

Abstract
Disruption of either intercellular or extracellular junctions involved in maintaining endothelial barrier function can result in increased endothelial permeability. Increased endothelial permeability, in turn, allows for the unregulated movement of fluid and solutes out of the vasculature and into the surrounding connective tissue, contributing to a number of disease states, including stroke and pulmonary edema (Ermert et al., 1995; Lee and Slutsky, 2010; van Hinsbergh, 1997; Waller et al., 1996; Warboys et al., 2010). Thus, a better understanding of the molecular mechanisms by which endothelial cell junction integrity is controlled is necessary for development of therapies aimed at treating such conditions. In this review, we will discuss the functions of three signaling molecules known to be involved in regulation of endothelial permeability: focal adhesion kinase (FAK), protein kinase C delta (PKCδ), and p190RhoGAP (p190). We will discuss the independent functions of each protein, as well as the interplay that exists between them and the effects of such interactions on endothelial function.
Keywords: FAK, PKC, p190RhoGAP, endothelium, permeability, focal adhesions
Introduction
The endothelium forms the innermost lining of all body vessels. This single layer of cells functions as a semipermeable barrier, serving to regulate the exchange of fluid and solutes between the vascular compartment and the interstitial space (Deanfield et al., 2005). The integrity of this barrier is dependent upon the adhesions between adjacent endothelial cells, as well as the adhesions of the endothelial cells to the underlying extracellular matrix (Dejana et al., 1995; Dejana and Del Maschio, 1995). Interendothelial cell tight junctions and adherens junctions prevent uncontrolled paracellular transport of substances, including plasma proteins and white blood cells, through the endothelium and into the surrounding connective tissue. At the same time, numerous transmembrane protein complexes, known as focal adhesions, function in endothelial barrier regulation by modulating the attachment of individual endothelial cells to the underlying basement membrane. However, recent studies indicate that focal adhesions can also affect the signaling pathways that control cell survival and differentiation, as well as those involved in cell migration. In order to mediate such a diverse array of functions, signaling at focal adhesions occurs in a dynamic nature. Several molecules controlling protein phosphorylation and organization of the actin cytoskeleton are particularly crucial in focal adhesion function; included in these are focal adhesion kinase and protein kinase C, as well as the RhoA regulatory protein, p190RhoGAP.
Regulation of Endothelial Permeability by FAK, PKCδ, and p190RhoGAP
Focal Adhesion Kinase (FAK)
Focal adhesion kinase (FAK) is a ubiquitously expressed cytoplasmic protein tyrosine kinase involved in the regulation of numerous endothelial cell functions. It serves a critical role in vascular endothelial growth factor (VEGF)-induced angiogenesis and vascular patterning; mice with endothelial-specific deletion of FAK die prior to birth (Eliceiri et al., 2002; Shen et al., 2005). FAK is also involved in endothelial cell apoptosis, in the response to a number of barrier agonists (Bellas et al., 2002; Claesson-Welsh et al., 1998; Kabir et al., 2002), and in the regulation of endothelial permeability (Guo et al., 2005; Harrington et al., 2005; Holinstat et al., 2006).
As its name implies, FAK was first characterized according to its role at focal adhesion complexes (Schlaepfer and Hunter, 1996). Focal adhesions are comprised of combinations of between 50–100 different proteins, giving the structures considerable heterogeneity and dynamic signaling properties. However, all focal adhesions contain two major components: integrins and FAK (Sieg et al., 1999). Integrins are transmembrane glycoproteins which serve as tethers between the intracellular cytoskeleton and the protein components of the extracellular matrix. Each integrin molecule is composed of a heterodimeric pairing of various α and β subunits. To date, 18 α and 8 β subunits have been identified, giving rise to 24 possible integrin pairs, each with distinct adhesion receptor properties (Hynes, 2002). The integrin cytoplasmic domains mediate binding to actin filaments, either directly or indirectly through adaptor proteins including paxillin, vinculin, talin, and α-actinin. Integrin adhesion to underlying matrix components, such as vitronectin and fibronectin, is transduced through the integrin extracellular domains. It was traditionally thought that integrin-matrix engagement and subsequent integrin receptor clustering at sites of cell-extracellular matrix adhesion serves as the stimulus for recruitment of FAK (Mitra and Schlaepfer, 2006). However, recent data also suggests that FAK promotes the activation of integrins, resulting in altered force generation at cell-extracellular matrix interactions (Michael et al., 2009).
Upon integrin engagement, FAK is directed from the cytoplasm to sites of focal adhesions through a focal adhesion targeting (FAT) sequence, within its 140 amino acid carboxyl-terminus (Hildebrand et al., 1993). The amino-terminal FERM (protein 4.1, ezrin, radizin, and moesin homology) domain functions to maintain FAK in an autoinhibited state by masking the catalytic domain (Lietha et al., 2007), as well as to facilitate the physical binding of FAK to integrin proteins, certain growth factor receptors, and the actin polymerizing protein complex, Arp2/3 (Chen and Chen, 2006; Serrels et al., 2007; Sieg et al., 2000). Integrin binding induces the autophosphorylation of FAK at tyrosine 397 (Hamadi et al., 2005; Hanks and Polte, 1997), within the carboxyl-terminal region, which in turn facilitates binding to the SH2 domains of Src or phosphatidylinositol 3-kinase (PI 3-kinase). The FAK protein complex then mediates the phosphorylation and activation of several key molecules important in cell adhesion and migration, including paxillin and p130Cas (Mitra and Schlaepfer, 2006; Schaller et al., 1994; Vuori, 1998). When FAK is molecularly inhibited or ablated, cell spreading and stress fiber formation is inhibited and nuclei become condensed and lobular (Almeida et al., 2000; Ilic et al., 1998; Ilic et al., 1995; Richardson et al., 1997; Sieg et al., 1999).
In addition to its role in cell-extracellular matrix adhesion, FAK has more recently been shown to contribute to endothelial cell-cell adhesion. The vascular barrier agonist, sphingosine 1-phosphate (S1P), a sphingolipid which has been shown to enhance vascular endothelial integrity (Garcia et al., 2001), induces a redistribution of FAK, and its binding partner paxillin, from internal sites of extracellular matrix adhesion to the cell periphery (Sun et al., 2009). Once at the cell periphery, FAK associates with VE-cadherin-mediated adherens junctions through its binding to β-catenin and p120-catenin (Knezevic et al., 2009). This interaction between FAK and adherens junction proteins fosters the establishment of the cortical actin ring. FAK was also shown to play a critical role in the reannealing of adherens junctions following settings of endothelial barrier dysfunction. PAR1 receptor activation via thrombin was shown to promote the release of the heterotrimeric Gβγ subunit from its sequestering protein, receptor for activated C kinase 1 (RACK1), and subsequent binding to and activation of Fyn and FAK (Knezevic et al., 2009). This sequence of events resulted in FAK binding to p120-catenin and reannealing of adherens junctions (Knezevic et al., 2009). By transiently overexpressing various forms of FAK, others have also demonstrated cross-talk between FAK and adherens junctions in the regulation of endothelial barrier function (Quadri and Bhattacharya, 2007; Usatyuk and Natarajan, 2005). In a recent study in which a kinase defective FAK protein that was conditionally knocked into the endothelium, it was shown that, while kinase dead FAK is protective against endothelial cell apoptosis, endothelial cell barrier function and the intercellular localization of VE-cadherin at the adherens junctions is dependent upon FAK kinase activity (Zhao et al., 2010). In addition, activation of the endothelial cell-specific, extracellular matrix bound integrin, αvβ3, caused the redistribution of this integrin to the cell periphery, diminution of VE-cadherin at inter-endothelial cell junctions, FAK activation, and concomitant barrier dysfunction (Alghisi et al., 2009). Thus FAK is a key regulator of endothelial barrier function at both integrin-mediated cell-extracellular matrix interactions, as well as intercellular adherens junctions.
Protein kinase C
Protein kinase C (PKC) is a family of serine/ threonine kinases consisting of ten known isoforms. These isoforms are divided into three subfamilies, based on their domain composition and corresponding cofactor requirements (Newton, 1995). All of the PKC enzymes possess a highly conserved carboxyl-terminal kinase domain which is autoinhibited by the binding of the amino-terminal pseudosubstrate domain within the substrate cleft of the catalytic domain. The PKC isoforms are catalytically activated upon: cofactor binding; sequential phosphorylation of select serine or threonine residues by phosphoinositide-dependent kinase-1 (PDK-1) and mammalian target of rapamycin (mTOR) complex 2 (mTORC2); and/ or allosteric interactions with key effector proteins (Newton, 2010; Rosse et al., 2010). The first subfamily, the conventional (c) PKC enzymes, is comprised of the α, βI, βII, and γ isoforms. Within the N-terminal regulatory region, these enzymes contain a C1 domain, which binds either diacylglycerol (DAG) or the pharmacologic analogue, phorbol ester. The C2 domain binds to the head groups of phospholipids, such as phosphatidylserine, in the presence of calcium. In order to be activated, both the C1 and C2 domains must be engaged, thus requiring the presence of calcium, diacylglycerol (DAG), and a phospholipid. The second group of PKC enzymes, the novel (n) family, is comprised of the δ, ε, η, and θ isoforms. Like the cPKC isoforms, activation of the nPKC enzymes requires binding to DAG/ phorbol esters, as well as to a phospholipid; however, due to a variation in the amino acid sequence of the C2 domain, the presence of calcium is not necessary. The third subfamily of PKC enzymes contains the atypical (a) isoforms, ζ and ι/λ. The aPKC enzymes, which lack functional C1 and C2 domains, do not require DAG or calcium for activation. Instead, aPKC are activated primarily by protein-protein interactions with the partitioning defective 6 (PAR6)-CDC42 protein complex (Suzuki et al., 2001). Much work has been done to elucidate signaling pathways upstream and downstream of the PKC isoforms since their discovery in the 1980s, however substrate specificity of each of the isoforms is still not known.
Inhibitor studies have suggested proteins associated with the cytoskeleton or adherens junction may serve as substrates for PKC and thus may be important in the modulation of thrombin-induced changes in endothelial monolayer permeability. Studies examining signaling molecules important in inducing lung edema showed that infusion of PKC activating agents induced edema formation, while infusion of PKC inhibitors blocked the effects of edemagenic agents in lung edema formation (Johnson et al., 1990; Johnson et al., 1989; Siflinger-Birnboim et al., 1992). More recently, phorbol ester-induced microvascular endothelial monolayer permeability was shown to require PKCδ, but not PKCα, βI, or ε (Tinsley et al., 2004). Others have demonstrated increased endothelial barrier dysfunction upon modulation of PKCα, PKCβI, or PKCζ activities and/or expression (Ferro et al., 2000; Huang et al., 2005; Li et al., 2004; Nagpala et al., 1996). PKCα activation promoted endothelial barrier disruption in response to a variety of edemagenic agents, including α-thrombin, TNF-α, and ROS (Aschner et al., 1997; Ferro et al., 2000; Konstantoulaki et al., 2003; Sandoval et al., 2001; Xiong et al., 2010). Intriguingly, we have shown that activation of PKCδ is critical for maintenance of basal barrier function and attenuated agonist-induced increases in permeability (Figure 1) (Harrington et al., 2003; Harrington et al., 2005; Klinger et al., 2007); these functions correlated with enhanced focal adhesion formation, actin filament stabilization, and RhoA activation. Similarly, using a siRNA approach, Carpenter and Alexander demonstrated that PKCδ activation within endothelial cells attenuated neutrophil transmigration across the monolayers (Carpenter and Alexander, 2008). Additional studies have suggested that diminished PKCδ expression and upregulation of PKCβII protein content in the endothelium may contribute to microvascular barrier dysfunction in settings of hyperglycemia, possibly through distinct subcellular compartmentalization of each isoform (Gaudreault et al., 2008; Yuan et al., 2000). Indeed, a recent study using a PKCδ selective fluorescence resonance energy transfer (FRET) reporter construct which reflected PKCδ enzymatic activity, showed that PKCδ activity was greatest at the plasma membrane in COS7 cells at both baseline and in agonist-induced states; data supportive of a role of PKCδ in modulating barrier function (Kajimoto et al., 2010). Interestingly, in a recent study, Geraldes and colleagues demonstrated an indirect effect of PKCδ in endothelial cell dysfunction via the induction of apoptosis. The investigators noted that hyperglycemia induced the upregulation of PKCδ dependent signaling in pericytes, which in turn led to pericyte apoptosis and microvascular dysfunction via increased endothelial permeability and cell proliferation, and the pathologic progression of diabetic retinopathy (Geraldes et al., 2009).
Figure 1. PKCδ inhibition promotes endothelial barrier dysfunction.
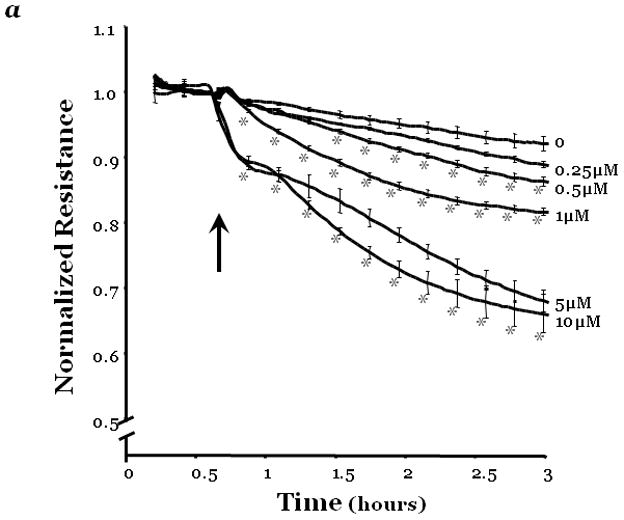
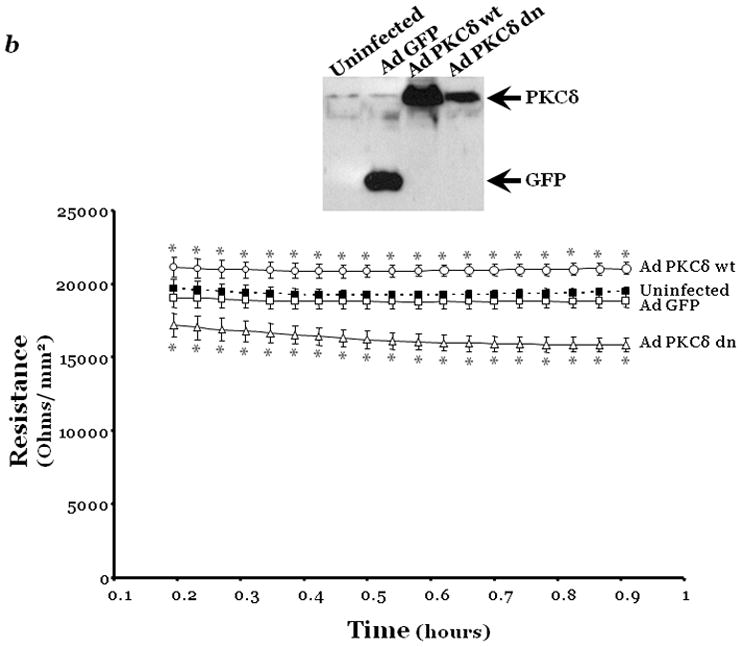
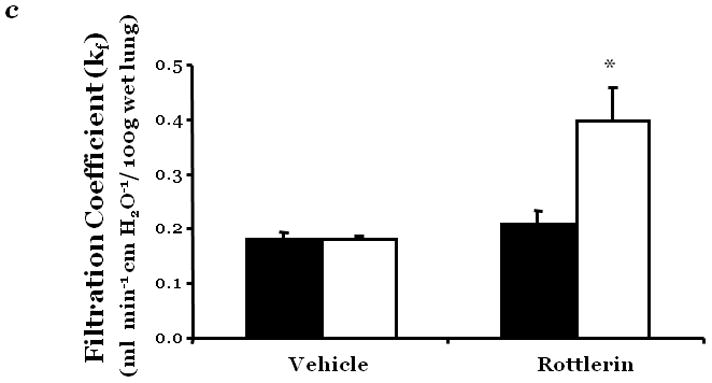
Changes in endothelial monolayer permeability were assessed in rat lung microvascular endothelial cells (LMVEC) (panel a) or endothelial cells derived from the epididymal fat pad (FPEC) (panel b) by assaying changes in resistance of endothelial monolayers grown on collagen coated gold electrodes using the electrical cell impedance system (ECIS); a drop in electrical resistance across the endothelial monolayers correlates with increased permeability. Panel a, vehicle (DMSO) or indicated concentration of rottlerin (a chemical inhibitor with specificity for PKCδ, relative to other PKC isoforms) was added to the monolayers, with arrows indicating time of addition. Panel b, endothelial monolayers containing equivalent numbers of endothelial cells were infected with indicated adenovirus. Protein overexpression was confirmed by immunoblot analyses (inset) and the effect of the overexpressed protein on monolayer permeability was determined by measuring the electrical resistance across the monolayers 24 hours post-infection. The mean±SE of the normalized electrical resistance are presented. Panels a, n=6–12; *p<0.05 vs. vehicle. Panel b, n=16; *p<0.05 vs. Ad GFP or uninfected. Panel c, pulmonary vascular permeability was measured by calculating the capillary filtration coefficients (Kf), using the Starling equation, from isolated, perfused rat lungs, which were fully recruited and in an isogravametric state. Kf was determined by measuring the lung weight gain following an increase in venous pressure divided by the change in capillary pressures and normalized to 100g wet lung mass at baseline (solid bars) and following a 45 minute exposure to vehicle (DMSO) or 50μM rottlerin (open bars). n=3–4, *p<0.05.
Panels a, c, and d: Reprinted from Klinger, J.R., et al., 2007. Rottlerin causes pulmonary edema in vivo: A possible role for PKCδ. Journal of Applied Physiology, 103:2084–2094. Panel b: Reprinted from Harrington, E.O., et al., 2005. PKCδ regulates endothelial basal barrier function through modulation of RhoA GTPase activity. Experimental Cell Research, 308:407–421.
Many agents that affect endothelial barrier function similarly modulate proliferation, migration, and angiogenesis. Thus it is not surprising that PKC isoforms are critical regulators of these endothelial functions (Anfuso et al., 2007; Graham et al., 2000; Hu and Fan, 1995; Spyridopoulos et al., 2002; Wong and Jin, 2005). PKCδ overexpression reduced endothelial cell proliferation via a diminished rate of progression through the G1/S-phase of the cell cycle and an attenuated level of expression of cyclin E (Ashton et al., 1999; Harrington et al., 1997). Further experiments have demonstrated that conditional overexpression of the PKCη pseudosubstrate motif, which inhibited PKCα, δ, ε, and η enzymatic activities in vitro, also attenuated the rate of endothelial cell proliferation, migration, and tubule formation (Harrington et al., 2000). We showed PKCδ overexpression enhanced endothelial cell adhesion to the extracellular matrix protein, vitronectin (Harrington et al., 1997). However, isoforms other than PKCδ have been shown to be necessary for the regulation of endothelial cell migration induced by various agents, including sphingosine-1-phosphate, hepatocyte growth factor, vascular endothelial growth factor (VEGF) (Gorshkova et al., 2008; Harrington et al., 1997; Wang et al., 2002).
Multiple studies have revealed a pro-apoptotic role for PKCδ in the progression of apoptosis in vitro and in vivo. PKCδ activation was shown to be associated with increased apoptosis in myocardial tissue of patients undergoing cardiopulmonary bypass and carioplegic arrest (Sodha et al., 2008). Furthermore, the PKCδ-specific translocation peptide inhibitor, KAI-9803, was shown to attenuate the infarct size in animal models and in humans with acute myocardial infarction (Direct Inhibition of δ-Protein Kinase C Enzyme to Limit Total Infarct Size in Acute Myocardial Infarction (DELTA MI) Investigators, 2008; Inagaki et al., 2003). Furthermore, the salivary glands of PKCδ null mice were resistant to ionizing radiation-induced apoptosis (Humphries et al., 2006). Interestingly, PKCδ has also been shown to protect against apoptosis (Kilpatrick et al., 2006). In epithelial cells, PKCδ promoted cell survival in response to acute hypoxia via activation of the autophagic response (Humphries et al., 2006). However, upon chronic hypoxic conditions, apoptosis ensues via a mechanism involving the catalytically-active form of PKCδ (PKCδCF) and caspase-3 (Humphries et al., 2006). Studies by Reyland and colleagues have demonstrated that upon exposure to apoptosis-inducing agents, PKCδ phosphorylation at tyrosine residues 64 and 115 causes a protein conformational change revealing a nuclear localization sequence, which in turn resulted in the translocation of PKCδ to the nucleus (DeVries-Seimon et al., 2002; DeVries-Seimon et al., 2007). Thus, it is likely that different subcellular compartments of PKCδ regulate distinct cellular functions; including monolayer permeability, proliferation, apoptosis, migration, adhesion, and angiogenesis. The maintenance of the vascular integrity requires a careful balance of signals regulating endothelial cell survival and apoptosis, thus it is possible that endothelial cell apoptosis plays a role in edema formation seen in settings of chronic obstructive pulmonary diseases and acute lung injury.
The expression pattern of the PKC isoforms varies in endothelial cells depending upon the organ or vascular bed from which the cells are isolated (Geraldes et al., 2009). For example, in the lung, we noted the PKCα, δ, ε, η, and λ isoforms in microvascular endothelial cells and only PKCα, δ, ε, and η in pulmonary artery derived endothelial cells (Fordjour and Harrington, 2009). Because PKC has been implicated in a variety of endothelial cell functions, including monolayer permeability, proliferation, apoptosis, migration, adhesion, and angiogenesis, it is likely that the PKC isoform profile of the endothelium may also influence the functional and pathologic response of the endothelial cell to environmental cues.
p190RhoGAP
The monomeric, small Rho GTPase proteins (RhoA, Rac-1, and Cdc-42) are members of the Ras superfamily and are key cytoskeletal regulators, functioning in cell motility and migration, cytokinesis, differentiation, polarity, and vesicular trafficking (Aspenstrom, 1999; Begum et al., 2004; Hall, 1998; Hall and Nobes, 2000; Hotchin and Hall, 1996; Ridley, 2001; Tapon and Hall, 1997; Wojciak-Stothard and Ridley, 2002a). These ubiquitous enzymes cycle between active, GTP-bound states and inactive, GDP-bound states, undergoing corresponding conformational changes. The exchange of GTP and GDP is regulated by the opposing actions of the guanine nucleotide exchange factors (GEF) and the GTPase activating proteins (GAP). The GEF proteins mediate the activation of RhoA, Rac-1, and Cdc-42 by stimulating the release and exchange of GDP for GTP. Conversely, the GAP proteins foster the inactivation of these proteins by activating their intrinsic GTPase activity, triggering the conversion from the active, GTP-bound state to the inactive, GDP-bound form. Another class of proteins, the Rho guanine nucleotide dissociation inhibitors (GDI) suppress RhoA signaling by sequestering the GDP-bound form of the small GTPases within the cytoplasm, preventing them from exposure to and activation by the GEF proteins (Fukumoto et al., 1990; Moon and Zheng, 2003).
The p190RhoGAP (p190) family of GTPase activating proteins is composed of two members: p190RhoGAP-A (p190-A) (Settleman et al., 1992) and p190RhoGAP-B (p190-B) (Burbelo et al., 1998). While both proteins function as GAP for the Rho family of small GTPases, they are encoded by different genes and share only about 50% sequence identity. The two isoforms do exhibit several overlapping functions, including the regulation of cortical actin assembly/disassembly and cell motility and invasion. p190-B, however, functions largely in regulation of development (Chakravarty et al., 2003; Heckman et al., 2007; Sordella et al., 2002) and cell-fate decisions (Sordella et al., 2003), while p190A is a critical regulator of cell migration and tumorigenesis in numerous cell types, including endothelial cells (Arthur et al., 2000; Wolf et al., 2003).
p190-A is comprised of three major domains: an N-terminal GTP-binding domain; a middle domain containing multiple protein-protein interaction sites, including several diphenylalanine (FF) motifs shown to interact with RNA-binding proteins (Jiang et al., 2005; Mammoto et al., 2009), numerous Src homology 3 (SH3)-domain binding sites, and a critical tyrosine residue, Tyr 1105, which, when phosphorylated, serves as the major binding site for p120RasGAP (Hu and Settleman, 1997; Roof et al., 1998); and a C-terminal GAP domain, which displays specificity for GTP-bound RhoA (Ridley et al., 1993).
p190-A plays a critical role in extracellular matrix dependent RhoA inhibition. Fibroblasts transiently overexpressing dominant negative p190 are unable to suppress RhoA during adhesion, leading to impaired cell spreading and migration (Arthur and Burridge, 2001). p190-A is also involved in regulation of adherens junction integrity, through its binding to p120-catenin. Activated RhoA disrupts adherens junction integrity by inducing myosin light chain-dependent actin stress fiber formation and initiating cytoskeleton retraction. While both p190-A and p120-catenin function independently to inhibit RhoA (p120-catenin functions as a modified GDI), the two also function in concert with one another to inhibit RhoA through Rac-1. Overexpression of dominant active Rac-1 induces a robust recruitment of p190 to adherens junctions, an effect which is blocked by depletion of p120-catenin. Likewise, p190-null cells lack adherens junction-associated p120-catenin, even in the presence of dominant active Rac-1 (Wildenberg et al., 2006).
In the endothelium, some evidence has suggested p190 is a key signaling modulator of endothelial barrier function. Angiopoietin-1 attenuation of lipopolysaccharide-induced endothelial barrier dysfunction in vitro and lung edema in vivo was shown to be blocked in settings of p190 protein suppression, suggesting that p190 signals through a barrier protective mechanism (Mammoto et al., 2007). In lung endothelial cells transiently overexpressing wild-type p190-A, while the endothelial cells remained adherent, we noted diminished actin stress fibers and significantly fewer focal adhesion complexes (Fordjour and Harrington, 2009). Interestingly, transient overexpression of dominant negative p190-A protein had no significant effect on endothelial stress fiber or focal adhesion complex formation and endothelial basal barrier function was unaffected upon siRNA suppression of p190-A and/or p190-B (Fordjour and Harrington, 2009), suggesting that other Rho GAP proteins may compensate for these cytoskeletal disruptions in p190 function within the endothelium. Thus, p190 may play a greater role in the regulation of endothelial monolayer permeability in the settings of barrier agonists and antagonists.
Functional Crosstalk between FAK, PKCδ, and p190
Despite the considerable phenotypic and functional heterogeneity between macrovascular and microvascular endothelial cells, numerous in vitro studies suggest that FAK, PKCδ, and p190-A play similar roles in regulation of barrier function in both cell types (Mehta, 2002; Harrington, 2005; Holinstat, 2006). However, the baseline permeability of macrovascular endothelium, such as that derived from the pulmonary artery, is considerably higher than that of microvascular endothelium, with macrovessel-derived endothelial monolayers exhibiting increased hydraulic conductance, relative to endothelial cells derived from microvessels (Parker, 2006). Several reasons have been implicated for this difference, including a differential response to intracellular Ca2+ flux and varying extracellular milieu (Kelly, 1998; Sisbarro, 2005). Interestingly, when pulmonary artery endothelial cells and lung microvascular endothelial cells were plated onto uncoated plastic, in order to mimic the microenvironment of injured tissue, macrovessel-derived cells displayed increased FAK activation and decreased RhoA activity, compared to microvascular endothelial cells (Sisbarro, 2005). Thus, it is likely that while the cellular function of these molecules is similar in macrovascular and microvascular endothelium, their relative levels of expression may differ depending on the specific microenvironment.
PKCδ and FAK are involved in the regulation of several common endothelial cell functions; thus it is perhaps not surprising that there exists a certain degree of crosstalk between the two. PKCδ, in addition to several other PKC isoforms, is activated upon integrin ligation, leading to translocation from the cytosol to the cell membrane (Besson et al., 2002; Chae et al., 2010). Inhibition of the PKC enzymes has been shown to prevent cell spreading and migration (Chae et al., 2010; Wang et al., 2002). In rat embryonic fibroblasts, PKCδ has been shown to be one of the first components recruited to newly formed focal adhesions, rapidly following recruitment of FAK (Barry and Critchley, 1994). Overexpression of PKCδ has been shown to enhance endothelial barrier integrity and to attenuate the degree to which thrombin induces barrier dysfunction (Harrington et al., 2003). These observations were accompanied by a significant increase in the number of FAK-based focal adhesion contacts (Harrington et al., 2003). Conversely, inhibition of PKCδ significantly reduced the number and size of focal adhesions and diminished level of filamentous actin; events which correlated with attenuation of FAK activity and diminished cell stiffness, respectively (Figure 2) (Harrington et al., 2005; Klinger et al., 2007). Interestingly, attenuation of FAK activity was not detectable until ten minutes after treatment with the PKCδ inhibitor, rottlerin, and overexpression of wild type FAK was unable to block rottlerin-induced effects on endothelial permeability and stress fiber disruption, strongly suggesting that the effects of PKCδ on FAK are mediated through at least one intermediate signaling molecule, and/ or that PKCδ itself serves as an signaling intermediate for activation of FAK. Autophosphorylation of FAK at tyrosine 397 has been shown to occur downstream of RhoA activation (Mukai et al., 2003); thus, it is possible that PKCδ activates RhoA, which in turn stimulates autophosphorylation and activation of FAK (Figure 4). Alternatively, under homeostatic conditions, PKCδ may serve as signaling intermediate for one of the established modulators of FAK autophosphorylation, such as EGF or c-Met (Kharait et al., 2006; Thors et al., 2003; Wang et al., 2009).
Figure 2. PKCδ inhibition blunts FAK activity and diminished cytoskeletal stiffness.
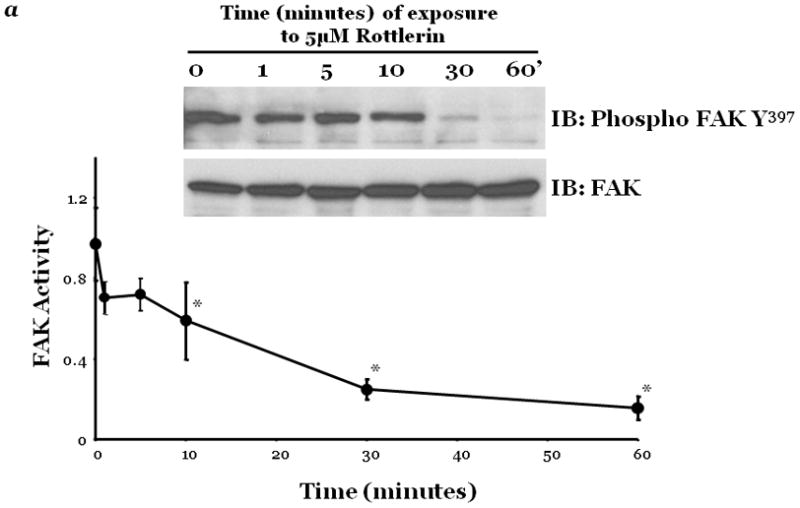
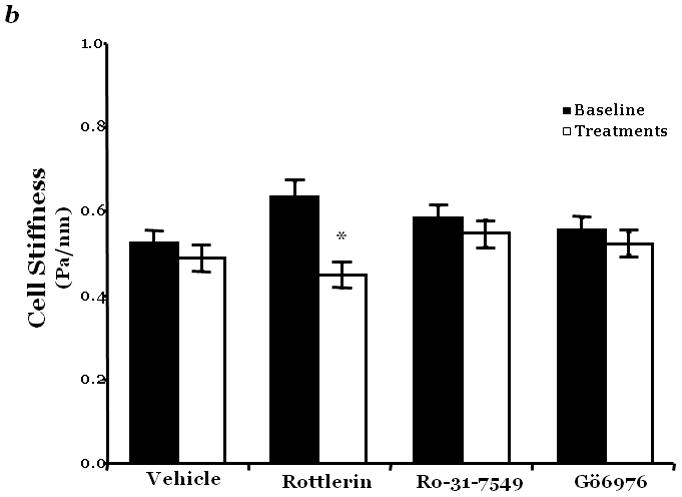
Panel a, FAK activity was determined by measuring the level of phosphorylation at FAK Y397 by immunoblot analysis at indicated times following incubation of endothelial cells derived from the epididymal fat pad (FPEC) with 5μM rottlerin. The immunoblotted membranes were subsequently stripped and reprobed for FAK. Immunoblot signals were quantitated by densitometry and the level of FAK activity is presented as the mean±SE of the ratio of FAK Y397 phosphorylation to total FAK. Panel b, barrier function is dictated by changes in both contractile and adhesive forces, thus to measure changes in the contractile forces, cytoskeletal stiffness was assessed in lung microvascular endothelial cells (LMVEC) which were overlaid with ferrimagnetic beads, coated with the integrin receptor-specific peptide sequence (Arg-Gly-Asp; RGD), forming apical focal adhesions between the LMVEC and the ferrimagnetic beads. The beads were then twisted, using a magnet, and the resistant force was measure both before treatment (i.e., baseline) and in the same cultures 30 minutes following exposure to vehicle, 250nM Ro-31-7549 (a chemical inhibitor with specificity for PKCα, β, γ, ε), 10nM Gö6976 (a chemical inhibitor with specificity for PKCα, β, γ), or 5μM rottlerin. Data are presented as mean±SE (n=280–520 cells). *p<0.05 vs. vehicle with respective treatment. Panel a: Reprinted from Harrington, E.O., et al., 2005. PKCδ regulates endothelial basal barrier function through modulation of RhoA GTPase activity. Experimental Cell Research, 308:407–421. Panel b: Reprinted from Klinger, J.R., et al., 2007. Rottlerin causes pulmonary edema in vivo: A possible role for PKCδ. Journal of Applied Physiology, 103:2084–2094.
Figure 4. Model of potential cross talk between PKCδ, p190, and FAK.
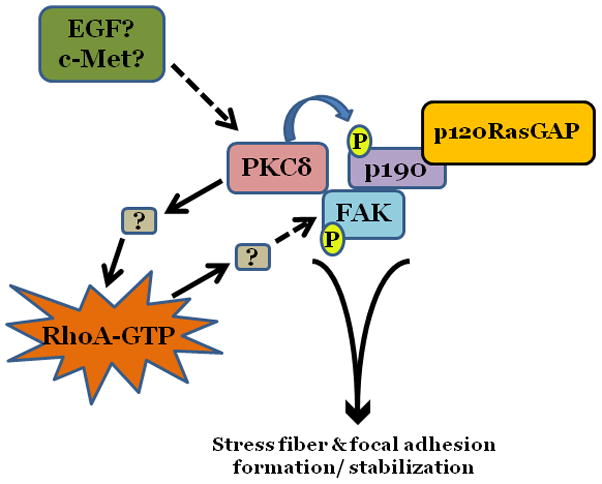
Under basal conditions, PKCδ functions to maintain endothelial barrier function through maintenance of a static level of RhoA activation and stimulation of FAK autophosphorylation. This allows for focal adhesion stabilization and organization of actin stress fibers. PKCδ-mediated activation of RhoA appears to occur independently of its effects on p190 phosphorylation.
Neutrophil transendothelial migration has been shown to induce changes in the phosphorylation of endothelial FAK, leading to an increase in vascular permeability (Guo et al., 2005). Treatment of endothelial cells with either phorbol 12-myristate 13-acetate (PMA), a known pan PKC activator, or bryostatin-1, an activator specific for PKCδ and PKCε, were shown to significantly attenuate neutrophil transendothelial migration, coincident with increased cell substrate adhesion and increased intracellular staining for the activated, phosphorylated form of FAK, phosphorylated tyrosine 397 (Y397) FAK (Carpenter and Alexander, 2008). Although the mechanism by which PKCδ may regulate FAK has not yet been elucidated, PKCδ activity has been shown to increase following phosphorylation by Fyn kinase, a known activator of FAK (Kronfeld et al., 2000). Chu et al. recently reported an increase in FAK serine 910 (S910) phosphorylation in rat ventricular myocytes transfected with constituitively active PKCδ (Chu et al., 2010). They also observed an inhibition of endothelin-1-induced FAK S910 phosphorylation in cells overexpressing dominant negative PKCδ. Endothelin is an important vasoactive mediator associated with numerous cell responses, including the release of nitric oxide from vascular endothelial cells. Thus, further investigation into the mechanism(s) by which PKCδ affects changes in FAK activation/ phosphorylation will serve to increase our understanding of the pathways involved in endothelial barrier regulation.
Coincident with their critical role in cytoskeletal organization and actin dynamics, the small Rho GTPase proteins are tightly regulated. Basal activation of RhoA is necessary for homeostatic endothelial function (Etienne-Manneville and Hall, 2002); increased RhoA activation, however, has been shown to correlate with endothelial cell migration and metastasis (Della Peruta et al., 2010; van Nieuw Amerongen et al., 2003; Zhao et al., 2006), increased neutrophil extravasation (Adamson et al., 1999; Wittchen et al., 2005; Worthylake and Burridge, 2003; Worthylake et al., 2001), and is the key mediator of endothelial cell contraction, coincident with increased vascular permeability (Carbajal and Schaeffer, 1999; McKenzie and Ridley, 2007; Partridge et al., 1992; Wojciak-Stothard and Ridley, 2002b). Thrombin, an established edemagenic agent, causes a very rapid and robust increase in endothelial RhoA activation, correlating with decreased endothelial barrier function (van Nieuw Amerongen et al., 2000).
Interestingly, thrombin also induces the recruitment of FAK to focal adhesions, where it has been implicated to function in the recovery of the endothelial barrier following disruption; endothelial monolayers in which FAK has been depleted are unable to restore barrier function following thrombin treatment (Mehta et al., 2002). Based on several independent observations that: 1) deletion of FAK in fibroblasts led to an increase in RhoA activation; 2) increased RhoA signaling correlated with increased endothelial permeability; 3) p190 co-localized with FAK at focal adhesions; and 4) p190 is activated by tyrosine phosphorylation, Hollinstat and colleagues (Holinstat et al., 2006) further investigated the specific role of FAK in regulation of endothelial RhoA activation. They noted increased tyrosine phosphorylation of p190 in response to thrombin, correlating with decreased RhoA activation. In addition, they demonstrated FAK phosphorylation of p190 in vitro, and showed that inhibition of FAK resulted in decreased p190 activation and a concomitant increase in RhoA activity and endothelial permeability. More recently, FAK/ p190 signaling was also implicated in mediating the anti-proliferative/ antiangiogenic effects of the combustion byproduct 3-methylcholanthrene (3MC), an aryl-hydrocarbon receptor agonist, in human umbilical vein endothelial cells (HUVEC) (Pang et al., 2008). Treatment with this compound resulted in a down-regulation of FAK, coincident with increased RhoA activation; this was in turn correlated with suppression of p190 activation by FAK (Chang et al., 2009). In addition, Tomar and colleagues demonstrated that a complex of FAK, p120RasGAP, and p190 functions to regulate the polarity and cell migration of fibroblasts, carcinoma cells, and endothelial cells (Tomar et al., 2009). In this study, they observed that following fibronectin-integrin engagement, activated FAK binds to p120RasGAP, through its SH2-SH3-SH2 region. Through this same region, p120RasGAP binds to p190-A, facilitating its activation by FAK. The FAK-p120RasGAP-p190A complex is then targeted to leading-edge focal adhesions, permitting spatially regulated RhoA suppression at cell protrusions. The authors also observed that overexpression of p190 mutants either lacking GAP activity (p190A-RA) or with key tyrosine residues mutated to phenylalanine (p190A-FF; Y1087F, Y1105F) blocked cell polarization, supporting the involvement of these p190-A domains in its interaction with FAK and p120RasGAP. Interestingly, PKCδ, which immunoprecipitates with p120RasGAP (Harrington et al., 2005), has been shown in vitro to bind to these two domains of p190-A (Fordjour and Harrington, 2009), suggesting a possible interplay of FAK, p190-A, and PKCδ.
p190-A also serves as a requisite binding partner for p120-catenin at intercellular adherens junctions, preventing its translocation to the cytoplasm (Wildenberg et al., 2006). Given the reported interaction between FAK and adherens junction associated p120-catenin (Sun et al., 2009), it appears that p190-A is essential for FAK function not only at focal adhesions but also at adherens junctions.
While both PKCδ and p190-A function in regulation of endothelial FAK signaling, it appears that the two may serve differential functions in settings of barrier maintenance versus settings of barrier recovery after insult. For example, it was previously reported that, in addition to causing diminished FAK activation, inhibition of PKCδ also caused a concomitant decrease in baseline RhoA activity, resulting in disruption of basal endothelial barrier function (Harrington et al., 2005). Furthermore, PKCδ has been shown to co-immunoprecipitate with both p120RasGAP and p190 (Harrington et al., 2005). However, while PKCδ is able to regulate p190 activity (Figure 3), suppression of p190-A or p190-B, either independently or in combination, was unable to attenuate the effects of PKCδ inhibition on RhoA activity, focal adhesion disruption, stress fiber formation, or endothelial permeability in unstimulated cells (Fordjour and Harrington, 2009); this suggests that, under baseline conditions, PKCδ functions to regulate RhoA activity independently of p190. However, Holinstat and colleagues demonstrated that FAK signaling through p190 and subsequent inhibition of RhoA was critical for restoration of the pulmonary endothelium after thrombin-induced injury (Holinstat, 2006). Thus, as outlined in Figure 4, it is likely that the activities of both PKCδ and p190 differ depending upon the state of the endothelium. PKCδ may function to maintain basal levels of RhoA activation independently of p190 when the endothelial barrier is intact. In this setting, PKCδ may phosphorylate p190, facilitating its association with FAK, p120RasGAP, and potentially p120-catenin, without affecting its ability to inhibit RhoA activity, which presumably should remain at homeostatic levels. However, when the barrier is disrupted, as occurs following exposure to thrombin, and RhoA activation is elevated to supraphysiologic levels, PKCδ signaling may be acutely suppressed, providing a feedback loop preventing further activation of RhoA and potentially alleviating serine/ threonine phosphorylation of p190.
Figure 3. PKCδ activity inversely affects p190 activity.
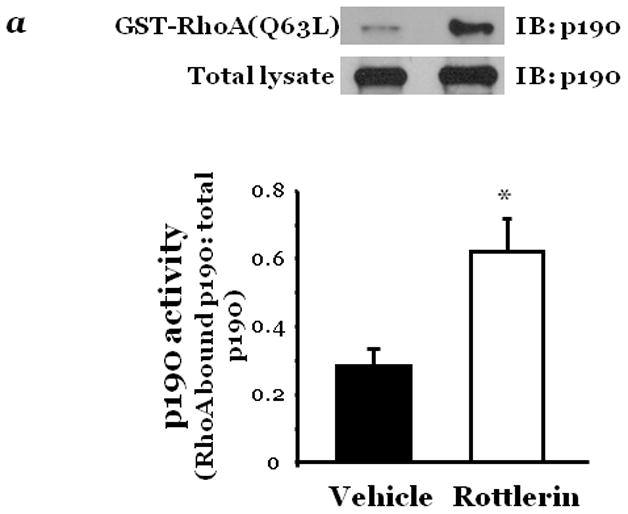
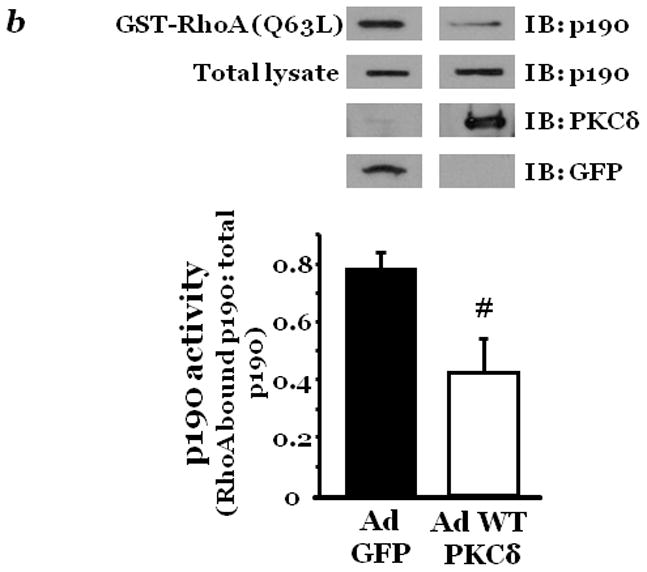
Confluent lung microvascular endothelial cells (LMVEC) were treated with vehicle (DMSO) or 10μM rottlerin for 30 minutes (panel a) or infected with adenoviral vectors encoding GFP or wild-type PKCδ cDNA (panel b). Cells were harvested and p190 activity determined as the level of p190 bound to GST-fused constitutively actvated RhoA. The level of active p190 relative to total p190 was determined by densitometry. In panel b, GFP and PKCδ overexpression was confirmed in the transfected endothelial cells by immunoblot analysis. Data are presented as the mean±SE. Panel a, n=4; *p<0.05 vs. vehicle. Panel b, n=7, #p<0.05 vs. GFP. Panels a and b: Reprinted from Fordjour, A.K. and Harrington, E.O. 2009. PKCδ influences p190 phosphorylation and activity: Events independent of PKCδ-mediated regulation of endothelial cell stress fiber and focal adhesion formation and barrier function. Biochimica et Biophysica Acta, 1790:1179–1190.
Given the critical role of FAK in regulating endothelial cell function, a greater understanding of the cellular proteins involved in FAK activation and downstream signaling, including PKCδ and p190, will help to gain insight into the mechanisms responsible for endothelial dysfunction in various disease states (Figure 4). Further investigation into the dynamic interactions of adherens junction-associated p120-catenin with FAK and its binding partner p120RasGAP may reveal a potential signaling axis along which signaling molecules, such as PKCδ and p190, serve to maintain the balance between endothelial cell-ECM adhesions and intercellular interactions necessary for vascular barrier function. Once the functions of these molecules with regard to FAK have been elucidated, as well as the temporal nature in which they are altered in different physiological settings, targeted therapies may be designed with the goal of attenuating endothelial injury and result barrier disruption.
RESEARCH HIGHLIGHTS
FAK, PKC, and p190RhoGAP exhibit crosstalk in their regulation of endothelial cell (EC) function.
Changes in PKCδ correlate with alterations in FAK activity and related effects on EC function.
FAK inhibition decreases p190RhoGAP activation; whereas PKCδ inihibition increases p190RhoGAP activation.
PKCδ and p190RhoGAP may regulate FAK differentially in settings of barrier maintenance versus recovery.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adamson P, Etienne S, Couraud PO, Calder V, Greenwood J. Lymphocyte migration through brain endothelial cell monolayers involves signaling through endothelial ICAM-1 via a rho-dependent pathway. Journal of Immunology. 1999;162(5):2964–2973. [PubMed] [Google Scholar]
- Alghisi GC, Ponsonnet L, Rüegg C. The integrin antagonist cilengitide activates αVβ3, disrupts VE-cadherin localization at cell junctions and enhances permeability in endothelial cells. PLoS One. 2009;4:e4449. doi: 10.1371/journal.pone.0004449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida EA, et al. Matrix survival signaling: from fibronectin via focal adhesion kinase to c-Jun NH(2)-terminal kinase. Journal of Cell Biology. 2000;149:741–754. doi: 10.1083/jcb.149.3.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anfuso CD, et al. Endothelial cell-pericyte cocultures induce PLA2 protein expression through activation of PKCα and the MAPK/ERK cascade. Journal of Lipid Research. 2007;48(4):782–793. doi: 10.1194/jlr.M600489-JLR200. [DOI] [PubMed] [Google Scholar]
- Arthur WT, Burridge K. RhoA inactivation by p190RhoGAP regulates cell spreading and migration by promoting membrane protrusion and polarity. Molecular Biology of the Cell. 2001;12(9):2711–2720. doi: 10.1091/mbc.12.9.2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arthur WT, Petch LA, Burridge K. Integrin engagement suppresses RhoA activity via a c-Src-dependent mechanism. Current Biology. 2000;10(12):719–722. doi: 10.1016/s0960-9822(00)00537-6. [DOI] [PubMed] [Google Scholar]
- Aschner JL, Lum H, Fletcher PW, Malik AB. Bradykinin- and thrombin-induced increases in endothelial permeability occur independently of phospholipase C but require protein kinase C activation. Journal of Cell Physiology. 1997;173(3):387–396. doi: 10.1002/(SICI)1097-4652(199712)173:3<387::AID-JCP11>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Ashton AW, et al. Protein kinase Cδ inhibition of S-phase transition in capillary endothelial cells involves the cyclin-dependent kinase inhibitor p27(Kip1) Journal of Biological Chemistry. 1999;274(30):20805–20811. doi: 10.1074/jbc.274.30.20805. [DOI] [PubMed] [Google Scholar]
- Aspenstrom P. The Rho GTPases have multiple effects on the actin cytoskeleton. Experimental Cell Research. 1999;246(1):20–25. doi: 10.1006/excr.1998.4300. [DOI] [PubMed] [Google Scholar]
- Barry ST, Critchley DR. The RhoA-dependent assembly of focal adhesions in Swiss 3T3 cells is associated with increased tyrosine phosphorylation and the recruitment of both pp125FAK and protein kinase C-δ to focal adhesions. Journal of Cell Science. 1994;107:2033–2045. doi: 10.1242/jcs.107.7.2033. [DOI] [PubMed] [Google Scholar]
- Begum R, Nur E, Kamal MS, Zaman MA. The role of Rho GTPases in the regulation of the rearrangement of actin cytoskeleton and cell movement. Experimental and Molecular Medicine. 2004;36(4):358–366. doi: 10.1038/emm.2004.47. [DOI] [PubMed] [Google Scholar]
- Bellas RE, et al. FAK blunts adenosine-homocysteine-induced endothelial cell apoptosis: requirement for PI 3-kinase. American Journal of Physiology. 2002;282(5):L1135–L1142. doi: 10.1152/ajplung.00174.2001. [DOI] [PubMed] [Google Scholar]
- Besson A, Wilson TL, Yong VW. The anchoring protein RACK1 links protein kinase Cepsilon to integrin beta chains. Requirements for adhesion and motility. Journal of Biological Chemistry. 2002;277(24):22073–22084. doi: 10.1074/jbc.M111644200. [DOI] [PubMed] [Google Scholar]
- Burbelo PD, Finegold AA, Kozak CA, Yamada Y, Takami H. Cloning, genomic organization and chromosomal assignment of the mouse p190-B gene. Biochimica et Biophysica Acta. 1998;1443(1–2):203–210. doi: 10.1016/s0167-4781(98)00207-3. [DOI] [PubMed] [Google Scholar]
- Carbajal JM, Schaeffer RC. RhoA inactivation enhances endothelial barrier function. American Journal of Physiology. 1999;277(5 Pt 1):C955–C964. doi: 10.1152/ajpcell.1999.277.5.C955. [DOI] [PubMed] [Google Scholar]
- Carpenter AC, Alexander JS. Endothelial PKCδ activation attenuates neutrophil transendothelial migration. Inflammation Research. 2008;57(5):216–229. doi: 10.1007/s00011-007-7031-4. [DOI] [PubMed] [Google Scholar]
- Chae YC, et al. PKCδ-mediated phosphorylation of phospholipase D controls integrin-mediated cell spreading. Molecular and Cellular Biology. 2010;30:5086–5098. doi: 10.1128/MCB.00443-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravarty G, Hadsell D, Buitrago W, Settleman J, Rosen JM. p190-B RhoGAP regulates mammary ductal morphogenesis. Molecular Endocrinology. 2003;17(6):1054–1065. doi: 10.1210/me.2002-0428. [DOI] [PubMed] [Google Scholar]
- Chang CC, et al. Aryl-hydrocarbon receptor-dependent alteration of FAK/RhoA in the inhibition of HUVEC motility by 3-methylcholanthrene. Cellular and Molecular Life Sciences. 2009;66(19):3193–3205. doi: 10.1007/s00018-009-0102-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SY, Chen HC. Direct interaction of focal adhesion kinase (FAK) with Met is required for FAK to promote hepatocyte growth factor-induced cell invasion. Molecular and Cellular Biology. 2006;26:5155–5167. doi: 10.1128/MCB.02186-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu M, Iyengar R, Koshman Y, Kim T, Samarel AM. Endothelin-1 induces Serine 910 phosphorylation of focal adhesion kinase via PKCδ-and Src-dependent signaling pathways. FASEB Journal. 2010;24 in press. [Google Scholar]
- Claesson-Welsh L, et al. Angiostatin induces endothelial cell apoptosis and activation of focal adhesion kinase independently of the integrin-binding motif RGD. Proceedings of the National Academies of Science. 1998;95(10):5579–5583. doi: 10.1073/pnas.95.10.5579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deanfield J, et al. Endothelial function and dysfunction. Part I: Methodological issues for assessment in the different vascular beds: a statement by the Working Group on Endothelin and Endothelial Factors of the European Society of Hypertension. Journal of Hypertension. 2005;23(1):7–17. doi: 10.1097/00004872-200501000-00004. [DOI] [PubMed] [Google Scholar]
- Dejana E, Corada M, Lampugnani MG. Endothelial cell-to-cell junctions. FASEB Journal. 1995;9(10):910–918. [PubMed] [Google Scholar]
- Dejana E, Del Maschio A. Molecular organization and functional regulation of cell to cell junctions in the endothelium. Thrombosis and Haemostasis. 1995;74(1):309–312. [PubMed] [Google Scholar]
- Della Peruta M, Giagulli C, Laudanna C, Scarpa A, Sorio C. RHOA and PRKCZ control different aspects of cell motility in pancreatic cancer metastatic clones. Molecular Cancer. 2010;9:61–68. doi: 10.1186/1476-4598-9-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVries-Seimon TA, Neville MC, Reyland ME. Nuclear import of PKCδ is required for apoptosis: identification of a novel nuclear import sequence. Embo J. 2002;21:6050–6060. doi: 10.1093/emboj/cdf606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVries-Seimon TA, Ohm AM, Humphries MJ, Reyland ME. Induction of Apoptosis Is Driven by Nuclear Retention of Protein Kinase Cδ. Journal of Biological Chemistry. 2007;282(31):22307–22314. doi: 10.1074/jbc.M703661200. [DOI] [PubMed] [Google Scholar]
- Direct Inhibition of δ-Protein Kinase C Enzyme to Limit Total Infarct Size in Acute Myocardial Infarction (DELTA MI) Investigators. Intracoronary KAI-9803 as an Adjunct to Primary Percutaneous Coronary Intervention for Acute ST-Segment Elevation Myocardial Infarction. Circulation. 2008;117(7):886–896. doi: 10.1161/CIRCULATIONAHA.107.759167. [DOI] [PubMed] [Google Scholar]
- Eliceiri BP, et al. Src-mediated coupling of focal adhesion kinase to integrin αvβ5 in vascular endothelial growth factor signaling. Journal of Cell Biology. 2002;157(1):149–160. doi: 10.1083/jcb.200109079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ermert L, et al. Role of endothelial cytoskeleton in high-permeability edema due to botulinum C2 toxin in perfused rabbit lungs. American Journal of Physiology. 1995;268(5 Pt 1):L753–L761. doi: 10.1152/ajplung.1995.268.5.L753. [DOI] [PubMed] [Google Scholar]
- Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002;420(6916):629–635. doi: 10.1038/nature01148. [DOI] [PubMed] [Google Scholar]
- Ferro T, Neumann P, Gertzberg N, Clements R, Johnson A. Protein kinase C-alpha mediates endothelial barrier dysfunction induced by TNFα. American Journal of Physiology. 2000;278(6):L1107–L1117. doi: 10.1152/ajplung.2000.278.6.L1107. [DOI] [PubMed] [Google Scholar]
- Fordjour AK, Harrington EO. PKCδ influences p190 phosphorylation and activity: events independent of PKCδ-mediated regulation of endothelial cell stress fiber and focal adhesion formation and barrier function. Biochimica et Biophysica Acta. 2009;1790(10):1179–1190. doi: 10.1016/j.bbagen.2009.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukumoto Y, et al. Molecular cloning and characterization of a novel type of regulatory protein (GDI) for the rho proteins, ras p21-like small GTP-binding proteins. Oncogene. 1990;5(9):1321–1328. [PubMed] [Google Scholar]
- Garcia JGN, et al. Sphingosine 1-phosphate promotes endothelial cell barrier integrity by Edg-dependent cytoskeletal rearrangement. The Journal of Clinical Investigation. 2001;108(5):689–701. doi: 10.1172/JCI12450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudreault N, et al. Counter Regulatory Effects of PKCβII and PKCδ on Coronary Endothelial Permeability. Arteriosclerosis, Thrombosis, and Vascular Biology. 2008;28(8):1527–1533. doi: 10.1161/ATVBAHA.108.166975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geraldes P, et al. Activation of PKCδ and SHP-1 by hyperglycemia causes vascular cell apoptosis and diabetic retinopathy. Nature Medicine. 2009;15(11):1298–1306. doi: 10.1038/nm.2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorshkova I, et al. Protein kinase C-ε regulates sphingosine 1-phosphate-mediated migration of human lung endothelial cells through activation of phospholipase D2, protein kinase C-ζ, and Rac1. Journal of Biological Chemistry. 2008;283:11794–11806. doi: 10.1074/jbc.M800250200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham MA, Rawe I, Dartt DA, Joyce NC. Protein kinase C regulation of corneal endothelial cell proliferation and cell cycle. Investigative Ophthalmological and Visual Science. 2000;41(13):4124–4132. [PubMed] [Google Scholar]
- Guo M, Wu MH, Granger HJ, Yuan SY. Focal adhesion kinase in neutrophil-induced microvascular hyperpermeability. Microcirculation. 2005;12(2):223–232. doi: 10.1080/10739680590905251. [DOI] [PubMed] [Google Scholar]
- Hall A. Rho GTPases and the actin cytoskeleton. Science. 1998;279(5350):509–514. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- Hall A, Nobes CD. Rho GTPases: molecular switches that control the organization and dynamics of the actin cytoskeleton. Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences. 2000;355(1399):965–970. doi: 10.1098/rstb.2000.0632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamadi A, et al. Regulation of focal adhesion dynamics and disassembly by phosphorylation of FAK at tyrosine 397. Journal of Cell Science. 2005;118(Pt 19):4415–4425. doi: 10.1242/jcs.02565. [DOI] [PubMed] [Google Scholar]
- Hanks SK, Polte TR. Signaling through focal adhesion kinase. Bioessays. 1997;19(2):137–145. doi: 10.1002/bies.950190208. [DOI] [PubMed] [Google Scholar]
- Harrington EO, et al. Role of protein kinase C isoforms in rat epididymal microvascular endothelial barrier function. American Journal of Respiratory Cell and Molecular Biology. 2003;28(5):626–636. doi: 10.1165/rcmb.2002-0085OC. [DOI] [PubMed] [Google Scholar]
- Harrington EO, Doyle KE, Brunelle JL, Ware JA. Endothelial Proliferation, Migration, and Differentiation Are Blunted by Conditionally Expressed Protein Kinase C Pseudosubstrate Peptides. Biochemical and Biophysical Research Communications. 2000;271(2):499–508. doi: 10.1006/bbrc.2000.2655. [DOI] [PubMed] [Google Scholar]
- Harrington EO, et al. Enhancement of migration by protein kinase Cα and inhibition of proliferation and cell cycle progression by protein kinase Cδ in capillary endothelial cells. Journal of Biological Chemistry. 1997;272:7390–7397. doi: 10.1074/jbc.272.11.7390. [DOI] [PubMed] [Google Scholar]
- Harrington EO, et al. PKCδ regulates endothelial basal barrier function through modulation of RhoA GTPase activity. Experimental Cell Research. 2005;308(2):407–421. doi: 10.1016/j.yexcr.2005.05.005. [DOI] [PubMed] [Google Scholar]
- Heckman BM, et al. Crosstalk between the p190-B RhoGAP and IGF signaling pathways is required for embryonic mammary bud development. Developmental Biology. 2007;309(1):137–149. doi: 10.1016/j.ydbio.2007.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrand JD, Schaller MD, Parsons JT. Identification of sequences required for the efficient localization of the focal adhesion kinase, pp125FAK, to cellular focal adhesions. Journal of Cell Biology. 1993;123:993–1005. doi: 10.1083/jcb.123.4.993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holinstat M, et al. Suppression of RhoA activity by focal adhesion kinase-induced activation of p190RhoGAP: role in regulation of endothelial permeability. Journal of Biological Chemistry. 2006;281(4):2296–2305. doi: 10.1074/jbc.M511248200. [DOI] [PubMed] [Google Scholar]
- Hotchin NA, Hall A. Regulation of the actin cytoskeleton, integrins and cell growth by the Rho family of small GTPases. Cancer Surveys. 1996;27:311–322. [PubMed] [Google Scholar]
- Hu DE, Fan TP. Protein kinase C inhibitor calphostin C prevents cytokine-induced angiogenesis in the rat. Inflammation. 1995;19(1):39–54. doi: 10.1007/BF01534379. [DOI] [PubMed] [Google Scholar]
- Hu KQ, Settleman J. Tandem SH2 binding sites mediate the RasGAP-RhoGAP interaction: a conformational mechanism for SH3 domain regulation. Embo Journal. 1997;16(3):473–483. doi: 10.1093/emboj/16.3.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang F, et al. Lysophosphatidylcholine increases endothelial permeability: Role of PKCα and RhoA cross talk. American Journal of Physiology. 2005;289(2):L176–L185. doi: 10.1152/ajplung.00003.2005. [DOI] [PubMed] [Google Scholar]
- Humphries MJ, et al. Suppression of Apoptosis in the Protein Kinase Cδ Null Mouse in Vivo. Journal of Biological Chemistry. 2006;281(14):9728–9737. doi: 10.1074/jbc.M507851200. [DOI] [PubMed] [Google Scholar]
- Hynes R. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110:673–687. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- Ilic D, et al. Extracellular matrix survival signals transduced by focal adhesion kinase suppress p53-mediated apoptosis. Journal of Cell Biology. 1998;143:547–560. doi: 10.1083/jcb.143.2.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilic D, et al. Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient mice. Nature. 1995;377:539–544. doi: 10.1038/377539a0. [DOI] [PubMed] [Google Scholar]
- Inagaki K, et al. Inhibition of δ-Protein Kinase C Protects Against Reperfusion Injury of the Ischemic Heart In Vivo. Circulation. 2003;108(19):2304–2307. doi: 10.1161/01.CIR.0000101682.24138.36. [DOI] [PubMed] [Google Scholar]
- Jiang W, et al. An FF domain-dependent protein interaction mediates a signaling pathway for growth factor-induced gene expression. Molecular Cell. 2005;17(1):23–35. doi: 10.1016/j.molcel.2004.11.024. [DOI] [PubMed] [Google Scholar]
- Johnson A, Hocking DC, Ferro TJ. Mechanisms of pulmonary edema induced by a diacylglycerol second messenger. American Journal of Physiology. 1990;258(1):H85–H91. doi: 10.1152/ajpheart.1990.258.1.H85. [DOI] [PubMed] [Google Scholar]
- Johnson A, Phillips P, Hocking DC, Tsan MF, Ferro TJ. Protein kinase inhibitor prevents pulmonary edema in response to H2O2. American Journal of Physiology. 1989;256:H1012–H1019. doi: 10.1152/ajpheart.1989.256.4.H1012. [DOI] [PubMed] [Google Scholar]
- Kabir J, Lobo M, Zachary I. Staurosporine induces endothelial cell apoptosis via focal adhesion kinase dephosphorylation and focal adhesion disassembly independent of focal adhesion kinase proteolysis. Biochemical Journal. 2002;367(Pt 1):145–155. doi: 10.1042/BJ20020665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajimoto T, Sawamura S, Tohyama Y, Mori Y, Newton AC. Protein Kinase C δ-specific Activity Reporter Reveals Agonist-evoked Nuclear Activity Controlled by Src Family of Kinases. Journal of Biological Chemistry. 2010;285(53):41896–41910. doi: 10.1074/jbc.M110.184028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilpatrick LE, et al. Regulation of TNF mediated antiapoptotic signaling in human neutrophils: role of δPKC and ERK1/2. Journal of Leukocyte Biology. 2006 doi: 10.1189/jlb.0406284. in press. [DOI] [PubMed] [Google Scholar]
- Klinger JR, et al. Rottlerin causes pulmonary edema in vivo: a possible role for PKCδ. Journal of Applied Physiology. 2007;103(6):2084–2094. doi: 10.1152/japplphysiol.00695.2007. [DOI] [PubMed] [Google Scholar]
- Knezevic N, Tauseef M, Thennes T, Mehta D. The G protein βγ subunit mediates reannealing of adherens junctions to reverse endothelial permeability increase by thrombin. Journal of Experimental Medicine. 2009;206(12):2761–2777. doi: 10.1084/jem.20090652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konstantoulaki M, Kouklis P, Malik AB. Protein kinase C modifications of VE-cadherin, p120, and β-catenin contribute to endothelial barrier dysregulation induced by thrombin. American Journal of Physiology. 2003;285(2):L434–L442. doi: 10.1152/ajplung.00075.2003. [DOI] [PubMed] [Google Scholar]
- Kronfeld I, et al. Phosphorylation of protein kinase Cδ on distinct tyrosine residues regulates specific cellular functions. Journal of Biological Chemistry. 2000;275(45):35491–35498. doi: 10.1074/jbc.M005991200. [DOI] [PubMed] [Google Scholar]
- Lee WL, Slutsky AS. Sepsis and endothelial permeability. New England Journal of Medicine. 2010;363(7):689–691. doi: 10.1056/NEJMcibr1007320. [DOI] [PubMed] [Google Scholar]
- Li X, et al. Role of protein kinase Cζ in thrombin-induced endothelial permeability changes: inhibition by angiopoietin-1. Blood. 2004;104(6):1716–1724. doi: 10.1182/blood-2003-11-3744. [DOI] [PubMed] [Google Scholar]
- Lietha D, et al. Structural basis for the autoinhibition of focal adhesion kinase. Cell. 2007;129:1177–1187. doi: 10.1016/j.cell.2007.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mammoto A, et al. A mechanosensitive transcriptional mechanism that controls angiogenesis. Nature. 2009;457(7233):1103–1108. doi: 10.1038/nature07765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mammoto T, et al. Angiopoietin-1 requires p190 RhoGAP to protect against vascular leakage in vivo. Journal of Biological Chemistry. 2007;282:23910–23918. doi: 10.1074/jbc.M702169200. [DOI] [PubMed] [Google Scholar]
- McKenzie JA, Ridley AJ. Roles of Rho/ROCK and MLCK in TNF-α-induced changes in endothelial morphology and permeability. Journal of Cell Physiology. 2007;213(1):221–228. doi: 10.1002/jcp.21114. [DOI] [PubMed] [Google Scholar]
- Mehta D, et al. Modulatory role of focal adhesion kinase in regulating human pulmonary arterial endothelial barrier function. Journal of Physiology. 2002;539(Pt 3):779–789. doi: 10.1113/jphysiol.2001.013289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michael KE, Dumbauld DW, Burns KL, Hanks SK, Garcia AJ. Focal Adhesion Kinase Modulates Cell Adhesion Strengthening via Integrin Activation. Molecular Biology of the Cell. 2009;20(9):2508–2519. doi: 10.1091/mbc.E08-01-0076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra SK, Schlaepfer DD. Integrin-regulated FAK-Src signaling in normal and cancer cells. Current Opinion in Cell Biology. 2006;18(5):516–523. doi: 10.1016/j.ceb.2006.08.011. [DOI] [PubMed] [Google Scholar]
- Moon SY, Zheng Y. Rho GTPase-activating proteins in cell regulation. Trends in Cell Biology. 2003;13(1):13–22. doi: 10.1016/s0962-8924(02)00004-1. [DOI] [PubMed] [Google Scholar]
- Nagpala PG, Malik AB, Vuong PT, Lum H. Protein kinase C β1 overexpression augments phorbol ester-induced increase in endothelial permeability. Journal of Cellular Physiology. 1996;166:249–255. doi: 10.1002/(SICI)1097-4652(199602)166:2<249::AID-JCP2>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Newton AC. Protein kinase C: structure, function, and regulation. Journal of Biological Chemistry. 1995;270(48):28495–28498. doi: 10.1074/jbc.270.48.28495. [DOI] [PubMed] [Google Scholar]
- Newton AC. Protein kinase C: Poised to signal. American Journal of Physiology. 2010;298:E395–E402. doi: 10.1152/ajpendo.00477.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang PH, et al. Molecular mechanisms of p21 and p27 induction by 3-methylcholanthrene, an aryl-hydrocarbon receptor agonist, involved in antiproliferation of human umbilical vascular endothelial cells. Journal of Cell Physiology. 2008;215(1):161–171. doi: 10.1002/jcp.21299. [DOI] [PubMed] [Google Scholar]
- Partridge CA, Horvath CJ, Del Vecchio PJ, Phillips PG, Malik AB. Influence of extracellular matrix in tumor necrosis factor-induced increase in endothelial permeability. American Journal of Physiology. 1992;263(6 Pt 1):L627–L633. doi: 10.1152/ajplung.1992.263.6.L627. [DOI] [PubMed] [Google Scholar]
- Quadri SK, Bhattacharya J. Resealing of endothelial junctions by focal adhesion kinase. American Journal of Physiology. 2007;292(1):L334–L342. doi: 10.1152/ajplung.00228.2006. [DOI] [PubMed] [Google Scholar]
- Richardson A, Malik RK, Hildebrand JD, Parsons JT. Inhibition of cell spreading by expression of the C-terminal domain of focal adhesion kinase (FAK) is rescued by coexpression of Src or catalytically inactive FAK: a role for paxillin tyrosine phosphorylation. Molecular and Cellular Biology. 1997;17(12):6906–6914. doi: 10.1128/mcb.17.12.6906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridley AJ. Rho GTPases and cell migration. Journal of Cell Science. 2001;114(Pt 15):2713–2722. doi: 10.1242/jcs.114.15.2713. [DOI] [PubMed] [Google Scholar]
- Ridley AJ, et al. rho family GTPase activating proteins p190, bcr and rhoGAP show distinct specificities in vitro and in vivo. Embo Journal. 1993;12(13):5151–5160. doi: 10.1002/j.1460-2075.1993.tb06210.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roof RW, et al. Phosphotyrosine (p-Tyr)-dependent and -independent mechanisms of p190 RhoGAP-p120 RasGAP interaction: Tyr 1105 of p190, a substrate for c-Src, is the sole p-Tyr mediator of complex formation. Molecular and Cellular Biology. 1998;18(12):7052–7063. doi: 10.1128/mcb.18.12.7052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosse C, et al. PKC and the control of localized signal dynamics. Nature Reviews: Molecular Cell Biology. 2010;11:103–112. doi: 10.1038/nrm2847. [DOI] [PubMed] [Google Scholar]
- Sandoval R, et al. Ca(2+) signalling and PKCα activate increased endothelial permeability by disassembly of VE-cadherin junctions. Journal of Physiology. 2001;533(Pt 2):433–445. doi: 10.1111/j.1469-7793.2001.0433a.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaller MD, et al. Autophosphorylation of the focal adhesion kinase, pp125FAK, directs SH2-dependent binding of pp60src. Molecular and Cellular Biology. 1994;14(3):1680–1688. doi: 10.1128/mcb.14.3.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlaepfer DD, Hunter T. Signal transduction from the extracellular matrix--a role for the focal adhesion protein-tyrosine kinase FAK. Cell Structure and Function. 1996;21(5):445–450. doi: 10.1247/csf.21.445. [DOI] [PubMed] [Google Scholar]
- Serrels B, et al. Focal adhesion kinase controls actin assembly via a FERM-mediated interaction with the Arp2/3 complex. Nature Cell Biology. 2007;9:1046–1056. doi: 10.1038/ncb1626. [DOI] [PubMed] [Google Scholar]
- Settleman J, Narasimhan V, Foster LC, Weinberg RA. Molecular cloning of cDNAs encoding the GAP-associated protein p190: implications for a signaling pathway from ras to the nucleus. Cell. 1992;69(3):539–549. doi: 10.1016/0092-8674(92)90454-k. [DOI] [PubMed] [Google Scholar]
- Shen TL, et al. Conditional knockout of focal adhesion kinase in endothelial cells reveals its role in angiogenesis and vascular development in late embryogenesis. Journal of Cell Biology. 2005;169(6):941–952. doi: 10.1083/jcb.200411155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieg DJ, et al. FAK integrates growth-factor and integrin signals to promote cell migration. Nature Cell Biology. 2000;2:249–256. doi: 10.1038/35010517. [DOI] [PubMed] [Google Scholar]
- Sieg DJ, Hauck CR, Schlaepfer DD. Required role of focal adhesion kinase (FAK) for integrin-stimulated cell migration. Journal of Cell Science. 1999;112:2677–2691. doi: 10.1242/jcs.112.16.2677. [DOI] [PubMed] [Google Scholar]
- Siflinger-Birnboim A, Goligorsky MS, Del Vecchio PJ, Malik AB. Activation of protein kinase C pathway contributes to hydrogen peroxide-induced increase in endothelial permeability. Laboratory Investigation. 1992;67:24–30. [PubMed] [Google Scholar]
- Sodha NR, Clements RT, Bianchi C, Sellke FW. Cardiopulmonary bypass with cardioplegic arrest activates protein kinase C in the human myocardium. Journal of American College of Surgeons. 2008;206:33–41. doi: 10.1016/j.jamcollsurg.2007.06.308. [DOI] [PubMed] [Google Scholar]
- Sordella R, et al. Modulation of CREB activity by the Rho GTPase regulates cell and organism size during mouse embryonic development. Developmental Cell. 2002;2(5):553–565. doi: 10.1016/s1534-5807(02)00162-4. [DOI] [PubMed] [Google Scholar]
- Sordella R, Jiang W, Chen GC, Curto M, Settleman J. Modulation of Rho GTPase signaling regulates a switch between adipogenesis and myogenesis. Cell. 2003;113(2):147–158. doi: 10.1016/s0092-8674(03)00271-x. [DOI] [PubMed] [Google Scholar]
- Spyridopoulos I, et al. Divergence of angiogenic and vascular permeability signaling by VEGF: inhibition of protein kinase C suppresses VEGF-induced angiogenesis, but promotes VEGF-induced, NO-dependent vascular permeability. Arteriosclerosis, Thrombosis, and Vascular Biology. 2002;22(6):901–906. doi: 10.1161/01.atv.0000020006.89055.11. [DOI] [PubMed] [Google Scholar]
- Sun X, et al. Enhanced interaction between focal adhesion and adherens junction proteins: involvement in sphingosine 1-phosphate-induced endothelial barrier enhancement. Microvascular Research. 2009;77(3):304–313. doi: 10.1016/j.mvr.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki A, et al. Atypical protein kinase C is involved in the evolutionarily conserved par protein complex and plays a critical role in establishing epithelia-specific junctional structures. Journal of Cell Biology. 2001;152:1183–1196. doi: 10.1083/jcb.152.6.1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapon N, Hall A. Rho, Rac and Cdc42 GTPases regulate the organization of the actin cytoskeleton. Current Opinion in Cell Biology. 1997;9(1):86–92. doi: 10.1016/s0955-0674(97)80156-1. [DOI] [PubMed] [Google Scholar]
- Tinsley JH, Teasdale NR, Yuan SY. Involvement of PKCδ and PKD in pulmonary microvascular endothelial cell hyperpermeability. American Journal of Physiology. 2004;286(1):C105–C111. doi: 10.1152/ajpcell.00340.2003. [DOI] [PubMed] [Google Scholar]
- Tomar A, Lim ST, Lim Y, Schlaepfer DD. A FAK-p120RasGAP-p190RhoGAP complex regulates polarity in migrating cells. J Cell Sci. 2009;122(Pt 11):1852–62. doi: 10.1242/jcs.046870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usatyuk PV, Natarajan V. Regulation of reactive oxygen species-induced endothelial cell-cell and cell-matrix contacts by focal adhesion kinase and adherens junction proteins. American Journal of Physiology. 2005;289(6):L999–L1010. doi: 10.1152/ajplung.00211.2005. [DOI] [PubMed] [Google Scholar]
- van Hinsbergh WM. Endothelial permeability for macromolecules. Mechanistic aspects of pathophysiological modulation. Arteriosclerosis, Thrombosis, and Vascular Biology. 1997;17(6):1018–1023. doi: 10.1161/01.atv.17.6.1018. [DOI] [PubMed] [Google Scholar]
- van Nieuw Amerongen GP, Koolwijk P, Versteilen A, van Hinsbergh VW. Involvement of RhoA/Rho kinase signaling in VEGF-induced endothelial cell migration and angiogenesis in vitro. Arteriosclerosis, Thrombosis, and Vascular Biology. 2003;23(2):211–217. doi: 10.1161/01.atv.0000054198.68894.88. [DOI] [PubMed] [Google Scholar]
- van Nieuw Amerongen GP, van Delft S, Vermeer MA, Collard JG, van Hinsbergh VW. Activation of RhoA by thrombin in endothelial hyperpermeability: role of Rho kinase and protein tyrosine kinases. Circulation Research. 2000;87(4):335–340. doi: 10.1161/01.res.87.4.335. [DOI] [PubMed] [Google Scholar]
- Vuori K. Integrin signaling: tyrosine phosphorylation events in focal adhesions. Journal of Membrane Biology. 1998;165(3):191–199. doi: 10.1007/s002329900433. [DOI] [PubMed] [Google Scholar]
- Waller DA, Keavey P, Woodfine L, Dark JH. Pulmonary endothelial permeability changes after major lung resection. Annals of Thoracic Surgery. 1996;61(5):1435–1440. doi: 10.1016/0003-4975(96)00103-8. [DOI] [PubMed] [Google Scholar]
- Wang A, Nomura M, Patan S, Ware JA. Inhibition of protein kinase Cα prevents endothelial cell migration and vascular tube formation in vitro and myocardial neovascularization in vivo. Circulation Research. 2002;90(5):609–616. doi: 10.1161/01.res.0000012503.30315.e8. [DOI] [PubMed] [Google Scholar]
- Warboys CM, Eric Berson R, Mann GE, Pearson JD, Weinberg PD. Acute and chronic exposure to shear stress have opposite effects on endothelial permeability to macromolecules. American Journal of Physiology. 2010;298(6):H1850–H1856. doi: 10.1152/ajpheart.00114.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wildenberg GA, et al. p120-catenin and p190RhoGAP regulate cell-cell adhesion by coordinating antagonism between Rac and Rho. Cell. 2006;127(5):1027–1039. doi: 10.1016/j.cell.2006.09.046. [DOI] [PubMed] [Google Scholar]
- Wittchen ES, van Buul JD, Burridge K, Worthylake RA. Trading spaces: Rap, Rac, and Rho as architects of transendothelial migration. Current Opinion in Hematology. 2005;12(1):14–21. doi: 10.1097/01.moh.0000147892.83713.a7. [DOI] [PubMed] [Google Scholar]
- Wojciak-Stothard B, Ridley AJ. Rho GTPases and the regulation of endothelial permeability. Vascul Pharmacol. 2002a;39(4–5):187–99. doi: 10.1016/s1537-1891(03)00008-9. [DOI] [PubMed] [Google Scholar]
- Wojciak-Stothard B, Ridley AJ. Rho GTPases and the regulation of endothelial permeability. Vascular Pharmacology. 2002b;39(4–5):187–199. doi: 10.1016/s1537-1891(03)00008-9. [DOI] [PubMed] [Google Scholar]
- Wolf RM, et al. p190RhoGAP can act to inhibit PDGF-induced gliomas in mice: a putative tumor suppressor encoded on human chromosome 19q13.3. Genes and Development. 2003;17(4):476–487. doi: 10.1101/gad.1040003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong C, Jin ZG. Protein kinase C-dependent protein kinase D activation modulates ERK signal pathway and endothelial cell proliferation by vascular endothelial growth factor. Journal of Biological Chemistry. 2005;280(39):33262–33269. doi: 10.1074/jbc.M503198200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worthylake RA, Burridge K. RhoA and ROCK promote migration by limiting membrane protrusions. Journal of Biological Chemistry. 2003;278(15):13578–13584. doi: 10.1074/jbc.M211584200. [DOI] [PubMed] [Google Scholar]
- Worthylake RA, Lemoine S, Watson JM, Burridge K. RhoA is required for monocyte tail retraction during transendothelial migration. Journal of Cell Biology. 2001;154(1):147–160. doi: 10.1083/jcb.200103048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong C, et al. The lectin-like domain of TNF protects from listeriolysin-induced hyperpermeability in human pulmonary microvascular endothelial cells - a crucial role for protein kinase Cα inhibition. Vascular Pharmacology. 2010;52(5–6):207–213. doi: 10.1016/j.vph.2009.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan SY, et al. Protein Kinase C Activation Contributes to Microvascular Barrier Dysfunction in the Heart at Early Stages of Diabetes. Circulation Research. 2000;87(5):412–417. doi: 10.1161/01.res.87.5.412. [DOI] [PubMed] [Google Scholar]
- Zhao L, et al. The effect of RhoA on human umbilical vein endothelial cell migration and angiogenesis in vitro. Oncology and Reproduction. 2006;15(5):1147–1152. [PubMed] [Google Scholar]
- Zhao X, Peng X, Sun S, Park AY, Guan JL. Role of kinase-independent and -dependent functions of FAK in endothelial cell survival and barrier function during embryonic development. Journal of Cell Biology. 2010;189:955–965. doi: 10.1083/jcb.200912094. [DOI] [PMC free article] [PubMed] [Google Scholar]