

Abstract
Purpose
There is substantial germline genetic variability within angiogenesis pathway genes, thereby causing inter-individual differences in angiogenic capacity and resistance to anti-angiogenesis therapy. We investigated germline polymorphisms in genes involved in VEGF-dependent and –independent angiogenesis pathways to predict clinical outcome and tumor response in metastatic colorectal cancer patients (mCRC) treated with bevacizumab (BV) and oxaliplatin-based chemotherapy.
Experimental Design
A total of 132 patients treated with first-line BV and FOLFOX or XELOX were included in this study. Genomic DNA was isolated from whole blood samples by PCR-RFLP or direct DNA-sequencing. The endpoints of the study were progression-free survival (PFS), overall survival (OS) and response rate (RR).
Results
The minor alleles of EGF rs444903 A>G and IGF-1 rs6220 A>G were associated with increased OS and remained significant in multivariate COX regression analysis (HR 0.52; 95%CI 0.31–0.87; adjusted-P=0.012 and HR 0.60; 95%CI 0.36–0.99; adjusted-P=0.046, respectively). The minor allele of HIF1α rs11549465 C>T was significantly associated with increased PFS, but lost its significance in multivariate analysis. CXCR1 rs2234671 G>C, CXCR2 rs2230054 T>C, EGFR rs2227983 G>A and VEGFR-2 rs2305948 C>T predicted tumor response, with CXCR1 rs2234671 G>C remaining significant in multiple testing (Pact=0.003).
Conclusion
In this study we identified common germline variants in VEGF-dependent and – independent angiogenesis genes predicting clinical outcome and tumor response in patients with mCRC receiving first-line BV and oxaliplatin-based chemotherapy.
Keywords: bevacizumab, oxaliplatin, colorectal cancer, angiogenesis, polymorphisms
Introduction
Angiogenesis is a universal requirement for the growth of solid tumors beyond the limits of oxygen diffusion from the existing vasculature.(1, 2) Inhibition of angiogenesis has proven to be beneficial in multiple types of malignancies, including colon cancer.(3) VEGF-A is one of the major regulators of both physiological and pathological angiogenesis. Rapid proliferation of tumor cells and poor blood flow suggest a hypoxia-conducive environment in different areas of tumors. Under hypoxic conditions, hypoxia inducible factor (HIF) binds to the hypoxia response element present in the VEGF-A gene, thus inducing the transcription of VEGF-A protein. Circulating VEGF-A binds to VEGF-receptor (VEGFR)-1 and VEGFR-2 stimulating the recruitment and proliferation of endothelial cells.(4–6) Bevacizumab (BV) is a humanized monoclonal antibody (mAB) directed against vascular endothelial growth factor (VEGF)-A.(7)
Treatment strategies incorporating BV have demonstrated efficacy in metastatic colorectal cancer (mCRC).(8, 9) The majority of previously untreated mCRC patients are treated with BV in combination with oxaliplatin and infusional 5-fluorouracil/leucovorin or capecitabine (FOLFOX or XELOX).(9) The addition of BV to first-line oxaliplatin-based chemotherapy in mCRC was shown to significantly prolong the median progression-free survival (PFS; from 8.0 to 9.4 months, corresponding to a hazard ratio (HR) of 0.83; P=0.0023). Median overall survival (OS) was 21.3 months in the BV group and 19.9 months in the placebo group (HR 0.89; P=0.077). The response rate (RR) was similar in both arms (38% vs 38%; odds ratio (OR) 1.00; P=0.99).(10)
The identification of biomarkers that may influence the efficacy of BV in combination with oxaliplatin-based chemotherapy is of considerable interest.(3, 7) Despite the effects of BV in unselected mCRC patients, the ability to target therapy towards well selected subgroups of patients would increase the likelihood of benefit and would improve cost-effectiveness and therapeutic outcomes. Current evidence indicates potential predictive value for germline polymorphisms affecting genes of the VEGF-pathway in patients receiving BV.(11–14) This is not surprising because angiogenesis depends largely on the response of the host. There is substantial inherited genetic variability within angiogenesis pathway genes, thereby causing inter-individual differences in angiogenic capacity and resistance to BV. In a pioneering study, Schneider et al. investigated five VEGF and two VEGFR-2 polymorphisms in a retrospective subset analyses of the E2100 trial cohort (paclitaxel±BV in metastatic breast cancer) and found two VEGF genotypes (VEGF 2578 A/A and VEGF 1154 A/A) predicting a superior OS for patients in the combination, but not in the control arm, thus indicating a predictive marker.(14)
Recent studies in several experimental models suggest that alternative angiogenic factors are potentially involved in resistance to anti-VEGF treatment.(15–17) Sustained tumor angiogenesis could occur through VEGF-independent mechanisms, thus indicating that these angiogenic factors may serve as predictors of BV efficacy. We recently reported a functional germline polymorphism in interleukin (IL)-8 (251 T/A, A-allele associated with increased IL-8 protein levels), a potent VEGF-independent pro-angiogenic factor, significantly associated with lower RR in a phase II trial in patients with ovarian cancer treated with BV and cyclophosphamide.(12)
In the present study, we investigated germline polymorphisms in a comprehensive panel of angiogenesis genes to predict clinical outcome and tumor response in mCRC patients treated with first-line BV and oxaliplatin-based chemotherapy. We analyzed VEGF-dependent genes such as VEGF-A, VEGFR-2, HIF1α, aryl hydrocarbon receptor nuclear translocator (ARNT) and neuropilin-1 (NRP1) and VEGF-independent angiogenesis genes such as IL-1β, IL-6, IL-8, interleukin receptor-1/2 (CXCR1 and CXCR2), leptin, tissue factor (TF), endostatin (ES), fibroblast growth factor receptor (FGFR)-4, insulin like growth factor (IGF)-1/2, insulin like growth factor receptor (IGFR1), nuclear factor-κB (NF-κB), epidermal growth factor (EGF), epidermal growth factor receptor (EGFR), cyclooxygenase (COX)-2, tumor necrosis factor (TNF)-α and β, inter-cellular adhesion molecule (ICAM)-1 and matrix metalloproteinases (MMP)-2 and 7.
Patients and methods
Eligible patients
A total of 132 patients with histopathologically confirmed mCRC and first-line treatment with FOLFOX or XELOX and BV were included in this retrospective study. These patients received first-line treatment with FOLFOX or XELOX and BV (5mg/kg day 1 of a 2-week cycle when given with FOLFOX, 7.5mg/kg on day 1 of a 3-week cycle for XELOX) between April 2004 and October 2009 at the Norris Comprehensive Cancer Center/University of Southern California (NCCC/USC) or the Los Angeles County/USC Medical Center (LAC/USCMC) and the Division of Clinical Oncology, Medical University of Graz (MUG), Austria. Patients included in the study were required to be ≥18 years old, have present one or more unidimensionally measurable lesion, response data available during at least 2 cycles of BV plus FOLFOX or XELOX, and have not received prior systemic therapy for mCRC or previous treatment with monoclonal antibodies. At the time of treatment initiation, the following criteria were used as contraindication for BV: brain metastases, high-dose NSAIDs, serious non-healing wound, prior pulmonary embolism or recent venous thromboembolic event, any arterial thromboembolic event, and/or baseline ≥ grade 2 proteinuria. Patient data were collected retrospectively through chart review by a medical oncologist (HS). For quality control purposes all clinical data were independently reviewed by a second medical oncologist (AE). Whole blood samples were collected at the time of diagnosis and stored at −80 degree Celsius. Blood samples from 119 patients were available for the current genetic analyses. This retrospective study was approved by the Institutional Review Boards of USC and MUG. All patients signed an informed consent for the analysis of molecular correlates. Baseline clinical examinations and staging CT-scans were performed within 4 weeks of starting treatment and repeated every 8 weeks until progression. The Response Evaluation Criteria in Solid Tumors (RECIST) were used to assess response.
Candidate polymorphisms
Genes and polymorphisms known to modulate VEGF-dependent and –independent angiogenesis have been selected based on public literature resources and databases. Stringent and pre-defined criteria were used and included: (a) credible scientific basis to support a gene’s involvement in angiogenesis signaling pathways; (b) polymorphism that could alter the function of the gene in a biologically relevant manner (either published data or predicted function using Functional-Single-Nucleotide-Polymorphism (F-SNP) database)(18, 19); (c) minor allele frequency ≥10% in Caucasians (for relative allelic frequencies of the polymorphisms in different ethnicities, we refer to the population genetics section in the Ensembl Genome Browser: http://uswest.ensembl.org/index.html). As it was not possible to select all angiogenesis signaling related genes and polymorphisms matching these criteria, this study focused on the most promising (Table 1 and 2).
Table 1.
Analyzed VEGF-dependent angiogenesis gene polymorphisms
| Gene | rs-number | Base exchange | Function | Genotyping |
|---|---|---|---|---|
| VEGF-A | rs2010963 | C>T | C lower VEGF plasma level | DS |
| rs3025039 | C>T | T lower VEGF plasma level | RE (NiaIII) | |
| rs1570360 | G>A | A lower VEGF plasma level | DS | |
| rs833061 | T>C | T lower BV associated hypertension | DS | |
| rs699947 | C>A | C higher VEGF serum level | RE (BgIII) | |
| VEGFR-2 | no rs-number | 11–14 CA repeat | 11/11 higher gene expression | γATP-labeled PCR |
| rs2305948 | C>T | T higher MVD | DS | |
| rs2071559 | T>C | C higher MVD | RE (BsmI) | |
| HIF1α | rs1154946 | C>T | T higher HIF1α protein expression | RE (Tsp451) |
| ARNT | rs2228099 | G>C | NA | DS |
| NRP1 | rs3750733 | C>T | NA | DS |
Abbreviations: DS, direct DNA sequencing; RE, restriction enzyme; BC, breast cancer; MVD, microvessel density; NA, not analyzed; OS, overall survival; VEGF, vascular endothelial growth factor; VEGFR, VEGF-receptor; HIF, hypoxia inducible factor; ARNT, aryl hydrocarbon receptor nuclear translocator; NRP, neuropilin
Table 2.
Analyzed VEGF-independent angiogenesis gene polymorphisms
| Gene | rs-number | Base exchange | Function | Genotyping |
|---|---|---|---|---|
| IL-1β | rs16944 | C>T | T higher IL-1β plasma level | RE (AvaI) |
| rs1143634 | C>T | T higher IL-1β plasma level | RE (TaqI) | |
| IL-6 | rs1800795 | G>C | C lower IL-6 plasma level | RE (NiaIII) |
| IL-8 | rs4073 | T>A | A increased IL-8 plasma level | RE (MfeI) |
| CXCR1 | rs2234671 | G>C | NA | DS |
| CXCR2 | rs2230054 | T>C | NA | DS |
| Leptin | rs7799039 | G>A | A higher Leptin serum level | RE (HhaI) |
| TF | rs1361600 | A>G | G higher TF gene expression | RE (BstNI) |
| ES | rs12483377 | G>A | A lower ES function | RE (MseI) |
| FGFR-4 | rs351855 | G>A | A higher FGFR4 gene expression | RE (BstNI) |
| IGF-1 | rs6214 | C>T | NA | NiaIII |
| rs6220 | A>G | G higher IGF plasma level | MnlI | |
| IGF-2 | rs10840452 | G>A | NA | DS |
| IGFR1 | rs2229765 | G>A | A associated with lower IGF-1 plasma levels | RE (MnlI) |
| NF-κB | no rs-number | 18–26 CA repeat | NA | γATP-labeled PCR |
| Cox-2 | rs5275 | T>C | C allele associated with lower promoter activity | DS |
| TNF-α | rs361525 | G>A | A higher TNF-α plasma level | DS |
| TNF-β | rs909253 | A>G | NA | RE (HinfI) |
| ICAM-1 | rs5498 | T>C | A lower ICAM-1 plasma level | RE (BstUI) |
| MMP-2 | rs243865 | C>T | T lower promoter activity | DS |
| MMP-7 | rs1156881 | A>G | G higher promoter activity | DS |
| EGF | rs4444903 | A>G | G higher EGF serum levels | RE (AluI) |
| EGFR | rs2227983 | G>A | A attenuated EGFR ligand binding | RE (BSTNI) |
Abbreviations: BC, breast cancer; CRC, colorectal cancer; TTR, time to tumor recurrence; IL, interleukin; CXCR, interleukin receptor; TF, tissue factor; ES, endostatin; FGFR, fibroblast growth factor receptor; IGF, insulin like growth factor; IGFR, insulin like growth factor receptor; NF-κB, nuclear factor-κB; EGF, epidermal growth factor; EGFR, epidermal growth factor receptor; COX, cyclooxygenase; TNF, tumor necrosis factor; ICAM, inter-cellular adhesion molecule; MMP, matrix metalloproteinases
Genotyping
Genomic DNA was extracted from peripheral blood using the QIAmp-kit (Qiagen). The majority of the samples were tested using PCR-based restriction fragment length polymorphism (PCR-RFLP) analysis. Briefly, forward and reverse primers were used for PCR amplification, PCR products were digested by restriction enzymes (New England Biolab), and alleles were separated on 4% NuSieve ethidium bromide stained agarose gel. If no matching restriction enzyme could be found, samples were analyzed by direct DNA-sequencing. The dinucleotide polymorphisms were determined by a 5′-end 33p γATP–labeled PCR protocol with a few modifications. In brief, DNA template, deoxynucleotide triphosphates, 5′-end 33p γATP-labeled primer, unlabeled complementary primer, Taq Polymerase (Perkin-Elmer), and PCR Buffer were used in PCR. The reaction products were separated on a 6% denaturing polyacrylamid DNA-sequencing gel, which was vacuum blotted for 1 hour at 80 degree Celsius and exposed to XAR-film (Eastman-Kodak) overnight. For quality control purposes, a total of 5% PCR-RFLP analyzed samples were re-analyzed by direct DNA-sequencing. Patients' characteristics and clinical outcome were unknown to the investigator performing the genetic analyses (AG).
Statistical analysis
The endpoints of the study were PFS, OS and RR. The PFS was calculated from the time of the first day of treatment until the first observation of disease progression or death from any cause. OS was defined as the time from the first day of treatment to death from any cause. If a patient was alive and had not progressed, PFS and OS were censored at the time of last follow-up. RR was categorized as two groups: complete or partial response and stable or progressive disease. With 119 patients, there was an 80% power to detect a minimum HR of 1.7 and 1.8, and 34% tumor response differences across the range of minor allele frequencies (0.1–0.5) using a dominant model for PFS, OS and RR, respectively. The NF-κB repeat polymorphism was analyzed by categorizing 3 groups: (a) carrying both alleles <24 repeats; (b) carrying one allele <24; and (c) carrying both alleles ≥24 repeats. For the VEGFR-2 repeat polymorphisms, we combined the 14 with the 13 CA repeats and categorized in 11/11, 11/13 and 13/13 CA repeats. Allelic distribution of the polymorphisms by ethnicity for deviation from Hardy-Weinberg equilibrium and the allelic frequencies of each polymorphism between different ethnic groups were tested using χ2-test. The associations between polymorphisms and baseline demographic and clinical characteristics and RR were examined using contingency tables and the Fisher’s exact test. The associations of polymorphisms with PFS and OS were analyzed using Kaplan-Meier curves and log-rank test. The true mode of inheritance of all polymorphisms tested is not established yet and we assumed a codominant, additive, dominant or recessive genetic model where appropriate. In the multivariate Cox regression analysis for PFS and OS and the logistic regression analysis for RR, the models were adjusted for sex, age, primary tumor site, histologic differentiation, metastatic site, number of metastatic sites, chemotherapy backbone (FOLFOX or XELOX) and study site (Hospital) stratified by ethnicity. P-values for all polymorphisms were adjusted for multiple testing using a modified test of Conneely and Boehnke (Pact) that was applied for the correlated tests due to linkage disequilibrium and different modes of inheritance considered.(20) Recursive partitioning (RP), including cross-validation, was used to explore and identify polymorphism interactions associated with RR, PFS and OS using the rPart-function in S-plus. Case-wise deletion for missing polymorphisms was used in univariate and multivariate analyses. In the RP-analysis, all patients with at least one polymorphism result available were included. All analyses were performed using the SAS statistical package version 9.2 (SAS-Institute Inc., USA).
Results
Baseline patient characteristics and the association with clinical outcome and RR are summarized in Table 3. There were no statistically significant differences in age and sex between the patients from USC and MUG, however, more MUG patients had rectosigmoid tumors (28% vs 6%) and received FOLFOX (80% vs 43%), compared to USC patients. The median age at time of diagnosis was 56 years (range 28–81). The median follow-up time was 3.5 years (range 0.3–6.5) and the median PFS and OS was 11.2 months (95%CI 8.4–13.7) and 27.9 months (95%CI 22.4–32.4), respectively. Sixty-four patients (48.5%) showed complete or partial tumor response, 63 patients (47.7%) had stable or progressive disease, and for 5 patients (3.8%) response data were not available. The genotyping quality control by direct DNA-sequencing provided a genotype concordance of ≥99%. Genotyping was successful in at least 90% of cases in each polymorphism analyzed, except the NF-κB repeat polymorphism with 84%. The allelic frequencies for all polymorphisms were within the probability limits of Hardy-Weinberg equilibrium. VEGF-A rs2010963, rs699947, rs1570360 and rs833061 were in linkage disequilibrium (D’ ranged from 0.79 to 1.0 and r2 ranged from 0.13 to 0.81). VEGF-A rs3025039 was not in linkage disequilibrium with the other four VEGF-A gene variants (D’ <0.16 and r2<0.01). The VEGFR-2 polymorphisms were not in strong linkage disequilibrium (D’=0.47 and r2=0.04). Genotype frequencies for all polymorphisms in each ethnicity are summarized in Supplementary Table 1.
Table 3.
Baseline patient characteristics and the association with clinical outcome and RR (Ethnicity was self-defined by the patients)
| RR | PFS | OS | |||||
|---|---|---|---|---|---|---|---|
| n | % | CR/PR | SD/PD | HR (95%CI) | HR (95%CI) | ||
| Sex | |||||||
| Female | 57 | 43 | 23(41%) | 33(59%) | 1 (Reference) | 1 (Reference) | |
| Male | 75 | 57 | 41 (58%) | 30 (42%) | 0.64 (0.44, 0.93) | 0.78 (0.50, 1.22) | |
| P value | 0.075 | 0.016 | 0.27 | ||||
| Age, years | |||||||
| <55 | 53 | 40 | 23 (44%) | 29 (56%) | 1 (Reference) | 1 (Reference) | |
| 55–64 | 48 | 36 | 24 (55%) | 20 (45%) | 0.73 (0.47, 1.12) | 0.80 (0.48, 1.33) | |
| 65+ | 31 | 23 | 17 (55%) | 14 (45%) | 0.90 (0.56, 1.46) | 0.96 (0.55, 1.68) | |
| P value | 0.51 | 0.33 | 0.66 | ||||
| Ethnicity | |||||||
| African American | 5 | 4 | 1 (20%) | 4 (80%) | 1.41 (0.56, 3.55) | 2.31 (0.80, 6.65) | |
| Asian | 27 | 20 | 14 (52%) | 13 (48%) | 1.04 (0.65, 1.68) | 1.31 (0.76, 2.27) | |
| Caucasian | 65 | 49 | 29 (48%) | 32 (52%) | 1 (Reference) | 1 (Reference) | |
| Hispanic | 35 | 27 | 20 (59%) | 14 (41%) | 0.83 (0.52, 1.30) | 0.70 (0.39, 1.25) | |
| P value | 0.41 | 0.65 | 0.086 | ||||
| Karnofsky performance status% | |||||||
| 100 | 17 | 13 | 7 (41%) | 10 (59%) | 1 (Reference) | 1 (Reference) | |
| 90 | 43 | 33 | 27 (64%) | 15 (36%) | 1.60 (0.84, 3.06) | 1.33 (0.64, 2.80) | |
| 80 | 22 | 17 | 10 (45%) | 12 (55%) | 1.98 (0.97, 4.02) | 2.38 (1.03, 5.53) | |
| Missing | 50 | 37 | |||||
| P value | 0.17 | 0.13 | 0.066 | ||||
| Primary tumor site | |||||||
| Colon | 87 | 66 | 45 (54%) | 38 (46%) | 1 (Reference) | 1 (Reference) | |
| Rectosigmoid | 13 | 10 | 3 (25%) | 9 (75%) | 1.28 (0.71, 2.31) | 0.96 (0.43, 2.11) | |
| Rectum | 32 | 24 | 16 (50%) | 16 (50%) | 1.37 (0.88, 2.11) | 1.21 (0.73, 2.01) | |
| P value | 0.18 | 0.32 | 0.74 | ||||
| Histologic differentiation | |||||||
| Well | 2 | 1 | 2 (100%) | 0 (0%) | |||
| Moderate | 94 | 72 | 47 (52%) | 44 (48%) | 1 (Reference)* | 1 (Reference)* | |
| Poor | 31 | 23 | 14 (48%) | 15 (52%) | 1.07 (0.68, 1.68) | 1.63 (0.98, 2.70) | |
| Missing | 5 | 4 | |||||
| P value | 0.60 | 0.76 | 0.056 | ||||
| Tumor stage at diagnosis Stage I | |||||||
| Stage I | 2 | 1 | 1 (50%) | 1 (50%) | |||
| Stage II | 6 | 5 | 3 (50%) | 3 (50%) | |||
| Stage III | 13 | 10 | 6 (46%) | 7 (54%) | 1 (Reference)* | 1 (Reference)* | |
| Stage IV | 110 | 83 | 53 (50%) | 52 (50%) | 1.39 (0.82, 2.36) | 2.11 (1.08, 4.10) | |
| Missing | 1 | 1 | |||||
| P value | 1.00 | 0.22 | 0.022 | ||||
| Metastatic site, n | |||||||
| 1 site | 76 | 58 | 44 (58%) | 31 (43%) | 1 (Reference) | 1 (Reference) | |
| 2+ sites | 56 | 42 | 20 (38%) | 32 (62%) | 1.38 (0.95, 2.01) | 1.72 (1.10, 2.70) | |
| P value | 0.031 | 0.085 | 0.013 | ||||
| Metastatic site, localization | |||||||
| Liver | 98 | 74 | 55 (57%) | 42 (43%) | 0.76 (0.50, 1.17) | 0.94 (0.56, 1.58) | |
| 0.012 | 0.21 | 0.82 | |||||
| Lung | 31 | 23 | 8 (26%) | 23 (74%) | 1.42 (0.92, 2.17) | 1.57 (0.98, 2.53) | |
| 0.002 | 0.10 | 0.059 | |||||
| Peritoneum | 23 | 17 | 7 (35%) | 13 (65%) | 1.18 (0.73, 1.90) | 1.43 (0.81, 2.51) | |
| 0.15 | 0.50 | 0.21 | |||||
| Other | 32 | 24 | 10 (34%) | 19 (66%) | 0.95 (0.62, 1.47) | 1.17 (0.70, 1.96) | |
| P value | 0.059 | 0.83 | 0.55 | ||||
| Chemotherapy backbone | |||||||
| FOLFOX-4 | 66 | 50 | 23 (37%) | 39 (63%) | 1 (Reference) | 1 (Reference) | |
| XELOX | 66 | 50 | 41 (63%) | 24 (37%) | 0.98 (0.68, 1.42) | 1.07 (0.69, 1.67) | |
| P value | 0.005 | 0.91 | 0.75 | ||||
| Hospital | |||||||
| USC | 107 | 81 | 57 (55%) | 46 (45%) | 1 (Reference) | 1 (Reference) | |
| MUG | 25 | 19 | 7 (29%) | 17 (71%) | 1.75 (1.09, 2.81) | 0.84 (0.44, 1.58) | |
| P value | 0.025 | 0.016 | 0.59 | ||||
Combined with the above categories for stable estimates.
Abbreviations: USC, University of Southern California; MUG: Medical University of Graz; n, number; CR, complete response; PR, partial response; SD, stable disease; PD, progressive disease; RR, response rate; PFS, progression-free survival; OS, overall survival; HR, hazard ratio; CI, confidence interval.
Gene variants and progression-free survival
In the univariate analysis, the minor allele of HIF1α rs11549465 C>T was significantly associated with increased PFS. Patients carrying at least one T allele showed a median PFS of 11.7 months. In contrast, patients homozygous C/C had a median PFS of 10.2 months (HR 0.55; 95%CI 0.31–0.99; P=0.038). None of the other tested polymorphisms were associated with PFS. In multivariate analysis stratified by ethnicity and multiple testing including all polymorphisms, HIF1α rs11549465 C>T did not remain significantly associated with PFS (HR 0.65; 95%CI 0.34–1.22; adjusted-P=0.18; Pact=0.362; Table 4).
Table 4.
Univariate analysis of polymorphisms for PFS, OS and RR. PFS and OS were calculated for dominant models (combining heterozygous and homozygous minor variants)
| n | RR | PFS | OS | ||||
|---|---|---|---|---|---|---|---|
| CR or PR | SD or PD | Median months (95%CI) |
HR (95%CI) | Median months (95%CI) |
HR (95%CI) | ||
| CXCR1 rs2234671 | |||||||
| G/G | 51 | 35 (71%) | 14 (29%) | 13.9 (11.0, 16.0) | 1 (Reference) | 28.6 (21.5, 36.9) | 1 (Reference) |
| G/C | 62 | 22 (37%) | 38 (63%) | 8.1 (7.3, 10.4) | 1.38 (0.93, 2.05) | 27.9 (21.6, 41.9) | 0.95 (0.60, 1.52) |
| C/C | 6 | 1 (17%) | 5 (83%) | ||||
| P | <0.001 | 0.10 | 0.83 | ||||
| CXCR2 rs2230054 | |||||||
| T/T | 49 | 18 (38%) | 29 (62%) | 8.1 (7.1, 10.4) | 1 (Reference) | 24.2 (17.2, 34.4) | 1 (Reference) |
| T/C | 53 | 29 (56%) | 23 (44%) | 12.4 (10.1, 15.0) | 0.89 (0.60, 1.33) | 28.6 (22.0, 42.7) | 0.88 (0.54, 1.42) |
| C/C | 15 | 11 (79%) | 3 (21%) | ||||
| P | 0.008 | 0.57 | 0.59 | ||||
| EGFR rs222798 | |||||||
| G/G | 64 | 27 (43%) | 36 (57%) | 8.3 (7.1, 11.2) | 1 (Reference) | 23.0 (19.3, 31.6) | 1 (Reference) |
| G/A | 43 | 22 (55%) | 18 (45%) | 12.4 (8.4, 17.0) | 0.74 (0.50, 1.10) | 32.4 (22.0, 43.3) | 0.74 (0.46, 1.19) |
| A/A | 11 | 9 (82%) | 2 (18%) | ||||
| P | 0.024 | 0.13 | 0.21 | ||||
| VEGFR-2 rs2305948 | |||||||
| C/C | 92 | 50 (57%) | 38 (43%) | 10.1 (7.9, 12.4) | 1 (Reference) | 31.0 (22.9, 42.7) | 1 (Reference) |
| C/T | 24 | 7 (29%) | 17 (71%) | 10.8 (7.6, 16.0) | 1.18 (0.74, 1.87) | 22.0 (17.1, 32.4) | 1.36 (0.79, 2.32) |
| T/T | 3 | 1 (33%) | 2 (67%) | ||||
| P | 0.024 | 0.48 | 0.26 | ||||
| HIF1α rs11549465 | |||||||
| C/C | 99 | 49 (52%) | 46 (48%) | 10.2 (7.9, 12.4) | 1 (Reference) | 27.9 (22.0, 34.4) | 1 (Reference) |
| C/T | 18 | 8 (44%) | 10 (56%) | 11.7 (6.3, 36.0+) | 0.55 (0.31, 0.99) | 32.4 (17.1, 60.0+) | 0.81 (0.44, 1.52) |
| T/T | 2 | 1 (50%) | 1 (50%) | ||||
| P | 0.68 | 0.038 | 0.51 | ||||
| EGF rs444903 | |||||||
| A/A | 34 | 14 (44%) | 18 (56%) | 7.1 (5.7, 13.2) | 1 (Reference) | 21.9 (13.5, 30.9) | 1 (Reference) |
| A/G | 55 | 32 (59%) | 22 (41%) | 11.1 (8.3, 13.7) | 0.72 (0.47, 1.10) | 32.4 (22.9, 43.3) | 0.54 (0.33, 0.88) |
| G/G | 30 | 12 (41%) | 17 (59%) | ||||
| P | 1.00 | 0.13 | 0.011 | ||||
| IGF-1 rs6220 | |||||||
| A/A | 52 | 24 (46%) | 28 (54%) | 8.4 (7.4, 13.2) | 1 (Reference) | 22.1 (16.4, 28.6) | 1 (Reference) |
| A/G | 51 | 29 (60%) | 19 (40%) | 11.6 (8.1, 14.2) | 0.92 (0.62, 1.37) | 32.4 (27.9, 47.2) | 0.51 (0.32, 0.83) |
| G/G | 12 | 4 (33%) | 8 (67%) | ||||
| P | 1.00 | 0.68 | 0.005 | ||||
Abbreviations: RR, response rate; PFS, progression-free survival; OS, overall survival; CXCR, interleukin receptor; EGF, epidermal growth factor; EGFR, epidermal growth factor receptor; VEGF, vascular endothelial growth factor; VEGFR, VEGF-receptor; IGF, insulin like growth factor; CR, complete response; PR, partial response; CI, confidence interval; HR, hazard ratio
Gene variants and overall survival
In the univariate analysis, the minor alleles of EGF rs444903 A>G and IGF-1 rs6220 A>G were significantly associated with increased OS. Patients carrying at least one G allele in EGF rs444903 A>G showed a median OS of 32.4 months. In contrast, patients homozygous A/A had a median OS of 21.9 months (HR 0.54; 95%CI 0.33–0.88; P=0.011). Patients harboring the minor allele of IGF-1 rs6220 A>G showed a median OS of 32.4 months compared to 22.1 months harboring homozygous A/A (HR 0.51; 95%CI 0.32–0.83; P=0.005; Table 4; Figure 1). None of the other tested polymorphisms demonstrated a statistically significant association with OS. In the multivariate analysis stratified by ethnicity, the minor alleles of EGF rs444903 A>G and IGF-1 rs6220 A>G remained significantly associated with increased OS (HR 0.52; 95%CI 0.31–0.87; adjusted-P=0.012 and HR 0.60; 95%CI 0.36–0.99; adjusted-P=0.046, respectively). In multiple testing, EGF rs444903 A>G and IGF-1 rs6220 A>G did not remain significantly associated with OS (Pact=0.664 and 0.780, respectively)
Figure 1.
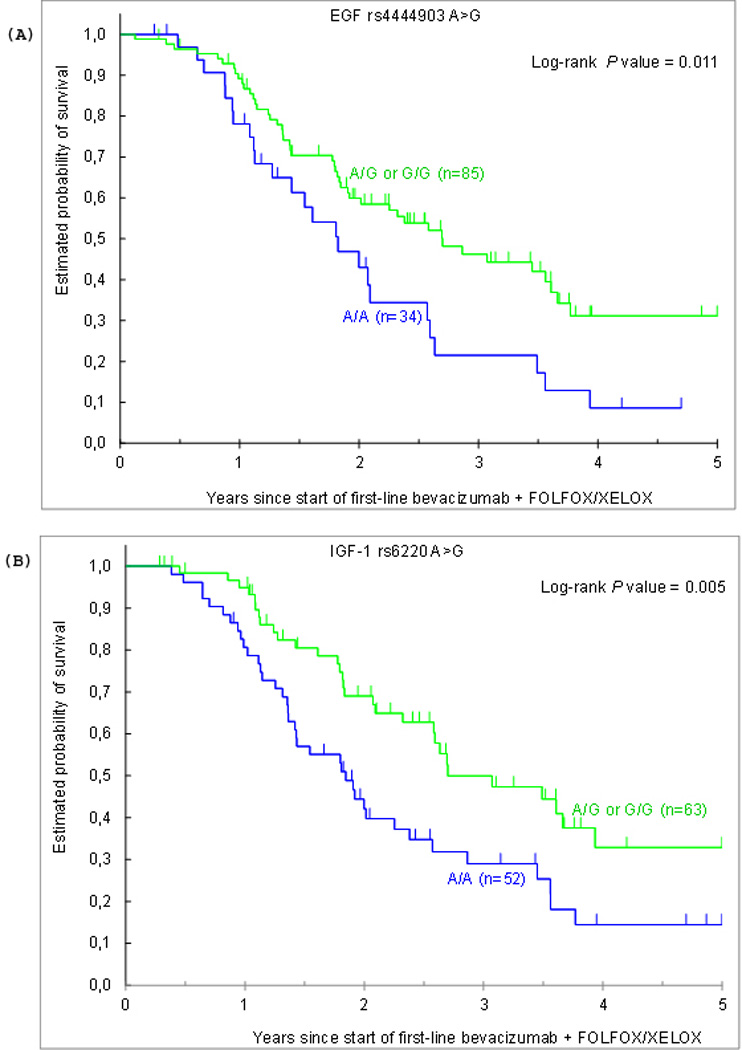
Overall survival by (A) EGF rs4444903 A>G and (B) IGF-1 rs6220 A>G
Gene variants and tumor response
CXCR1 rs2234671 G>C, CXCR2 rs2230054 T>C, EGFR rs2227983 G>A and VEGFR-2 rs2305948 C>T were significantly associated with RR. Patients homozygous for the wild-type allele of CXCR1 rs2234671 G>C and VEGFR-2 rs2305948 C>T were more likely to show higher tumor response (71% and 57%, respectively), compared to patients carrying one (37% and 29%, respectively) or two minor alleles (17% and 33%; P<0.001 and P=0.024, respectively). In CXCR2 rs2230054 T>C and EGFR rs2227983 G>A, patients harboring the wild-type genotype had a significantly lower RR (38% and 43%, respectively), compared to patients heterozygous (56% and 55%, respectively) or homozygous for the minor allele (79% and 82%; P=0.008 and P=0.024, respectively; Table 4). None of the other analyzed polymorphisms predicted RR. In logistic regression analysis, CXCR1 rs2234671 G>C remained significantly associated with RR (P<0.001). In multiple testing including all polymorphisms, CXCR1 rs2234671 G>C remained statistically significant for RR (Pact=0.003).
Gene variant interactions by recursive partitioning
When RP was utilized to construct decision-trees as predictive models for PFS, OS and RR to classify patients based on the gene variants, high- and low-risk patient subgroups were identified. In the resultant tree for PFS, the most important factor that determined PFS in our study cohort was HIF1α rs11549465 C>T. Patients carrying the combination of HIF1α rs11549465 C>T wild-type and the minor allele of VEGF rs699947 C>A and EGFR rs2227983 G>A wild-type demonstrated a PFS of 7.8 months compared to 11.7 months in patients harboring at least one minor allele of HIF1α rs11549465 C>T (HR 2.66; 95%CI 1.30–5.42; P<0.001). The resultant tree for OS showed that patients heterozygous or homozygous for the minor alleles of IGF-1 rs6220 A>G and IL6 rs1800795 G>C demonstrated an OS of 60 months compared to 21.7 months in patients harboring the IGF-1 rs6220 A>G wild-type (HR 2.67; 95%CI 1.25–5.66; P<0.001). For RR, CXCR1 rs2234671 G>C was the main split criteria in the decision tree, but no other gene variants were shown to improve the prediction success.
Clinical outcome and tumor response by ethnicity
When the allelic frequencies of each polymorphism, which was significantly associated with clinical outcome or tumor response were tested between ethnic groups, we found a significant difference for CXCR2 rs2230054 T>C, VEGFR-2 rs2305948 and EGF rs444903 A>G (P=0.017, P=0.018 and P=0.005, respectively). The minor allele of EGF rs444903 A>G remained significantly associated with increased OS (HR 0.47; 95%CI 0.24–0.93; P=0.021) in Caucasians, but not in Asians and Hispanics (HR 0.65; 95%CI 0.21–1.98; P=0.42 and HR 0.50; 95%CI 0.17–1.50; P=0.21, respectively). African Americans were excluded from sub-analyses because of the small number in our study cohort (n=5). CXCR2 rs2230054 T>C remained significantly associated with RR in Caucasians (wild-type: 35%, heterozygous for the minor allele: 60%, homozygous for the minor allele: 100%; P=0.035), but not in Asians and Hispanics (P=0.351 and P=0.191, respectively). Patients homozygous for the wild-type allele of VEGFR-2 rs2305948 C>T were more likely to show higher tumor response compared to patients carrying one or two minor alleles, but this effect did not remain significant in sub-analyses for ethnicity (Caucasian: P=0.17, Asian: P=0.055, Hispanic: P=0.86).
Discussion
Even though thousands of patients have been enrolled in randomized clinical trials of BV, only few insights are available about specific subgroups of patients who may actually benefit.(21) At the same time, no biomarkers are currently available to quantify the contribution of BV to the activity of cytotoxic drugs. Since the efficacy of BV may vary between different chemotherapy backbones, the current priority in translational research is focused on biomarkers to the response or resistance to combined therapies. In this investigation, we focused on host-related VEGF-dependent and –independent angiogenic biomarkers to predict survival and tumor response of mCRC patients treated with BV and oxaliplatin-based chemotherapy. This study has been performed to draw biological observations to be validated in biomarker-embedded trials.
In mCRC, Loupakis et al. found no association between VEGF and VEGFR-2 polymorphisms and clinical outcome in 57 patients enrolled in a phase II trial of FOLFOXIRI plus BV as first-line treatment.(22) One report by Formica et al. suggested that VEGF rs1570360 and VEGF rs2010963 genotypes predict PFS and RR in patients receiving BV and FOLFIRI.(13) A genetic interaction profile by Pander et al. including VEGF rs2010963 was associated with PFS in mCRC patients treated with XELOX plus BV.(23) While this study did not confirm these findings, we identified two other VEGF-dependent gene polymorphisms associated with PFS and RR in mCRC patients treated with BV and oxaliplatin-based chemotherapy. The minor allele of HIF1α rs11549465 C>T, which has been recently associated with higher HIF1α protein expression, predicted an increased PFS in the univariate analysis.(24) The VEGF-A promoter contains a hypoxic response element that can bind HIF1α, and initiate transcriptional activation of the VEGF-A gene.(25) Although the association found in our study is biologically plausible and in concordance with several studies showing that patients with VEGF-A activation more likely benefit from BV treatment, HIF1α rs11549465 C>T did not remain significant in the multivariate analysis.(26, 27) In VEGFR-2 rs2305948 C>T, the minor allele predicted a lower RR in the univariate analysis when analyzed in all study patients. However, this effect did not remain significant in sub-analyses for ethnicity. VEGFR-2 rs2305948 C>T is associated with microvessel density (MVD) in CRC with the C/T and T/T genotypes showing a significantly higher MVD compared to the wild-type.(28) Taking into account the functional effect of this polymorphism and the association found in our study, we suggest that the variant allele in the VEGFR-2 gene may drive angiogenesis independent of VEGF-A ligand binding.
Since alternative angiogenic mechanisms are potentially involved in resistance to BV, we investigated a comprehensive panel of VEGF-independent gene polymorphisms. CXCR1 rs2234671 G>C represented the most promising polymorphism in our study, predicting RR in multivariate analysis and multiple testing, while CXCR2 rs2230054 C>T lost its significance in multivariate analysis and multiple testing. IL-8 exerts its angiogenic properties on endothelial cells through interaction with its cognate receptors CXCR1 and CXCR2.(29) Induction of IL-8 preserved the angiogenic response in HIF1α-deficient colon cancer cells, suggesting that IL-8 dependent angiogenesis is independent of VEGF.(30) The detailed molecular mechanisms involved in how the CXCR1 rs2234671 G>C and CXCR2 rs2230054 C>T polymorphisms exert effects on mCRC and predict RR in BV and FOLFOX or XELOX treated patients are unclear. We used the F-SNP database to predict the functional effects of these gene variants. F-SNP gathers computationally predicted functional information about polymorphisms, particularly aiming to facilitate identification of disease-related polymorphisms in association studies. Specifically, it provides information about potential deleterious effects of polymorphisms with respect to major molecular functions.(18, 19) When used for the CXCR gene variants, F-SNP predicted changes in splicing regulation and post-translation for CXCR1 rs2234671 G>C, and changes in splicing regulation and transcriptional regulation for CXCR2 rs2230054 C>T, thus supporting a biological function and the effects seen in our study.
In a recent study by Cascone et al. using mouse xenograft models of human lung adenocarcinomas, EGFR activation in stromal cells, but not in tumor cells has been shown to be involved in BV resistance, indicating a host-regulated process of VEGF-independent angiogenesis.(31) We found a functional germline polymorphism in EGFR associated with RR in univariate analysis in our study cohort. Patients harboring at least one minor allele of EGFR rs2227983 G>A were more likely to show a higher tumor response, compared to patients homozygous for the wild-type allele. The minor allele was previously found to be associated with attenuated EGFR ligand binding.(32) Our finding is therefore consistent with the functional effect of this polymorphism, as decreased EGFR-signaling may attenuate the VEGF-independent angiogenic capacity.
Another finding of our study was that EGF rs4444903 A>G and IGF-1 rs6220 A>G predict OS in the univariate and multivariate analysis. The minor allele of EGF rs4444903 A>G is transcriptionally more active than the wild-type and has been associated with higher EGF serum levels.(33) In a recent study by Pander et al., EGF rs4444903 A>G was not associated with PFS in mCRC patients treated with XELOX plus BV.(34) EGF-signaling contributes to cell proliferation and angiogenesis; therefore, the favorable effect of the EGF rs4444903 minor allele seen in our study may be counterintuitive. EGF-signaling promotes angiogenesis not only exerting direct effects on endothelial cells, but also by upregulating VEGF mRNA expression.(35) This partly VEGF-dependent mechanism may be responsible for the increased OS survival in BV-treated patients harboring the minor allele. Similar potential mechanisms may also apply for IGF-1 rs6220 A>G, since the minor allele shows higher IGF-1 plasma levels, which is associated with increased VEGF mRNA expression.(36, 37) On the other hand, it has been shown that at specific concentrations that vary between experimental model, EGF induces apoptosis and growth inhibition, rather than cell proliferation.(38–40) According to such findings and the fact that EGF rs4444903 A>G predicts OS but not PFS or RR, this polymorphism could represent a prognostic rather than a predictive biomarker, which is in concordance with several studies showing that the EGF upregulating genotype predicts significantly better survival rates.(41)
We performed multiple testing because of the large number of independent genetic variants evaluated. Application of a modified test of Conneely and Boehnke for correlated tests resulted in a significant Pact-value only for CXCR1 rs2234671 G>C in predicting RR. Nevertheless, the biological plausibility and our translational findings hold promise for future investigations and warrant validation in a larger cohort. Investigating gene variant interactions using RP, we found that combinations of gene variants may improve the prediction success for PFS and OS, but not for RR in our study cohort. Interestingly, VEGF rs699947 C>A and IL6 rs1800795 G>C were included in the decision tree by RP for PFS and OS, respectively. The minor allele of VEGF rs699947 C>A is associated with lower VEGF serum levels and predicted the low PFS subgroup in RP analysis. This association is in concordance with several studies showing that patients with VEGF-A activation more likely benefit from BV treatment.(26, 27) The minor allele of IL6 rs1800795 G>C is associated with lower IL6 plasma levels and may therefore contribute to the high OS subgroup predicted in RP analysis. The fact that allelic frequencies of polymorphisms can differ between ethnic groups and influence the clinical effect as seen in our study points out the importance of ethnicity specific analyses in future studies. Because of the combined treatment in our study and the lack of an appropriate control group, the results are not directly attributable to single-agent administration of BV, but should be referred to the combined treatment of BV with FOLFOX or XELOX.
This study provides evidence that functional germline polymorphisms in VEGF-dependent and –independent angiogenesis genes predict tumor response und survival in mCRC patients treated with BV and FOLFOX or XELOX. Biomarker-embedded translational trials are warranted to validate these findings.
Translational relevance.
Germline variants in VEGF-dependent angiogenesis genes have been linked to bevacizumab resistance in recent studies. Here we investigated germline polymorphisms in genes involved in VEGF-dependent and -independent angiogenesis pathways to predict clinical outcome and tumor response in metastatic colorectal cancer patients treated with bevacizumab and oxaliplatin-based chemotherapy. We hypothesized that these functional gene variants cause inter-individual differences in angiogenic capacity and bevacizumab resistance. Our findings suggest that overall survival and tumor response vary according to EGF, IGF-1 and CXCR1 genotypes, thus representing valuable biomarkers to validate in larger prospective cohorts.
Supplementary Material
Acknowledgments
Grant support: This work was funded by the NIH grant P30CA and the Dhont Foundation; AG is supported in part by a research grant from the Austrian Society of Hematology and Oncology, the Bank Austria Visiting Scientists Program and the “Verein fuer Krebskranke” of the Medical University Graz; TW is supported in part by a research grant of the Austrian Society of Hematology and Oncology and the “Kurt und Senta-Herrmann” foundation; PB is supported in part by a research grant from the “Ligue Suisse contre le cancer”
Footnotes
Conflict of interest: AE, speaker’s bureau and consultant for Roche; H-JL, advisory board and consultant for Roche
References
- 1.Folkman J, Shing Y. Angiogenesis. J Biol Chem. 1992;267:10931–10934. [PubMed] [Google Scholar]
- 2.Carmeliet P. Angiogenesis in health and disease. Nat Med. 2003;9:653–660. doi: 10.1038/nm0603-653. [DOI] [PubMed] [Google Scholar]
- 3.Gerger A, Labonte M, Lenz HJ. Molecular predictors of response to antiangiogenesis therapies. Cancer J. 2011;17:134–141. doi: 10.1097/PPO.0b013e318212db3c. [DOI] [PubMed] [Google Scholar]
- 4.Ferrara N. VEGF-A: a critical regulator of blood vessel growth. Eur Cytokine Netw. 2009;20:158–163. doi: 10.1684/ecn.2009.0170. [DOI] [PubMed] [Google Scholar]
- 5.Ferrara N. Vascular endothelial growth factor. Arterioscler Thromb Vasc Biol. 2009;29:789–791. doi: 10.1161/ATVBAHA.108.179663. [DOI] [PubMed] [Google Scholar]
- 6.Ferrara N. Vascular endothelial growth factor: basic science and clinical progress. Endocr Rev. 2004;25:581–611. doi: 10.1210/er.2003-0027. [DOI] [PubMed] [Google Scholar]
- 7.Jubb AM, Harris AL. Biomarkers to predict the clinical efficacy of bevacizumab in cancer. Lancet Oncol. 2010;11:1172–1183. doi: 10.1016/S1470-2045(10)70232-1. [DOI] [PubMed] [Google Scholar]
- 8.Cunningham D, Atkin W, Lenz HJ, et al. Colorectal cancer. Lancet. 2010;375:1030–1047. doi: 10.1016/S0140-6736(10)60353-4. [DOI] [PubMed] [Google Scholar]
- 9.Welch S, Spithoff K, Rumble RB, Maroun J. Bevacizumab combined with chemotherapy for patients with advanced colorectal cancer: a systematic review. Ann Oncol. 2010;21:1152–1162. doi: 10.1093/annonc/mdp533. [DOI] [PubMed] [Google Scholar]
- 10.Saltz LB, Clarke S, Diaz-Rubio E, et al. Bevacizumab in combination with oxaliplatin-based chemotherapy as first-line therapy in metastatic colorectal cancer: a randomized phase III study. J Clin Oncol. 2008;26:2013–2019. doi: 10.1200/JCO.2007.14.9930. [DOI] [PubMed] [Google Scholar]
- 11.Steffensen KD, Waldstrom M, Brandslund I, Jakobsen A. The relationship of VEGF polymorphisms with serum VEGF levels and progression-free survival in patients with epithelial ovarian cancer. Gynecol Oncol. 2010;117:109–116. doi: 10.1016/j.ygyno.2009.11.011. [DOI] [PubMed] [Google Scholar]
- 12.Schultheis AM, Lurje G, Rhodes KE, et al. Polymorphisms and clinical outcome in recurrent ovarian cancer treated with cyclophosphamide and bevacizumab. Clin Cancer Res. 2008;14:7554–7563. doi: 10.1158/1078-0432.CCR-08-0351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Formica V, Palmirotta R, Del Monte G, et al. Predictive value of VEGF gene polymorphisms for metastatic colorectal cancer patients receiving first-line treatment including fluorouracil, irinotecan, and bevacizumab. Int J Colorectal Dis. 2011;26:143–151. doi: 10.1007/s00384-010-1108-1. [DOI] [PubMed] [Google Scholar]
- 14.Schneider BP, Wang M, Radovich M, et al. Association of vascular endothelial growth factor and vascular endothelial growth factor receptor-2 genetic polymorphisms with outcome in a trial of paclitaxel compared with paclitaxel plus bevacizumab in advanced breast cancer: ECOG 2100. J Clin Oncol. 2008;26:4672–4678. doi: 10.1200/JCO.2008.16.1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ferrara N. Pathways mediating VEGF-independent tumor angiogenesis. Cytokine Growth Factor Rev. 2010;21:21–26. doi: 10.1016/j.cytogfr.2009.11.003. [DOI] [PubMed] [Google Scholar]
- 16.Waugh DJ, Wilson C. The interleukin-8 pathway in cancer. Clin Cancer Res. 2008;14:6735–6741. doi: 10.1158/1078-0432.CCR-07-4843. [DOI] [PubMed] [Google Scholar]
- 17.Cao Y, Zhong W, Sun Y. Improvement of antiangiogenic cancer therapy by understanding the mechanisms of angiogenic factor interplay and drug resistance. Semin Cancer Biol. 2009;19:338–343. doi: 10.1016/j.semcancer.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 18.Lee PH, Shatkay H. F-SNP: computationally predicted functional SNPs for disease association studies. Nucleic Acids Res. 2008;36:D820–D824. doi: 10.1093/nar/gkm904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee PH, Shatkay H. An integrative scoring system for ranking SNPs by their potential deleterious effects. Bioinformatics. 2009;25:1048–1055. doi: 10.1093/bioinformatics/btp103. [DOI] [PubMed] [Google Scholar]
- 20.Conneely KN, Boehnke M. So Many Correlated Tests, So Little Time! Rapid Adjustment of P Values for Multiple Correlated Tests. Am J Hum Genet. 2007;81 doi: 10.1086/522036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hayes DF. Bevacizumab treatment for solid tumors: boon or bust? JAMA. 2011;305:506–508. doi: 10.1001/jama.2011.57. [DOI] [PubMed] [Google Scholar]
- 22.Loupakis F, Cremolini C, Fioravanti A, et al. Pharmacodynamic and pharmacogenetic angiogenesis-related markers of first-line FOLFOXIRI plus bevacizumab schedule in metastatic colorectal cancer. Br J Cancer. 2010;104:1262–1269. doi: 10.1038/bjc.2011.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pander J, Wessels JA, Gelderblom H, van der Straaten T, Punt CJ, Guchelaar HJ. Pharmacogenetic interaction analysis for the efficacy of systemic treatment in metastatic colorectal cancer. Ann Oncol. 2011;22:1147–1153. doi: 10.1093/annonc/mdq572. [DOI] [PubMed] [Google Scholar]
- 24.Kim HO, Jo YH, Lee J, Lee SS, Yoon KS. The C1772T genetic polymorphism in human HIF-1alpha gene associates with expression of HIF-1alpha protein in breast cancer. Oncol Rep. 2008;20:1181–1187. [PubMed] [Google Scholar]
- 25.Pugh CW, Ratcliffe PJ. Regulation of angiogenesis by hypoxia: role of the HIF system. Nat Med. 2003;9:677–684. doi: 10.1038/nm0603-677. [DOI] [PubMed] [Google Scholar]
- 26.Yang SX, Steinberg SM, Nguyen D, Wu TD, Modrusan Z, Swain SM. Gene expression profile and angiogenic marker correlates with response to neoadjuvant bevacizumab followed by bevacizumab plus chemotherapy in breast cancer. Clin Cancer Res. 2008;14:5893–5899. doi: 10.1158/1078-0432.CCR-07-4762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dowlati A, Gray R, Sandler AB, Schiller JH, Johnson DH. Cell adhesion molecules, vascular endothelial growth factor, and basic fibroblast growth factor in patients with non-small cell lung cancer treated with chemotherapy with or without bevacizumab--an Eastern Cooperative Oncology Group Study. Clin Cancer Res. 2008;14:1407–1412. doi: 10.1158/1078-0432.CCR-07-1154. [DOI] [PubMed] [Google Scholar]
- 28.Hansen TF, Sorensen FB, Spindler KL, et al. Microvessel density and the association with single nucleotide polymorphisms of the vascular endothelial growth factor receptor 2 in patients with colorectal cancer. Virchows Arch. 2010;456:251–260. doi: 10.1007/s00428-009-0878-8. [DOI] [PubMed] [Google Scholar]
- 29.Zeng Y, Sun HR, Yu C, et al. CXCR1 and CXCR2 are novel mechano-sensors mediating laminar shear stress-induced endothelial cell migration. Cytokine. 2011;53:42–51. doi: 10.1016/j.cyto.2010.09.007. [DOI] [PubMed] [Google Scholar]
- 30.Mizukami Y, Jo WS, Duerr EM, et al. Induction of interleukin-8 preserves the angiogenic response in HIF-1alpha-deficient colon cancer cells. Nat Med. 2005;11:992–997. doi: 10.1038/nm1294. [DOI] [PubMed] [Google Scholar]
- 31.Cascone T, Herynk MH, Xu L, et al. Upregulated stromal EGFR and vascular remodeling in mouse xenograft models of angiogenesis inhibitor-resistant human lung adenocarcinoma. J Clin Invest. 2011 doi: 10.1172/JCI42405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moriai T, Kobrin MS, Hope C, Speck L, Korc M. A variant epidermal growth factor receptor exhibits altered type alpha transforming growth factor binding and transmembrane signaling. Proc Natl Acad Sci U S A. 1994;91:10217–10221. doi: 10.1073/pnas.91.21.10217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lanuti M, Liu G, Goodwin JM, et al. A functional epidermal growth factor (EGF) polymorphism, EGF serum levels, and esophageal adenocarcinoma risk and outcome. Clin Cancer Res. 2008;14:3216–3222. doi: 10.1158/1078-0432.CCR-07-4932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pander J, Gelderblom H, Antonini NF, et al. Correlation of FCGR3A and EGFR germline polymorphisms with the efficacy of cetuximab in KRAS wild-type metastatic colorectal cancer. Eur J Cancer. 2010;46:1829–1834. doi: 10.1016/j.ejca.2010.03.017. [DOI] [PubMed] [Google Scholar]
- 35.van Cruijsen H, Giaccone G, Hoekman K. Epidermal growth factor receptor and angiogenesis: Opportunities for combined anticancer strategies. Int J Cancer. 2005;117:883–888. doi: 10.1002/ijc.21479. [DOI] [PubMed] [Google Scholar]
- 36.Verheus M, McKay JD, Kaaks R, et al. Common genetic variation in the IGF-1 gene, serum IGF-I levels and breast density. Breast Cancer Res Treat. 2008;112:109–122. doi: 10.1007/s10549-007-9827-x. [DOI] [PubMed] [Google Scholar]
- 37.Jiang ZY, He Z, King BL, et al. Characterization of multiple signaling pathways of insulin in the regulation of vascular endothelial growth factor expression in vascular cells and angiogenesis. J Biol Chem. 2003;278:31964–31971. doi: 10.1074/jbc.M303314200. [DOI] [PubMed] [Google Scholar]
- 38.Zhao X, Dai W, Zhu H, et al. Epidermal growth factor (EGF) induces apoptosis in a transfected cell line expressing EGF receptor on its membrane. Cell Biol Int. 2006;30:653–658. doi: 10.1016/j.cellbi.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 39.Tikhomirov O, Carpenter G. Ligand-induced, p38-dependent apoptosis in cells expressing high levels of epidermal growth factor receptor and ErbB-2. J Biol Chem. 2004;279:12988–12996. doi: 10.1074/jbc.M311655200. [DOI] [PubMed] [Google Scholar]
- 40.Chiu B, Mirkin B, Madonna MB. Mitogenic and apoptotic actions of epidermal growth factor on neuroblastoma cells are concentration-dependent. J Surg Res. 2006;135:209–212. doi: 10.1016/j.jss.2006.04.018. [DOI] [PubMed] [Google Scholar]
- 41.Graziano F, Ruzzo A, Loupakis F, et al. Pharmacogenetic profiling for cetuximab plus irinotecan therapy in patients with refractory advanced colorectal cancer. J Clin Oncol. 2008;26:1427–1434. doi: 10.1200/JCO.2007.12.4602. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.