

Abstract
Access to genetically encoded data depends on the dynamics of DNA-binding proteins searching for specific target sites in the genome. This search process is thought to occur by facilitated diffusion—a combination of three-dimensional diffusion and one-dimensional sliding. Although facilitated diffusion is capable of significantly speeding up the search in vitro, the importance of this mechanism in vivo remains unclear. We use numeric simulations and analytical theory to model the target-search dynamics of DNA-binding proteins under a wide range of conditions. Our models reproduce experimental measurements of search-rate enhancement within bulk in vitro experiments, as well as the target search time for transcription factors measured in vivo. We find that facilitated diffusion can accelerate the search process only for a limited range of parameters and only under dilute DNA conditions. We address the role of DNA configuration and confinement, demonstrating that facilitated diffusion does not speed up the search on coiled versus straight DNA. Furthermore, we show that, under in vivo conditions, the search process becomes effectively diffusive and is independent of DNA configuration. We believe our results cast in a new light the role of facilitated diffusion in DNA targeting kinetics within the cell.
Introduction
For the information encoded in a genome to serve its biological function, relevant regions of DNA must be bound by site-specific proteins, including restriction enzymes in bacteria, chromatin-remodeling proteins in eukarya, and transcription factors across all domains of life. These proteins generally find their target sites through random exploration of the eukaryotic nucleus or prokaryotic cytoplasm, and they must do so sufficiently quickly to keep up with the metabolic, regulatory, reproductive, and defensive needs of the cell. This feat is particularly remarkable given the low copy numbers of individual DNA-binding proteins in vivo. For example, Escherichia coli contains ∼20 copies of the LacI repressor protein (1) that must search through 4.6 million basepairs to reach a single target.
In vitro measurements have shown that proteins can locate their binding sites at rates far above the inherent limit for encounters via three-dimensional diffusion (2–4). It has been postulated that nonspecific affinity of DNA-binding proteins can accelerate the target search process through facilitated diffusion (5,6). This search model assumes that a protein locates its target by a series of one-dimensional slides along nonspecific DNA, interspersed by three-dimensional excursions that can result in reattachment to a sequentially proximal DNA segment (microhop), rebinding to a distal segment (macrohop), or escape into bulk solution. As in many biological processes, insights provided by in vitro experiments must be translated to the relevant conditions in vivo, necessitating a flexible quantitative platform that is capable of addressing behavior over a broad range of conditions. Our goal in this work is to incorporate the facilitated diffusion mechanism into a computational and theoretical model to reveal the range of behaviors that are relevant to in vitro and in vivo target-site search dynamics.
A number of experiments support the facilitated diffusion model and help quantify the key dynamic parameters. Single molecule fluorescent tracking has been used to measure off-rates and one-dimensional diffusion coefficients on stretched DNA strands in vitro (7–13). Microhops on extended DNA have been directly observed with fluorescent tracking (9) and inferred from observations of proteins bypassing bound obstacles on DNA (14). The role of nonspecific DNA has been further elucidated by bulk experiments that measure search rates as a function of DNA length (4), processivity between multiple target sites (15,16), and localization to targets on catenated DNA rings (17). Fluorescent tracking studies in vivo have allowed the measurement of kinetic and transport coefficients for transcription factors undergoing facilitated diffusion in a living cell (8).
Theoretical efforts at quantifying facilitated diffusion were pioneered with the classic analytical model of Berg et al. (3,4), which considered the target search as a multistage process of binding and unbinding from nonspecific DNA. A simple model consisting of a series of bulk excursions and local explorations was later proposed (6). Other analytical approaches considered alternating on- and off-cycles required to reach the target (18–22). Reaction-diffusion approaches have been employed to systematically examine scaling regimes (23) and DNA coiling effects (24). Several studies have focused on the transport properties of a protein undergoing facilitated diffusion, both in a crowded three-dimensional environment (25,26) and as an effective one-dimensional motion (27). A number of these analytical approaches reveal an optimum target-search rate when a protein spends approximately equal time on and off the DNA (18,23,27,28). Several numerical simulation studies examined the role of binding strength in facilitated diffusion (29–31).
In this work, we present simulations that directly model in vitro and in vivo target site search through facilitated diffusion to highlight the different roles played by this process in various regimes. Our simulations reproduce the nonmonotonic dependence of the search rate on binding affinity that has been previously seen in experimental (4), analytical (3), and computational (28) studies. We examine the role of DNA configuration in facilitated diffusion by simulating target search on extended versus coiled DNA, with comparison to analytical models. Finally, we model the search process in vivo by using parameters that are relevant to LacI diffusion in E. coli. Our results, which closely match experimentally measured target search times, validate a simple effective description of facilitated diffusion in a living cell.
Methods
Simulation overview
We numerically model facilitated diffusion using a hybrid dynamic Monte Carlo and Brownian dynamics simulation. The DNA is treated as a discretized semiflexible chain, which is packed into a cubical box of appropriate volume to give a specific concentration. Except where otherwise noted, we neglect steric exclusion and dynamics of the DNA chain; the validity of neglecting these effects is tested explicitly in the Results and Discussion. Details of the methods for generating DNA conformations are provided in Section S1A in the Supporting Material. The protein undergoing facilitated diffusion is treated as a point particle that can be either bound or unbound to the DNA.
The simulation begins with the protein in the unbound state at a random position, undergoing three-dimensional diffusive motion with diffusion coefficient D3. Whenever the protein is within the capture radius a = 6 nm of some segment of the DNA chain, it is capable of binding to the chain at the rate konLavail/(4/3πa3), where kon (units of M−1 bp−1 s−1) is the second-order binding rate constant per unit length, and Lavail is the length of DNA within the capture radius. We note that the capture radius was selected to approximate the distance at which a LacI protein and DNA chain would come into steric contact. At the salt concentrations discussed in this article, the length-scale for electrostatic interactions lies in the 0.5–2.5 nm range, and is thus encompassed by the capture region. As soon as the protein moves >1 nm away from any part of the DNA capture region, its motion is propagated by an alternate method.
Specifically, we consider the largest sphere surrounding the protein that does not overlap the capture region of any DNA segment. We select an exit time from the first-passage distribution for diffusive exit from a sphere (32) and then allow the protein to leave the sphere at a uniformly random position on its surface. We note that for randomly coiled 50 kbp DNA, the distance between segments is ∼100 nm, so this alternate propagation technique allows for much larger steps and consequently significant savings in CPU time, even when the protein is within the heart of the DNA coil.
Once the protein binds to the DNA it diffuses in one dimension along the DNA contour with a diffusion constant D1. Unbinding times are exponentially distributed with a rate constant koff. A bound propagation step culminates in the protein either encountering an absorbing boundary at a target site, or returning to the unbound state. The protein continues to cycle through bound and unbound states until it slides into a target-site while bound. A schematic of the simulation steps and an example protein trajectory are illustrated in Fig. 1. Section S1 in the Supporting Material describes the numerical algorithms for both one-dimensional and three-dimensional propagation, as well as the extraction of an overall target search rate constant from the simulation results. The simulation parameters for each of the calculations described in this article are summarized in Table S1 in the Supporting Material.
Figure 1.
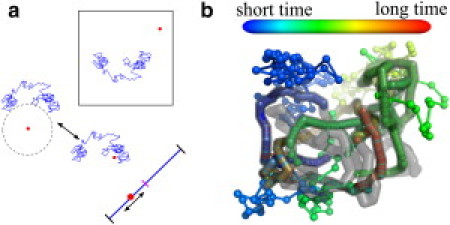
(a) Schematic of protein propagation within our facilitated diffusion simulations. The protein (red) diffuses within a box containing a single DNA chain (blue) using either spherical Green's function methods or Brownian dynamics steps to simulate three-dimensional motion. Upon binding, the protein slides along the DNA contour until it either unbinds or hits the target site (magenta). (b) Example simulation trajectory. (Spheres) Protein positions at equispaced time intervals with color proportional to time. (Gray) DNA.
Analytical models
We compare our simulation results to approximate analytical models for coiled, straight, and confined DNA. For coiled DNA in vitro we use the classic model developed by Berg et al. (3) and Winter et al. (4). This model treats the association and dissociation of the protein with nonspecific DNA as a series of stages (Fig. 2 a). It approximates the segments of a single DNA coil as being uniformly distributed throughout a sphere of radius rg, the radius of gyration for the chain.
Figure 2.
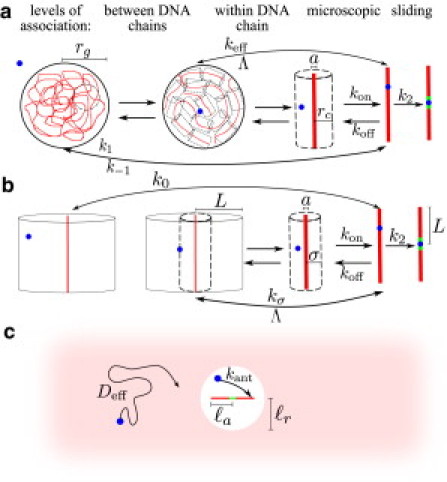
Schematic of analytical models with multiple association levels for: (a) Coiled DNA(3). Sliding to target after nonspecific binding occurs at rate k2. Microscopic dissociation and association rate constants are kon and koff. Association and dissociation from the vicinity of a local DNA segment within the coil occurs at rates keff, Λ. Interchange between the entire coil and empty solution occurs with association rate constant k1 and dissociation rate k–1. (b) Straight DNA. Λ is the rate of dissociation from a localized region within which transport is effectively one-dimensional, and kσ is the rate of rebinding from the edge of this region. Initial binding rate from bulk is k0. (c) Effective diffusion to an antenna region in vivo. In the vicinity of the target site, binding occurs at effective rate kant.
The protein binds nonspecifically to the DNA coil as a second-order kinetic process with an effective rate constant k1 (units of M−1 s−1). The binding position is assumed to be uniformly distributed on the coil. Once bound, the protein can either slide to the target site (at rate k2) or unbind from the DNA (at rate koff). If the protein does not move far enough away to encounter any DNA segments other than the one it just left, it can rebind to the same segment with rate constant kon (units of M−1 bp−1 s−1). The on- and off-rate constants kon and koff constitute the microscopic level of association, which includes microhops and slides. At this level, the protein effectively carries out a one-dimensional motion along the DNA chain, and we assume it remains within a cylinder of radius rc, the typical distance between DNA segments. The rate for a bound protein to leave this region and begin diffusing throughout the DNA coil is defined as Λ, whereas the corresponding effective binding rate within the coil is keff (units of s−1). At this level of association, the protein loses memory of its position along the DNA chain, but remains within the domain of a single coil. The final level of association corresponds to a protein leaving the domain of the DNA molecule entirely and diffusing into bulk solution, which it does with rate k–1. The on- and off-rates at each level of binding are related to Keq, the equilibrium binding constant per unit length,
| (1) |
where 2L is the contour length of the DNA.
Given this multistep picture for protein and DNA association, the overall rate constant for finding the target site is given by
| (2) |
where V is the volume per DNA molecule and the rate constant ka has units of M−1 s−1. Details for calculating the individual rate constants at the different levels of association are provided in Section S2A in the Supporting Material.
We use a similar approach in developing an approximate analytical model for facilitated diffusion on straight DNA. Unlike previous exact solutions of this problem (33), our model has the advantage of yielding a closed-form target-search rate constant, which can be directly compared with simulation results across different parameter regimes. A schematic of the model is illustrated in Fig. 2 b. We begin by considering the protein bound to a DNA chain, which is enclosed in a reflective cylindrical box of radius L, or half the chain length. The protein can hop on and off the DNA (with rate constants kon and koff) while still executing an effective one-dimensional motion, so long as it does not move beyond a predefined distance from the chain. We define this distance σ such that microhops within this radius do not lose track of the protein's position on the DNA. After leaving σ, the protein is assumed to rebind uniformly on the DNA chain. The rate Λ of a bound protein leaving this cylinder, as well as the overall rate of rebinding (kσ, units of s−1) for a protein starting at radius σ can be calculated by solving the diffusion equation in cylindrical coordinates. The initial binding rate k0 (units of s−1) for a protein that starts uniformly distributed within the large reflective cylinder can be similarly calculated. The sliding rate k2 is found in the same manner as for the coiled chain. Finally, to compare with simulations in a box of arbitrary size, we treat binding processes beginning outside the σ radius as second-order reactions, so that the rates kσ and k0 are scaled by the volume per DNA molecule. The overall rate constant of reaching the target site when the DNA concentration is [DNA] = 1/V is given by
| (3) |
Details of the calculations are provided in Section S2B in the Supporting Material.
The previously described classic model for coiled DNA can easily be modified for DNA under confinement. Specifically, the outermost association level in Fig. 2 a is no longer included because a single DNA molecule fills up the entire available volume. The radius rc of the cylinder around individual DNA segments can be approximated so that such cylinders fill up the volume, 2πLrc2 = V. The overall rate for a single protein to find the target site is then given by
| (4) |
For the parameter range relevant to LacI-facilitated diffusion in E. coli, the target search process can be approximated by a simple model of effective diffusion throughout the cell, followed by binding within an antenna region of the target site. In this antenna model, illustrated in Fig. 2 c, the rate to hit the target is approximated as
| (5) |
where Deff is the effective diffusion rate constant through the cell, [P] is the protein concentration, kant (units of s−1) is the effective binding rate in the antenna region, ℓa is the size of the antenna region, and phit is the probability of sliding to the target site before unbinding from the DNA antenna. Equation 5 stems from the solution of the reaction-diffusion equation in spherical coordinates, with a central sphere that absorbs uniformly at rate kant. In this model, the antenna DNA has length and the antenna capture region has radius ℓr = ℓa + a. Binding within the region occurs at the rate kant = kon/(4/3πa3) 〈Lavail〉, where the average length of available antenna DNA is 〈Lavail〉 = 2ℓaa3/ℓ r3. The probability of sliding to a target when starting distributed over a length ℓa before unbinding at rate koff is , independent of the parameters (see Eq. S7 in the Supporting Material). This approximate model is applicable when there is rapid equilibration between unbound and nonspecifically bound states of the protein, when unbinding is sufficiently fast that the antenna can be approximated as straight, and when the binding rate constant kon is low enough that dissociation from the antenna results in the protein resuming bulk search throughout the cell.
Results and Discussion
Target search acceleration in vitro
A number of experimental (2,3,34) and theoretical (3,6,23) studies determined that, under certain conditions, facilitated diffusion can allow a target search rate significantly above the limit that can be achieved by ordinary three-dimensional diffusion. One of the key results supporting this notion comes from the experiments of Winter et al. (4), which measured the time required for LacI to find a single target site on dilute 50-kbp DNA as a function of binding strength (modulated by salt concentration). We find that simulations with our two-state transport model can approximately reproduce the search acceleration observed in these experiments, as well as the nonmonotonic dependence of search rate on binding strength. In these simulations, we use a three-dimensional diffusion constant (D3) appropriate for a LacI-sized protein in water, and a binding rate constant (kon) that is sufficiently large for binding to be entirely diffusion-limited. The equilibrium binding constant (Keq = kon/koff) is expressed as a previously-measured function of salt concentration (4,35).
The results of these simulations, illustrated in Fig. 3, show three regimes for the target site search process as a function of nonspecific binding. In the case of large Keq, the search consists of a rapid initial binding event followed by a slow one-dimensional slide toward the target site. In this regime, the overall rate for reaching the target site can be approximated as the rate for sliding to an absorbing barrier via one-dimensional diffusion from a uniform initial distribution. The corresponding rate constant is given by ka/V = 3D1/L2, where D1 is the sliding diffusion constant, V is the volume per DNA molecule, and 2L is the total length of DNA. This limit corresponds to the dashed line in Fig. 3. We note that the one-dimensional diffusion constant for our simulations was selected specifically to match the experimental data in this regime.
Figure 3.
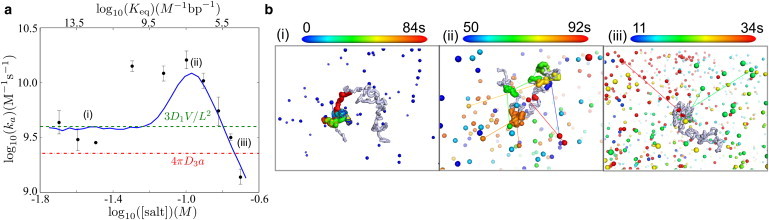
Simulation of facilitated diffusion on 50 kbp DNA at 1 pM concentration. (Left) Salt dependence of rate constant for reaching target site (ka). (Black dots) Experimental data (4); (blue solid line) simulation results; (green dashed line) pure sliding limit; and (red dash-dotted line) three-dimensional diffusion limit. (Right) Representative target-search trajectories in regimes of (i) strong binding, (ii) intermediate binding, and (iii) weak binding. Protein position is shown every 10 ms with color proportional to time. (Lines) Unbinding and rebinding events. (Gray) Chain configuration.
In the weak binding regime (low Keq), the search is dominated by the time required to initially bind the DNA and the high probability of diffusing away upon unbinding. Because nonspecific binding is required in our model to recognize the target site, the rate of finding the target slows down as the nonspecific binding strength decreases. This drop-off in rate can be explained in large part as a decrease in the size of the antenna surrounding the target site, within which the protein must bind so that it can slide into the target before unbinding (6,23).
Intermediate between these two regimes lies the region where facilitated diffusion results in the greatest acceleration of the search. In this regime, the protein performs a sufficient number of short- and long-range hops to take advantage of rapid three-dimensional transport while also having a long enough sliding length to significantly widen the effective target for binding. The peak in the target search rate occurs at approximately Keq = V/(2L). At this optimal binding strength, the protein spends equal times on and off the DNA, a condition that has been previously shown via scaling arguments to optimize the target search rate (18,23). We note that the width of the peak is primarily determined by the dependence of equilibrium binding strength Keq on the salt concentration. This dependence has been proposed to become weaker in the dilute salt regime (4), potentially explaining the discrepancy in peak width between simulations and experiments.
In addition to the well-known effect of increasing the effective target size by widening the antenna of DNA within sliding range, facilitated diffusion can accelerate the search process by essentially tethering the protein to the DNA—that is, by increasing the effective protein concentration in the vicinity of the chain. The magnitude of this tethering effect is dependent on the concentration of DNA, with acceleration above the diffusion-limited rate observed only if the DNA is sufficiently dilute (Fig. 4).
Figure 4.
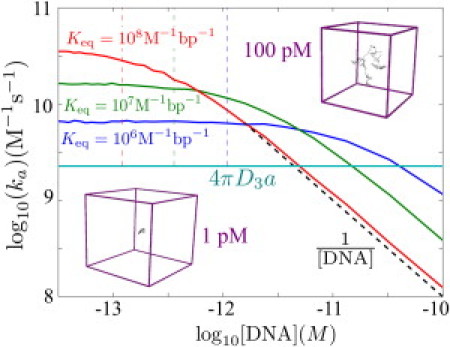
Effect of DNA concentration on target search. Simulated search rate constants (ka) are shown for three different binding strengths. (Cyan line) Diffusion-limited rate constant. (Dashed black line) Scaling at high DNA concentrations. (Dashed vertical lines) Concentration at which the time to find a DNA coil and the search time within the coil are equal, according to analytical model for coiled DNA. (Insets) Size of the simulation box for two different concentration values.
In Fig. 4, we see that the second-order rate constant ka for finding the target site is concentration-independent at low [DNA], whereas at high concentrations it varies inversely with [DNA]. Equivalently, the overall rate to find the target varies linearly with [DNA] at low concentrations but becomes independent of [DNA] for high concentrations. This effect is a manifestation of the transition from a second-order to a first-order reaction. When the DNA is dilute, the search is dominated by the time required to find individual DNA chains, whereas when the volume per DNA chain approaches the size of the chain itself the search is dominated by intramolecular transport within a single chain.
The target search rate drops well below the diffusion limit once the DNA becomes sufficiently concentrated. The tethering effect only has a meaningful role to play when the DNA takes up a small fraction of the volume available to the protein. For densely packed DNA in vivo, facilitated diffusion would therefore not be expected to provide significant acceleration above the diffusion-limited rate.
Role of DNA configuration in vitro
One of the key questions that arises in analyzing target site search by facilitated diffusion concerns the role played by the DNA conformation (23,24,36). In the simplest extreme, we can compare the rate of finding the target site on relaxed, coiled DNA to that on DNA that has been stretched out into a straight configuration. Prior studies (24) indicate that coiling of the DNA should speed up the search by allowing hops between sequentially distal DNA segments that are proximal in space. We use simulations to explore whether such an acceleration on coiled versus straight DNA is consistent with the picture here afforded by the facilitated diffusion model.
Using an approximate analytical theory, we calculate the search rate as a function of kon and koff for a straight 50-kbp chain of DNA confined in a box just large enough to enclose the chain (Fig. 5 a). According to this model, the coiled DNA speeds up the search by at most 20% for the parameters used here (Fig. 5 b). We see that even without the coiled DNA configuration required for macrohops, the search rate depends nonmonotonically on the equilibrium binding strength (Keq = kon/koff). For high on- and off-rates, where the binding is diffusion-limited and the sliding length is much shorter than the full length of the DNA, the rate is determined entirely by Keq and is maximized at 2LKeq = 1/[DNA], the binding strength that allows the protein to spend equal times on and off the DNA.
Figure 5.
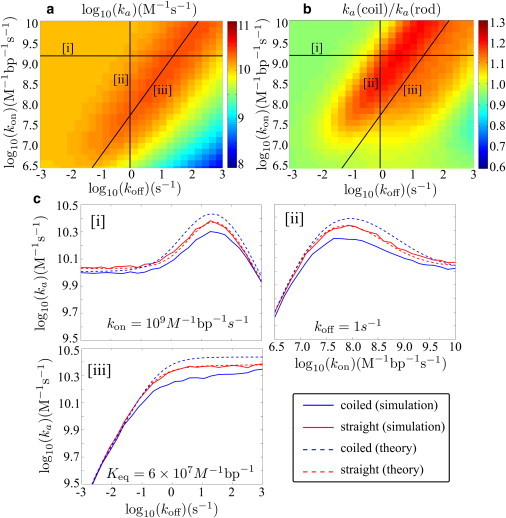
Facilitated diffusion on coiled versus straight 50 kbp DNA. (a) Target search rate constant (ka) as a function of on- and off-rates, from analytical model for straight DNA. (b) Ratio of search rates using analytical models for coiled and straight DNA. (c) Slices through the surface plots at (i) constant kon, (ii) constant koff, and (iii) constant Keq, comparing simulation results with analytical theory.
We note that the protein slides an average distance of before unbinding and that there is no single sliding length corresponding to optimum search. However, we can define an effective sliding length for the coil as the typical distance traveled without encountering a distal DNA segment. In the limit of high kon, this effective length is given by . This expression is analogous to the sliding length described in previous studies (6,28), where it was used as a measure of the length traveled effectively in one dimension along the DNA contour before embarking on a three-dimensional search. The optimum effective sliding length for the parameters used here is ξ ≈ 1400 bp, a much larger value than expected for crowded DNA in vivo (28) but comparable to the lengths measured in dilute solution (37).
To check the accuracy of the approximate analytical models, we run simulations for several slices through the surface plot using both straight and coiled DNA (Fig. 5 c). The simulations indicate that the coiled DNA model somewhat overpredicts the target search rate, although the qualitative features of the kon and koff dependence are well modeled. As a result, we find that target search through facilitated diffusion actually occurs at least as fast or faster on stretched rather than coiled DNA. Similar results are seen for more dilute DNA where the stretched configuration no longer spans the entire length of the simulation box (see Fig. S2 in the Supporting Material).
The slightly greater target search rates on stretched DNA may be explained by the interchange of protein between bulk solution and nonspecific DNA. A protein in solution will more rapidly locate a long, thin chain of DNA stretched across the length of the box than a dense coil occupying a small volume in three-dimensional space. This effect speeds up target search on a straight rod in the case of intermediate binding strength, where the process is still dominated by three-dimensional excursions but the protein spends sufficient time bound to make its ability to find nonspecific DNA useful. In the limiting case of very strong binding, there is a very slight preference for stretched DNA due to the distribution of binding positions for a protein approaching from the bulk. On coiled DNA, the protein has a greater probability of binding toward the edges of the chain so that a longer time is required to slide in toward the target site, which is located in the center for these simulations. With very weak binding the search problem is reduced to finding an antenna region around the target site by three-dimensional diffusion, and the DNA conformation on length scales much longer than the antenna makes no difference to the overall rate.
The comparable acceleration of the search process on straight as well as coiled DNA implies that the tethering effect keeping the protein in the vicinity of the DNA has a much greater role to play than the intrachain macrohops commonly held to be the keystone feature of facilitated diffusion, a concept that has been recently called into question (38). We note that our results differ qualitatively from prior theory that implied faster target site search on coiled DNA (24,36). Whereas these prior studies focused primarily on protein motion within the DNA coil, our theory and simulations take into account protein excursions into bulk solution and the concomitant tethering effect that arises at low DNA concentrations. Our model does not, however, include direct intersegmental transfers for proteins with multiple DNA binding sites; a significant configuration-dependent speedup of the search may be possible for such proteins (39).
Recent single-molecule experiments found that target search occurs up to twice as fast on coiled as on straight DNA (36) at high salt concentrations. A simulation using relevant parameters for this system yields rates that are close in magnitude to the measured results but show no difference between stretched and coiled DNA (see Fig. S3). Although the authors argue that the acceleration is caused by the increased availability of macrohops in the coiled DNA, our calculations imply that the classic facilitated diffusion process, in and of itself, is insufficient to explain the configuration dependence of the search rates. Instead, either the experimental details or alternate physical effects that are not encompassed by our model must be responsible for the observations.
One physical effect neglected in our simulations is the dynamics of the coiled DNA, which would help randomize the distribution of macrohops over time. To test the magnitude of this effect on the reported results, we run simulations where the configuration of the full DNA chain is reequilibrated at each unbinding event. Although we find that the target search rates on coiled DNA do increase for the 50-kbp chain, they remain no higher than the rates on stretched DNA except in the case of very rapid unbinding (Fig. S4). For the parameter regime relevant to the single-molecule experiments (36), reequilibration of the DNA had no discernible effect on target search rates (Fig. S3). Full reequilibration at each hop serves as the opposite extreme to a frozen DNA chain, and the similarity of the results implies that a more careful model of the DNA dynamics is unlikely to change the overall conclusion that facilitated diffusion occurs no faster on coiled than on stretched DNA.
Target-site search in vivo
To study the role of facilitated diffusion in living prokaryotes, we run simulations designed to model lac repressor (LacI) searching for its target site in E. coli. We obtain our simulation parameters from in vivo single-molecule observations (8) (see Table S1). The simulation (with no free fitting parameters) accurately reproduces the experimentally measured mean time, τ ≈ 60 s, for one of three LacI proteins to find one of two target sites (Fig. 6).
Figure 6.
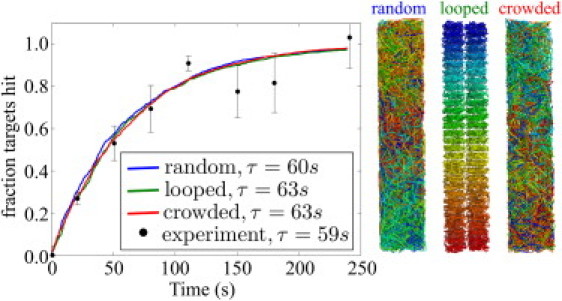
Cumulative time distributions for one of three LacI proteins to hit one of the two target sites in a simulated E. coli system with different genome packing arrangements. Simulation parameters and experimental data were drawn from Elf et al. (8). The time constant τ is determined by fitting to a single exponential. (Right) Sample configurations of DNA. The crowded packing includes steric exclusion among DNA segments and between DNA and protein.
It is worth noting that the rate to reach the target site at in vivo conditions is slower than would be expected through pure three-dimensional diffusion. For the parameters used here, the pure diffusion-limited rate is given by 4πD3a/V × 2 × 3 ≈ 1.4 s−1, nearly two orders-of-magnitude faster than the measured 0.017 s−1 rate. This result is a manifestation of the high nonspecific DNA concentration in vivo, which eliminates the tethering effect that is in large part responsible for search acceleration via facilitated diffusion.
We find the same approximate rate of target site search for three different arrangements of DNA within the cell. Specifically, we compare a confined random coil to an arrangement with 60 sequentially connected loops that has previously been proposed for E. coli genome packing (40). We also consider a crowded system that incorporates steric exclusion effects by forbidding the protein to approach within 5 nm of the DNA chain and packing the DNA chain so that no two segments come within 5 nm of each other. The similar results for these different arrangements indicate that the precise geometry of DNA within a cell has little effect on the facilitated diffusion process.
The reason behind this lack of sensitivity can be understood by considering the displacement of the protein over time (Fig. 7). We find that within a few milliseconds the protein begins to experience an effective diffusive motion, with the diffusion coefficient given by Deff = fbD1/3 + (1 – fb)D3, where fb = 2LKeq/(V + 2LKeq) is the fraction of time spent bound, as expected for a rapidly equilibrating system (8). For biologically relevant kinetic parameters, the details of the DNA geometry are thus washed out in the effective diffusion at timescales much shorter than that required to find the target site.
Figure 7.
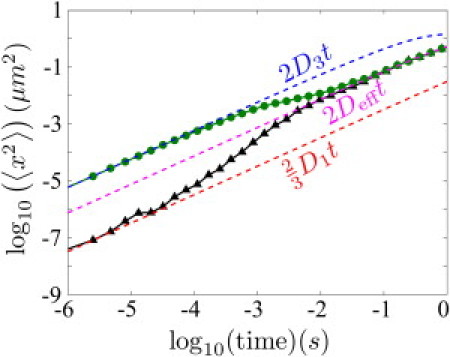
Mean-square displacement along longest cell axis for tracked proteins undergoing simulated facilitated diffusion in E. coli. Proteins start either bound (▴) or unbound (•) to the DNA. (Dashed lines, top to bottom) Effective motion for unbound diffusion, overall effective three-dimensional diffusion, and sliding (color online), respectively (color online).
Prior theoretical studies have considered the role of DNA conformation on target site search in vivo by treating the overall transport process as diffusion on a fractal (41,42) or through simulations that included subdiffusive three-dimensional transport induced by crowded conditions in the nucleus (26). We emphasize that the insensitivity to DNA arrangement observed in our study requires the DNA to be straight on the scale of the sliding length and the unbound protein motion to be diffusive. Although large RNA-protein particles and genomic loci move subdiffusively in the E. coli cytoplasm (43,44), analyses of in vivo transport of proteins comparable in size to LacI suggest diffusive motion (8,45). For the parameters used to model the transport of LacI in E. coli, the sliding length (∼20 nm) is smaller than the persistence length of naked DNA. However, in the case of the eukaryotic genome, which is tightly packed on comparable length scales, it may be that the effective three-dimensional motion of some DNA-binding proteins ceases to be classically diffusive.
The target search process on a confined genome can be approximated with the classic model for coiled DNA (3), modified as described in Methods. Although this model explicitly takes into account the interchange between sliding and three-dimensional hops, a much simpler analytic approximation can be made for the parameter regime in this study. Specifically, we treat the target search process as effective diffusion to an antenna region around the target site whose size is defined by the sliding length. We find that both models match the simulation results near the experimentally relevant parameters for this system, and that the former model provides a good approximation to the results in the entire range of parameters studied (Fig. 8).
Figure 8.
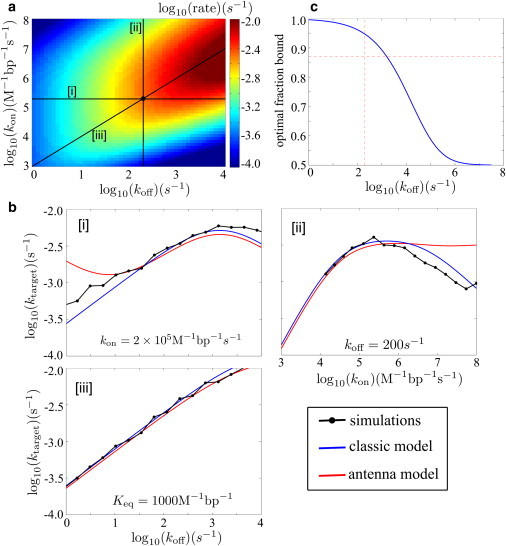
(a) Rate (ktarget) for a single LacI protein to find one target site in E. coli, as a function of kon and koff, calculated with modified classic model for confined DNA. (Central black dot) Experimentally measured parameters (8). (b) Slices through the surface plot comparing simulation results, classic model, and effective diffusion to an antenna at (i) fixed kon, (ii) fixed koff, and (iii) fixed Keq. (c) Optimal fraction of time spent bound for different off-rates, calculated with the classic model. (Dashed lines) Experimental values of the off-rate and fraction bound.
Much as in the in vitro results (Fig. 5), the target search rate depends nonmonotonically on both kon and koff. However, this system is not in a regime of diffusion-limited binding (i.e., kon < D3). Consequently, raising the on- and off-rates at a constant binding affinity Keq steadily increases the target search rate (Fig. 8 (iii)) in the vicinity of the experimental parameters. The maximum search rate would be reached when both kon and koff are several orders-of-magnitude larger and the protein spends half of its time bound to the DNA (Keq ≈ 140 M−1 bp−1). However, if there are inherent physical limits on the rate of nonspecific unbinding, then it becomes advantageous for the protein to spend a greater fraction of its time bound to the DNA (Fig. 8 c). This effect is a direct consequence of the crowded nature of the in vivo system.
Conclusions
Our work uses a combination of analytical theory and simulations of hopping and sliding along nonspecific DNA to model target-site search through facilitated diffusion in vitro and in vivo. Our detailed treatment of the multiscale transport processes reveals the essential mechanisms that dictate the overall target-site search rate, providing a clear identification of the conditions where facilitated diffusion accelerates the search kinetics. We reproduce the rapid search rates that are a hallmark feature of facilitated diffusion in vitro (4). However, the crowded environment in vivo ensures that the target search is reduced to an effective diffusion process that is slower than pure three-dimensional diffusion.
Our simulations help to highlight two mechanisms—an antenna effect and a tethering effect—through which facilitated diffusion can speed up target site search above the nominal diffusion limit. The antenna effect, which has previously been described by several groups (6,23), extends the effective size of the target by allowing the protein to bind nonspecifically to a stretch of DNA equal to a typical sliding length and then find the target rapidly in one dimension. Effectively, this is a short-range effect that is independent of the DNA configuration on length scales greater than the antenna length itself. The tethering effect corrals proteins in the vicinity of the DNA via nonspecific binding, thereby increasing the effective protein concentration near the target site. This effect plays an important role under dilute in vitro conditions, where the DNA chains take up a small fraction of the volume available to wandering proteins.
We find that the antenna and the tether effect together are sufficient to produce target search rates significantly above the diffusion limit on both straight and coiled DNA. One of the key conclusions from our study is that rapid target search through facilitated diffusion does not require macrohops that span sequentially distant regions of DNA looped close to each other in space. Although it has been proposed that the juxtaposition of distal DNA regions should enhance the search on coiled versus straight DNA (24), we find this effect is overshadowed by protein interchange between nonspecific DNA and bulk solution. Based on our simulations, target search occurs at least as fast when the DNA is stretched out into a straight rod as when it is allowed to form a random coil.
The DNA configuration also seems to make little difference in how quickly a protein finds its target site under in vivo conditions. For LacI in E. coli, protein transport through a combination of one-dimensional sliding and three-dimensional jumps reduces to an effective three-dimensional diffusion on timescales much shorter than the time required to find the target site. The overall target-site search process in this regime behaves as effective diffusion to a short antenna of DNA around the target site followed by rapid sliding to the target. The packing arrangement of the DNA washes out in the overall diffusive transport.
Our results highlight the different roles played by facilitated diffusion in vitro and in vivo. For dilute DNA molecules in vitro, the tethering effect can yield target search rates significantly faster than those that would be achieved by pure three-dimensional diffusion throughout the large available volume. Within a living cell, however, nonspecific binding to the highly crowded genomic DNA primarily slows down protein transport, leading to search rates that are slower than the diffusion limit. In both cases, there is a local antenna effect that modulates the size of the effective target region leading to a nonmonotonic dependence of the target search rate on binding strength. Although in vitro search rates are generally optimized at the equilibrium binding strength that enables the protein to spend half its time bound to the DNA, the optimal binding strength in vivo depends on the unbinding rate and, for the parameters studied here, appears to require that the protein spend a significantly higher fraction of its time nonspecifically bound.
The focus of this work is on the role of DNA configuration, concentration, and confinement in facilitated diffusion within in vitro and in vivo systems. Our basic simulation model can be adapted to incorporate other effects such as stationary and mobile obstacles on the DNA, colocalization of prokaryotic transcription factors and their target sites (19), and processivity between multiple targets (15). This work, which directly and quantitatively models facilitated diffusion under in vivo conditions, forms the foundation for building a physically motivated theory of genome processing kinetics in living cells.
Acknowledgments
E.F.K. acknowledges funding from the National Science Foundation Graduate Student Fellowship Program and the Hertz Foundation. M.A.D. is supported by a Bio-X Graduate Student Fellowship. A.J.S. acknowledges funding from the National Science Foundation CAREER Award Program. This research was carried out in part with TeraGrid resources provided by the Purdue Condor pool under grant No. TG-MCB090041.
Supporting Material
References
- 1.Gilbert W., Müller-Hill B. Isolation of the lac repressor. Proc. Natl. Acad. Sci. USA. 1966;56:1891–1898. doi: 10.1073/pnas.56.6.1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Riggs A.D., Bourgeois S., Cohn M. The lac repressor-operator interaction. 3. Kinetic studies. J. Mol. Biol. 1970;53:401–417. doi: 10.1016/0022-2836(70)90074-4. [DOI] [PubMed] [Google Scholar]
- 3.Berg O.G., Winter R.B., von Hippel P.H. Diffusion-driven mechanisms of protein translocation on nucleic acids. 1. Models and theory. Biochemistry. 1981;20:6929–6948. doi: 10.1021/bi00527a028. [DOI] [PubMed] [Google Scholar]
- 4.Winter R.B., Berg O.G., von Hippel P.H. Diffusion-driven mechanisms of protein translocation on nucleic acids. 3. The Escherichia coli lac repressor-operator interaction: kinetic measurements and conclusions. Biochemistry. 1981;20:6961–6977. doi: 10.1021/bi00527a030. [DOI] [PubMed] [Google Scholar]
- 5.von Hippel P.H., Berg O.G. Facilitated target location in biological systems. J. Biol. Chem. 1989;264:675–678. [PubMed] [Google Scholar]
- 6.Halford S.E., Marko J.F. How do site-specific DNA-binding proteins find their targets? Nucleic Acids Res. 2004;32:3040–3052. doi: 10.1093/nar/gkh624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang Y.M., Austin R.H., Cox E.C. Single molecule measurements of repressor protein 1D diffusion on DNA. Phys. Rev. Lett. 2006;97:048302. doi: 10.1103/PhysRevLett.97.048302. [DOI] [PubMed] [Google Scholar]
- 8.Elf J., Li G.W., Xie X.S. Probing transcription factor dynamics at the single-molecule level in a living cell. Science. 2007;316:1191–1194. doi: 10.1126/science.1141967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bonnet I., Biebricher A., Desbiolles P. Sliding and jumping of single EcoRV restriction enzymes on non-cognate DNA. Nucleic Acids Res. 2008;36:4118–4127. doi: 10.1093/nar/gkn376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blainey P., van Oijen A., Xie X. A base-excision DNA-repair protein finds intrahelical lesion bases by fast sliding in contact with DNA. Proc. Natl. Acad. Sci. USA. 2006;103:5752–5757. doi: 10.1073/pnas.0509723103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gorman J., Chowdhury A., Greene E.C. Dynamic basis for one-dimensional DNA scanning by the mismatch repair complex Msh2-Msh6. Mol. Cell. 2007;28:359–370. doi: 10.1016/j.molcel.2007.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim J.H., Larson R.G. Single-molecule analysis of 1D diffusion and transcription elongation of T7 RNA polymerase along individual stretched DNA molecules. Nucleic Acids Res. 2007;35:3848–3858. doi: 10.1093/nar/gkm332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim J., Dukkipati V., Larson R. Stretching and immobilization of DNA for studies of protein-DNA interactions at the single-molecule level. Nanoscale Res. Lett. 2007;2:185–201. [Google Scholar]
- 14.Gorman J., Plys A., Greene E. Visualizing one-dimensional diffusion of eukaryotic DNA repair factors along a chromatin lattice. Nat. Struct. Mol. Biol. 2010;17:932–938. doi: 10.1038/nsmb.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stanford N.P., Szczelkun M.D., Halford S.E. One- and three-dimensional pathways for proteins to reach specific DNA sites. EMBO J. 2000;19:6546–6557. doi: 10.1093/emboj/19.23.6546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gowers D.M., Wilson G.G., Halford S.E. Measurement of the contributions of 1D and 3D pathways to the translocation of a protein along DNA. Proc. Natl. Acad. Sci. USA. 2005;102:15883–15888. doi: 10.1073/pnas.0505378102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gowers D.M., Halford S.E. Protein motion from non-specific to specific DNA by three-dimensional routes aided by supercoiling. EMBO J. 2003;22:1410–1418. doi: 10.1093/emboj/cdg125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Slutsky M., Mirny L.A. Kinetics of protein-DNA interaction: facilitated target location in sequence-dependent potential. Biophys. J. 2004;87:4021–4035. doi: 10.1529/biophysj.104.050765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wunderlich Z., Mirny L.A. Spatial effects on the speed and reliability of protein-DNA search. Nucleic Acids Res. 2008;36:3570–3578. doi: 10.1093/nar/gkn173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kolesov G., Wunderlich Z., Mirny L.A. How gene order is influenced by the biophysics of transcription regulation. Proc. Natl. Acad. Sci. USA. 2007;104:13948–13953. doi: 10.1073/pnas.0700672104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cherstvy A.G., Kolomeisky A.B., Kornyshev A.A. Protein-DNA interactions: reaching and recognizing the targets. J. Phys. Chem. B. 2008;112:4741–4750. doi: 10.1021/jp076432e. [DOI] [PubMed] [Google Scholar]
- 22.Kolomeisky A. Physics of protein-DNA interactions: mechanisms of facilitated target search. Phys. Chem. Chem. Phys. 2010;13:2088–2095. doi: 10.1039/c0cp01966f. [DOI] [PubMed] [Google Scholar]
- 23.Hu T., Grosberg A.Y., Shklovskii B.I. How proteins search for their specific sites on DNA: the role of DNA conformation. Biophys. J. 2006;90:2731–2744. doi: 10.1529/biophysj.105.078162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lomholt M.A., van den Broek B., Metzler R. Facilitated diffusion with DNA coiling. Proc. Natl. Acad. Sci. USA. 2009;106:8204–8208. doi: 10.1073/pnas.0903293106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wedemeier A., Merlitz H., Langowski J. How proteins squeeze through polymer networks: a Cartesian lattice study. J. Chem. Phys. 2009;131:064905. doi: 10.1063/1.3205100. [DOI] [PubMed] [Google Scholar]
- 26.Wedemeier A., Zhang T., Langowski J. The role of chromatin conformations in diffusional transport of chromatin-binding proteins: Cartesian lattice simulations. J. Chem. Phys. 2008;128:155101. doi: 10.1063/1.2895048. [DOI] [PubMed] [Google Scholar]
- 27.Díaz de La Rosa M., Koslover E., Spakowitz A. Target-site search of DNA-binding proteins. Biophys. J. 2010;98:221. doi: 10.1016/j.bpj.2010.02.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Klenin K.V., Merlitz H., Wu C.X. Facilitated diffusion of DNA-binding proteins. Phys. Rev. Lett. 2006;96:018104. doi: 10.1103/PhysRevLett.96.018104. [DOI] [PubMed] [Google Scholar]
- 29.Das R.K., Kolomeisky A.B. Facilitated search of proteins on DNA: correlations are important. Phys. Chem. Chem. Phys. 2010;12:2999–3004. doi: 10.1039/b921303a. [DOI] [PubMed] [Google Scholar]
- 30.Merlitz H., Klenin K.V., Langowski J. Facilitated diffusion of DNA-binding proteins: efficient simulation with the method of excess collisions. J. Chem. Phys. 2006;124:134908. doi: 10.1063/1.2178806. [DOI] [PubMed] [Google Scholar]
- 31.Florescu A.M., Joyeux M. Description of nonspecific DNA-protein interaction and facilitated diffusion with a dynamical model. J. Chem. Phys. 2009;130:015103. doi: 10.1063/1.3050097. [DOI] [PubMed] [Google Scholar]
- 32.Redner S. Cambridge University Press; Nyack, NY: 2001. A Guide to First-Passage Processes. [Google Scholar]
- 33.Berg O.G., Ehrenberg M. Association kinetics with coupled three- and one-dimensional diffusion. Chain-length dependence of the association rate of specific DNA sites. Biophys. Chem. 1982;15:41–51. doi: 10.1016/0301-4622(82)87015-4. [DOI] [PubMed] [Google Scholar]
- 34.Halford S.E., Szczelkun M.D. How to get from A to B: strategies for analyzing protein motion on DNA. Eur. Biophys. J. 2002;31:257–267. doi: 10.1007/s00249-002-0224-4. [DOI] [PubMed] [Google Scholar]
- 35.deHaseth P.L., Lohman T.M., Record M.T., Jr. Nonspecific interaction of lac repressor with DNA: an association reaction driven by counterion release. Biochemistry. 1977;16:4783–4790. doi: 10.1021/bi00641a004. [DOI] [PubMed] [Google Scholar]
- 36.van den Broek B., Lomholt M.A., Wuite G.J. How DNA coiling enhances target localization by proteins. Proc. Natl. Acad. Sci. USA. 2008;105:15738–15742. doi: 10.1073/pnas.0804248105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jeltsch A., Wenz C., Pingoud A. Linear diffusion of the restriction endonuclease EcoRV on DNA is essential for the in vivo function of the enzyme. EMBO J. 1996;15:5104–5111. [PMC free article] [PubMed] [Google Scholar]
- 38.Halford S.E. An end to 40 years of mistakes in DNA-protein association kinetics? Biochem. Soc. Trans. 2009;37:343–348. doi: 10.1042/BST0370343. [DOI] [PubMed] [Google Scholar]
- 39.Sheinman M., Kafri Y. The effects of intersegmental transfers on target location by proteins. Phys. Biol. 2009;6:016003. doi: 10.1088/1478-3975/6/1/016003. [DOI] [PubMed] [Google Scholar]
- 40.Képès F. Periodic transcriptional organization of the E. coli genome. J. Mol. Biol. 2004;340:957–964. doi: 10.1016/j.jmb.2004.05.039. [DOI] [PubMed] [Google Scholar]
- 41.Bénichou O., Chevalier C., Voituriez R. Geometry-controlled kinetics. Nat. Chem. 2010;2:472–477. doi: 10.1038/nchem.622. [DOI] [PubMed] [Google Scholar]
- 42.Bénichou O., Chevalier C., Voituriez R. Facilitated diffusion of proteins on chromatin. Phys. Rev. Lett. 2011;106:038102. doi: 10.1103/PhysRevLett.106.038102. [DOI] [PubMed] [Google Scholar]
- 43.Weber S.C., Spakowitz A.J., Theriot J.A. Bacterial chromosomal loci move subdiffusively through a viscoelastic cytoplasm. Phys. Rev. Lett. 2010;104:238102. doi: 10.1103/PhysRevLett.104.238102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Golding I., Cox E.C. Physical nature of bacterial cytoplasm. Phys. Rev. Lett. 2006;96:098102. doi: 10.1103/PhysRevLett.96.098102. [DOI] [PubMed] [Google Scholar]
- 45.Elowitz M.B., Surette M.G., Leibler S. Protein mobility in the cytoplasm of Escherichia coli. J. Bacteriol. 1999;181:197–203. doi: 10.1128/jb.181.1.197-203.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.