

Abstract
A systematic investigation of the regioselectivities and stereoselectivities of (4+3) cycloadditions between unsymmetrical furans and a chiral oxazolidinone-substituted oxyallyl is presented. Cycloadditions were performed using an oxyallyl containing a (R)-4-phenyl-2-oxazolidinone auxiliary (2Ph), under either thermal or ZnCl2-catalyzed conditions. Reactions of 2Ph with 2-substituted furans gave syn cycloadducts selectively, while cycloadditions with 3-substituted furans gave selectively anti cycloadducts. The stereoselectivities were in favor of a single diastereoisomer (I) in all but one case (2-CO2R). Density functional theory calculations were performed to explain the selectivities. The results support a mechanism in which all cycloadducts are formed from the E isomer of the oxyallyl (in which the oxazolidinone C=O and oxyallyl oxygen are anti to each other) or the corresponding E ZnCl2 complex. The major diastereomer is derived from addition of the furan to the more crowded face of the oxyallyl. Crowded transition states are favored because they possess a stabilizing CH–π interaction between the furan and the Ph group.
INTRODUCTION
The (4+3) cycloadditions of oxyallyl (1) with dienes represent a valuable synthetic route to 7-membered carbocycles.1 We recently reported the cycloadditions shown in Scheme 1, where a chiral oxazolidinone-stabilized oxyallyl (2) is generated by oxidation of an allenamide, and reacts with a cyclopentadiene, pyrrole, or furan.2 This sequence provides access to a diverse range of cycloadducts having endo stereochemistry.3 Regioselective reactions with unsymmetrical furans have also been achieved. During our initial studies, we were surprised to find that the use of a 4-phenyl-2-oxazolidinone auxiliary (2, R = Ph) (“2Ph”) led to unusual selectivities in reactions involving several furans. We have therefore carried out a detailed survey of the regio- and stereochemical features of the oxyallyl protocol using this auxiliary. Experimental and computational results, presented here, support a novel mode of stereoinduction for these reactions, in which the furan adds preferentially to the more crowded face of the oxyallyl. The crowded transition state is stabilized by an attractive CH–π interaction between the furan and the Ph group.
Scheme 1.
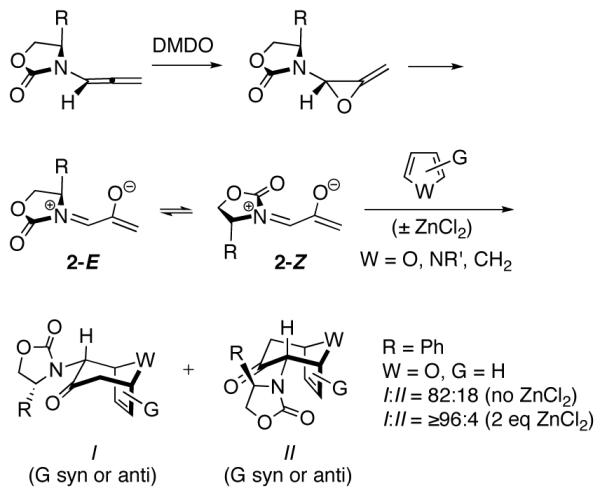
(4+3) Cycloadditions of Oxazolidinone-Substituted Oxyallyls
Cycloaddition of 2Ph with furan led selectively to the cycloadduct I with a diastereomer ratio (I:II) of 82:18.2a In the presence of ZnCl2, the dr increased to ≥96:4. Originally, it was thought2 that the stereoselectivities of cycloadditions involving 2 arose through steric control, as depicted in Scheme 2. The presence of the bulky R group was thought to block one face of the oxyallyl from the approaching furan. Addition of the furan to the less hindered face of 2-Z (or its ZnCl2 chelate) would lead to the major diastereomer of the cycloadduct (I), while addition to the more hindered face would lead to the minor diastereomer (II).
Scheme 2.
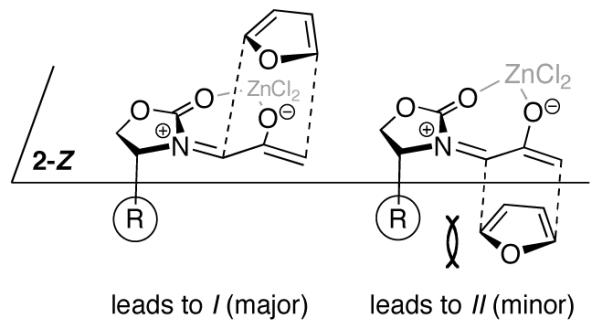
Steric Model Originally Proposed for (4+3) Cycloadditions of 2
Computational studies, however, revealed an altogether different mechanism.4 Computations showed that only cycloadditions involving the E isomer of 2Ph are energetically feasible (Scheme 3). The O-O repulsion is very destabilizing in conformations previously proposed (Scheme 2). This is true even in the presence of ZnCl2. Furan actually adds to the more crowded face of 2Ph-E, where a favorable CH–π interaction between the furan and the Ph group stabilizes the transition state by approximately 0.2 kcal/mol over the uncrowded TS.
Scheme 3.
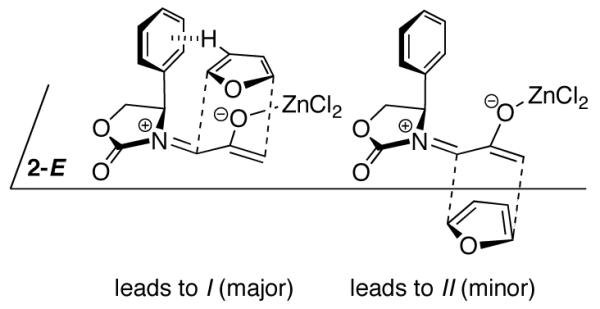
CH–π-Directed Stereoinduction
We present here experimental and computational studies of the cycloadditions of 2Ph with a range of monosubstituted furans, chosen to represent all available variations of electronic character and position of substitution. The data support the anti disposition of oxygens and attractive CH–π model, and also indicate situations where the CH–π interaction can be overcome by substituents on the furan, resulting in altered selectivities.
BACKGROUND
Previous Reports on Regioselectivities
Numerous regioselective cycloadditions between furans and N- or O-substituted oxyallyls have previously been reported.1 Some representative examples are shown in Scheme 4. In principle, the reactions of achiral heteroatom-substituted oxyallyls with unsymmetrical furans can yield two regioisomeric endo adducts; we denote these by the terms syn and anti, in reference to the position ultimately adopted by the substituents on the furan relative to the heteroatom.
Scheme 4.

(4+3) Cycloadditions of Achiral Oxygen- and Nitrogen-Stabilized Oxyallyl Intermediates5–8
Walters5 generated nitrogen-substituted oxyallyl intermediates from the α,α’-dihalide 3, by treatment with either Et3N in trifluoroethanol or Et3N/LiClO4 in acetonitrile. Both reacted with 2-methylfuran or 2-methoxyfuran to give predominantly anti adducts (4b). Föhlisch6 generated an oxygen-stabilized oxyallyl cation by treatment of the α-haloketone 5 with Et3N/LiClO4, and its cycloaddition with 2-methylfuran gave predominantly the anti cycloadduct 6b.7 By contrast, when Albizati8 treated the dimethylacetal 7 with a Lewis acid (TMSOTf, SnCl4, TiCl4, or BCl3), only the syn cycloadduct (6a) was obtained. In the reactions of both 3 and 5, detectable amounts of a third type of adduct resulting from the “sickle” form of the oxyallyl intermediate (rather than the “W” form) were also produced.9
We recently reported the regioselectivities of cycloadditions involving the parent achiral oxazolidinone-substituted oxyallyl (Scheme 5).10 Reactions with 2-methyl- or 2-CO2Me-furan led predominantly to syn cycloadducts, while anti adducts were obtained from 3-methyl- and 3-CO2Et-furan. Inclusion of ZnCl2 increased the yields, but had little effect on the regioselectivity.
Scheme 5.
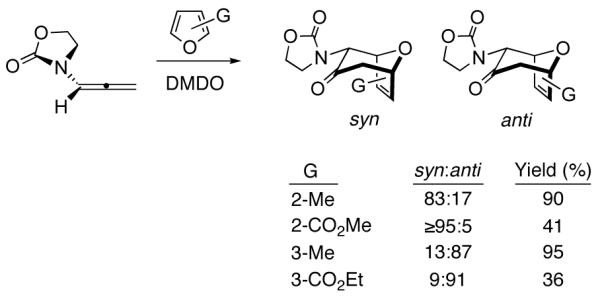
(4+3) Cycloadditions of An Achiral Oxazolidinone-Stabilized Oxyallyl10
Previous Reports on Stereoselectivities
Incorporation of a chiral auxiliary on the oxyallyl increases the number of possible endo cycloadducts to four. For each of the syn and anti regioisomers, there are two diastereomers, I and II, which differ according to the face of the oxyallyl intermediate to which the furan binds. Selected literature examples involving chiral nitrogen- and oxygen-substituted oxyallyls are shown in Scheme 6.
Scheme 6.

(4+3) Cycloadditions of Chiral Oxygen- and Nitrogen-Stabilized Oxyallyl Intermediates11,12
Walters generated a chiral nitrogen-stabilized oxyallyl intermediate by treatment of the α-haloketone 8 with Et3N/trifluoroethanol, and it reacted with 3-bromofuran to give selectively the anti-II adduct 9.11 Hoffmann studied the asymmetric (4+3) cycloadditions of oxygen-stabilized oxyallyl cations generated from 10 by treatment with TMSOTf; their cycloadditions with 3-substituted furans led predominantly to anti-I adducts.12 These results have recently been shown to conform to a CH–π mode of stereoinduction similar to that proposed for our oxyallyls 2.13
RESULTS AND DISCUSSION
We have now investigated the asymmetric cycloadditions of 2Ph with a range of 2- and 3-substituted furans. Methyl and acyloxymethyl-substituted furans were used as electron-rich substrates,14 while ester and cyano-substituted furans were used as electron-poor substrates. The experimentally measured regio- and stereoselectivities are given in Tables 1–3.15
Table 1.
Cycloadditions of 2Ph with 2-Substituted Furans in the Absence of ZnCl2a
| Entry | Furan | syn-endo-I | anti-endo-I | syn-endo-II | anti-endo-II | Yield (%) |
|---|---|---|---|---|---|---|
| 1 |
|
|
|
|
|
54 |
| 2 |
|
– | – |
|
– | 60 |
| 3 |
|
– | – |
|
– | 40 |
Isolated yields. Isomer distributions are given in parentheses, and were determined by 1H and/or 13C NMR.
Table 3.
Cycloadditions of 2Ph with 3-Substituted Furans in the Presence of ZnCl2a
| Entry | Furan | syn-endo-I | anti-endo-I | syn-endo-IIb | anti-endo-II | Yield (%) |
|---|---|---|---|---|---|---|
| 1 |
|
– |
|
|
– | 62 |
| 2 |
|
– |
|
|
– | 60 |
| 3 |
|
– |
|
|
– | 49 |
| 4 |
|
– |
|
– | – | 53 |
| 5 |
|
– |
|
|
– | 50 |
| 6 |
|
– |
|
– | – | 45 |
Isolated yields. Isomer distributions are given in parentheses, and were determined by 1H and/or 13C NMR.
The regiochemistry of all minor isomers were assigned as syn by COSY and NMR coupling patterns, but the stereochemistry (I or II) was not determined; they are listed as II here by comparison with the computational data.
1. Cycloadditions of 2Ph with 2-Substituted Furans
1.1 Cycloadditions in the absence of ZnCl2
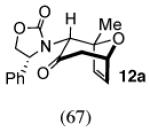
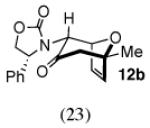
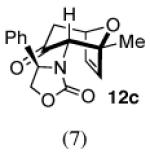
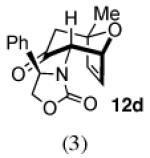
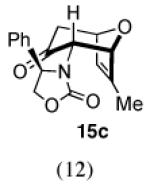
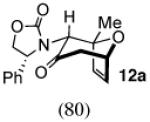
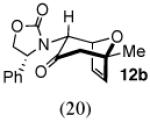
The cycloaddition of 2Ph with 2-methylfuran (Table 1) gave a 54% total yield of the endo cycloadducts 12a–d, in which the diastereomer ratio (dr) of I:II was 90:10. The regioisomer ratios were equivalent for the two diastereomers: I was obtained with a syn:anti ratio of 67:23 (12a:12b), while II was obtained with a syn:anti ratio of 70:30 (12c:12d). Stereochemical assignments were made on the basis of NMR coupling patterns and COSY spectra. The olefinic protons in both of the I isomers (12a and 12b) are shifted unusually upfield, due to anisotropic shielding by the nearby Ph ring on the oxazolidinone. Similar shielding effects were also observed with the other I isomers described below. The II isomers 12c and 12d showed no such upfield shifting. For the syn-I isomer (12a), the 1H NMR spectrum had to be collected at high temperature in order to discern the coupling patterns (Supporting Information).
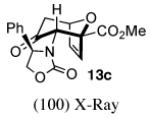
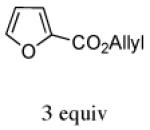
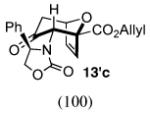
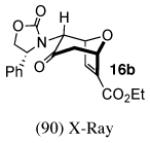
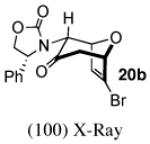
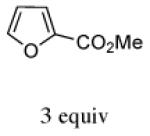
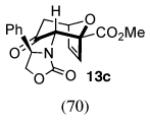
In contrast, the cycloadditions of 2Ph with 2-EWG-substituted furans produced diastereomer II but no observable I. Methyl 2-furoate and allyl 2-furoate gave the syn-II isomers 13c and 13’c as the sole cycloadducts in 40–60% yield. The stereochemistry of 13c was unambiguously assigned by X-ray crystallography (Figure 1).
Figure 1.

X-Ray Crystal Structures of Cycloadducts.
1.2 Cycloadditions in the presence of ZnCl2
Inclusion of ZnCl2 in the reaction mixture for cycloaddition with 2-methylfuran raised the diastereomer ratio from 90:10 to 100:0 (Table 2). The syn:anti ratio increased to 80:20.
Table 2.
Cycloadditions of 2Ph with 2-Substituted Furans in the Presence of ZnCl2a
| Entry | Furan | syn-endo-I | anti-endo-I | syn-endo-II | anti-endo-II | Yield (%) |
|---|---|---|---|---|---|---|
| 1 |
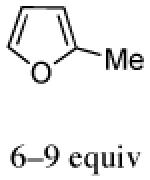
|
|
|
– | – | 47 |
| 2 |
|
|
– |
|
– | 63 |
| 3 |
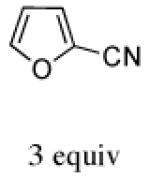
|
|
– |
|
– | 47 |
Isolated yields. Isomer distributions are given in parentheses, and were determined by 1H and/or 13C NMR.
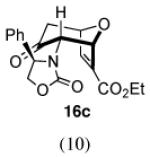
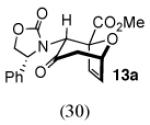
With methyl 2-furoate, the major product was still the syn-II isomer 13c, but it was now accompanied by a small amount of syn-I (13a). The structure of 13a was assigned spectroscopically; anisotropic shielding was again evident.2d Somewhat surprisingly, on going from the furoate ester to its nitrile analogue, a return in stereoselectivity back to I was observed! The syn-I isomer 14a was obtained from 2-cyanofuran with a dr of 83:17. The regioselectivity for both the ester and the nitrile was completely in favor of syn.
The regioselectivities observed here parallel those observed previously for the parent oxazolidinone-substituted oxyallyl (Scheme 5),10 although for 2-methylfuran the syn:anti ratio is lower with 2Ph than with the parent oxyallyl.
2. Cycloadditions of 2Ph with 3-Substituted Furans
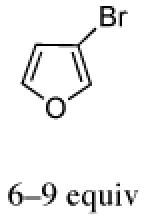
The cycloadditions of 2Ph with 3-substituted furans were studied under ZnCl2-catalyzed conditions. Our results are listed in Table 3. The stereochemistry of the major cycloadducts was unambiguously assigned by means of COSY and NOESY analysis and, in the cases of ethyl 3-furoate and 3-bromofuran, by X-ray single crystal structures (Figure 1). In all of the major products, the olefinic proton showed the typical anisotropic shielding by the Ph ring on the oxazolidinone. For the minor isomers, the regiochemistry could be firmly assigned as syn by COSY and NMR coupling patterns, but the stereochemistry was not determined. The syn regiochemistry ruled out the use of Ph shielding as a stereochemical diagnostic. We have tentatively assigned the II stereochemistry to the minor isomers, on the basis of our computational results reported below.
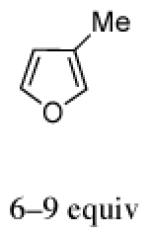
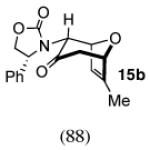
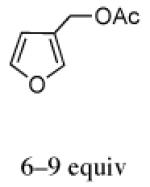
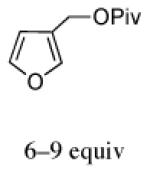
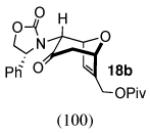
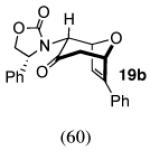
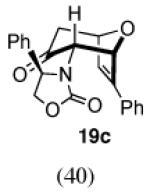
The regio- and stereoselectivities of the cycloadditions involving 3-substituted furans differ markedly from those of 2-substituted furans. All of the 3-substituted furans gave selectively the anti-I cycloadduct. For 3-methylfuran and methyl 3-furoate, the anti:syn ratio was approximately 90:10. 3-phenylfuran and 3-acetoxymethylfuran gave more modest anti:syn ratios (60:40 and 70:30, respectively), but replacement of the acetyl group in the latter by a pivaloyl group increased the anti selectivity to 100:0. 3-bromofuran also gave 100:0 anti selectivity.
The regioselectivities of these reactions mirror those of the parent oxazolidinone-substituted oxyallyl with 3-substituted furans (Scheme 5),10 both in direction and magnitude. Similar anti-I selectivity was reported by Hoffmann for the cycloadditions of 3-substituted furans with related oxygen-substituted oxyallyl cations (Scheme 6).12
3. Computational Studies
Numerous theoretical studies of oxyallyl cations and their cycloadditions have been reported previously.5,16–18 Cramer17b,c showed that the cycloadditions can take place by various mechanisms; the highly electrophilic hydroxyallyl cation was calculated to react with furan by a two-step process commencing with electrophilic attack at C-2 of the furan. We have found4 that the less electrophilic, neutral oxyallyls 2 undergo concerted, asynchronous cycloadditions with furans.
Our earlier findings4 are summarized in Scheme 7. At the B3LYP/6-31G(d) level, only the E conformer of 2Ph was an energy minimum. Attempts to locate the Z conformer led to the isomeric cyclopropanone. ZnCl2 complexes were located for both the E and the Z conformers, but the Z complex was 6.2 kcal/mol less stable. Transition states were located for cycloadditions involving both the E and the Z conformers, but those involving the Z isomer were disfavored by ≥15.4 kcal/mol (≥5.2 kcal/mol for the ZnCl2 complexes). Electrostatic repulsion between the oxygen atoms destabilizes all of the Z structures.
Scheme 7.

Previous Calculations4 on the Oxyallyl 2Ph, its ZnCl2 Complexes, and its Cycloadditions with Furan. Energies are in kcal/mol
The concerted transition state geometries are asynchronous such that the more advanced bonding interaction involves the more nucleophilic (CH2) carbon of the oxyallyl. This is the case also for reactions with substituted furans, shown in Scheme 5.10 On the other hand, the calculated Mulliken charges in the transition states indicate net charge transfer of 0.05–0.17e from the furan to the oxyallyl. Overall, the oxyallyls 2 are best characterized as ambiphilic toward furans. The activation barriers for reactions with either Me- or CO2Me-substituted furans (Scheme 5) are lower than those for furan itself. Methyl 2-furoate has the lowest barrier (ΔH‡ 3.2 kcal/mol lower than for furan).
To model the reactions of 2Ph with substituted furans, we calculated transition states involving addition to the E oxyallyl only. Calculations were performed at the B3LYP/6-31G(d) level (including LANL2DZ+ECP for Zn) in Gaussian 03.19 In Figure 2 are shown the four isomeric transition structures calculated for the reaction of 2Ph with 2-methylfuran. These are representative of the transition states calculated for the other furans also. Concerted, asynchronous transition states were found for all of the cycloadditions.
Figure 2.

Transition structures for the (4+3) cycloadditions of 2Ph with 2-methylfuran, calculated at the B3LYP/6-31G(d) level. Bond lengths in Å, energies in kcal/mol. ΔH‡ and ΔG‡ (in parentheses) are given below the TSs.
3.1 Cycloadditions of 2Ph with 2-Substituted Furans
The calculated activation energies for reactions of 2Ph-E with 2-substituted furans are listed in Table 4. The experimental selectivities are also included in the table for comparison.
Table 4.
Calculated Activation Energies and Experimental Cycloadduct Ratios for the (4+3) Cycloadditions of 2Ph with 2-Substituted Furansa
| syn-I | anti-I | syn-II | anti-II | |
|---|---|---|---|---|
| Reactions in the absence of ZnCl2 | ||||
| 2-Methylfuran | ||||
| Δ H ‡ | 6.2 | 7.5 | 6.7 | 7.8 |
| Δ G ‡ | 20.4 | 22.0 | 20.9 | 22.2 |
| Experimental | 67% | 23% | 7% | 3% |
| Methyl 2-furoate | ||||
| Δ H ‡ | 5.3 | 8.7 | 3.4 | 9.3 |
| Δ G ‡ | 20.5 | 23.2 | 19.3 | 23.3 |
| Experimental | 100% | |||
| Reactions in the presence of ZnCl2 | ||||
| 2-Methylfuran | ||||
| Δ H ‡ | −2.9 | −1.7 | −1.9 | −0.6 |
| Δ G ‡ | 11.7 | 13.6 | 11.7 | 13.4 |
| Experimental | 80% | 20% | ||
| Methyl 2-furoate | ||||
| Δ H ‡ | −1.1 | 4.1 | −1.7 | 5.4 |
| Δ G ‡ | 14.1 | 18.5 | 13.9 | 18.4 |
| Experimental | 30% | 70% | ||
| 2-Cyanofuran | ||||
| Δ H ‡ | 2.3 | 6.1 | 3.4 | 7.6 |
| Δ G ‡ | 17.0 | 20.9 | 17.7 | 21.4 |
| Experimental | 83% | 17% | ||
B3LYP/6-31G(d), 298.15 K, kcal/mol; Experimental isomer ratios given as percentages.
The calculations correctly predict the major product for each 2-substituted furan. Some differences in the minor isomers are found, which for the ZnCl2-catalyzed cycloadditions may be due to the use of excess furan and ZnCl2 in the experiment. For example, a furan molecule may coordinate to the Zn centre of 2Ph-E·ZnCl2, or ZnCl2 may coordinate to the furan that undergoes cycloaddition. These factors would lead to small differences in the proportions of the minor isomers but are unlikely to affect the overall selectivity.
The oxyallyl 2Ph is more nucleophilic at the CH2 terminus and more electrophilic at the CHN terminus. Yet, cycloadditions of 2Ph with both 2-EWG and 2-EDG-substituted furans favor the syn cycloadducts. This regioselectivity is predictable for the EWG-substituted furans, since the substituent renders the 5-position of the furan more electrophilic than the 2-position. The regioselectivity for the donor-substituted furan (2-Me-furan) is unexpected. Here electronic effects are small, and the regioselectivity instead arises from a preference for the transition state that allows the more advanced bonding interaction to be made to the less-substituted carbon (C-5) of the furan (Figure 2).
The stereoselectivities observed with 2-methylfuran and 2-cyanofuran conform to the CH–π model (Scheme 3), and have a magnitude somewhat smaller than that observed with furan itself (82:18 uncatalyzed, ≥96:4 with ZnCl2). The proton involved in the CH–π interaction is H-3 or H-4 of the furan (for the syn and anti isomers, respectively), which points roughly towards the centre of the Ph ring in the I transition states. The stabilizing role of the CH–π interaction has both electrostatic and dispersive components. To estimate the role of the latter, the dispersion energies in the transition states were calculated by means of Grimme’s 2006 empirical B3LYP-D formula, which sums the dispersion energies between each pair of atoms in the structure.20 A comparison between the dispersion energies of the syn-I and syn-II transition states for each 2-substituted furan is shown in Figure 3. Dispersive stabilization is 0.02–2.9 kcal/mol stronger in the I transition states than the II transition states.
Figure 3.
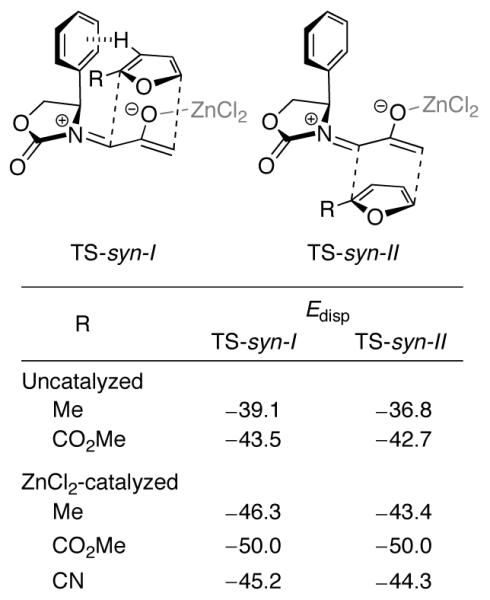
Total dispersion energies of the syn-I and syn-II transition states for cycloadditions of 2Ph with 2-substituted furans, calculated at the B3LYP-D/6-31G(d) level (kcal/mol).
The reversal in stereoselectivity for 2-CO2Me-furan was at first suprising, but it is indeed predicted by theory. The syn-II transition state is favored over the other three TSs by 1.9 kcal/mol in the uncatalyzed reaction and by 0.6 kcal/mol in the ZnCl2-catalyzed reaction (ΔΔH‡). A top-down view of the syn-I transition state for 2-CO2Me-furan under ZnCl2-catalyzed conditions is shown in Figure 4a. A destabilizing electrostatic interaction takes place between the ester carbonyl group and the Ph π-cloud. The distance between the carbonyl oxygen and the nearby C-2 of the Ph ring (red line: 3.22 Å) is equal to the sum of the van der Waals radii for C and O. This interaction outweighs the stabilizing effect of the CH–π interaction, and the uncrowded (II) transition state is instead favored.
Figure 4.
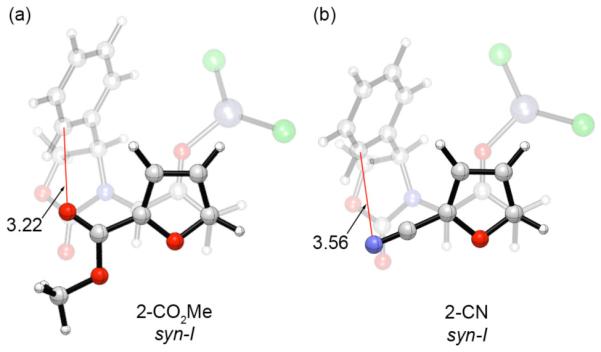
Syn-I transition structures for the ZnCl2-catalyzed (4+3) cycloadditions of 2Ph with 2-CO2Me- and 2-CN-furan, calculated at the B3LYP/6-31G(d) level. Distances in Å.
The calculations also correctly predict that the seemingly small change from a 2-CO2Me to a 2-CN group should induce a return to syn-I selectivity. The syn-I transition state for 2-CN-furan is shown in Figure 4b. The CN group is positioned further from the Ph ring (N–C 3.56 Å), and therefore does not present such severe electrostatic repulsion as the CO2Me group.
3.2 Cycloadditions of 2Ph with 3-Substituted Furans
The calculated activation energies for ZnCl2-catalyzed reactions of 2Ph with 3-substituted furans are listed in Table 5, together with the experimental selectivities.
Table 5.
Calculated Activation Energies and Experimental Cycloadduct Ratios for the ZnCl2-catalyzed (4+3) Cycloadditions of 2Ph with 3-Substituted Furansa
| syn-I | anti-I | syn-II | anti-II | |
|---|---|---|---|---|
| 3-Methylfuran | ||||
| Δ H ‡ | −1.4 | −2.8 | −2.0 | −1.9 |
| Δ G ‡ | 12.7 | 11.6 | 11.8 | 10.9 |
| Experimental | 88% | 12% | ||
| Methyl 3-furoate | ||||
| Δ H ‡ | 0.9 | −4.9 | −1.9 | −3.2 |
| Δ G ‡ | 16.7 | 11.7 | 13.2 | 12.8 |
| Experimentalb | 90% | 10% |
B3LYP/6-31G(d), 298.15 K, kcal/mol; Experimental isomer ratios given as percentages.
Experimental data for ethyl 3-furoate.
The calculations correctly predict the regio- and stereochemistry of the major product for methyl 3-furoate. For 3-methylfuran, the free energies of activation do not capture the correct stereoselectivity, but the enthalpies do predict both the regio- and stereoselectivity. As with the 2-EWG-substituted furans, the regioselectivity for 3-CO2Me-furan is under electronic control, since C-2 of the furan is more electrophilic than C-5. For 3-Me-furan, the regioselectivity is instead steric in origin, arising through avoidance of clashing between the Me group and the Ph group. This steric effect also influences the reaction of 3-CO2Me-furan: indeed, a concerted syn-I transition state could not be located, due to severe clashing.
Both 3-substituted furans favor the I stereoisomer, as do the four other 3-substituted furans in Table 3. The anti arrangement enables the CH–π interaction in the crowded TS to take place without interference from the substituent. The calculations predict that for both 3-substituted furans, the minor (syn) isomer should have the II stereochemistry, as the syn-I transition state is subject to unfavorable interaction between the Ph group and the furan 3-substituent.
CONCLUSIONS
A summary of the origins of the regio- and stereoselectivities of the (4+3) cycloadditions is given in Scheme 8. The oxyallyls 2 undergo (4+3) cycloadditions with furans through concerted transition states in most cases. Generally, bond development at the TS is most advanced at the nucleophilic (CH2) terminus of 2. The regioselectivities of (4+3) cycloadditions between 2 and furans arise through steric effects, but these are complemented by electronic effects when the sub-stituent is an electron-withdrawing group. For 2-substituted furans, syn cycloadducts are formed selectively, because this arrangement enables the stronger bonding interaction in the TS to involve the less-hindered (C-5) carbon. For 2-EWG-substituted furans, C-5 is also more electrophilic than C-2, enhancing regiocontrol. 3-Substituted furans undergo cycloaddition preferentially in the anti geometry, in order to avoid steric clashing between the 3-substituent and the Ph ring. Similar anti selectivity has even been observed when the Ph group is absent,10 indicating that the pseudoaxial group at the 4-position of the oxazolidinone has a steric influence even when it is only an H atom. For 3-EWG-substituted furans, the anti selectivity is complemented by the electronic effect of the substituent, which renders C-2 more electrophilic than C-5.
Scheme 8.
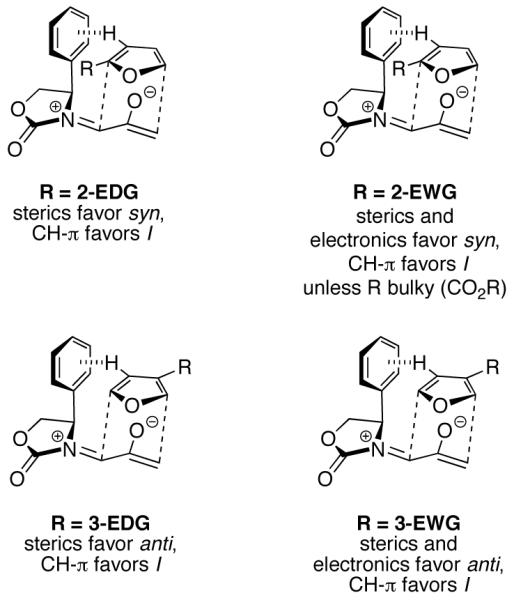
Summary of Factors Responsible for Regio- and Stereocontrol in the (4+3) Cycloadditions of 2Ph with Furans
All but two of the furans studied (Tables 1–3) lead selectively to the cycloadduct I. Only for 2-CO2R-furans (R = Me, allyl) does the unexpected II isomer predominate. The CH–π model (Scheme 3) therefore appears to have broad applicability for monosubstituted furans. When the oxazolidinone Ph group is replaced by a non-aromatic group, the stabilizing CH–π interaction is lost, and the stereoselectivity reverts to favor II, as shown by our earlier studies with a 4-Bn-oxazolidinone and with Seebach et al.’s21 4-iPr-5,5-Ph2-oxazolidinone auxiliary.4 Reversals of stereoselectivity have also previously been observed to occur in hetero-Diels–Alder reactions22 and conjugate radical additions23 catalyzed by chiral Lewis acid/bis(oxazoline) complexes, when the Ph groups on the ligand were replaced by tBu groups.
Chiral oxazolidinone auxiliaries have been used to achieve a wide range of other asymmetric transformations, including Diels–Alder reactions,24 aldol condensations,25 enolate alkylation,26 hydroxylation,27 and amination,28 Michael additions,29 epoxide synthesis,30 and resolutions of α-halo carbonyl compounds.31 The results described herein for the (4+3) cycloadditions of 2Ph likely hold relevance to stereocontrol in these other oxazolidinone-directed reactions.
EXPERIMENTAL METHODS
Typical Procedure for (4+3) Cycloadditions
To a solution of the allenamide in CH2Cl2 (0.1 M) were added the appropriate furan (3–9 equiv) and 4Å powder molecular sieves (0.5 g). The solution was cooled to −78 °C, and ZnCl2 (1.0 M in ether, 2.0 equiv) was added. DMDO (4.0–6.0 equiv, in acetone) was then added as a chilled solution via syringe pump (at −78 °C) over 3–4 h. The reaction mixture was stirred for a further 14 h, then quenched with saturated aqueous NaHCO3, filtered through Celite, concentrated in vacuo, extracted with CH2Cl2, dried over Na2SO4, and concentrated in vacuo. The crude residue was purified via column chromatography on silica gel (gradient eluent: 10% to 75% ethyl acetate in hexane).
THEORETICAL CALCULATIONS
Density functional theory calculations were performed at the B3LYP/6-31G(d) level32 in Gaussian 03.19 The LANL2DZ basis set and effective core potential were used for Zn.33 Species were characterized as minima or transition states on the basis of vibrational frequency analysis and, where appropriate, IRC calculations.34 Zero-point energy and thermal corrections were derived (unscaled) from the B3LYP/6-31G(d) frequencies. Conformational searching was performed for each species in order to identify the lowest-energy conformer. Enthalpies and free energies are reported at 298.15 K, with a standard state of 1 atm.
Supplementary Material
ACKNOWLEDGMENT
We thank the NIH (GM36700 to K.N.H. and GM-66055 to R.P.H.) and Australian Research Council (DP0985623 to E.H.K.) for generous financial support, and the NCSA, UCLA ATS, UCLA IDRE, and NCF NI (Australia) for computer resources. E.H.K. also thanks the Australian–American Fulbright Commission for a postdoctoral scholarship and the ARC Centre of Excellence for Free Radical Chemistry and Biotechnology for financial support. We thank Dr Vic Young and Dr Ben Kucera of the University of Minnesota for X-ray crystallography.
Footnotes
Supporting Information. Full experimental procedures, NMR spectra and characterizations for all new compounds, X-ray data, computed geometries and energies, and a complete citation for ref. 19. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1(a).For reviews, see: Lohse AG, Hsung RP. Chem. Eur. J. 2011:17, 3812–3822. doi: 10.1002/chem.201100260. Harmata M. Chem. Commun. 2010;46:8886–8903. doi: 10.1039/c0cc03620j. Harmata M. Adv. Synth. Catal. 2006;348:2297–2306. Battiste MA, Pelphrey PM, Wright DL. Chem. Eur. J. 2006;12:3438–3447. doi: 10.1002/chem.200501083. Antoline JE, Hsung RP. ChemTracts. 2005;18:562–568. Hartung IV, Hoffmann HMR. Angew. Chem. Int. Ed. 2004;43:1934–1949. doi: 10.1002/anie.200300622. Harmata M, Rashatasakhon P. Tetrahedron. 2003;59:2371–2395. Harmata M. Acc. Chem. Res. 2001;34:595–605. doi: 10.1021/ar000064e. Davies HML. In: Advances in Cycloaddition. Harmata M, editor. Vol. 5. JAI; Stamford, CT: 1999. pp. 119–164. West FG. In: Advances in Cycloaddition. Lautens M, editor. Vol. 4. JAI; Greenwich, CT: 1997. pp. 1–40. Rigby JH, Pigge FC. Org. React. 1997;51:351–478. Harmata M. Tetrahedron. 1997;53:6235–6280. Harmata M. Recent Res. Devel. Org. Chem. 1997;1:523–535. Katritzky AR, Dennis N. Chem. Rev. 1989;89:827–861.
- 2(a).Xiong H, Hsung RP, Berry CR, Rameshkumar C. J. Am. Chem. Soc. 2001;123:7174–7175. doi: 10.1021/ja0108638. [DOI] [PubMed] [Google Scholar]; (b) Rameshkumar C, Xiong H, Tracey MR, Berry CR, Yao LJ, Hsung RP. J. Org. Chem. 2002;67:1339–1345. doi: 10.1021/jo011048d. [DOI] [PubMed] [Google Scholar]; (c) Xiong H, Huang J, Ghosh SK, Hsung RP. J. Am. Chem. Soc. 2003;125:12694–12695. doi: 10.1021/ja030416n. [DOI] [PubMed] [Google Scholar]; (d) Rameshkumar C, Hsung RP. Angew. Chem. Int. Ed. 2004;43:615–618. doi: 10.1002/anie.200352632. [DOI] [PubMed] [Google Scholar]; (e) Antoline JE, Hsung RP, Huang J, Song Z, Li G. Org. Lett. 2007;9:1275–1278. doi: 10.1021/ol070103n. [DOI] [PubMed] [Google Scholar]; (f) Antoline JE, Hsung RP. Synlett. 2008:739–744. [Google Scholar]
- 3.We use the term “endo” in reference to the relationship between the diene unit and the oxyallyl oxygen in the cycloadducts and the TSs leading to them. Hoffmann has used the term “compact” to describe the same geometry: Hoffmann HMR. Angew. Chem. Int. Ed. 1973:12, 819–835.
- 4.Krenske EH, Houk KN, Lohse AG, Antoline JE, Hsung RP. Chem. Sci. 2010;1:387–392. doi: 10.1039/C0SC00280A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5(a).Walters MA, Arcand HR, Lawrie DJ. Tetrahedron Lett. 1995;36:23–26. [Google Scholar]; (b) Walters MA, Arcand HR. J. Org. Chem. 1996;61:1478–1486. [Google Scholar]
- 6.Föhlisch B, Krimmer D, Gehrlach E, Käshammer D. Chem. Ber. 1988;121:1585–1594. [Google Scholar]
- 7.For related work, see: Harmata M, Rashatasakhon P. Synlett. 2000:1419–1422.
- 8.Murray DH, Albizati KF. Tetrahedron Lett. 1990;31:4109–4112. [Google Scholar]
- 9.“W” and “sickle” oxyallyl configurations are defined in ref. 3.
- 10.Lohse AG, Krenske EH, Antoline JE, Houk KN, Hsung RP. Org. Lett. 2010;12:5506–5509. doi: 10.1021/ol1023745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.MaGee DI, Godineau E, Thornton PD, Walters MA, Sponholtz D. J. Eur. J. Org. Chem. 2006:3667–3680. [Google Scholar]
- 12.Beck H, Stark CBW, Hoffmann HMR. Org. Lett. 2000;2:883–886. doi: 10.1021/ol991386p. [DOI] [PubMed] [Google Scholar]
- 13.Krenske EH, Houk KN, Harmata M. Org. Lett. 2010;12:444–447. doi: 10.1021/ol902591k. [DOI] [PubMed] [Google Scholar]
- 14.Attempts to perform the cycloadditions with more electron-rich furans such as 2-methoxyfuran were hampered by competing oxidation of the furan: see ref 2f.
- 15.Several earlier results on cycloadditions involving 2-substituted furans under other conditions have previously been reported: see ref. 2.
- 16(a).Coolidge MB, Yamashita K, Morokuma K, Borden WT. J. Am. Chem. Soc. 1990;112:1751–1754. [Google Scholar]; (b) Ichimura AS, Lahti PM, Matlin AR. J. Am. Chem. Soc. 1990;112:2868–2875. [Google Scholar]; (c) Janoschek R, Kalcher J. Int. J. Quantum Chem. 1990;38:653–664. [Google Scholar]; (d) Turecek F, Drinkwater DE, McLafferty FW. J. Am. Chem. Soc. 1991;113:5950–5958. [Google Scholar]; (e) Lim D, Hrovat DA, Borden WT, Jorgensen WL. J. Am. Chem. Soc. 1994;116:3494–3499. [Google Scholar]; (f) Goodman JM, Hoffmann HMR, Vinter JG. Tetrahedron Lett. 1995;36:7757–7760. [Google Scholar]; (g) Ichino T, Villano SM, Gianola AJ, Goebbert DJ, Velarde L, Sanov A, Blanksby SJ, Zhou X, Hrovat DA, Borden WT, Lineberger WC. Angew. Chem. Int. Ed. 2009;48:8509–8511. doi: 10.1002/anie.200904417. [DOI] [PubMed] [Google Scholar]; (h) Kuzmanich G, Spänig F, Tsai C-K, Um JM, Hoekstra RM, Houk KN, Guldi DM, Garcia-Garibay MA. J. Am. Chem. Soc. 2011;133:2342–2345. doi: 10.1021/ja109494b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17(a).Noyori R, Shimizu F, Fukuta K, Takaya H, Hayakawa Y. J. Am. Chem. Soc. 1977;99:5196–5198. [Google Scholar]; (b) Cramer CJ, Barrows SE. J. Org. Chem. 1998;63:5523–5532. [Google Scholar]; (c) Cramer CJ, Barrows SE. J. Phys. Org. Chem. 2000;13:176–186. [Google Scholar]; (d) Cramer CJ, Harmata M, Rashatasakhon P. J. Org. Chem. 2001;66:5641–5644. doi: 10.1021/jo015695g. [DOI] [PubMed] [Google Scholar]; (e) Harmata M, Schreiner PR. Org. Lett. 2001;3:3663–3665. doi: 10.1021/ol016611t. [DOI] [PubMed] [Google Scholar]; (f) Sáez JA, Arnó M, Domingo LR. Org. Lett. 2003;5:4117–4120. doi: 10.1021/ol035652h. [DOI] [PubMed] [Google Scholar]; (g) Arnó M, Picher MT, Domingo LR, Andrés J. Chem. Eur. J. 2004;10:4742–4749. doi: 10.1002/chem.200400277. [DOI] [PubMed] [Google Scholar]; (h) Sáez JA, Arnó M, Domingo LR. Tetrahedron. 2005;61:7538–7545. [Google Scholar]
- 18.Fernández I, Cossío FP, de Cózar A, Lledós A, Mascareñas JL. Chem. Eur. J. 2010;16:12147–12157. doi: 10.1002/chem.201001714. [DOI] [PubMed] [Google Scholar]
- 19.Frisch MJ, et al. Gaussian 03. Revision E.01 Gaussian, Inc.; Wallingford CT: 2004. [Google Scholar]
- 20.Grimme SJ. Comput. Chem. 2006;27:1787–1799. doi: 10.1002/jcc.20495. [DOI] [PubMed] [Google Scholar]
- 21(a).Hintermann T, Seebach D. Helv. Chim. Acta. 1998;81:2093–2126. [Google Scholar]; (b) Gaul C, Schweizer BW, Seiler P, Seebach D. Helv. Chim. Acta. 2002;85:1546–1566. [Google Scholar]
- 22(a).Yao S, Johannsen M, Audrain H, Hazell RG, Jørgensen KA. J. Am. Chem. Soc. 1998;120:8599–8605. [Google Scholar]; (b) Evans DA, Johnson JS, Olhava EJ. J. Am. Chem. Soc. 2000;122:1635–1649. [Google Scholar]
- 23.Sibi MP, Ji J, Wu J. Hongliu, Gürtler S, Porter NA. J. Am. Chem. Soc. 1996;118:9200–9201. [Google Scholar]
- 24(a).Evans DA, Chapman KT, Bisaha J. J. Am. Chem. Soc. 1984;106:4261–4263. [Google Scholar]; (b) Evans DA, Chapman KT, Bisaha J. Tetrahedron Lett. 1984;25:4071–4074. [Google Scholar]; (c) Evans DA, Chapman KT, Hung DT, Kawaguchi AT. Angew. Chem. Int. Ed. 1987;26:1184–1186. [Google Scholar]; (d) Evans DA, Chapman KT, Bisaha J. J. Am. Chem. Soc. 1988;110:1238–1256. [Google Scholar]; (e) Evans DA, Ripin D. H. Brown, Johnson JS, Shaughnessy EA. Angew. Chem. Int. Ed. 1997;36:2119–2121. [Google Scholar]
- 25.Evans DA, Bartroli J, Shih TL. J. Am. Chem. Soc. 1981;103:2127–2129. [Google Scholar]
- 26.Evans DA, Ennis MD, Mathre DJ. J. Am. Chem. Soc. 1982;104:1737–1739. [Google Scholar]
- 27.Evans DA, Morrissey MM, Dorow RL. J. Am. Chem. Soc. 1985;107:4346–4348. [Google Scholar]
- 28.Evans DA, Britton TC, Dorow RL, Dellaria JF. J. Am. Chem. Soc. 1986;108:6395–6397. [Google Scholar]
- 29.Le Coz S, Mann A. Synth. Commun. 1993;23:165–171. [Google Scholar]
- 30.Abdel-Magid A, Pridgen LN, Eggleston DS, Lantos I. J. Am. Chem. Soc. 1986;108:4595–4602. [Google Scholar]
- 31.Caddick S, Jenkins K, Treweeke N, Candeias SX, Afonso CAM. Tetrahedron Lett. 1998;39:2203–2206. [Google Scholar]
- 32(a).Lee C, Yang W, Parr RG. Phys. Rev. B. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]; (b) Becke AD. J. Chem. Phys. 1993;98:1372–1377. [Google Scholar]; (c) Becke AD. J. Chem. Phys. 1993;98:5648–5652. [Google Scholar]
- 33.Hay PJ, Wadt WR. J. Chem. Phys. 1985;82:270–283. 299–310. [Google Scholar]
- 34.Gonzalez C, Schlegel HB. J. Chem. Phys. 1989;90:2154–2161. [Google Scholar]; (b) Gonzalez C, Schlegel HB. J. Phys. Chem. 1990;94:5523–5527. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.