

Abstract
Many estrogen receptor-positive breast cancers respond well initially to endocrine therapies, but often develop resistance during treatment with selective estrogen receptor modulators (SERMs) such as tamoxifen. We have reported that the 14-3-3 family member and conserved protein, 14-3-3ζ, is up-regulated by tamoxifen and that high expression correlated with an early time to disease recurrence. However, the mechanism by which tamoxifen up-regulates 14-3-3ζ and may promote the development of endocrine resistance is not known. Our findings herein reveal that the tamoxifen up-regulation of 14-3-3ζ results from its ability to rapidly down-regulate miR-451 that specifically targets 14-3-3ζ. The levels of 14-3-3ζ and miR-451 were inversely correlated, with 14-3-3ζ being elevated and miR-451 being at a greatly reduced level in tamoxifen-resistant breast cancer cells. Of note, down-regulation of miR-451 was selectively elicited by tamoxifen but not by other SERMs such as raloxifene or ICI182,780 (Fulvestrant). Increasing the level of miR-451 by overexpression, which decreased 14-3-3ζ, suppressed cell proliferation and colony formation, markedly reduced activation of HER2, EGFR, and MAPK signaling, increased apoptosis, and importantly, restored the growth inhibitory effectiveness of SERMs in endocrine-resistant cells. Opposite effects were elicited by miR-451 knock-down. Thus, we identify tamoxifen down-regulation of miR-451, and consequent elevation of the key survival factor 14-3-3ζ, as a mechanistic basis of tamoxifen-associated development of endocrine resistance. These findings suggest that therapeutic approaches to increase expression of this tumor suppressor-like microRNA should be considered to down-regulate 14-3-3ζ and enhance the effectiveness of endocrine therapies. Furthermore, the selective ability of the SERM tamoxifen but not raloxifene to regulate miR-451 and 14-3-3ζ may assist in understanding differences in their activities, as seen in the STAR breast cancer prevention trial and in other clinical trials.
Keywords: breast cancer, tamoxifen, endocrine resistance, 14-3-3ζ, miR-451, tumor suppressor
INTRODUCTION
Tamoxifen has proven to be one of the most successful agents in the management of estrogen receptor (ER) -positive breast cancers, and it has been a mainstay of endocrine therapy for breast cancer over the last 30 years. Large studies have shown its value in the improvement of survival in early breast cancer (EBCTCG 2005, Osborne 1998) as well as an improvement in the quality of life for patients with metastatic disease (Jaiyesimi et al 1995). However, despite the remarkable benefits of using ER- targeted therapies, including selective ER modulators (SERMs) such as tamoxifen in the ca. 70% of women with hormone-sensitive breast cancers expressing ERs, many will eventually experience disease progression, due to the development of resistance to the therapy. The mechanisms that underlie endocrine resistance are complex and are not fully understood, despite significant advances in defining some of the players involved (Arpino et al 2008, Creighton et al 2006, Green and Carroll 2007, Massarweh et al 2008).
In a previous study, based on genome-wide gene expression analyses in human breast tumors and in estrogen receptor-positive breast cancer cells, we found 14-3-3ζ (also known as YWHAZ) to be up-regulated by tamoxifen and we observed a poor clinical outcome on tamoxifen treatment for patients with high levels of 14-3-3ζ in their breast tumors (Frasor et al 2006). This correlation between overexpression of 14-3-3ζ and early onset of recurrence was also observed by us in data from other large microarray gene expression studies, implying 14-3-3ζ to be a marker of a poor prognosis in women with ER-positive breast cancers (Bergamaschi et al 2009).
14-3-3ζ is a member of the highly conserved family of seven 14-3-3 proteins, all encoded by different genes (Tzivion et al 2006). 14-3-3ζ serves as a pivotal factor that binds and stabilizes key proteins involved in signal transduction, cell proliferation and apoptosis (Ando et al 2004, Costa 2005), including EGFR, HER2, PKC, β-catenin, and RAF-1 (Filipowicz et al 2008, McPherson et al 1999, Oksvold et al 2004).
In view of the very detrimental role of 14-3-3ζ in promoting cancer cell survival and endocrine resistance, we were interested in understanding how 14-3-3ζ is regulated, and in particular, how cellular levels of 14-3-3ζ might be down-regulated to enhance responsiveness to endocrine therapies. Since growing evidence links microRNAs (miRs) to the control of many crucial processes in cancer including proliferation, differentiation and apoptosis (Croce 2009), we have herein explored the involvement of miRs in the regulation of 14-3-3ζ. The great importance of miRs is underscored by the fact that they are estimated to play roles in regulating the expression of more than one-third of all human genes. miRs, which are small, ca. 22 nucleotide RNA sequences, bind principally to the 3’UTR region of mRNAs of protein-coding target genes to direct their repression by destabilizing target mRNAs and decreasing mRNA translational efficiency (Bartel 2004, Guo et al 2010, Harfe 2005).
In the present study, to investigate the mechanism underlying tamoxifen up-regulation of 14-3-3ζ, we first identified, by bioinformatic analysis, potential miRs that might regulate 14-3-3ζ, and we then went on to show tamoxifen down-regulation of miR-451 and to delineate the role of miR-451 in regulating 14-3-3ζ and its impact on the aggressiveness, cell survival properties, and endocrine sensitivity of ER-positive breast cancer cells. The findings suggest that increasing the cellular level of miR-451 might be a useful therapeutic approach for reversing endocrine resistance and enhancing the efficacy of endocrine therapies in the greater than 40% of breast tumors that overexpress 14-3-3ζ.
RESULTS
Tamoxifen down-regulates miR-451 and there is an inverse relationship between miR-451 and 14-3-3ζ levels
Although we have reported 14-3-3ζ to be up-regulated by the antiestrogen (SERM) tamoxifen in breast cancer (Frasor et al 2006), the mechanism regulating tamoxifen-driven stimulation of 14-3-3ζ is not known. Given the growing evidence for the importance of miRNAs in regulating gene expression, we envisioned that miRNAs might play an important role in modulating 14-3-3ζ expression. Using two different target prediction algorithms, TargetScan (http://www.targetScan.org/) and miRanda (http://www.microrna.org/microrna/home.do), we found nine miRNAs that were predicted by both algorithms to target 14-3-3ζ. From these, we chose to investigate miR-451 because it showed relatively few potential targets, and 14-3-3ζ was the only predicted target from the 14-3-3 family. We then examined whether tamoxifen might regulate miR-451. As shown in Fig.1A, where we monitored the effects of different estrogen receptor ligands on miR-451 expression, we observed that only Tam had the ability to down-regulate miR-451 and that down-regulation occurred as early as 4 h, whereas the estrogen estradiol (E2), the SERM raloxifene (Ral), and the pure antiestrogen ICI182,780 (ICI, Fulvestrant) had no effect on miR-451. The downregulation of miR-451 by Tam was reversed by excess ICI implying mediation of the Tam effect by estrogen receptor. Likewise, depletion of ERα by ca. 90% by cell treatment with siRNA targeting ERα (Chang et al 2008) eliminated the down-regulation of miR-451 by tamoxifen (Fig. 1A).
Fig. 1. miR-451 is down-regulated by tamoxifen in ER-positive breast cancer cells and its expression is inversely correlated with 14-3-3ζ in MCF7 and tamoxifen resistant (TamR) cells.
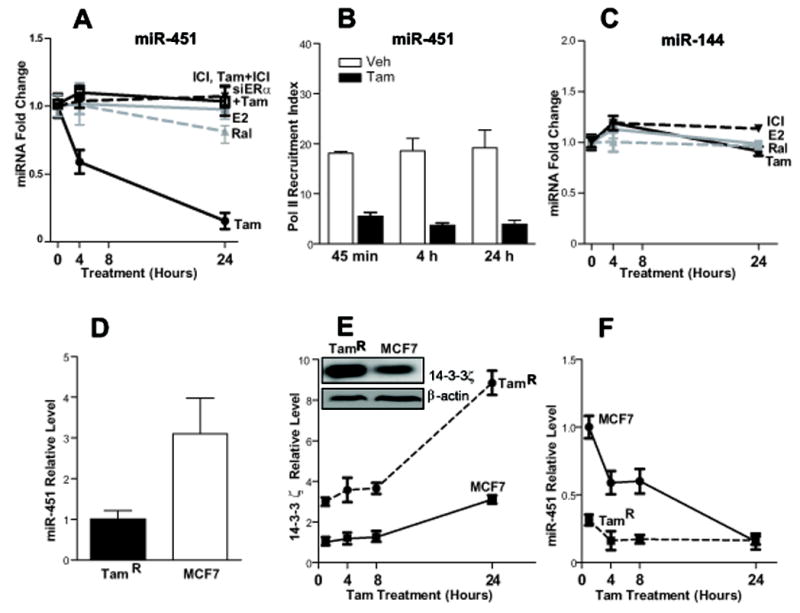
A) Regulation of miR-451 in MCF7 cells by the estrogen receptor ligands Tamoxifen (Tam, 1μM), Raloxifene (Ral, 1μM), estradiol (E2, 10nM), and ICI 182,780 (ICI Fulvestrant, 1μM) or 0.1μM Tam plus 3μM ICI, after 0, 4, and 24 h of treatment. ERα was also depleted from cells by treatment with siRNA for ERα prior to Tam treatment for 0, 4, and 24h. B) RNA Polymerase II (Pol II) recruitment to the transcription start site (TSS) of miR-451 in the presence of Veh or Tam for the times indicated. C) Absence of regulation of the adjacent miR-144 by the four estrogen receptor ligands. D) Levels of miR-451 in MCF7 and TamR breast cancer cells detected by qPCR. E-F) Change in 14-3-3ζ and miR-451 levels analyzed by quantitative RT-PCR in MCF7 and TamR cells after treatment with Tam. Values are fold expression compared with vehicle control. Each experiment was performed in triplicate with three experimental replicates. Insert in panel E shows 14-3-3ζ detected by Western blot.
To further explore the regulation of miR-451 by tamoxifen, we analyzed the presence over time of RNA polymerase II (Pol II) at the transcription start site (TSS) of miR-451 (TSS derived from data of Welboren et al. 2009) in cells treated with control vehicle or Tam. Interestingly, we found that Tam treatment markedly reduced the level of Pol II at the TSS of miR-451 (Fig.1B), consistent with the down-regulation of miR-451 expression we observed upon treatment with tamoxifen.
The highly conserved miR-451 gene is located on chromosome 17, only 100 bp upstream of another miR, miR-144. We therefore examined if this nearby miR-144 was also regulated by Tam or other ER ligands. As shown in Fig.1C, we observed that Tam had no effect on the expression of miR-144, indicating the selectivity of tamoxifen for miR-451 regulation.
We next examined the levels of miR-451 in parental MCF7 and in tamoxifen resistant MCF7 (TamR) cells and found that parental cells expressed 3-times more miR-451 than did TamR cells (Fig. 1D), whereas TamR cells had higher levels of 14-3-3ζ (Fig. 1E), suggesting an inverse relationship between these two factors. Further, upon treatment of both MCF7 and TamR cells with tamoxifen, we observed a time-dependent decrease in miR-451 and an increase in 14-3-3ζ over time (Fig. 1E and 1F). A similar inverse relationship between miR-451 and 14-3-3ζ was observed in other tamoxifen-resistant MCF7 cells selected in our laboratory (data not presented).
Overexpression or knock-down of miR-451 impacts 14-3-3ζ, cell proliferation and colony formation, apoptosis and sensitivity to SERMs
To further investigate miR-451 regulation of 14-3-3ζ, we overexpressed miR-451 by transfecting pri-miRNA into MCF7 and TamR cells and monitored mRNA and protein levels of 14-3-3ζ by qPCR and Western blot. As shown in Figure 2A and 2B, overexpression of miR-451 greatly reduced the level of 14-3-3ζ in both cell lines. We also examined the specificity of miR-451 action for targeting only 14-3-3ζ and not other 14-3-3 family members, and found that only 14-3-3ζ was significantly affected by expression of miR-451 (Fig. 2C).
Fig. 2. Regulation of 14-3-3ζ by miR-451 overexpression in MCF7 and TamR cells.
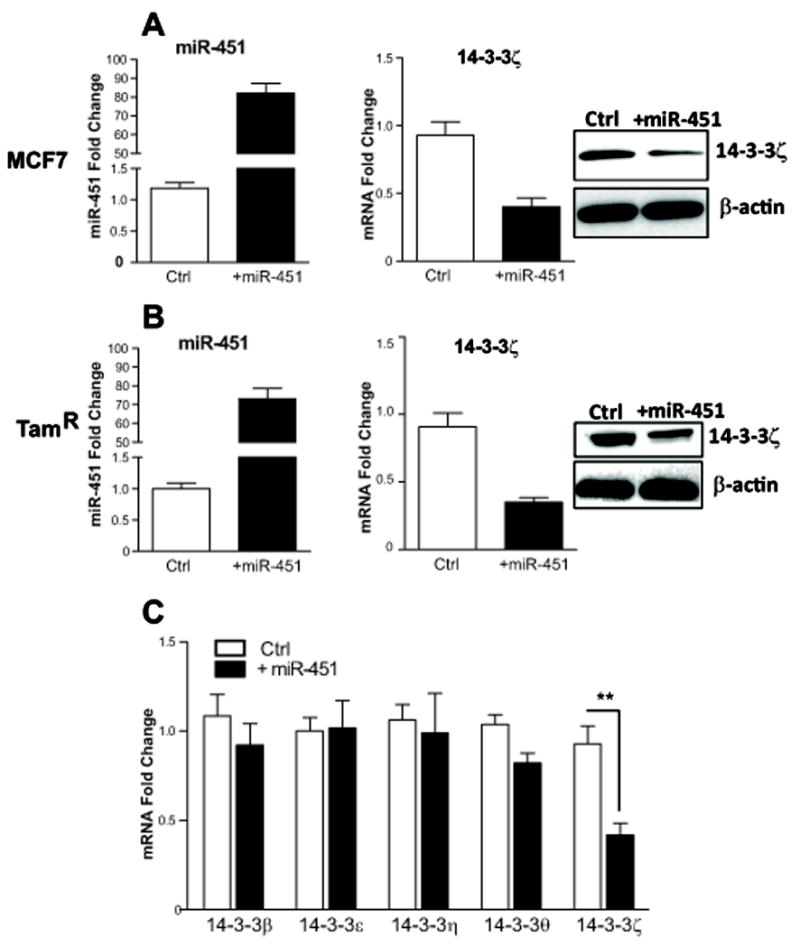
A-B) Overexpression of miR-451 resulted in the downregulation of 14-3-3ζ mRNA (quantified by RT-qPCR) and protein (by Western blot). Control (Ctrl) cells received vector only. Each experiment was performed in triplicate and mean ± SD is shown. C) Expression of five 14-3-3 family members in MCF7 cells without and with miR-451 overexpression. **, p<0.01.
We next examined the impact of miR-451 status on proliferation and apoptosis of breast cancer cells resistant to Tam, by either overexpressing or knocking-down miR-451. As shown in Fig. 3A, Tam acted as an agonist in TamR cells, enhancing the proliferation of these cells over vehicle control. Cells transfected with miR-451 showed greatly reduced proliferation and notably, this suppressive effect of miR-451 on proliferation was reversed by expression of 14-3-3ζ (Fig. 3A). Also, knock-down of miR-451 (miR-451 KD) by treatment of cells with miR-451 antagomir, which suppressed endogenous miR-451 and increased 14-3-3ζ levels (Fig. 3B), increased cell proliferation (Fig. 3C). To determine the impact of 14-3-3ζ alone on cell viability, we down-regulated its expression by siRNA and observed a significant decrease in basal and tamoxifen-stimulated cell proliferation. (Fig 3D). Overexpression of miR-451, which reduced 14-3-3ζ without affecting ERα protein or mRNA levels (Fig. 3F), resulted in suppression of the growth of TamR cells in the presence of the SERMs tamoxifen or Raloxifene, or Fulvestrant (ICI 182,780) (Fig. 3E). After overexpression of miR-451, we also observed a marked reduction in anchorage-independent colony formation of TamR cells, i.e. a significant decrease in number and size of colonies in the vehicle and Tam-treated cells, suggesting a potential tumor suppressor-like activity of this miR (Fig. 3G).
Fig. 3. The cellular level of miR-451 affects growth and colony formation.
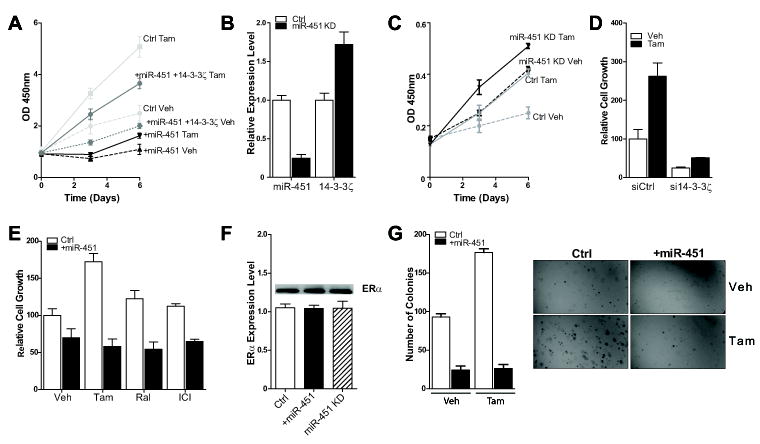
A) Growth of TamR cells in the presence of Tam or Veh was followed for times up to 6 days after transfection of pri-miR-451 (+miR-451) or Ctrl vector (Ctrl) or pri-miR-451 and 14-3-3ζ (+miR-451 + 14-3-3ζ). The growth index was assessed at day 0, 3, and 6. B) Effect of antagomiR-451transfection (denoted miR-451 KD) on miR-451 and 14-3-3ζ expression levels. C) Effect of miR-451 KD on viability of TamR cells. Cell viability was assessed at day 0, 3, and 6 of Tam or Veh treatment. D) After 14-3-3ζ knockdown by siRNA, growth of TamR cells in the presence of Tam or Veh was evaluated at day 6. E) Sensitivity to SERMs was assessed after 72 h treatment with control vehicle, or 1μM Tam, Raloxifene or ICI in cells with control plasmid (Ctrl) or with miR-451 overexpression. F) miR-451 overexpression or miR-451 downregulation has no impact on ERα mRNA or protein level. G) TamR cells were assayed for soft agar colony formation after overexpression of miR-451 or control plasmid and exposure to Tam or Veh. Number of colonies and colony size were assessed and photography of colonies is also presented.
To understand the basis of the reduced cell viability and colony formation in cells overexpressing miR-451, we monitored 14-3-3ζ, CASP7, and cleaved CASP7 protein upon Tam treatment. As seen in Fig. 4A, we observed greatly reduced 14-3-3ζ protein with miR-451 overexpression, as expected, and also elevated levels of CASP7 and cleaved CASP7. When 14-3-3ζ was re-expressed in cells overexpressing miR-451, the CASP7 and cleaved CASP7 levels were greatly decreased, becoming comparable to the low levels seen in control cells, indicating that miR-451, through altering intracellular14-3-3ζ, not only inhibits proliferation as shown in Fig. 3, but also stimulates the apoptotic cascade. These changes were also reflected in the marked increase in the proportion of cells in the sub-G1 phase of the cell cycle with miR-451 overexpression (Fig. 4B). There also was a great increase in the percent of apoptotic cells, rising from 2 to 13% and from 5 to 24% in vehicle or Tam treated cells, respectively (Fig.4B). The proportion of cells in the sub-G1 phase of the cell cycle, and the percent of apoptotic cells were significantly reduced when 14-3-3ζ was re-expressed in cells overexpressing miR-451 (Fig. 4B and C).
Fig. 4. Increasing the expression of miR-451 reduces 14-3-3ζ and stimulates apoptosis, and the effects of miR-451 are reversed by expression of 14-3-3ζ.
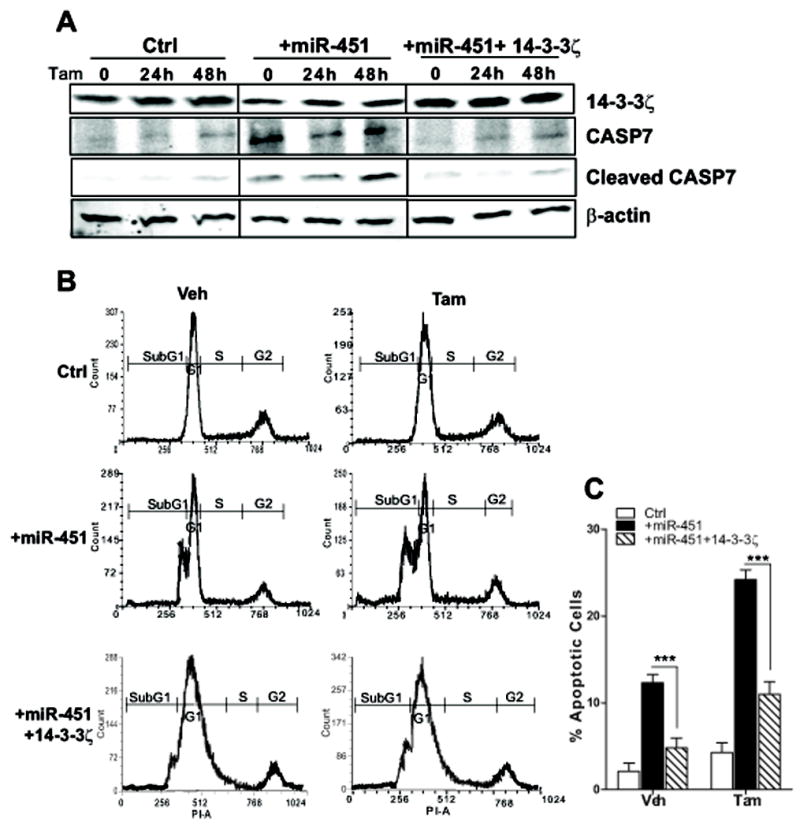
A) Western blots of whole cell extracts from vector, miR-451 alone, or miR-451 and 14-3-3ζ transfected MCF7 cells treated with Tam for 0, 24, or 48h and probed with antibodies against 14-3-3ζ and caspase-7 to detect intact and cleaved CASP7 products. β-actin was used as loading control. B) Control vector, miR-451, or miR-451 and 14-3-3ζ-expressing TamR cells were treated with 1μM Tam or control vehicle for 72 h, then fixed and stained with propidium iodide, and cell cycle distribution was monitored by flow cytometry. C) The percent of apoptotic cells (sub-G1 peak) in control vector (Ctrl), miR-451, and miR-451 and 14-3-3ζ -expressing cells is presented. ***, P < 0.001.
Elevated levels of miR-451 reduce the activation of growth factor receptor tyrosine kinases and protein kinases
Because endocrine resistance is often associated with the up-regulation of growth factor receptor and protein kinase signaling, we evaluated the effects of 14-3-3ζ and miR-451 on the activation of some growth factor receptors and kinases. We observed a significant increase in phosphorylation of HER2, EGFR, AKT and MAPK upon 14-3-3ζ overexpression (Fig. 5A). We also examined the impact of miR-451 on these receptors and downstream signaling pathways, and observed that levels of phosphoHER2 and phosphoEGFR were greatly reduced in cells overexpressing miR-451, and pMAPK was also decreased, although only slight reduction was observed in pAKT (Fig. 5B).
Fig. 5. miR-451 and 14-3-3ζ affect the activation of receptor tyrosine kinases and protein kinases.
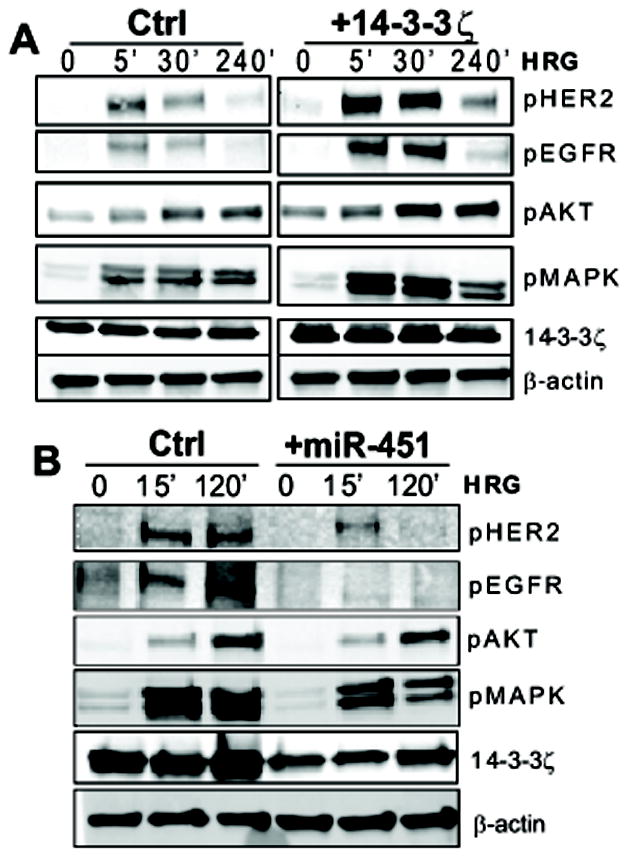
Immunoblotting for pHer2, pEGFR, pAKT, pMAPK, and 14-3-3ζ in TamR cells treated with heregulin (HRG) for the minutes indicated after cells were transfected with A) control or 14-3-3ζ expressing polylysine-coated adenovirus or B) control or pre-miR-451 vector to overexpress miR-451. β-actin was used as a loading control.
Selective action of miR-451 on 14-3-3ζ controls cell proliferation
To further delineate the importance of 14-3-3ζ in the actions of miR-451, we used a selective 14-3-3ζ target protector to examine the specificity of miR-451 action. Of the 14 targets for miR-451 predicted by the TargetScan algorithm, we found eight to be expressed in MCF7 cells. Knockdown of endogenous miR-451 increased only the level of 14-3-3ζ (Fig. 6A). However, with high overexpression of miR-451, 14-3-3ζ was down-regulated along with two other factors, O-GlcNAc transferase (OGT) and cyclin dependent N-kinase 2D (CDKN2D) (Fig. 6A).
Fig. 6. Effects of miR-451 knock-down or overexpression on potential target genes.
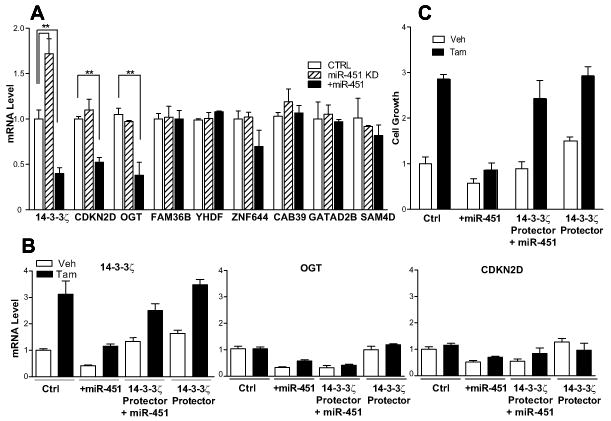
A) Expression levels of predicted target genes of miR-451, after knock-down of endogenous miR-451 or overexpression of miR-451 (**, P < 0.01). B) qPCR detection of expression levels of 14-3-3ζ, OGT or CDKN2D in vehicle or 1 μM Tam treated cells, after control vector (Ctrl) or after miR-451 overexpression and/or 14-3-3ζ target protector exposure. C) Growth of TamR cells, with vehicle or 1 μM Tam treatment, after control vector (Ctrl) or after miR-451 overexpression and/or 14-3-3ζ target protector exposure.
As shown in Fig. 6B, in control cells, tamoxifen only upregulated 14-3-3ζ, and had no effect on OGT or CDKN2D. These observations suggest that OGT and CDKN2D are less sensitive to miR-451 and, unlike 14-3-3ζ, are not suppressed by endogenous levels of this miR. To examine whether 14-3-3 ζ was primarily responsible for the impact of miR-451 on cellular behavior, we utilized an RNA binding antisense oligonucleotide specific for the interaction between miR-451 and the 3’UTR of 14-3-3ζ (target protector), so as to disrupt only this interaction. We monitored the levels of 14-3-3ζ, OGT, and CDKN2D in cells overexpressing miR-451, or 14-3-3ζ protector alone, or both combined (Fig. 6B). Overexpression of miR-451 reduced the expression of all three, but the addition of the 14-3-3ζ protector in miR-451 overexpressing cells restored the basal level and tamoxifen response of only 14-3-3ζ, reversing the effect of miR-451 overexpression. By contrast, there was no effect of the protector on OGT and CDKN2D with miR-451 overexpression. In cells exposed to 14-3-3ζ protector alone, there was an increase in the basal (Veh) level of 14-3-3ζ but no effect on OGT or CDKN2D, as would be expected from reduction in the effect of endogenous miR-451 on 14-3-3ζ.
We then examined the effect of these perturbations on the growth of TamR cells (Fig. 6C). As shown previously in Fig. 3, miR-451 knock-down increased 14-3-3ζ and cell proliferation whereas miR-451 overexpression suppressed both basal and tamoxifen-stimulated proliferation, and these were restored to the levels in control (Ctrl) cells by co-presence of the 14-3-3ζ protector (Fig. 6C). The protector alone raised the proliferation rate of vehicle (Veh) treated cells, consistent with its effect on the endogenous 14-3-3ζ level, shown in Fig. 6B, left panel. Collectively, these results support the hypothesis that the effects of both up and down regulation of miR-451 on cell proliferation and response to tamoxifen are mediated principally by miR-451 regulation of 14-3-3ζ levels. Our overall findings, schematically depicted in the model in Fig. 7, show that tamoxifen decreases endogenous miR-451, thereby increasing the level of 14-3-3ζ. 14-3-3ζ promotes breast cancer cell proliferation and survival and receptor tyrosine kinase (EGFR, HER2) activation and protein kinase signaling while suppressing apoptosis, all of which support the progression to endocrine resistance.
Fig. 7. Schematic representation of the effect of tamoxifen on miR-451 and 14-3-3ζ regulation and their impact on breast cancer cell phenotypic properties leading to tamoxifen resistance.
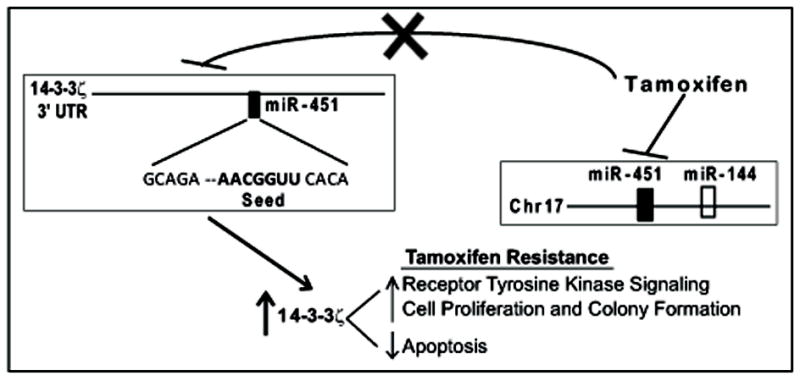
Tamoxifen down-regulates miR-451, resulting in the up-regulation of 14-3-3ζ, with consequent increased receptor tyrosine kinase signaling, increased cell proliferation and colony formation, and reduced apoptosis, thereby leading to tamoxifen resistance.
DISCUSSION
The development of resistance to endocrine therapy is a severe limitation in the treatment of hormone-receptor positive breast tumors. In this study, we provide evidence for a novel mechanism by which tamoxifen controls 14-3-3ζ levels through its regulation of the microRNA, miR-451. It is becoming increasingly clear that miRNAs have a profound impact on many pathologic and physiologic processes, including proliferation, differentiation, and apoptosis (Bartel 2004, Harfe 2005), by dampening the expression of target genes and thereby affording finely tuned cellular regulation. Lowered mRNA levels appear to be the predominant mode of miR regulation, although decreased translational efficiency often contributes to reduced protein output as well (Guo et al 2010).
Our previous studies described the up-regulation of 14-3-3ζ by tamoxifen and revealed that high expression of 14-3-3ζ in primary breast cancers was associated with a poor clinical outcome on tamoxifen (Frasor et al 2006). 14-3-3ζ, which is harbored in a region of frequent genomic gain (8q23), is overexpressed in greater than 40% of breast tumors (Neal et al 2009) and is overexpressed also in other types of aggressive solid tumors, such as lung cancer (Li et al 2008). Thus, 14-3-3ζ has properties of an oncogene, yet surprisingly, its regulation in breast cancer has been largely unknown.
It is of note that the regulation of 14-3-3ζ and miR-451 is selective for tamoxifen and is not brought about by other ER ligands tested, including the estrogen estradiol or the antiestrogens raloxifene and ICI 182,780, highlighting the remarkable ability of distinct ER-ligand complexes to selectively affect the transcription of specific genes (Frasor et al 2004, Frasor et al 2006, Katzenellenbogen and Katzenellenbogen 2002, Katzenellenbogen et al 1996, Shang and Brown 2002). In all cases we examined, we observed an inverse relationship between miR-451 and 14-3-3ζ, implying the importance of this miRNA in the regulation of 14-3-3ζ. This regulation of miR-451 by tamoxifen was also highly selective, as tamoxifen did not affect the nearby miR-144. The differential regulation of miR-451 and 14-3-3ζ by tamoxifen vs. raloxifene is reminiscent of other differences also observed by us previously when genome-wide gene expression comparison analyses of these two SERMs were performed (Frasor et al 2004). These differential effects of the two SERMs in regulation of miR-451 and 14-3-3ζ may underlie some differences in their activities observed in the STAR (Study of Tamoxifen and Raloxifene) breast cancer prevention trial (Dunn and Ford 2001, Vogel et al 2006, Vogel et al 2010).
Previous reports have noted that miR-451 is very highly conserved among vertebrates, and that its maturation is independent of Dicer (Yang et al 2010). Moreover, miR-451 has been shown recently to play a crucial role in erythrocyte differentiation by targeting 14-3-3ζ (Dore et al 2008, Masaki et al 2007, Patrick et al 2010, Yu et al 2010). Expression of miR-451 has also been detected in normal gastric mucosa and found to be decreased in gastric cancer (Bandres et al 2009). These findings imply that miR-451 is required for the development and maintenance of normal tissues (Williams et al 2007) and may be down-regulated during the progression to cancer. Of note, we found that our less aggressive cell line (MCF7) showed higher expression of miR-451 compared to the more aggressive tamoxifen resistant cell line. We have also observed up-regulation of 14-3-3ζ and down-regulation of miR-451 by tamoxifen in ER-positive, HER2-positive BT474 cells (data not presented). Therefore, the loss of miR-451 induced by Tam treatment appears to be an underlying mechanism by which ER-positive breast cancer cells become resistant to tamoxifen therapy by upregulation of 14-3-3ζ. As supporting evidence for this, we show that reduction of miR-451 by tamoxifen treatment or knock-down of miR-451 by a specific antagomir increased 14-3-3ζ and cell survival, whereas increasing miR-451 could resensitize TamR cells to SERMs, as observed by the decrease in cell viability and suppression of anchorage-independent growth that correlated with increased apoptosis and reduced EGFR, HER2 and MAPK signaling in tamoxifen treated miR-451 overexpressing cells. This sensitization of tamoxifen resistant cells to the growth inhibitory effects of Tam and other antiestrogens by increased expression of miR-451 suggests that restoration of expression of this miRNA in endocrine therapy-resistant cancer cells might have important implications for effective breast cancer treatment.
In summary, we document miR-451 to be a potent suppressor of the progression to tamoxifen resistance through its ability to target 14-3-3ζ, a key proliferative and antiapoptotic factor in breast cancer. Modulation of miR-451 status impacted growth, apoptosis, receptor tyrosine kinase activity and sensitivity to SERMs. We identify miR-451 as a target of the ER-tamoxifen complex and show an inverse correlation between cellular miR-451 and 14-3-3ζ levels. Collectively, these data imply that increasing the level of miR-451 may have beneficial therapeutic effects in the large proportion of breast cancers expressing high levels of 14-3-3ζ. Progress in microRNA-directed therapeutic approaches (Lowery et al 2008, Shan et al 2008) offers hope that such strategies might prove useful in reversing endocrine resistance and reducing breast cancer recurrence.
MATERIAL AND METHODS
Cell cultures, constructs and cell transfections
MCF7 cells, obtained from the American Type Culture Collection (Manassas, VA)and tamoxifen resistant MCF7 cells (TamR cells) described previously (Herman and Katzenellenbogen 1996), were cultured in MEM (Sigma-Aldrich Corp., St. Louis, MO) supplemented with 5% calf serum (HyClone, Logan, UT), 100 μg/ml penicillin/streptomycin (Invitrogen, Carlsbad, CA), and 25 μg/ml gentamicin (Invitrogen). Four days before control vehicle or ligand treatments, cells were seeded in phenol red-free MEM containing 5% charcoal-dextran-treated calf serum. Medium was changed on day 2 and 4 of culture before treatments. Pri-miRNA-451 was purchased from Origene, and antagomiR-451, antagomiR-144 and negative control antagomiRs were purchased from Applied Biosystems. A 40nt single-stranded RNA binding antisense oligonucleotide specific for the interaction between miR-451 and the 3’UTR of 14-3-3ζ (target protector) was purchased from Qiagen. SiRNA targeting ERα was as described previously (Chang et al 2008). Ectopic overexpression of 14-3-3ζ used lysine-coated adenovirus or control adenovirus (Allgood et al 1997). Cell transfections were performed with Fugene 6 (Roche) or Dharmafect (Dharmacon).
RT-PCR and Quantitative PCR
Total RNA was isolated from cells using TRIzol (Invitrogen), RNA samples were reverse transcribed by SuperScript II reverse transcriptase (Invitrogen), and real-time PCR was carried out on the ABI Prism 7900HT using SYBR Green PCR Master Mix (Applied Biosystems) as described previously (Frasor et al 2006).
Western blot analysis
Whole-cell extracts were prepared using 1X RIPA lysis buffer (Upstate/Chemicon) supplemented with 1X complete protease inhibitor (Roche). Proteins were separated on 15% SDS-PAGE gels and transferred to nitrocellulose membranes. Western blotting used antibodies against 14-3-3ζ (Santa Cruz Biotechnology, Santa Cruz, CA), β-actin (Sigma-Aldrich Corp., St. Louis, MO), pEGFR, pHER2, pMAPK and pAKT (Cell Signaling).
Cell proliferation assays
WST-1 assay (Roche, Basel, Switzerland) was used to quantify cell viability. Absorbance was measured at 450nm using a BioRad 680 Microplate Reader, and all assays were performed in triplicate.
Apoptosis assays
Apoptosis was measured based on DNA content and analyzed by flow cytometry using BD-FACS Canto. Cells were fixed in 70% ethanol, stained for 30 min with 20ug/ml propidium iodide (PI, Molecular Probe) in Triton-X (Sigma) in presence of DNAse-free RNAse A (Sigma), and PI staining was measured as previously reported (Riccardi and Nicoletti 2006).
Soft-agar colony formation assays
A 1.5-ml base layer of agar (0.5% agar in phenol red-free DMEM with 5% charcoal stripped-FCS) was allowed to solidify in a six-well flat-bottomed plate before the addition of 1.5 ml of cell suspensions containing 4,000 cells in 0.35% agar in phenol red-free DMEM with 5% charcoal stripped-FCS. The cell-containing layer was then solidified at 4°C for 20 min. Colonies were allowed to grow for 13 days at 37°C before imaging.
Acknowledgments
This research was supported by grants from The Breast Cancer Research Foundation (B.S.K.) and the NIH (P01AG024387 and P50 AT006268, B.S.K.) and a Postdoctoral Fellowship from the Department of Defense (W81XWH-09-1-0398, A.B.). We thank Dr. Nancy Weigel, Baylor College of Medicine, for providing lysine-coated adenovirus.
Abbreviations
- ER
estrogen receptor
- ICI
ICI 182,780, fulvestrant
- miR
microRNA
- Ral
raloxifene
- SERM
selective estrogen receptor modulator
- Tam
tamoxifen
Footnotes
CONFLICT OF INTEREST The authors declare no conflict of interest.
References
- Allgood VE, Zhang Y, O’Malley BW, Weigel NL. Analysis of chicken progesterone receptor function and phosphorylation using an adenovirus-mediated procedure for high-efficiency DNA transfer. Biochemistry. 1997;36:224–232. doi: 10.1021/bi961125c. [DOI] [PubMed] [Google Scholar]
- Ando K, Ozaki T, Yamamoto H, Furuya K, Hosoda M, Hayashi S, et al. Polo-like kinase 1 (Plk1) inhibits p53 function by physical interaction and phosphorylation. The Journal of biological chemistry. 2004;279:25549–25561. doi: 10.1074/jbc.M314182200. [DOI] [PubMed] [Google Scholar]
- Arpino G, Wiechmann L, Osborne CK, Schiff R. Crosstalk between the estrogen receptor and the HER tyrosine kinase receptor family: molecular mechanism and clinical implications for endocrine therapy resistance. Endocrine reviews. 2008;29:217–233. doi: 10.1210/er.2006-0045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandres E, Bitarte N, Arias F, Agorreta J, Fortes P, Agirre X, et al. microRNA-451 regulates macrophage migration inhibitory factor production and proliferation of gastrointestinal cancer cells. Clin Cancer Res. 2009;15:2281–2290. doi: 10.1158/1078-0432.CCR-08-1818. [DOI] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- Bergamaschi A, Frasor J, Kieser K, Wiley E, Katzenellenbogen B. A breast cancer gene signature associated with high expression of 14-3-3z predicts poor clinical outcome on endocrine therapy. Proceedings Amer Assoc Cancer Research. 2009:2854. 2_Annual_Meeting. [Google Scholar]
- Chang EC, Charn TH, Park SH, Helferich WG, Komm B, Katzenellenbogen JA, et al. Estrogen Receptors alpha and beta as determinants of gene expression: influence of ligand, dose, and chromatin binding. Mol Endocrinol. 2008;22:1032–1043. doi: 10.1210/me.2007-0356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa RH. FoxM1 dances with mitosis. Nat Cell Biol. 2005;7:108–110. doi: 10.1038/ncb0205-108. [DOI] [PubMed] [Google Scholar]
- Creighton CJ, Hilger AM, Murthy S, Rae JM, Chinnaiyan AM, El-Ashry D. Activation of mitogen-activated protein kinase in estrogen receptor alpha-positive breast cancer cells in vitro induces an in vivo molecular phenotype of estrogen receptor alpha-negative human breast tumors. Cancer Res. 2006;66:3903–3911. doi: 10.1158/0008-5472.CAN-05-4363. [DOI] [PubMed] [Google Scholar]
- Croce CM. Causes and consequences of microRNA dysregulation in cancer. Nat Rev Genet. 2009;10:704–714. doi: 10.1038/nrg2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dore LC, Amigo JD, Dos Santos CO, Zhang Z, Gai X, Tobias JW, et al. A GATA-1-regulated microRNA locus essential for erythropoiesis. Proc Natl Acad Sci U S A. 2008;105:3333–3338. doi: 10.1073/pnas.0712312105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn BK, Ford LG. From adjuvant therapy to breast cancer prevention: BCPT and STAR. Breast J. 2001;7:144–157. doi: 10.1046/j.1524-4741.2001.007003144.x. [DOI] [PubMed] [Google Scholar]
- EBCTCG. Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. Lancet. 2005;365:1687–1717. doi: 10.1016/S0140-6736(05)66544-0. [DOI] [PubMed] [Google Scholar]
- Filipowicz W, Bhattacharyya SN, Sonenberg N. Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat Rev Genet. 2008;9:102–114. doi: 10.1038/nrg2290. [DOI] [PubMed] [Google Scholar]
- Frasor J, Stossi F, Danes JM, Komm B, Lyttle CR, Katzenellenbogen BS. Selective estrogen receptor modulators: discrimination of agonistic versus antagonistic activities by gene expression profiling in breast cancer cells. Cancer Res. 2004;64:1522–1533. doi: 10.1158/0008-5472.can-03-3326. [DOI] [PubMed] [Google Scholar]
- Frasor J, Chang EC, Komm B, Lin CY, Vega VB, Liu ET, et al. Gene expression preferentially regulated by tamoxifen in breast cancer cells and correlations with clinical outcome. Cancer Res. 2006;66:7334–7340. doi: 10.1158/0008-5472.CAN-05-4269. [DOI] [PubMed] [Google Scholar]
- Green KA, Carroll JS. Oestrogen-receptor-mediated transcription and the influence of co-factors and chromatin state. Nature reviews. 2007;7:713–722. doi: 10.1038/nrc2211. [DOI] [PubMed] [Google Scholar]
- Guo H, Ingolia NT, Weissman JS, Bartel DP. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature. 2010;466:835–840. doi: 10.1038/nature09267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harfe BD. MicroRNAs in vertebrate development. Curr Opin Genet Dev. 2005;15:410–415. doi: 10.1016/j.gde.2005.06.012. [DOI] [PubMed] [Google Scholar]
- Herman ME, Katzenellenbogen BS. Response-specific antiestrogen resistance in a newly characterized MCF-7 human breast cancer cell line resulting from long-term exposure to trans-hydroxytamoxifen. J Steroid Biochem Mol Biol. 1996;59:121–134. doi: 10.1016/s0960-0760(96)00114-8. [DOI] [PubMed] [Google Scholar]
- Jaiyesimi IA, Buzdar AU, Decker DA, Hortobagyi GN. Use of tamoxifen for breast cancer: twenty-eight years later. J Clin Oncol. 1995;13:513–529. doi: 10.1200/JCO.1995.13.2.513. [DOI] [PubMed] [Google Scholar]
- Katzenellenbogen BS, Katzenellenbogen JA. Biomedicine. Defining the “S” in SERMs. Science. 2002;295:2380–2381. doi: 10.1126/science.1070442. [DOI] [PubMed] [Google Scholar]
- Katzenellenbogen JA, O’Malley BW, Katzenellenbogen BS. Tripartite steroid hormone receptor pharmacology: interaction with multiple effector sites as a basis for the cell- and promoter-specific action of these hormones. Mol Endocrinol. 1996;10:119–131. doi: 10.1210/mend.10.2.8825552. [DOI] [PubMed] [Google Scholar]
- Li Z, Zhao J, Du Y, Park HR, Sun SY, Bernal-Mizrachi L, et al. Down-regulation of 14-3-3zeta suppresses anchorage-independent growth of lung cancer cells through anoikis activation. Proc Natl Acad Sci U S A. 2008;105:162–167. doi: 10.1073/pnas.0710905105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowery AJ, Miller N, McNeill RE, Kerin MJ. MicroRNAs as prognostic indicators and therapeutic targets: potential effect on breast cancer management. Clin Cancer Res. 2008;14:360–365. doi: 10.1158/1078-0432.CCR-07-0992. [DOI] [PubMed] [Google Scholar]
- Masaki S, Ohtsuka R, Abe Y, Muta K, Umemura T. Expression patterns of microRNAs 155 and 451 during normal human erythropoiesis. Biochem Biophys Res Commun. 2007;364:509–514. doi: 10.1016/j.bbrc.2007.10.077. [DOI] [PubMed] [Google Scholar]
- Massarweh S, Osborne CK, Creighton CJ, Qin L, Tsimelzon A, Huang S, et al. Tamoxifen resistance in breast tumors is driven by growth factor receptor signaling with repression of classic estrogen receptor genomic function. Cancer Res. 2008;68:826–833. doi: 10.1158/0008-5472.CAN-07-2707. [DOI] [PubMed] [Google Scholar]
- McPherson RA, Harding A, Roy S, Lane A, Hancock JF. Interactions of c-Raf-1 with phosphatidylserine and 14-3-3. Oncogene. 1999;18:3862–3869. doi: 10.1038/sj.onc.1202730. [DOI] [PubMed] [Google Scholar]
- Neal CL, Yao J, Yang W, Zhou X, Nguyen NT, Lu J, et al. 14-3-3{zeta} Overexpression Defines High Risk for Breast Cancer Recurrence and Promotes Cancer Cell Survival. Cancer Res. 2009;69:3425–3432. doi: 10.1158/0008-5472.CAN-08-2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oksvold MP, Huitfeldt HS, Langdon WY. Identification of 14-3-3[zeta] as an EGF receptor interacting protein. FEBS Letters. 2004;569:207–210. doi: 10.1016/j.febslet.2004.05.068. [DOI] [PubMed] [Google Scholar]
- Osborne CK. Tamoxifen in the treatment of breast cancer. The New England journal of medicine. 1998;339:1609–1618. doi: 10.1056/NEJM199811263392207. [DOI] [PubMed] [Google Scholar]
- Patrick DM, Zhang CC, Tao Y, Yao H, Qi X, Schwartz RJ, et al. Defective erythroid differentiation in miR-451 mutant mice mediated by 14-3-3{zeta} Genes Dev. 2010;24:1614–1619. doi: 10.1101/gad.1942810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riccardi C, Nicoletti I. Analysis of apoptosis by propidium iodide staining and flow cytometry. Nat Protocols. 2006;1:1458–1461. doi: 10.1038/nprot.2006.238. [DOI] [PubMed] [Google Scholar]
- Shan G, Li Y, Zhang J, Li W, Szulwach KE, Duan R, et al. A small molecule enhances RNA interference and promotes microRNA processing. Nat Biotechnol. 2008;26:933–940. doi: 10.1038/nbt.1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang Y, Brown M. Molecular determinants for the tissue specificity of SERMs. Science. 2002;295:2465–2468. doi: 10.1126/science.1068537. [DOI] [PubMed] [Google Scholar]
- Tzivion G, Gupta VS, Kaplun L, Balan V. 14-3-3 proteins as potential oncogenes. Semin Cancer Biol. 2006;16:203–213. doi: 10.1016/j.semcancer.2006.03.004. [DOI] [PubMed] [Google Scholar]
- Vogel VG, Costantino JP, Wickerham DL, Cronin WM, Cecchini RS, Atkins JN, et al. Effects of tamoxifen vs raloxifene on the risk of developing invasive breast cancer and other disease outcomes: the NSABP Study of Tamoxifen and Raloxifene (STAR) P-2 trial. JAMA. 2006;295:2727–2741. doi: 10.1001/jama.295.23.joc60074. [DOI] [PubMed] [Google Scholar]
- Vogel VG, Costantino JP, Wickerham DL, Cronin WM, Cecchini RS, Atkins JN, et al. Update of the National Surgical Adjuvant Breast and Bowel Project Study of Tamoxifen and Raloxifene (STAR) P-2 Trial: Preventing Breast Cancer. Cancer Prevention Research. 2010;3:696–706. doi: 10.1158/1940-6207.CAPR-10-0076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams AE, Perry MM, Moschos SA, Lindsay MA. microRNA expression in the aging mouse lung. BMC Genomics. 2007;8:172. doi: 10.1186/1471-2164-8-172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JS, Maurin T, Robine N, Rasmussen KD, Jeffrey KL, Chandwani R, et al. Conserved vertebrate mir-451 provides a platform for Dicer-independent, Ago2-mediated microRNA biogenesis. Proc Natl Acad Sci U S A. 2010;107:15163–15168. doi: 10.1073/pnas.1006432107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu D, Dos Santos CO, Zhao G, Jiang J, Amigo JD, Khandros E, et al. miR-451 protects against erythroid oxidant stress by repressing 14-3-3{zeta} Genes Dev. 2010;24:1620–1633. doi: 10.1101/gad.1942110. [DOI] [PMC free article] [PubMed] [Google Scholar]