

Abstract
We use a 6.8-fs laser as the light source for broad-band femtosecond pump-probe real-time vibrational spectroscopy to investigate both electronic relaxation and vibrational dynamics of the Qy-band of Chl-a at 293 K. More than 25 vibrational modes coupled to the Qy transition are observed. Eleven of them have been clarified predominantly due to the excited state, and six of them are concluded to be nearly exclusively resulting from the ground-state wave-packet motion. Moreover, thanks to the broad-band detection over 5000 cm−1, the modulated signals due to the excited state vibrational coherence are observed on both sides of the 0-0 transition with equal separation. The corresponding nonlinear process has been studied using a three-level model, from which the probe wavelength dependence of the phase of the periodic modulation can be calculated. The probe wavelength dependence of the vibrational amplitude is interpreted in terms of the interaction between the “pump” or “laser,” Stokes, and anti-Stokes field intermediated by the molecular vibrations. In addition, an excited state absorption peak at ∼709 nm has been observed. To the best of our knowledge, this is the first study of broad-band real-time vibrational spectroscopy in Chl-a.
Introduction
Chlorophyll a (Chl-a) is the most abundant pigment among the photosynthesis in nature, which plays an essential role in light harvesting and conversion processes in green plants and in various algae. Vibrational properties of the chlorophylls have been investigated extensively by optical spectroscopic techniques, such as resonance Raman (RR) spectroscopy (1–8), spectral hole-burning (HB), and fluorescence line-narrowing (FLN) techniques (9–16). Specifically, the data achieved using HB and FLN techniques are corresponding to the vibrations in the excited and ground electronic state, respectively; however, these techniques can only be operated at very low temperatures (usually ∼4 K). As for the RR spectroscopy, both the Soret-resonant and Q-resonant Raman spectra have been successfully obtained and have reported a wealth of detailed information on the vibrational modes of the electronic ground state of Chl-a molecules. However, nearly all of thus-far reported Raman spectra observed at room temperature are only corresponding to the Soret band in Chl-a. It is difficult to measure the Q-resonant (especially the Qy-band) Raman spectra of Chl-a at room temperature because of the disturbance by intense fluorescence in Chl-a solution. In contrast, the fluorescence is significantly quenched and red-shifted in the films. Therefore, the Qy-excitation RR spectra of Chl-a can only be acquired in a solid film environment by surface-enhanced Raman scattering spectroscopy which also requires the use of very low temperature (5–8).
The above-mentioned methods are powerful for studying vibrational structure of Chl-a, and plenty of results have been reported; however, none of them can provide the straightforward vibrational information related to the electronic transition of the lowest excited state of the Chl-a Qy-band at room temperature. Information at room temperature is especially physiologically important because it is the usual temperature of the physiological condition in native complexes.
The featureless Qy absorption band displays a limited structure even on lowering the sample temperature. To discover the presence of hidden characteristics of the absorption band at physiologically relevant temperatures, the absorption band shape for the Q transition region of Chl-a is usually calculated by using the vibrational frequency modes and Franck-Condon factors obtained from HB and FLN spectroscopies as a function of temperature (13,17) because of the scarcity of vibrational information at room temperature. However, it was reported by Krawczyk (18) using RR spectra of Chl-a at 77 K that the pentacoordinated Chl-a at room temperature would convert into hexacoordinated one at 77 K. Rather recently, Rätsep et al. (16) reported that while cooling from 295 K to 4.5 K, the Chl-a in 1-propanol and diethyl ether was converted into a hexacoordinated state, but the one in 2-propanol mainly retained its pentacoordinated status.
Due to the complexity of the temperature-dependent characteristics of Chl-a, there is no current consensus among researchers on how well the data achieved at low temperatures will satisfy the situation at physiologically relevant temperatures. Peterman et al. (13) simulated the temperature dependence of the low-energy part of the Qy absorption spectrum of light-harvesting complex II very well up to 220 K using the phonon wing from the 4 K emission spectra; however, the simulated and experimental results started to deviate above 220 K. Zucchelli et al. (17) calculated the spectral band shape of Chl-a in the Q-absorption region as a function of temperature to study the Chl-a in penta- and hexacoordinated states, respectively. They concluded that vibrational modes in the range 540–850 cm−1 were highly Mg-coordinate-dependent; however, Rätsep et al. (16) denied this conclusion in their recent study, and suggested that the vibrational degrees of freedom were not sensitive to the temperature-dependent absorption spectra change as the electronic one was.
To clarify the above-mentioned controversial issues, the most straightforward first step seems to be to obtain the vibrational information coupled to Q-transition of Chl-a at room temperature directly. Shiu et al. (19) has studied the vibrational frequencies within 98–791 cm−1 of the Qy excited state of Chl-a in ethanol solution. In their experiment, however, they use an excitation pulse with the duration of 25 fs. Only when the duration of the pump pulse is shorter than the vibration period can the coherent molecular vibration of the corresponding mode be excited; consequently, the highest detectable vibrational frequency in their research was limited to ∼1300 cm−1. Moreover, the samples were probed at only one wavelength in Shiu et al. (19), and broad-band information was not obtained.
In this study, we used a 6.8 fs laser pulse in the broadband femtosecond pump-probe real-time vibration spectroscopy, which has been proved to be a powerful tool for the observation of dynamical processes in molecules (20–22), to obtain both electronic and vibrational dynamics of Chl-a Qy-band in solution at 293 K. After the excitation by the ultrashort pump pulse, a wave-packet is prepared in the form of linear superposition of several vibrational levels of different quantum numbers. Then the wave-packet moves on the potential energy surface, and the motion modifies the transition spectra and intensities composed of contributions from various vibronic transitions between the vibronic levels in the initial and final electronic states. The contributions of the vibrational levels are proportional to the product of the Franck-Condon factor, which determines the absorption spectrum in the case of Condon approximation is satisfied (23) as well as the spectral intensity of the pump pulse with the vibronic transition energy. Thus, the wave-packet motion of some vibrational mode can be detected by measuring the time-dependent difference absorbance with the weaker probe pulse.
There is another technique to detect the electronic relaxation and vibrational wave-packet dynamics in real time—the time- and frequency-gated spontaneous emission method. However, it has much less sensitivity because of the low efficiency in either upconversion sum-frequency gate (second-order nonlinear process) or Kerr gate (third-order nonlinear process) in the detection scheme, and it is also difficult to achieve the high temporal resolution which is limited by geometrical path difference in the former and Kerr response time in the latter. This method may also be affected by the elongation of relaxation time due to self-absorption (24). By using our method, we are not suffering from these effects. In addition, compared with other conventional vibrational spectroscopy, this method has the following advantages:
-
1.
This technique is not limited by the measurement temperatures, so that it can provide both the electronic and vibrational characteristics of Chl-a at physiologically relevant temperatures.
-
2.
RR signals are very frequently overwhelmed by the fluorescence signal, especially in the case of highly fluorescent molecules (e.g., Chl-a). In contrast, the contamination effect of spontaneous fluorescence can be almost totally avoided in real-time vibrational spectroscopy because of much more intense, highly directional probe-light probe beam than spontaneous fluorescence.
-
3.
RR spectroscopy is not capable of reliably detecting the presence of very low-frequency modes due to intense Rayleigh scattering of the excitation beam (25). However, the low-frequency modes can easily be studied by pump-probe experiment as long as a few quanta of the modes can be covered by the broad laser spectrum with nearly constant phase.
-
4.
Very small instantaneous frequency change can be detected in real-time spectroscopy; hence, molecular structural change information, for the transition state, can be detected.
-
5.
The dynamics of vibrational modes coupled to the electronic transition can be studied in relation to the decay dynamics of the electronic excited states at the same time, and under exactly the same experimental conditions.
-
6.
The probe-dependent vibrational amplitude can be detected as a function of pump-probe delay time. Therefore, the corresponding vibrational phase information can be obtained.
In this study, using the ultrafast excitation and broad-band detection, the dynamics of vibrational modes coupled to the electronic transition of Chl-a Q-band has been studied by the real-time vibrational spectroscopy. Both pump and probe pulses possess a broadband of 200 nm, which can cover the whole Qy absorption band of Chl-a simultaneously. The vibrational modes due to both ground-state and excited-state wave-packet motions have been observed and they are assigned according to the vibrational phase. The data containing both electronic relaxation and vibrational dynamics have been analyzed to obtain a more reliable relaxation mechanism of the excitations than a combination of individual studies of electronic and vibrational relaxations.
To the best of our knowledge, this is the first direct broad-band study of Chl-a vibrational signal coupled with the Qy-band electronic transition at room temperature.
Materials and Methods
In this experiment, both pump and probe pulses are generated from a noncollinear optical parametric amplifier (NOPA) laser system which is seeded by a white-light continuum (26–28), as described in the Supporting Material. The pulse duration of the NOPA output is 6.8 fs and covered the spectral range extending from 539 nm to 738 nm, as shown in Fig. 1. After sample, the pump-probe signal is detected by the combination detection system of the polychromator and multichannel lock-in amplifier (see the Supporting Material for further details). The spectral resolution of the total system is ∼1.5 nm. The wavelength-dependent difference absorbance of the probe at 128 wavelengths is measured by changing the pump-probe delay times from −200 to 1800 fs with a 0.8-fs step. All the experiments are performed at a constant temperature (293 K).
Figure 1.
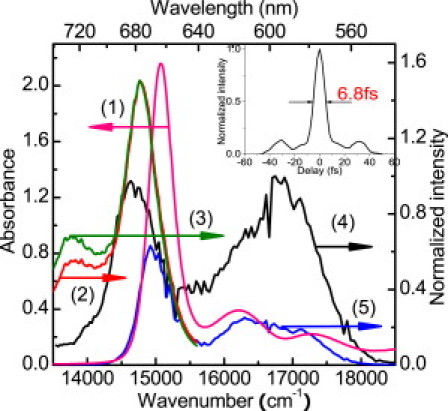
The absorption spectrum (1), fluorescence spectrum (2), and stimulated emission spectrum (3) of Chl-a, the laser spectrum (4), and the absorbed laser photon energy distribution spectrum by Chl-a with 0.5-mm thickness (5). (Inset) Temporal intensity profile of the compressed laser pulse.
Chl-a used in this study is extracted from spinach leaves and subsequently purified using chromatography, according to the method described in Strain and Svec (29). The solvent used here is a mixture of petroleum ether and 2-propanol with a ratio of 100:5. Optical density is measured to be OD1mm = 2.16 at the Qy(0,0) transition peak of 664-nm using a 1-mm cell. The stationary absorption and the fluorescence spectra of Chl-a solution were measured with an absorption spectrometer (UV-3101PC; Shimadzu, Kyoto, Japan) and a fluorophotometer (F-4500; Hitachi, Tokyo, Japan), respectively.
Results and Discussion
Stationary absorption and fluorescence spectra and time-resolved difference absorption spectrum
The stationary absorption, fluorescence, and stimulated emission spectra of Chl-a, together with the NOPA output laser spectrum and the absorbed laser spectrum (0.5-mm flow cell is used in real-time spectroscopy; see the Supporting Material for further details), are shown in Fig. 1. There are three absorption peaks in the laser spectrum range, located at 664, 618, and 580 nm, respectively. There is no doubt that the peak at longest wavelength belongs to the Qy(0-0) band of the Chl-a, and the observed absorption spectrum with a Qy(0,0) transition peak at 664 nm indicates that the sample used in this work consists of the monomer structure.
Fig. 2 a shows the two-dimensionally plotted difference absorption ΔA against the probe delay time and probe photon energy. On top of it, we plot the probe photon energy dependence of time-resolved spectra ΔA probed from 100 to 1200 fs with an integration time width of 100 fs. Near the two absorption bands at ∼617 nm and 580 nm, a weak bleaching signal modulates the positive signal due to excited state absorption. These two bleaching signals are much weaker because of the strong induced absorption in this range. In addition, we note that the most intense negative peak signal at ∼664 nm has an asymmetric profile, and the negative signal in the shorter wavelength side is much steeper than the longer side. It can be explained in the following way. The transient signal observed at a probe wavelength longer than 664 nm is due to the mixed contribution of both the ground state bleaching and the stimulated emission, while the ΔA at a probe wavelength shorter than 664 nm is dominated by the bleaching and the induced absorption.
Figure 2.
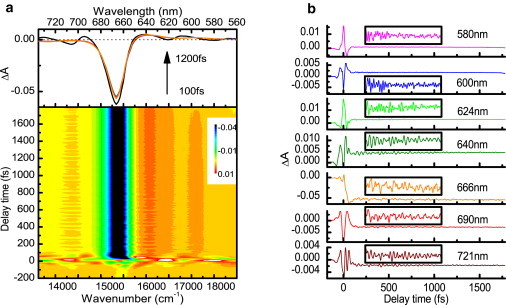
(a) Two-dimensional pseudo-color display of the time dependence of the absorbance changes (probe photon energy versus probe delay time, bottom figure) together with the time-resolved difference absorption spectrum probed from 100 to 1200 fs with an integration time-width of 100 fs (top figure). (b) Real-time traces at seven typical wavelengths. (Inset boxes) Enlarged signals between 100 and 1000 fs.
Fig. 2 b shows the real-time ΔA traces at seven typical wavelengths. The sharp and intense peaks around zero probe-delay time are due to pump-probe coupling induced by the nonlinear process of the pump-probe-pump time-ordering interaction of the laser fields and the interference between the scattered pump pulses and the probe pulses. The ΔA signal observed in the detection time of −200 fs to 1800 fs is nearly constant because of the nanosecond lifetime of the Qy excited state (30,31). It is obvious that the time-dependent ΔA traces shown in Fig. 2 b consist of the slow relaxation component due to electronic decay and the highly oscillating component due to molecular vibrations. The enlarged oscillating signals between 100 and 1000 fs are illustrated in the inset boxes of Fig. 2 b. It is noteworthy that the oscillations are especially clearly observed around the Qy band.
Ultrafast dynamics of vibrational modes
As shown in Fig. 2, predominant periodic modulation could be observed in the time-dependent difference absorption, which is caused by the transition probability change induced by molecular vibrations. To gain a better understanding of the vibronic coupling mechanisms, the vibrational mode frequencies and its corresponding amplitudes have been realized by the fast Fourier transform (FFT) analysis of the time-resolved difference absorption spectra.
The FFT analysis is performed in the range from 50 fs to 1800 fs. (Note that, to avoid possible interference effects between the scattered pump and probe pulses, the difference absorption signals ranging from delay 0 to 50 fs are not included.) The slow decay dynamics of relevant electronic states is removed by subtraction of averaged data over 200-fs time window from the raw data. After that, the FFT power spectra are calculated by using a Hanning window after zero-padding procedure to increase the frequency resolution. The two-dimensional plot of the Fourier amplitude against the molecular vibration frequency and probe photon energy is presented in Fig. 3 a. The FT amplitude spectra at several typical wavelengths are shown in Fig. 3 b. Various strong vibrational modes are highly concentrated at the bleaching band at ∼664 nm. Twenty-seven different frequencies of 214, 259, 300, 346, 407, 519, 565, 621, 667, 744, 799, 915, 982, 1043, 1084, 1124, 1175, 1252, 1353, 1415, 1460, 1516, 1582, 1613, 1648, 1699, and 1770 cm−1 are obtained. Most of them are similar to those observed by Raman and/or the HB spectroscopy by many groups; however, the modes of 300, 621, 1582, 1648, and 1770 cm−1 are observed (to our knowledge) for the first time in this study. Most of the intense frequency signals below 1000 cm−1 are assigned to vibrations of the tetrapyrrole skeleton and deformations of the peripheral substituent groups (6,7), while those higher than 1000 cm−1 are mainly assigned to the CH3 bending, CH bending, and CO, CC, and CN stretching vibrations (5,8,32).
Figure 3.
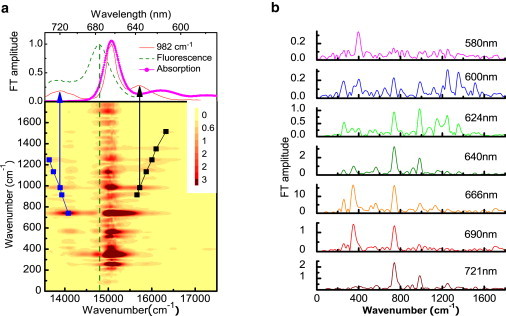
(a) Two-dimensional contour plot of FT amplitude spectra of the pump-probe signal. (Solid line) Mass centers of FT amplitude spectra at side bands. (Top) FT amplitude of the 982 cm−1 mode together with the stationary fluorescence and absorption spectra. (b) FT amplitude spectra of the corresponding traces in Fig. 2b.
In Chl-a, there are only small differences in frequency of the same vibrational modes between the ground and excited states (11,13,14). Therefore, it is difficult to distinguish whether they are due to the wave-packet motion in the ground or excited state due to our limited frequency resolution. Thanks to the broad probe spectral band detection of real-time traces, we can calculate the probe wavelength dependence of the initial phases of the molecular vibrations. It provides information for identifying the wave-packet motion either in the ground state or in the excited state (33–35). If the time dependence of the absorbance change is due to the wave-packet prepared on the ground-state potential energy surface, the initial vibrational phase will be ±π/2. If the time-dependent amplitude modulation is induced by the excited state wave-packet motion, the phase will be 0 or ±π. Otherwise, it will be due to the mixed contributions from wave-packet motions in the ground and excited states. Using this simple criterion, we can easily assign the vibrational modes of Qy band. Vibrational modes with frequencies of 214, 259, 300, 346, 407, 565, 1084, 1175, 1460, 1613, and 1770 cm−1 result from the excited-state wave-packet motion, while those at 519, 621, 799, 1415, 1582, and 1648 cm−1 are corresponding to the ground state wave-packet motion; the other modes with 667, 1043, and 1699 cm−1 are contributed from wave-packet motions in both ground and excited states. As examples, Fig. 4 shows the initial phases of three different values together with their vibrational amplitudes.
Figure 4.
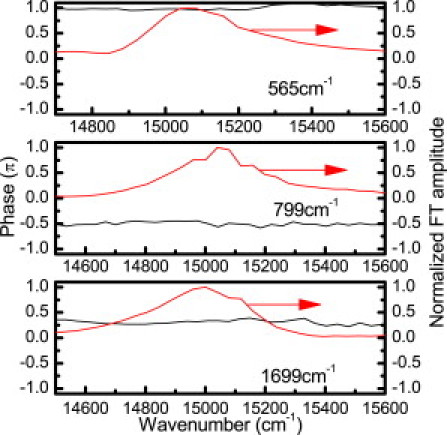
Phases of the vibrational modes with frequencies of 565 (π), 799 (−π/2), and 1699 (neither π nor ±π/2) cm−1 together with their normalized amplitudes.
The Fourier power line-width is inversely proportional to the dephasing time including effects of temperature in-sensitive inhomogeneity and temperature-sensitive homogeneous dephasing. With increasing temperature, the homogeneous contribution increases because of increased mode-mode coupling. In the Raman spectrum at 15 K (7), the width can be considered to be dominated by inhomogeneous width, and it is calculated to be ∼15 cm−1. It is found that the line-widths in the real-time spectroscopy in our experiment are ∼3 times as broad as those measured in Raman experiment at low temperature, which are due to the homogeneous origin. Even though the excited-state lifetime of Chl-a is in the nanosecond time range, the electronic relaxation will be affected by the fast vibrational relaxation through vibronic coupling, which will dissipate some of the electronic energy and hence compete with the Chl-a involved energy transfer process. Special attention should be paid to the low-frequency modes (<250 cm−1), which have been classified as intermolecular vibrations (16,36). Because the rate of the resonance energy transfer through dipole-dipole interaction depends on the distance between the energy donor and acceptor, the energy transfer between Chl-a and its neighboring molecule is expected to be periodically modulated by the low-frequency intermolecular vibrations in their dephasing times of <1 ps.
The high-frequency modes at 744, 915, 982, 1124, 1251, 1353, and 1516 cm−1 exhibit more complex spectral distributions than those discussed above, and the simple criterion cannot be performed any more. As shown in Fig. 3 a, side bands are clearly observed at both sides of the most intense vibrational band for these modes (only one side for 1353 and 1516 cm−1 mode because of probe range). The probe photon energy dependence of FT amplitude for the 982 cm−1 mode is shown at the top of Fig. 3 a as an example. In order to investigate the spectral position and the profile of this sideband in detail, the peak position of each vibrational mode has been recorded as a function of the probe photon energy. To minimize the possible error, the side-band mass centers are defined by the first moment given by
| (1) |
where Fv(ω) is the probe photon frequency (ω)-dependent FT amplitude spectra of the vibrational modes with a vibrational frequency of ωv.
The calculation results are displayed in Fig. 3 a and connected by black and blue lines in the higher and lower probe energy ranges, respectively. These two fitted lines have slopes of −0.96 ± 0.03 and 0.97 ± 0.01, which are very close to ±1. It means that the side bands of each vibrational mode are spectrally distributed according to a linear function (slope K = ±1) of their own vibrational frequencies. Therefore, they are symmetric with respect to a center axis. Because the Huang-Rhys factors in Chl-a are very small (<0.05) (11), the displacement in the coordinate space from the potential minimum of the ground to that of the excited states is small. In the experiment, what we observed is the modulation of the electronic transition probability induced by a wave-packet motion through vibronic coupling.
Therefore, if the vibrational modes appearing at side bands are due to the wave-packet motion in the ground state, the vibrational amplitudes are related to the absorption spectrum from the ground to excited state transition. The peaks of the vibrational amplitudes are expected to symmetrically appear on both sides of transition energy from the ground to excited state, displayed as Stokes and anti-Stokes bands. If the wave-packet motion on the excited-state potential surface modulates the stimulated emission intensity, the observed vibrational amplitude at side bands will have peaks symmetric with respect to the stimulated emission peak, also displaying as Stokes and anti-Stokes shifted structures. As shown in Fig. 3 a, the measured vibrational signals at side bands are spectrally symmetric with respect to the axis of the stimulated emission peak, so they were assigned to vibrational modes coupling to the stimulated emission from the Qy electronic excited state. However, the signals at the central band are surprisingly weak; in fact, they nearly vanish in this spectral range.
Why do the vibrational signals disappear in the symmetric center and those at sidebands have a linear spectral distribution? Here, to analyze the complicated results, we use a coupled three-level model to explain the experimental result by the theory of nonlinear ultrafast spectroscopy in the following discussion. In this study, all the vibrational modes in the discussion of the following section showing clear side bands are much larger than 200 cm−1. Because of the experimental temperature of 293 K, the initial population on the vibrational levels with vibrational quantum numbers larger than zero can be neglected for all these involved in the vibrational modes. That means only the lowest vibrational level of the electronic ground state, |g0〉, could be regarded as the initial state in the discussion of the following section.
Theory and Discussion
It is well known that the wave-packet can be generated either via the simultaneous coherent excitation of the vibronic polarization of several vibrational levels in the excited state or via impulsive stimulated Raman scattering in the ground state. The former case is known as a split excited state, which can be represented in terms of V-type. Analogously, the latter case is referred to as a split ground state, which is expressed in terms of Λ-type interaction. In the previous article (37), the coherent excitation of the vibrations on the excited state was prevented so that only the ground-state vibrations were excited. Hence, the Λ-type interaction could be used. In our experiment, due to the good overlap of the broad-band laser and the absorption band, the vibrations in the excited states can be coherently excited, as shown in Fig. 5, where ω0 denotes the Qy (0-0) emission (central band) or the one with emission energy equal to it.
Figure 5.
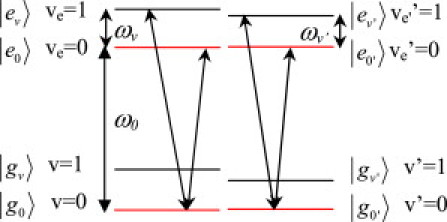
Three-level system describing the excitation processes of the vibrational coherence generation in the excited state. The lowest vibrational state in the ground and excited states is represented by g0 and e0; gv and ev represent where the vibrational coherence exists, where v and v′ denote vibrational mode with different frequency. The value ω0 denotes the Qy (0-0) emission (central band) or the one with emission energy equal to it. The values ωv and ωv′ represent different vibrational frequencies.
Because there are only small frequency differences of the same vibrational mode between the ground state and that of the excited state (11,13,14), we assume the vibrational energy (ωv) in the ground and excited states to be identical. In addition, because the Huang-Rhys factors of Chl-a are very significantly small (S < 0.05), most of the Franck-Condon intensity is concentrated in the 0-0 transition. Therefore, we only considered the 0-0 and 0-1 transition in the following. As mentioned above, only a stimulated emission process should be considered in the following discussions. In total, there are four related NL processes related to our experimental results. As defined in the Supporting Material, they are NL process A (Stokes band, located at the lower probe photon energy range); NL process B (located around the symmetric center); NL process A′ (anti-Stokes band, located at the higher probe photon energy range); and NL process B′ (located around the symmetric center), respectively.
The observed pump-probe signals, represented by the difference absorption for NL process A and NL process B, can be expressed by (37–44)
| (2) |
| (3) |
respectively, and the dephasing rate γ2 ≡ 1/T2. and denote the Fourier transforms of Epr(t) and P(3)(t), respectively. The exact expression of the amplitude of the probe field Epr(t) and the third-order NL polarization P(3)(t) are listed in the Supporting Material. The spectral widths in Eqs. 2 and 3 can be determined using the electronic dephasing time T2, whereas the vibrational dephasing time is given by the bandwidth of the FT amplitude of each mode. Therefore, the electronic dephasing time and the vibrational dephasing time can be independently determined by this principle. Thanks to the measurement of real-time resolved vibrational amplitude, the dependence of vibrational initial phase upon impulsive excitation on the probe photon energy can also be determined. The total observed difference absorption signal can be expressed by
| (4) |
Here, the first term characterizes the slow dynamic component of the real-time ΔA traces, and τe is the electronic decay time depending on the sample. The second term represents the molecular vibration, which is a sum of all the contributions of vibrational modes, v, with frequency ωvi and corresponds to the oscillation component in the ΔA traces. For a certain vibrational mode with a frequency of ωv, δΔA is given by
| (5) |
where τv is the vibrational dephasing time for the vibrational mode v including both homogeneous and inhomogeneous contributions. The corresponding phase for NL processes A and B of this vibrational mode can be derived from Eqs. 2, 3, and 5 by
| (6) |
and
| (7) |
Similarly, the corresponding phase for NL processes A′ and B′ of vibrational mode ωv can be derived as
| (8) |
and
| (9) |
Because it is easy to satisfy the conditions of |ω−(ω0−ωv)| and |ω−ω0| >> γ2, the difference of the phases should be π between the higher-energy region (ω > ω0±ωv for process A and A′, ω > ω0 for process B and B′) and the lower-energy region (ω < ω0±ωv for process A and A′, ω < ω0 for process B and B′). Specifically, phase change from −π/2 to +π/2 is expected at ∼ω = ω0−ωv with the increasing of the laser photon energy because of excited state wave-packet motion in the case of NL process A; and change from +π/2 to −π/2 is expected at ∼ω = ω0+ωv for NL process A′ with the increasing of the photon energy ω from ω < ω0+ωv to ω > ω0+ωv because the signs of the phases are opposite in both cases. Similarly, the pump-probe signals due to NL processes B and B′ also have antiphase relations.
The physical meaning of the phase relation is interpreted as follows. After excitation, a pump could generate molecular vibrational coherence in the Qy excited state between v = 0 and v = >1 levels. Then a probe comes to the sample and interacts with the vibrational coherent polarization between v = 0 and v = >1, resulting in the possibility of laser-Stokes energy exchange as well as laser-anti-Stokes exchange energy. The energy exchange process between the probe and the molecular vibration is then dependent on the molecular vibrational phase, which determines whether the spectra in the range of ω = ω0 (±ωv) are amplified and/or deamplified. For the antiphase relation between the oscillation signals around the spectral range ω0±ωv, the signal around ω0−ωv is amplified if the one around ω0+ωv is deamplified at the same time. As for the signals resulting from NL process B and B′, because both of them share the same spectral range ω=ω0 and are satisfied with antiphase relation, their oscillations are expected to take place concomitantly and then cancel each other out. It is consistent with the experimental result shown in Fig. 3 that, despite the clear vibrational signals on both the lower and higher energy sides due to NL processes A and A′, respectively, the vibrations in the center position (straight dashed line) have nearly disappeared.
The phase of the vibrational modes with the frequencies of 982 cm−1 and 1251 cm−1 at ω0−ωv range are shown in Fig. 6. We chose these two modes as examples because they show the clearest sidebands around their corresponding ω0±ωv range. Both of the vibrational phases have shown a clear phase jump from −π/2 to +π/2 (∼13,800cm−1 for the 1252 cm−1 mode and 14,020 cm−1 for 982 cm−1), as expected. The electronic dephasing time T2 is determined to be 41 ± 2 fs from the real-time traces of absorbance change in the negative delay time range (45), which is comparable with the one of 54 fs obtained for bacteriochlorophyll (46). However, the dephasing time is calculated to be ∼150 fs from Eq. 6, which is rather longer than the experimental value. This disagreement may be explained as due to the deviation from the simple three-level model. In a real system, there are other possible contributions to vibration amplitude through modulation of electronic transitions from the excited Qy to other states, which are not included in the simple three-level model. For example, the induced absorption from the lowest electronic excited state to a higher one may couple with the same vibrational modes and affect the calculation result.
Figure 6.
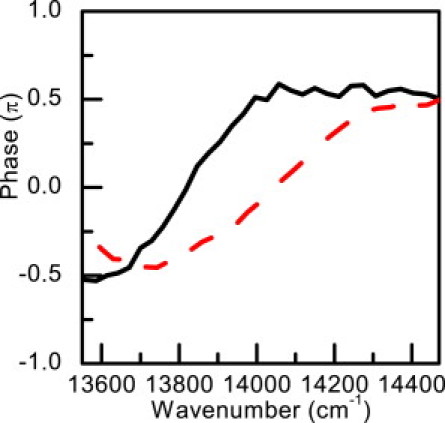
Phase spectra of the mode of 982 cm−1 (dashed line) and 1251 cm−1 (solid line) observed in the real-time vibration spectra probed at lower energy region.
As shown in Fig. 2 a, there is another highly oscillating band centered at 14,100 cm−1 with a full bandwidth of ∼300 cm−1, which gives rise to a concentrated vibrational band in addition to those coupled with ground-state bleaching and those symmetrically distributed due to the stimulated emission in Fig. 3. Even though stimulated emission provides the largest contribution in the signal in the ω0−ωv range, induced absorption at ∼14,100 cm−1 is still intense enough to compete with it because the total negative signal is close to zero. In addition, considering the very small spectral overlap between these vibrations and the stimulated emission peak, it is reasonable to assign these vibrations coupled to the transition from the Qy excited state to a higher electronic state by induced absorption, which is peaked at ∼14,100 cm−1 (∼709 nm). Recently, the excited-state absorption spectrum of Chl-a has been studied by Z-scan technique using the white-light continuum, but the excited-state absorption could only be observed in the wavelength region below the ground-state absorption (47). In this study, the excited-state absorption peak at ∼709 nm can be recognized through the evidence from both the electronic and vibrational dynamics simultaneously. Because of the coexistence of stimulated emission and the induced absorption, even though the phase jump from −π/2 to +π/2 still maintains, the jump position and width predicted by the two-electronic level theory has been affected.
The phase characteristic at ω0+ωv range has also been studied, but it is too complicated (not shown here) to be compared with the calculation. That might be due to the much stronger influence of the induced absorption in this spectral range. As shown in Fig. 2 a, the positive ΔA signal due to induced absorption is most intense in the higher spectral range (15,500–16,500 cm−1), and all the possible electronic processes, the ground-state bleaching, stimulated emission, and induced absorption, can take place in this photon energy range. Even though the vibrational amplitude position is not much affected, the vibrational phase is much more sensitive, and the influence from other processes is too intense to be neglected for the phase calculation and hence it must be explored with a model far beyond the simple three-level (two electronic levels and one vibrational level) system.
Conclusions
A femtosecond pump-probe experiment is performed on Chl-a using a 6.8-fs visible pulse. Electronic relaxation and vibrational dynamics are simultaneously observed by real-time vibrational spectroscopy with a 0.8-fs delay step at room temperature. Wave-packet motions in the excited state coupled to the stimulated emission are observed over a broad detection from 539 nm to 738 nm. The modulation signals are found to nearly vanish around the stimulated emission peak; however, they are symmetrically distributed with respect to the emission peak as a function of their own vibrational frequencies at both sides. The corresponding nonlinear process has been studied using a three-level model, from which the probe wavelength dependence of the phase of the periodic modulation and the spectral distribution of vibrational amplitude has been explained in detail. Because of the scarcity of Qy-band vibrational information in Chl-a at a physiologically relevant temperature, low-temperature data are generally used in the calculation of the absorption profile at room temperature.
Due to the complexity of the temperature-dependent characteristic of Chl-a, there is no current consensus as to how well the low-temperature data will satisfy the physiologically relevant temperatures situation, and many controversial issues have arisen in these calculations. The Qy-band vibrational information obtained in this study seems the most straightforward initial step for resolving these, and holds promise to reveal hidden features in the absorption band shape of Chl-a with limited structure at physiologically relevant temperature.
Acknowledgments
This work was supported by the Core Research for Evolutional Science and Technology program of the Japan Science and Technology Agency, National Science Council of the Republic of China, Taiwan (NSC No. 98-2112-M-009-001-MY3), and a grant from the Ministry of Education, Aiming for Top University Program at National Chiao-Tung University. A part of this work was performed under the joint research project of the Institute of Laser Engineering, Osaka University under contract No. A3-01.
Supporting Material
References
- 1.Lutz M. Resonance Raman spectra of chlorophyll in solution. J. Raman Spectrosc. 1974;2:497–516. [Google Scholar]
- 2.Fujiwara M., Tasumi M. Meta-sensitive bands in the Raman and infrared spectra of intact and metal-substituted chlorophyll a. J. Phys. Chem. 1986;90:5646–5650. [Google Scholar]
- 3.Pascal A.A., Caron L., Robert B. Resonance Raman spectroscopy of a light-harvesting protein from the brown alga Laminaria saccharina. Biochemistry. 1998;37:2450–2457. doi: 10.1021/bi9719657. [DOI] [PubMed] [Google Scholar]
- 4.Koyama Y., Umemoto Y., Akamatsu A. Raman spectra of chlorophyll forms. J. Mol. Struct. 1986;146:273–287. [Google Scholar]
- 5.Thomas L.L., Kim J.H., Cotton T.M. Comparative study of resonance Raman and surface-enhanced resonance Raman chlorophyll a spectra using Soret and red excitation. J. Am. Chem. Soc. 1990;112:9378–9386. [Google Scholar]
- 6.Diers J.R., Zhu Y., Bocian D.F. Qy-excitation resonance Raman spectra of chlorophyll a and bacteriochlorophyll c/d aggregates. Effects of peripheral substituents on the low-frequency vibrational characteristics. J. Phys. Chem. 1996;100:8573–8579. doi: 10.1021/jp953544+. [DOI] [PubMed] [Google Scholar]
- 7.Zhou C., Diers J.R., Bocian D.F. Qy-excitation resonance Raman spectra of chlorophyll a and related complexes. Normal mode characteristics of the low-frequency vibrations. J. Phys. Chem. B. 1997;101:9635–9644. doi: 10.1021/jp953544+. [DOI] [PubMed] [Google Scholar]
- 8.Woolley P.S., Keely B.J., Hester R.E. Surface-enhanced resonance Raman spectroscopic identification of chlorophyll a allomers. J. Chem. Soc., Perkin Trans. 1997;2:1731–1734. [Google Scholar]
- 9.Rebane K.K., Avarmaa R.A. Sharp line vibronic spectra of chlorophyll and its derivatives in solid solutions. Chem. Phys. 1982;68:191–200. [Google Scholar]
- 10.Avarmaa R.A., Rebane K.K. High-resolution optical spectra of chlorophyll molecules. Spectrochim. Acta [A] 1985;41A:1365–1380. [Google Scholar]
- 11.Gillie J.K., Small G.J., Golbeck J.H. Nonphotochemical hole burning of the native antenna complex of photosystem I (PSI-200) J. Phys. Chem. 1989;93:1620–1627. [Google Scholar]
- 12.Pascal A., Peterman E., Robert B. Structure and interactions of the chlorophyll a molecules in the higher plant Lhcb4 antenna protein. J. Phys. Chem. B. 2000;104:9317–9321. [Google Scholar]
- 13.Peterman E.J.G., Pullerits T., van Amerongen H. Electron-phonon coupling and vibronic fine structure of light-harvesting complex II of green plants: temperature dependent absorption and high-resolution fluorescence spectroscopy. J. Phys. Chem. 1997;101:4448–4457. [Google Scholar]
- 14.Pieper J., Voigt J., Small G.J. Chlorophyll a Franck-Condon factors and excitation energy transfer. J. Phys. Chem. B. 1999;103:2319–2322. [Google Scholar]
- 15.Renge I., Mauring K., Avarmaa R. Vibrationally resolved optical spectra of Chlorophyll derivatives in different solid media. J. Phys. Chem. 1986;90:6611–6616. [Google Scholar]
- 16.Rätsep M., Linnanto J., Freiberg A. Mirror symmetry and vibrational structure in optical spectra of chlorophyll a. J. Chem. Phys. 2009;130:194501. doi: 10.1063/1.3125183. [DOI] [PubMed] [Google Scholar]
- 17.Zucchelli G.R., Jennings R.C., Cremonesi O. The calculated in vitro and in vivo chlorophyll a absorption bandshape. Biophys. J. 2002;82:378–390. doi: 10.1016/S0006-3495(02)75402-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krawczyk S. The effects of hydrogen bonding and coordination interaction in visible absorption and vibrational spectra of chlorophyll a. Biochim. Biophys. Acta. 1989;976:140–149. [Google Scholar]
- 19.Shiu Y.J., Shi Y., Lin S.H. Femtosecond spectroscopy study of electronically excited states of chlorophyll a molecules in ethanol. Chem. Phys. Lett. 2003;378:202–210. [Google Scholar]
- 20.Megerle U., Pugliesi I., Riedle E. Sub-50 fs broadband absorption spectroscopy with tunable excitation: putting the analysis of ultrafast molecular dynamics on solid ground. Appl. Phys. B. 2009;96:215–231. [Google Scholar]
- 21.Polli D., Lüer L., Cerullo G. High-time-resolution pump-probe system with broadband detection for the study of time-domain vibrational dynamics. Rev. Sci. Instrum. 2007;78:103108. doi: 10.1063/1.2800778. [DOI] [PubMed] [Google Scholar]
- 22.Yabushita A., Lee Y.H., Kobayashi T. Development of a multiplex fast-scan system for ultrafast time-resolved spectroscopy. Rev. Sci. Instrum. 2010;81:063110. doi: 10.1063/1.3455809. [DOI] [PubMed] [Google Scholar]
- 23.Kobayashi T., Zhang J., Wang Z. Non-Condon vibronic coupling of coherent molecular vibration in MEH-PPV induced by a visible few-cycle pulse laser. N. J. Phys. 2009;11:013048. [Google Scholar]
- 24.Kobayashi T., Nagakura S. Reabsorption and high density excitation effects on the time-resolved fluorescence spectra of anthracene crystal. Molec. Cryst. Liq. Cryst. 1974;26:33–43. [Google Scholar]
- 25.Cupane A., Leone M., Schweitzer-Stenner R. Dynamics of various metal-octaethylporphyrins in solution studied by resonance Raman and low-temperature optical absorption spectroscopies. Role of the central metal. J. Phys. Chem. B. 1998;102:6612–6620. [Google Scholar]
- 26.Shirakawa A., Kobayashi T. Noncollinearly phase-matched femtosecond optical parametric amplification with a 2000 cm−1 bandwidth. Appl. Phys. Lett. 1998;72:147–149. [Google Scholar]
- 27.Shirakawa A., Sakane I., Kobayashi T. Pulse-front-matched optical parametric amplification for sub-10-fs pulse generation tunable in the visible and near infrared. Opt. Lett. 1998;23:1292–1294. doi: 10.1364/ol.23.001292. [DOI] [PubMed] [Google Scholar]
- 28.Baltuška A., Fuji T., Kobayashi T. Visible pulse compression to 4 fs by optical parametric amplification and programmable dispersion control. Opt. Lett. 2002;27:306–308. doi: 10.1364/ol.27.000306. [DOI] [PubMed] [Google Scholar]
- 29.Strain H.H., Svec W.A. Extraction, separation, estimation and isolation of the chlorophylls. In: Vernon L.P., Seeley G.R., editors. The Chlorophylls. Academic Press; New York: 1966. pp. 21–66. [Google Scholar]
- 30.Martinsson P., Oksanen J.A.I., Åkesson E. Dynamics of ground and excited state chlorophyll a molecules in pyridine solution probed by femtosecond transient absorption spectroscopy. Chem. Phys. Lett. 1999;309:386–394. [Google Scholar]
- 31.Linke M., Lauer A., Heyne K. Three-dimensional orientation of the Qy electronic transition dipole moment within the chlorophyll a molecule determined by femtosecond polarization resolved VIS pump-IR probe spectroscopy. J. Am. Chem. Soc. 2008;130:14904–14905. doi: 10.1021/ja804096s. [DOI] [PubMed] [Google Scholar]
- 32.Cai Z.L., Zeng H., Larkum A.W. Raman spectroscopy of chlorophyll d from Acaryochloris marina. Biochim. Biophys. Acta. 2002;1556:89–91. doi: 10.1016/s0005-2728(02)00357-2. [DOI] [PubMed] [Google Scholar]
- 33.Kumar A.T.N., Rosca F., Champion P.M. Investigations of amplitude and phase excitation profiles in femtosecond coherence spectroscopy. J. Chem. Phys. 2001;114:701–724. [Google Scholar]
- 34.Kumar A.T.N., Rosca F., Champion P.M. Investigations of ultrafast nuclear response induced by resonant and nonresonant laser pulses. J. Chem. Phys. 2001;114:6795–6815. [Google Scholar]
- 35.Ikuta M., Yuasa Y., Kobayashi T. Phase analysis of vibrational wave packets in the ground and excited states in polydiacetylene. Phys. Rev. B. 2004;70:214301. [Google Scholar]
- 36.Rätsep M., Pieper J., Freiberg A. Excitation wavelength-dependent electron-phonon and electron-vibrational coupling in the CP29 antenna complex of green plants. J. Phys. Chem. B. 2008;112:110–118. doi: 10.1021/jp075170d. [DOI] [PubMed] [Google Scholar]
- 37.Ishii N., Tokunaga E., Kobayashi T. Optical frequency- and vibrational time-resolved two-dimensional spectroscopy by real-time impulsive resonant coherent Raman scattering in polydiacetylene. Phys. Rev. A. 2004;70:023811. [Google Scholar]
- 38.Mukamel S. Oxford University Press; New York: 1995. Principles of Nonlinear Optical Spectroscopy. [Google Scholar]
- 39.Parson W.W. Springer-Verlag; Berlin: 2007. Modern Optical Spectroscopy: With Examples from Biophysics and Biochemistry. [Google Scholar]
- 40.Mukamel S. Multidimensional femtosecond correlation spectroscopies of electronic and vibrational excitations. Annu. Rev. Phys. Chem. 2000;51:691–729. doi: 10.1146/annurev.physchem.51.1.691. [DOI] [PubMed] [Google Scholar]
- 41.Pollard W.T., Lee S.-Y., Mathies R.A. Wave packet theory of dynamic absorption spectra in femtosecond pump-probe experiments. J. Chem. Phys. 1990;92:4012–4029. [Google Scholar]
- 42.Walmsley I.A., Tang C.L. The determination of electronic dephasing rates in time-resolved quantum beat spectroscopy. J. Chem. Phys. 1990;92:1568–1574. [Google Scholar]
- 43.Walmsley I.A., Mitsunaga M., Tang C.L. Theory of quantum beats in optical transmission-correlation and pump-probe experiments for a general Raman configuration. Phys. Rev. A. 1988;38:4681–4689. doi: 10.1103/physreva.38.4681. [DOI] [PubMed] [Google Scholar]
- 44.Mitsunaga M., Tang C.L. Theory of quantum beats in optical transmission-correlation and pump-probe measurements. Phys. Rev. A. 1987;35:1720–1728. doi: 10.1103/physreva.35.1720. [DOI] [PubMed] [Google Scholar]
- 45.Kobayashi T., Yabushita A. Dynamics of vibrational and electronic coherences in the electronic excited state studied in a negative-time range. Chem. Phys. Lett. 2009;482:143–147. [Google Scholar]
- 46.Cherepy N.J., Shreve A.P., Mathies R.A. Electronic and nuclear dynamics of the accessory bacteriochlorophylls in bacterial photosynthetic reaction centers from resonance Raman intensities. J. Phys. Chem. B. 1997;101:3250–3260. [Google Scholar]
- 47.De Boni L., Correa D.S., Mendonça C.R. Excited state absorption spectrum of chlorophyll a obtained with white-light continuum. J. Chem. Phys. 2007;126:165102. doi: 10.1063/1.2722755. 165102–165104. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.