared with sham-treated rats (n = 5), nor was there an effect on the mRNA levels for NR1 and NR2A and NR2B subunits (C, n = 6). The Western blotting data are given as the relative expression of the phosphorylated subunit per total subunit (means ± SE). The levels of mRNA are plotted as 2−ΔCT, which gives a linear estimate of gene expression relative to the reference gene. Data were compared by Student's t-test.
Effect of Chronic WA Stress on Spinal Astrocytes, Glial Glu Transporters, and GS
Elevated extracellular levels of Glu could cause persistent stimulation of NMDARs in the spinal cord, leading to hyperalgesia in the absence of NMDAR upregulation. Elevated extracellular levels of Glu can result from decreased reuptake or decreased conversion to glutamine or both. Western blotting of spinal cord extracts showed a significant decrease in the level of the key astrocyte cytoskeletal protein, GFAP, in WA rats compared with controls (0.91 ± 0.07 vs. 1.43 ± 0.19, P < 0.05). Similarly, protein levels for GLT1 (a Glu transporter primarily expressed on glia cells) were reduced in WA compared with controls (0.33 ± 0.02 vs. 0.24 ± 0.04, P < 0.05). However, the expression of the other glial Glu transporter GLAST was increased in WA compared with controls (0.49 ± 0.06 vs. 0.74 ± 0.06, P < 0.05). Finally, there was a striking fivefold reduction in the expression of glutamine synthetase in WA compared with controls (0.386 ± 0.100 vs. 0.073 ± 0.006, P < 0.01). These data are presented along with original representative photographs of the blots in Fig. 3.
Fig. 3.
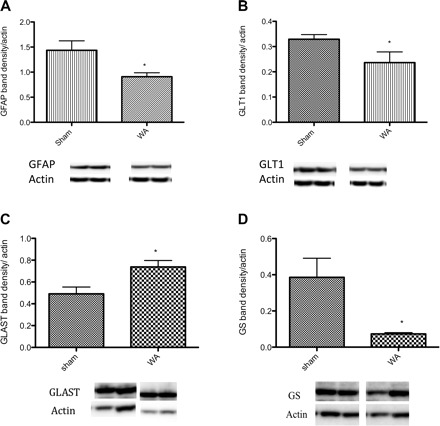
Stress-induced change in expression of spinal glial fibrillary acidic protein (GFAP), the glutamate (Glu) transporter 1 (GLT-1), l-Glu-l-aspartate transporter (GLAST), and glutamine synthetase (GS). Protein levels for GFAP (A), GLT1 (B), and GS (D) are reduced whereas GLAST (C) is increased in WA compared with sham WA (n = 4–7/group). Data are expressed as the mean band density relative to actin (means ± SE) *P < 0.05 significantly different from sham, Student's t-test. Representative Western blots showing the bands corresponding to GFAP, GLT1, GS, and GLAST and actin for each group.
The changes in glial Glu transporters and metabolizing enzymes promoted us to examine GFAP, an astrocyte intermediate filament protein whose expression is increased in various models of inflammation and nerve injury (35). GFAP immunostaining was found mainly in the gray matter in the superficial laminae of the dorsal horn. We performed a quantitative analysis of the density of GFAP staining and a measurement of the number of GFAP positive cells per field as a means to provide qualitative and semiquantitative analysis of the effect of WA stress on GFAP staining in the spinal cord. The density of GFAP staining was reduced in sections from WA rats compared with controls, measured in 38 × 38 μm regions in the superficial laminae by use of the NIH Image J program (P < 0.05), whereas the number of GFAP-positive cells were not different (Fig. 4). Although blinded observations of the sections suggested smaller size astrocytes in the WA group compared with controls, quantitative measurement and comparison of exact cell size could not be performed because of experimental limitations.
Fig. 4.
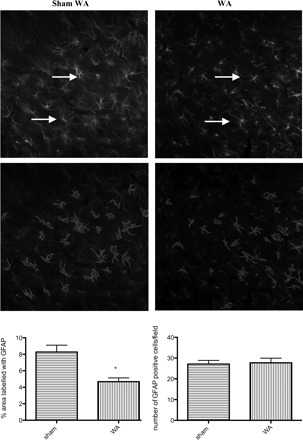
Photographs representative of immunofluorescent labeling for GFAP in selected areas of the dorsal horn in sham WA (left) and WA (right) (38 × 38 μm). Photographs are converted to grayscale and GFAP-positive cell count was performed by using the NIH Image software with individual setting adjustment (bottom images showing outlines GFAP+ cells). The density of GFAP staining was reduced in sections from WA rats compared with controls, measured in 38 × 38 μm regions in the superficial laminae (bar graph at left; P < 0.05). However, there was no significant change of the number of GFAP-positive cells in WA compared with controls (bar graph at right; n = 4, 5–8 sections counted for each n).
Effect of the Glia Modulating Agent Propentofylline on Visceral Nociceptive Response in WA and Control Rats
We tested the effect of the glia modulating agent propentofylline in our stress model. Rats that received sustained intrathecal delivery of propentofylline (10 μg in 12 μl per day; 0.03 μM) during the 10 days of WA stress exposure failed to develop stress-induced visceral hyperalgesia measured at day 11 (Fig. 5). In contrast, vehicle-treated rats exposed to chronic WA stress showed increased EMG response to CRD at day 11 (P < 0.05). The mean difference from baseline was significantly decreased in stressed rats treated with propentofylline compared with vehicle (P < 0.05). In the control group exposed to sham WA and treated with propentofylline, a lower EMG response to CRD was observed compared with baseline (P < 0.05), suggesting an analgesic effect. This response was significantly different from the response observed in sham WA rats treated with saline (P < 0.05) (Fig. 5).
Fig. 5.
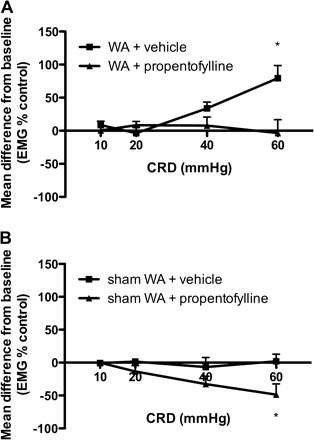
Effect of chronic treatment with propentofylline on stress-induced visceral hyperalgesia. A: stressed rats treated with intrathecal propentofylline (10 μg/day for 10 days) failed to develop visceral hyperalgesia at day 11. The mean change from baseline in propentofylline-treated stressed rats was significantly decreased compared with vehicle-treated rats that showed hyperalgesia at day 11 (n = 5–8). B: control animals treated with propentofylline showed decreased EMG response at day 11 compared with baseline and the decrease was significant compared with vehicle (n = 5–7). Data are given as means ± SE. *P < 0.05 significantly different from vehicle (2-way ANOVA followed by Bonferroni posttest).
Effect of Selective Inhibition of GLT1 on Basal Visceral Nociception and the Role of Astrocytes in This Effect
To investigate whether decreased Glu reuptake and recycling could account for the development of visceral hyperalgesia following chronic stress, we tested the effect of a GLT1 inhibitor on the EMG response to CRD in nonstressed rats. Rats treated with the selective GLT1 inhibitor DHK (20 mM in saline, 10 μl it) showed significantly increased EMG response to CRD compared with rats receiving vehicle only (P < 0.001), suggesting a pronociceptive effect of DHK (Fig. 6). EMG responses were significantly increased in DHK-treated rats at pressures of 40 and 60 mmHg, indicating the development of visceral hyperalgesia.
Fig. 6.
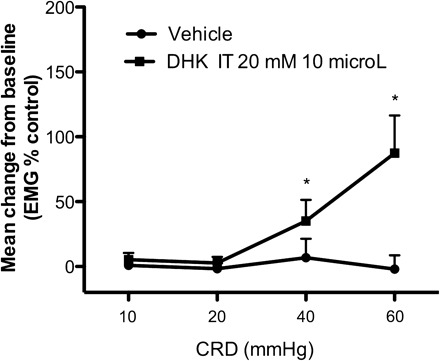
Effect of dihydrokainic acid (DHK) on electromyographic (EMG) response in control rats. Acute intrathecal injection of DHK (20 mM in saline, 10 μl) increased the EMG response compared with baseline (n = 11). The increase was significant compared with vehicle injection (n = 15), consistent with visceral hyperalgesia, P < 0.05. Data are means ± SE. * P < 0.05 significantly different from vehicle (2-way ANOVA followed by Bonferroni posttest).
DISCUSSION
The present study provides evidence for stress-induced modulation of glia-controlled spinal glutamatergic neurotransmission and its involvement in chronic stress-induced visceral hyperalgesia. We demonstrate stress-induced changes in the expression of several components of astrocyte Glu metabolism cycle in the lumbar spinal cord including the Glu transporters GLT1 and GLAST and GS.
We hypothesize that the greater accumulation of extracellular Glu at excitatory synapses induced by chronic stress may result in greater depolarization of postsynaptic neurons and activation of NMDA receptors. We show that stress-induced visceral hyperalgesia is blocked by intrathecal administration of the NMDAR antagonist MK-801, whereas it had no effect on visceral nociceptive response in nonstressed, control animals as previously demonstrated (14). These results are consistent with a role of spinal NMDARs in chronic stress-induced visceral hyperalgesia but not to baseline nociceptive responses (11, 14). However, in contrast with prior reports showing increased spinal NR1 expression and phosphorylation in a model of inflammatory pain facilitation (44), we did not find any changes in the mRNA and protein expression nor in the level of phosphorylation of the NR1/2 subunits. It should be noted that our measurements were performed in stressed animals not exposed to painful stimulation, and one may hypothesize that increased phosphorylation of NMDAR subunits occurs in situation of spontaneous pain or evoked nociceptive response (to CRD for example). Other components of the Glu signaling system, including intracellular scaffolding proteins (42) and metabotropic Glu receptors that could contribute to the stress-induced visceral hyperalgesia, were not assessed in the present study.
Astrocytic clearance of Glu from the synaptic space constitutes an important component in the control of the glutamatergic transmission. In the CNS, the high-affinity Glu transporters GLT1 and GLAST, which are primarily expressed by astroglia, have a major role in this process (reviewed in Ref. 7). We observed a stress-induced reduction in the spinal expression of GLT1 that is consistent with previous reports showing decreased spinal Glu transporter expression in various chronic pain models (28, 39, 45) as well as in a rat model of maternal separation stress (16). However, in the present study, the reduction in GLT1 expression was associated with an increased expression of GLAST. Differential changes in GLT1 and GLAST expression have been described (10), suggesting that each Glu transporter type is under different regulatory control. For example, in primary murine astrocytes cultures, cell surface expression of GLAST can be rapidly regulated by extracellular Glu concentration whereas GLT1 expression is not affected (10). In addition, although GLT1 and GLAST are mostly located on astrocytes, there is evidence of plasticity in expression of GLT1 and GLAST on microglia in the spinal dorsal horn in models of chronic pain induced by partial sciatic nerve ligation (49). It is therefore possible that microglia Glu transporter expression is affected in our study, possibly as a counterregulatory mechanism. Unfortunately, we could not test this possibility in our model because the antibodies for GLT1 and GLAST were unsuitable for immunohistochemistry. Further studies are required to better characterize the complex regulatory mechanisms engaged in microglia-astrocyte interactions during chronic stress.
It has been previously reported that dysfunction in Glu transport produces marked changes in spinal processing of nociceptive inputs (33). Inhibition of Glu transporters causes an elevation in spinal extracellular Glu concentrations, associated with spontaneous nociceptive behaviors and hypersensitivity to mechanical and thermal stimuli (37). Of particular interest, coadministration of a Glu transporter blocker with the NMDAR antagonist, MK-801, blocks evoked spontaneous pain behaviors as well as thermal and mechanical hyperalgesia (23). Conversely, gene transfer of GLT-1 into the spinal cord attenuates inflammatory and neuropathic pain in rats (27). Therefore we hypothesize that although GLAST is upregulated, possibly on microglia as a regulatory mechanism in response to decreased GLT1 or increased extracellular Glu, the net balance of Glu in the extracellular space is positive. Even though measurement of Glu levels in the lumbar CSF was not performed, we found that DHK, a potent GLT1 inhibitor, injected intrathecally in control rats, induced a marked visceral hyperalgesia. This finding supports the concept that reduced synaptic Glu reuptake leads to increased visceral nociception. It is also consistent with results from a recent report using transgenic mice overexpressing human EAAT2 (GLT1), which exhibited a twofold enhanced Glu uptake and attenuated visceral pain response to intraperitoneal acetic acid (24). In addition, EAAT2 transgenic mice showed a 53–64% reduction in visceromotor response to CRD, suggesting that enhanced Glu uptake provides protective effects against visceral pain.
Although Glu transporters are important in the homeostasis of the extracellular space, intracellular Glu metabolic enzymes, including GS, play an equally important role in the regulation of glutamatergic signaling. The main role of GS is to keep intracellular Glu levels low by converting it to the less neurotoxic amino acid glutamine. Glutamine is released to the extracellular space, from where it either enters the bloodstream or is taken up by neurons and converted back to Glu for storage in synaptic vesicles. Our finding of a marked stress-induced reduction of spinal GS expression suggests a lower conversion rate of Glu to glutamine in astrocytes, which in turn may reduce their capacity to capture extracellular Glu (38). The reason for the reduced GS expression in astroglial processes could be a response to a change in the number of Glu transporters and/or low astrocytic capacity for GS synthesis (38). In the chronic stress model, decreased GS expression may indicate the failure of astrocytes to counteract excessive synaptic Glu release and increase the activation of neuronal Glu receptors. Previous studies showed an increased extracellular level of Glu in the CNS in several animal models of stress (26, 32).
Astrocytes are the predominant glial cell type in the adult mammalian central nervous system, where they modulate synaptic transmission by regulating the levels of Glu in brain and spinal cord synapses. Astrocytes are a heterogeneous group of cells, the transcriptional control of their phenotype being determined by the local environment (reviewed in Ref. 47). GFAP, a key cytoskeletal intermediate filament in astrocytes, becomes expressed in mature, fully differentiated astrocytes, and its increased expression is commonly used as a marker of hypertrophic, reactive astrocytes. Such a phenotype is characteristic of spinal astrogliosis, a phenomenon reported in various models of neuropathic and inflammatory chronic pain state (12, 35). In the present study, we found a decrease in GFAP protein level and immunostaining in the spinal cord. The decreased density of immunostaining for GFAP in chronically stressed animals was not associated with a reduction in the number of GFAP+ cells but instead is more consistent with a stress-induced decreased gene expression of GFAP. Our findings contrast with astrogliosis reported in other types of chronic pain; however, they are consistent with findings in high-anxiety rat strains and in various rat models of chronic stress, which also show decreased GFAP in several CNS regions (13, 15, 25). A number of studies also demonstrated a reduction in GFAP+ cells in postmortem brain samples of patients with depression (17). The fact that GFAP plays a key role in astrocytic Glu transporter trafficking and function (19) suggests that reduced GFAP observed in our stress model may alter GLT1/GLAST-mediated clearance of Glu from the synaptic space, contributing to increased excitatory glutamatergic neurotransmission. When viewed together with previously published results, our study suggests that chronic psychological stress is associated with a specific pattern of phenotypic changes of astrocytes within the CNS that differs from the patterns seen in other types of pain (Fig. 7).
Fig. 7.
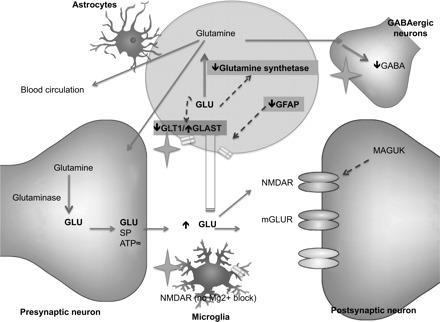
Schematic representation of the proposed model of regulatory mechanisms in the spinal glutamatergic synapse in response to chronic WA stress: Glu released from presynaptic terminals (Glu) is cleared from the synaptic space via the astrocytic glutamate transporters GLT1 and GLAST. Intracellular Glu in astrocytes is converted by GS into glutamine, which is either transported out and back into presynaptic neurons (to regenerate the glutamate pool after conversion by glutaminase) or to GABAergic neurons or to the blood vessels nearby. The activity of GS, as well as the activity of GLT1 and GLAST, is modulated by the intracellular concentration of Glu. Their trafficking to the cell membrane and activity can be modulated by GFAP. Glu interacts with GLUT receptors on postsynaptic neurons, including NMDARs. Sustained postsynaptic NMDAR activation results in central sensitization, and increased expression and phosphorylation of the NR1/NR2 subunits have been described in models of chronic pain. Their activity can also be modulated by membrane-associated guanylate kinase (MAGUK). NMDAR have been described on microglia on which their activation does not depend on the release of the magnesium block. However, the role of glial NMDAR in central sensitization is not known. In our study, we found decreased GLT1 expression combined with increased GLAST after chronic WA stress. We hypothesize that the effect of stress results in net decrease of Glu reuptake from astrocytes. Also, we hypothesize that stress-induced change in expression of GLT1 and GLAST is related to the decreased expression of GFAP. We propose that decreased GFAP and GS expression combined with changes in Glu transporters expression converge toward increased concentration of Glu in the synaptic space. We found no change in NMDAR expression or phosphorylation after chronic stress. Text in gray boxes represents the findings from our study. The star represents the potential sites of action of propentofylline. mGLUR, metabotropic glutamate receptor.
Propentofylline has been widely used to interfere with astrocyte activity, and although its mechanisms of action on astrocytes remains unclear, several studies have confirmed its ability to prevent the development of neuropathic pain in various animal models (40). In the present study, we found that chronic intrathecal treatment with propentofylline blocks the development of stress-induced visceral hyperalgesia, suggesting that the observed stress-induced change in astrocytic phenotype (decreased GFAP and altered GLUT reuptake) is involved in visceral pain sensitization. Previous in vivo and in vitro studies have demonstrated the ability of propentofylline to modulate the expression of Glu transporters on astrocytes, for instance to increase GLT1 expression or reverse nerve injury-induced decreased level of GLT1 (reviewed in Ref. 40), which may account for the effect of propentofylline observed in our study. Furthermore, it has been proposed that the anti-hyperalgesic/anti-allodynic effect of propentofylline is mediated by inhibition of glial proinflammatory cytokine production. Alternatively or perhaps in addition, the observed analgesic effect of the propentofylline in nonstressed rats may be related to an increase in the production of the inhibitory neurotransmitter GABA, thereby increasing endogenous inhibitory tone.
In conclusion, the findings reported in this study demonstrate a unique pattern of stress-induced changes in spinal Glu signaling and metabolism associated with enhanced nociceptive responses to visceral distension. A better understanding of the mechanisms involved in the modulation of glia activation may have translational implications for the development of new therapeutic perspectives for the treatment of some patients with stress-sensitive persistent visceral pain disorders, such as irritable bowel syndrome and interstitial cystitis/painful bladder syndrome.
GRANTS
This research was supported by National Institutes of Health Grants RO1 DA026597-01A2 (S. Bradesi) P50 DK064539 (E. A. Mayer).
DISCLOSURES
The authors have no financial interests related to material presented in this paper.
REFERENCES
- 1. Banasr M, Chowdhury GM, Terwilliger R, Newton SS, Duman RS, Behar KL, Sanacora G. Glial pathology in an animal model of depression: reversal of stress-induced cellular, metabolic and behavioral deficits by the glutamate-modulating drug riluzole. Mol Psychiatry 15: 501–511, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Basbaum AI, Bautista DM, Scherrer G, Julius D. Cellular and molecular mechanisms of pain. Cell 139: 267–284, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bradesi S, Schwetz I, Ennes HS, Lamy CM, Ohning G, Fanselow M, Pothoulakis C, McRoberts JA, Mayer EA. Repeated exposure to water avoidance stress in rats: a new model for sustained visceral hyperalgesia. Am J Physiol Gastrointest Liver Physiol 289: G42–G53, 2005 [DOI] [PubMed] [Google Scholar]
- 4. Bradesi S, Schwetz I, Ennes HS, Lamy CM, Ohning G, Fanselow M, Pothoulakis C, McRoberts JA, Mayer EA. Repeated exposure to water avoidance stress in rats: a new model for sustained visceral hyperalgesia. Am J Physiol Gastrointest Liver Physiol 289: G42–G53, 2005. [DOI] [PubMed] [Google Scholar]
- 5. Bradesi S, Svensson CI, Steinauer J, Pothoulakis C, Yaksh TL, Mayer EA. Role of spinal microglia in visceral hyperalgesia and NK1R up-regulation in a rat model of chronic stress. Gastroenterology 136: 1339–1348, e1–2, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Broer S, Brookes N. Transfer of glutamine between astrocytes and neurons. J Neurochem 77: 705–719, 2001 [DOI] [PubMed] [Google Scholar]
- 7. Bunch L, Erichsen MN, Jensen AA. Excitatory amino acid transporters as potential drug targets. Expert Opin Ther Targets 13: 719–731, 2009 [DOI] [PubMed] [Google Scholar]
- 8. Cao J, Yang X, Liu YN, Suo ZW, Shi L, Zheng CR, Yang HB, Li S, Hu XD. GABAergic disinhibition induced pain hypersensitivity by upregulating NMDA receptor functions in spinal dorsal horn. Neuropharmacology 60: 921–929, 2011 [DOI] [PubMed] [Google Scholar]
- 9. Daikhin Y, Yudkoff M. Compartmentation of brain glutamate metabolism in neurons and glia. J Nutr 130: 1026S–1031S, 2000 [DOI] [PubMed] [Google Scholar]
- 10. Duan S, Anderson CM, Stein BA, Swanson RA. Glutamate induces rapid upregulation of astrocyte glutamate transport and cell-surface expression of GLAST. J Neurosci 19: 10193–10200, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dubner R, Ruda MA. Activity-dependent neuronal plasticity following tissue injury and inflammation. Trends Neurosci 15: 96–103, 1992 [DOI] [PubMed] [Google Scholar]
- 12. Eng LF, Ghirnikar RS, Lee YL. Glial fibrillary acidic protein: GFAP-thirty-one years (1969–2000). Neurochem Res 25: 1439–1451, 2000 [DOI] [PubMed] [Google Scholar]
- 13. Fuchs E, Czeh B, Kole MH, Michaelis T, Lucassen PJ. Alterations of neuroplasticity in depression: the hippocampus and beyond. Eur Neuropsychopharmacol 14, Suppl 5: S481–490, 2004 [DOI] [PubMed] [Google Scholar]
- 14. Gaudreau GA, Plourde V. Involvement of N-methyl-d-aspartate (NMDA) receptors in a rat model of visceral hypersensitivity. Behav Brain Res 150: 185–189, 2004 [DOI] [PubMed] [Google Scholar]
- 15. Gosselin RD, Gibney S, O'Malley D, Dinan TG, Cryan JF. Region specific decrease in glial fibrillary acidic protein immunoreactivity in the brain of a rat model of depression. Neuroscience 159: 915–925, 2009 [DOI] [PubMed] [Google Scholar]
- 16. Gosselin RD, O'Connor RM, Tramullas M, Julio-Pieper M, Dinan TG, Cryan JF. Riluzole normalizes early-life stress-induced visceral hypersensitivity in rats: role of spinal glutamate reuptake mechanisms. Gastroenterology 138: 2418–2425, 2009 [DOI] [PubMed] [Google Scholar]
- 17. Halassa MM, Haydon PG. Integrated brain circuits: astrocytic networks modulate neuronal activity and behavior. Annu Rev Physiol 72: 335–355, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hertz L. Autonomic control of neuronal-astrocytic interactions, regulating metabolic activities, and ion fluxes in the CNS. Brain Res Bull 29: 303–313, 1992 [DOI] [PubMed] [Google Scholar]
- 19. Hughes EG, Maguire JL, McMinn MT, Scholz RE, Sutherland ML. Loss of glial fibrillary acidic protein results in decreased glutamate transport and inhibition of PKA-induced EAAT2 cell surface trafficking. Brain Res Mol Brain Res 124: 114–123, 2004 [DOI] [PubMed] [Google Scholar]
- 20. Jacobsson J, Persson M, Hansson E, Ronnback L. Corticosterone inhibits expression of the microglial glutamate transporter GLT-1 in vitro. Neuroscience 139: 475–483, 2006 [DOI] [PubMed] [Google Scholar]
- 21. Kugler P, Schmitt A. Glutamate transporter EAAC1 is expressed in neurons and glial cells in the rat nervous system. Glia 27: 129–142, 1999 [DOI] [PubMed] [Google Scholar]
- 22. Latremoliere A, Woolf CJ. Central sensitization: a generator of pain hypersensitivity by central neural plasticity. J Pain 10: 895–926, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liaw WJ, Stephens RL, Jr, Binns BC, Chu Y, Sepkuty JP, Johns RA, Rothstein JD, Tao XY. Spinal glutamate uptake is critical for maintaining normal sensory transmission in rat spinal cord. Pain 115: 60–70, 2005 [DOI] [PubMed] [Google Scholar]
- 24. Lin Y, Tian G, Roman K, Handy C, Travers JB, Lin CL, Stephens RL., Jr Increased glial glutamate transporter EAAT2 expression reduces visceral nociceptive response in mice. Am J Physiol Gastrointest Liver Physiol 296: G129–G134, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liu Q, Li B, Zhu HY, Wang YQ, Yu J, Wu GC. Clomipramine treatment reversed the glial pathology in a chronic unpredictable stress-induced rat model of depression. Eur Neuropsychopharmacol 19: 796–805, 2009 [DOI] [PubMed] [Google Scholar]
- 26. Lowy MT, Wittenberg L, Yamamoto BK. Effect of acute stress on hippocampal glutamate levels and spectrin proteolysis in young and aged rats. J Neurochem 65: 268–274, 1995 [DOI] [PubMed] [Google Scholar]
- 27. Maeda S, Kawamoto A, Yatani Y, Shirakawa H, Nakagawa T, Kaneko S. Gene transfer of GLT-1, a glial glutamate transporter, into the spinal cord by recombinant adenovirus attenuates inflammatory and neuropathic pain in rats. Mol Pain 4: 65, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mao J, Sung B, Ji RR, Lim G. Chronic morphine induces downregulation of spinal glutamate transporters: implications in morphine tolerance and abnormal pain sensitivity. J Neurosci 22: 8312–8323, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Marvizón JC, McRoberts JA, Ennes HS, Johansson T, Corneliussen B, Jinton L, Mayer EA. Expression of NMDA receptor subunits in different types of primary afferent neurons (Abstract). Gastroenterology 120: A331, 2001. [Google Scholar]
- 30. Mates JM, Segura JA, Campos-Sandoval JA, Lobo C, Alonso L, Alonso FJ, Marquez J. Glutamine homeostasis and mitochondrial dynamics. Int J Biochem Cell Biol 41: 2051–2061, 2009 [DOI] [PubMed] [Google Scholar]
- 31. Miller KE, Hoffman EM, Sutharshan M, Schechter R. Glutamate pharmacology and metabolism in peripheral primary afferents: physiological and pathophysiological mechanisms. Pharmacol Ther 130: 283–309, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Moghaddam B, Bolinao ML, Stein-Behrens B, Sapolsky R. Glucocorticoids mediate the stress-induced extracellular accumulation of glutamate. Brain Res 655: 251–254, 1994 [DOI] [PubMed] [Google Scholar]
- 33. Nie H, Weng HR. Glutamate transporters prevent excessive activation of NMDA receptors and extrasynaptic glutamate spillover in the spinal dorsal horn. J Neurophysiol 101: 2041–2051, 2009 [DOI] [PubMed] [Google Scholar]
- 34. O'Shea RD. Roles and regulation of glutamate transporters in the central nervous system. Clin Exp Pharmacol Physiol 29: 1018–1023, 2002 [DOI] [PubMed] [Google Scholar]
- 35. Raghavendra V, Tanga FY, DeLeo JA. Complete Freunds adjuvant-induced peripheral inflammation evokes glial activation and proinflammatory cytokine expression in the CNS. Eur J Neurosci 20: 467–473, 2004. [DOI] [PubMed] [Google Scholar]
- 37. Ramos KM, Lewis MT, Morgan KN, Crysdale NY, Kroll JL, Taylor FR, Harrison JA, Sloane EM, Maier SF, Watkins LR. Spinal upregulation of glutamate transporter GLT-1 by ceftriaxone: therapeutic efficacy in a range of experimental nervous system disorders. Neuroscience 169: 1888–1900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Suarez I, Bodega G, Fernandez B. Glutamine synthetase in brain: effect of ammonia. Neurochem Int 41: 123–142, 2002 [DOI] [PubMed] [Google Scholar]
- 39. Sung B, Lim G, Mao J. Altered expression and uptake activity of spinal glutamate transporters after nerve injury contribute to the pathogenesis of neuropathic pain in rats. J Neurosci 23: 2899–2910, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sweitzer S, De Leo J. Propentofylline: glial modulation, neuroprotection, and alleviation of chronic pain. Hand Exp Pharmacol 200: 235–250, 2011 [DOI] [PubMed] [Google Scholar]
- 41. Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, Iwama H, Nishikawa T, Ichihara N, Kikuchi T, Okuyama S, Kawashima N, Hori S, Takimoto M, Wada K. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science 276: 1699–1702, 1997 [DOI] [PubMed] [Google Scholar]
- 42. Tao YX, Raja SN. Are synaptic MAGUK proteins involved in chronic pain? Trends Pharmacol Sci 25: 397–400, 2004 [DOI] [PubMed] [Google Scholar]
- 43. Wang S, Lim G, Yang L, Sung B, Mao J. Downregulation of spinal glutamate transporter EAAC1 following nerve injury is regulated by central glucocorticoid receptors in rats. Pain 120: 78–85, 2006 [DOI] [PubMed] [Google Scholar]
- 44. Wang S, Lim G, Zeng Q, Sung B, Yang L, Mao J. Central glucocorticoid receptors modulate the expression and function of spinal NMDA receptors after peripheral nerve injury. J Neurosci 25: 488–495, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Weng HR, Aravindan N, Cata JP, Chen JH, Shaw AD, Dougherty PM. Spinal glial glutamate transporters downregulate in rats with taxol-induced hyperalgesia. Neurosci Lett 386: 18–22, 2005 [DOI] [PubMed] [Google Scholar]
- 46. Weng HR, Chen JH, Cata JP. Inhibition of glutamate uptake in the spinal cord induces hyperalgesia and increased responses of spinal dorsal horn neurons to peripheral afferent stimulation. Neuroscience 138: 1351–1360, 2006 [DOI] [PubMed] [Google Scholar]
- 47. White RE, Jakeman LB. Don't fence me in: harnessing the beneficial roles of astrocytes for spinal cord repair. Restor Neurol Neurosci 26: 197–214, 2008 [PMC free article] [PubMed] [Google Scholar]
- 48. Woolf CJ, Salter MW. Neuronal plasticity: increasing the gain in pain. Science 288: 1765–1769, 2000 [DOI] [PubMed] [Google Scholar]
- 49. Xin WJ, Weng HR, Dougherty PM. Plasticity in expression of the glutamate transporters GLT-1 and GLAST in spinal dorsal horn glial cells following partial sciatic nerve ligation. Mol Pain 5: 15, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zou J, Wang YX, Dou FF, Lu HZ, Ma ZW, Lu PH, Xu XM. Glutamine synthetase down-regulation reduces astrocyte protection against glutamate excitotoxicity to neurons. Neurochem Int 56: 577–584, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]