

Abstract
Presently, most simulations of ion channel function rely upon nonatomistic Brownian dynamics calculations, indirect interpretation of energy maps, or application of external electric fields. We present a computational method to directly simulate ion flux through membrane channels based on biologically realistic electrochemical gradients. In close analogy to single-channel electrophysiology, physiologically and experimentally relevant timescales are achieved. We apply our method to the bacterial channel PorB from pathogenic Neisseria meningitidis, which, during Neisserial infection, inserts into the mitochondrial membrane of target cells and elicits apoptosis by dissipating the membrane potential. We show that our method accurately predicts ion conductance and selectivity and elucidates ion conduction mechanisms in great detail. Handles for overcoming channel-related antibiotic resistance are identified.
Introduction
Ion channels play an essential role in cellular homeostasis and signaling. The study of their function is crucial both for an understanding of intercellular communication and to develop drugs against a plethora of channel-induced diseases (1,2). At present, Brownian dynamics (BD) simulations are the most established technique to study ion flux through channels on experimentally relevant timescales (3). Their efficiency, however, is achieved through neglect of specific molecular interactions, channel structural detail, and macromolecular dynamics. By using more accurate atomistic equilibrium molecular dynamics (MD) simulations, transfer rates and selectivities of individual ions can be deduced from the reconstruction of potential-of-mean-force (PMF) landscapes. This approach, however, rests on considerable approximations such as a predefined reaction pathway, the application of (gas-phase) transition state theory on biological systems, and a limited coverage of correlated motions and multiparticle interactions. As a consequence, wide pores are especially difficult to treat (3).
More recently, MD simulations with applied external electric fields have become a popular tool for the study of ion channels (4–6). Despite initial concerns about a certain degree of artificiality (7,8), it has recently been shown that a constant external electric field is a valid representation of the influence of an electromotive force, exerted by a voltage difference between two electrodes (4). Nonetheless, it is desirable to simulate sustained ionic concentration gradients such as Nernst gradients across the membrane, for instance to model reversal potential experiments on ion channels. Moreover, in biological cells and in experiments, the local effect of an electromotive force on the lipid bilayer, driving ion flux across channels, is actually conferred by a small charge imbalance across the membrane (4,9).
We present a computational method that controls both an ionic concentration gradient and a potential difference across the membrane—evoked by small charge imbalances in two aqueous compartments (7)—during atomistic MD simulations. The compartments are separated by a two-bilayer setup, as used in earlier studies on the simulation of osmotic pressure acting on membranes (10), and of osmotically driven water transport through carbon nanotubes (11).
In these pioneering studies, the simulation system was separated into two compartments of unequal ionic strength, modeled as electrically neutral in each case. A similar setup was later used to establish potentials across nonpermeable membranes by a small charge imbalance between the two compartments (7), and to model short-term, unsustained flux bursts through nanopores (9). In this study, the charge imbalance across the double-membrane configuration, as used before (7,10,11), generates a sustained internal electric field. This closely resembles the situation in biological cells. The potential exerted by different ion concentrations in the two aqueous environments can be directly assessed by integration of the charge density, yielding a well-defined transmembrane voltage condition. We developed a particle interchange method, which exchanges ion/water pairs between buffer regions in both compartments to maintain a given concentration gradient and/or charge imbalance. In some respects, this concept is similar to the effect of ion-exchanging electrodes on a membrane/aqueous system, e.g., a pair of AgCl/Cl− electrodes, commonly used to establish transmembrane potentials, which remove or release Cl− from the bulk solution (4).
We tested the method by applying it to the bacterial porin PorB from Neisseria meningitidis (Nme). The atomic structure of Nme PorB has recently been determined at 2.3 Å resolution (12). Porins are trimeric β-barrels in the outer membrane of Gram-negative bacteria, acting as the principal passageways for ions and small molecules across the outer bacterial membrane (13–15). The bacterial genus Neisseria contains two highly pathogenic species, Neisseria meningitidis (Nme) and Neisseria gonorrheae (Ngo), which cause bacterial meningitis and gonorrhea, respectively (16). PorB is a particularly interesting channel because it is instrumental in serious infections such as bacterial meningitis: In the course of host infection, PorB inserts into the inner mitochondrial membrane of target cells, resulting in uncontrolled ion flux that dissipates the membrane potential and ultimately leads to cell death (17). Porins such as PorB also form the major entryways for antibiotic molecules into Gram-negative bacteria (18). PorB exhibits relatively large conductance and selectivity for anions (12). As small changes in porin sequences contribute to bacterial antibiotic resistance, and an increasing number of multiresistant Neisseria strains have been reported (19,20), a markedly better understanding of ionic and molecular transfer through these channels is needed for the development of improved drugs.
Our method is not restricted to the study of wide porins, but applicable to membrane channels of all conductance levels, e.g., K+ channels. We implemented our computational scheme into the efficient MD software GROMACS (21) (see Methods for performance details). Most importantly, the timescales that can be attained in this way are of the same order as biological signaling processes; for instance, opening events of nicotinic acetylcholine receptors in synaptic transmission (4 μs) (22). As experimental electrophysiology achieves a similar time resolution (6 μs) (22), direct comparison of simulations with electrophysiological experiments is possible.
Methods
All simulations were carried out using GROMACS 4.5 (21), for which the described ion/water exchange protocols were implemented.
Control of ionic concentrations
The studied simulation systems each contain two lipid bilayers separating aqueous compartments of similar size (Fig. 1, A and B). Periodic boundary conditions are used in all three dimensions, and thus two distinct compartments are formed, labeled α and β in the figure. The compartments are defined as the volumes between the xy planes running through the center of mass (marked with a cross in Fig. 1 A) of each channel or channel multimer.
Figure 1.
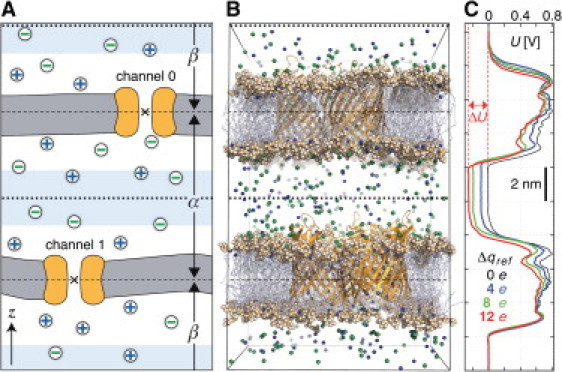
(A) Sketch of the simulation system. (Gray) The two lipid bilayers. (Orange) Channels; (Green) Anions; (Blue) Cations. (Dashed lines) Boundaries between compartments α and β. (Dotted lines) Compartment midplanes around which the buffer volumes (light blue) are centered. (B) Snapshot of a simulation of wild-type PorB at 200 mM NaCl concentration at t = 22 ns. Water molecules are omitted for clarity. (C) Time average of the electrostatic potential U along the z axis (0–50 ns) for wild-type PorB at 1 M NaCl, arising from imbalances Δq between 0 and 12 elementary charges (color-coded). See Fig. 3 for the corresponding ionic currents.
Under biologically relevant conditions, transfer of water molecules and solvated ions between compartments exclusively occurs through channels in the bilayer, if present in at least one of the membranes. To sample positive and negative potential differences at the same time, we use two channels, one residing in each membrane, oriented as shown in the figure with respect to the z axis.
The ion concentration in each compartment, and thus the charge imbalance Δq(t) and the potential difference ΔU(t), is regulated by exchanges of ions from one compartment with water molecules from the other compartment. Whether an exchange is performed and how exactly an exchange is carried out is controlled by one of the two following protocols.
Deterministic protocol
In the deterministic protocol, the number of anions n− and cations n+ is determined at regular, short intervals for each compartment, yielding in α and in β. The total number of anions and cations is and , respectively. The compartments can either be electrically neutral, or a charge imbalance Δqref across the membrane can be imposed by setting reference numbers nref for each ion type in each compartment, with and . The deterministic protocol is constructed such that these reference values are restored.
At a given time step t, the current charge imbalance is calculated from the number of ions in each compartment as
| (1) |
with ionic valences z. (Note that inserting the reference values nref in Eq. 1 yields the imposed charge imbalance Δqref.)
If the number of anions or cations in a compartment differs by one or more from its reference value, n − nref ≥ 1, each excess ion is immediately exchanged with a water molecule from the other compartment, such that the reference concentration is restored. The new ionic position is the center of mass of the water molecule, while the water molecule's center is shifted to the former ion position. Particle velocities are not carried over to the next compartment. To prevent that ions fluctuating around the compartment boundaries (in the channel, for example) lead to back-and-forth exchanges, the ion concentrations are averaged over a brief time span, such that the deviation from the reference concentration has to continue for a certain time to actually lead to an exchange event.
To keep the disturbance of the membrane/protein system by an exchange event as small as possible, we select particles that maximize the distance to the compartment boundaries for the exchange (see also Results and Discussion). Those are particles located near the central planes of the compartments, which are marked by dotted lines in Fig. 1, A and B. One could also pick these particles from a broader buffer layer around the compartment midplanes (as in the switching protocol) at the cost of a somewhat larger disturbance.
Our deterministic protocol is similar to the BD reinjection method of Corry et al. (23). In bulk systems, ion release or removal, for instance at electrodes, takes place far from the membrane, at least compared with usual MD system sizes. This scheme thus does not fulfill the criteria for a grand-canonical treatment of the whole compartment, as this would require Monte Carlo-based exchange steps. However, as Corry et al. (23) have shown for BD simulations, realistic fluctuations of ion concentrations indeed occur near the points of interest, the membrane channels, also by using the simple reinjection method, as long as the buffer regions are chosen at sufficient distance. We observe similar ion fluctuations, also leading to realistic fluctuations in the locally acting transmembrane potential using this scheme. We have also compared the results obtained by this simple scheme to the use of a Monte Carlo (MC) nonequilibrium method below. For PorB, we applied the deterministic scheme with a moving average time window of 1 ps length.
In a short-circuited situation, the maximum current supported by this scheme was found to exceed that of the widest channels studied (porin trimers) by a factor of 100–1000. This demonstrates that the resistance introduced by the creation/annihilation buffers is by far smaller than that of membrane channels, and so the flux characteristics are governed by the channel resistance only.
Nonequilibrium switching protocol
Here, at regular, short time intervals ti, an ion is randomly selected from the buffer volume, defined as two layers centered around the compartment midplanes. The layers, which have a fixed thickness d and thus a fixed volume, are shaded light blue in Fig. 1 A. A random water molecule is selected simultaneously from the opposite buffer. Following Jarzynski and co-worker (24,25), we perform a short switching simulation of length τ for this ion/water pair, from which we derive the work Wτ needed to get from the initial state A to state B with exchanged ion/water positions. The states are expressed by the Hamiltonians HA and HB, respectively. In the switching process, the coupling parameter λ (26) progresses from 0 to 1, while the state evolves from A to B using the Hamiltonian Hλ = (1 – λ)HA + λHB. In state A, the chosen water molecule has all the properties of the applied water model, whereas in state B, it mimics its counterpart ion (Na+ or Cl–) by carrying no charges at the hydrogen positions but the ionic charge (+le/−le) at the oxygen position. The counterpart ion is treated like the switchable water molecule, just with exchanged A and B states. The work associated with a single ion/water swap is
| (2) |
The exchange is accepted with a probability according to the Metropolis Monte Carlo (MC) criterion
where β = kBT is the reciprocal thermal energy. The energy offset term W0, if nonzero, produces and maintains a charge imbalance between the buffer volumes. W0 has a fixed absolute value, but its sign depends on the type of ion and direction of the swap attempt:
In this scheme, W0 is the adjustable parameter, determining the target energy difference between the two compartments and thus the resulting transmembrane potential gradient. Fig. 6 B, found later in this article, shows the charge imbalance obtained from setting W0 to 125 and 175 kJ/mol in the PorB system, respectively.
Figure 6.
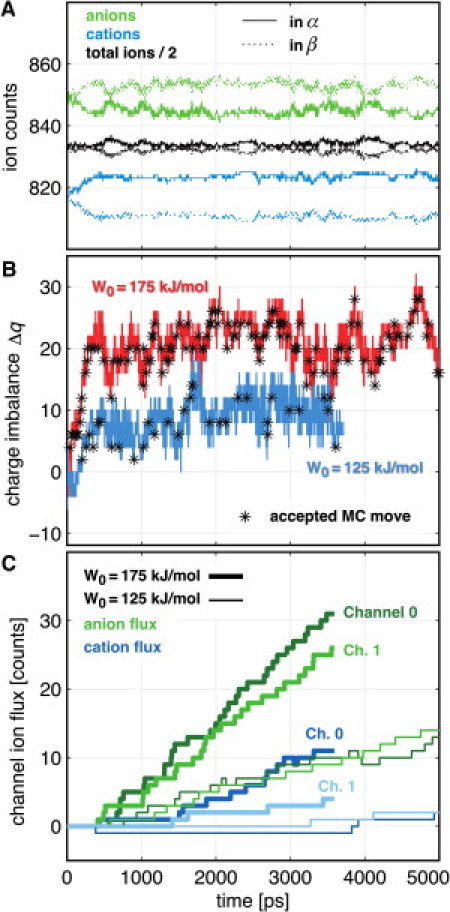
Sustaining ionic current with the nonequilibrium switching MC protocol. (A) Ion counts in the compartments in response to an energy offset of W0 = 175 kJ/mol, starting from an equilibrated system at t = 0 ps with neutral compartments. (B) Charge imbalance Δq(t), resulting from W0 = 125 kJ/mol (red), and W0 = 175 kJ/mol (cyan). Accepted MC moves (marked by black stars) quickly build up a charge imbalance starting from Δq = 0 e at t = 0 ps. (C) Ionic currents for each channel recorded in response to the applied energy offset of W0 = 125 kJ/mol (thick lines), and W0 = 175 kJ/mol (thin lines).
If the MC exchange attempt is rejected, the MD system is reset to time ti before the exchange attempt, or, if the MC move is accepted, the MD system continues to evolve from time ti + τ with the exchanged positions of the selected ion/water pair. After a certain time span of undisturbed MD, a new exchange is attempted with another ion/water pair selected from the buffer volumes. To keep the total ionic concentration within a small range around the desired concentration on both sides of the membrane, the MC anion or cation to be exchanged is selected from the compartment with a positive concentration difference. Any desired concentration ratio between the compartments can be retained with this protocol.
In contrast to the deterministic protocol, no force discontinuities arise from the position exchanges, because each swap is mediated by a smooth λ-transition between ion and water.
Transmembrane potential
Nonbalanced charge distributions Δq between the compartments give rise to a transmembrane potential difference ΔU(t), according to the membrane capacitance C (ΔU(t) = Δq(t)/C). We calculated ΔU(t) by using the implementation of the Poisson equation in the GROMACS tool g_potential (27) including a correction term to ensure that the potential is the same on both sides of the box (Fig. 1 C). This is—to good approximation—true for large double-membrane systems composed of symmetric bilayers, even in the case of small asymmetries introduced by membrane proteins. It also holds for systems containing two antiparallel bilayers with asymmetric leaflets, as the dipole moment over the entire system is zero in both cases. It is important to note that, as Gurtovenko and Vattulainen have recently shown (28), to accurately calculate the electrostatic potential in such a setup, one has to ensure that the centers of mass of each membrane/protein system do not show large fluctuations in z direction. Thus, in practice, a position restraint is applied on each bilayer/protein center during our calculations.
Simulation details
The membranes consisted of equilibrated and fully hydrated dimyristoylphosphatidylcholine (DMPC) lipid bilayers, each comprising 367 DMPC molecules. PorB trimers (Protein Data Bank entry 3a2r) were inserted into the bilayers using the tool g_membed (29) included in GROMACS 4.5. The aqueous compartments contained ∼26,000 water molecules each. The NaCl concentration was set to 1.0 mol/L, corresponding to 817 Na+ and 849 Cl− ions in each of the neutralized compartments, and, in a second set of simulations, to 0.2 mol/L, i.e., 155 Na+ and 187 Cl− ions in each neutral compartment. The volume of the membrane was accounted for in determining the ion concentration, as the membrane proved to attract and immobilize approximately the same number of ions a similar volume of water would contain. The AMBER99sb force field was used for the protein and ions (30). Parameters for DMPC were derived from Berger et al. (31), and water was modeled using the SPC/E water model (32). Water-bond distances and bond angles were constrained using the SETTLE algorithm (33), whereas all other bonds were constrained using LINCS (34,35). The temperature was kept constant by weakly (t = 0.1 ps) coupling the lipid molecules, protein, and solvent separately to a temperature bath of 320 K using the velocity rescaling thermostat of Bussi et al. (36). The slightly raised temperature was chosen to remain well above the temperature at which DMPC enters into gel formation. Modeling hydrogen atoms as virtual sites (37) allowed for an integration time step of 4 fs.
Simulations using the deterministic exchange protocol
Electrostatic interactions were computed with the smooth particle mesh Ewald method (38,39) using a short-range Coulomb cutoff of 1.0 nm and a reciprocal grid with 90 × 90 × 160 points, corresponding to a spacing of <0.132 nm. Lennard-Jones interactions were calculated with a cutoff at 1.0 nm. The system pressure was kept constant by semiisotropic Berendsen (40) coupling to a reference value of 1 bar. On a Linux cluster comprised of two quad-core Intel Xeon E5540@2.53GHz CPUs per node, connected by a ConnectX DDR Infiniband network, we achieved a performance of 38.2 ns/day on 192 cores, yielding >1 μs of simulation time per month.
After an initial equilibration time of 10 ns length, during which position restraints were applied on the protein heavy atoms, imbalances of 4–12 elementary charges between the compartments were established and sustained by using the deterministic particle exchange protocol. In short intervals of 0.2 ps, checks were performed to determine whether the ion exchange condition is met and ion/water pairs were exchanged as needed. In this way, voltages of −300..300 mV were applied across the membrane (Fig. 1 C and see also Fig. 3 B). A further equilibration period of 20 ns was used to allow the system to adapt to the electrochemical gradient. Then, trajectories of 50–250 ns length were computed for the various scenarios (Fig. 2). Note that the voltage gradient across the lower membrane is the inverse of that across the upper bilayer (Fig. 1 C).
Figure 3.
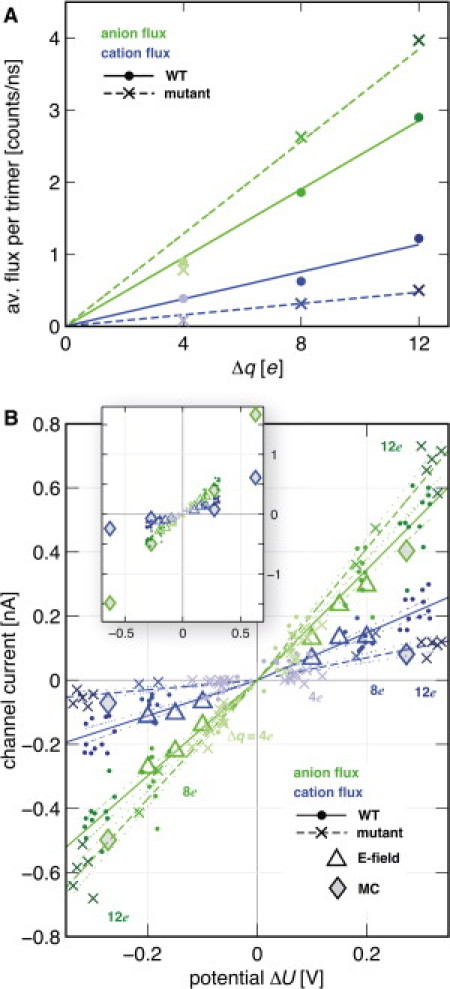
Ionic current is determined from the slopes of the flux curves shown in Fig. 2. (A) Net ion flux accumulated during the simulation for charge imbalances Δq/e = 4, 8, and 12. (B) Ionic current as a function of the potential difference determined for trajectory slices of length 20 ns.
Figure 2.

Ion flux recorded in response to different charge imbalances comparing wild-type (WT) and G103K PorB mutant at Δq = 4 e, 8 e, and 12 e. Different shading indicates different Δq. For each setting two flux curves are recorded, one for each membrane polarization.
Simulations using MC nonequilibrium switching
For a high accuracy, especially when determining the work Wτ associated with the position exchanges, the particle-mesh Ewald grid spacing was set to ≤0.120 nm, and the direct-space Coulomb potential was smoothly switched to zero between 0.9 and 1.0 nm. Lennard-Jones interactions were explicitly calculated up to 1.0 nm, and then smoothly switched to zero between 1.0 and 1.33 nm. Instead of Berendsen pressure coupling we used a constant volume in these simulations. Starting from the above described MD system after the initial 10-ns equilibration time, we let the system equilibrate for another 1 ns to adapt to the changed settings. Then we used the nonequilibrium switching protocol by attempting after every ti = 1 ps of normal MD an ion/water exchange between the buffer volumes (see Fig. 1 A) of thickness d = 2 nm each, using a switching time of τ = 1 ps and work offsets of and 175 kJ/mol.
To test our protocol, we used a smaller double-membrane MD system without channels, consisting of 424 DMPC lipids separated in two membranes, and ≈25,000 water molecules, and 770 Na+ and 770 Cl− ions in total. Here, starting from neutral compartments, charge imbalances can exclusively occur due to accepted MC moves. For this test, the ion/water buffer volumes were extended to include the whole compartment each.
The average work associated with the exchanges was Wτ ≈ 170(30), 52(14), and 30(7) kJ/mol for Na+/water exchanges and 200(44), 67(15), and 40(9) kJ/mol for Cl−/water exchanges for switching lengths τ = 1, 10, and 50 ps, respectively (bracketed values give the standard deviation). For all three τ-values we find that within 1–5 ns of simulation time for W0 = 0, 125, 250, and 375 kJ/mol, charge imbalances of 0.0, 3.9, 7.4, and 11.3 e are established, respectively (data not shown). The instantaneous Δq values are integer numbers varying around the averages, which were calculated from the last 20% of each trajectory. Whereas the equilibrium state based on W0 is reached quickly, only about every 1000th exchange attempt is actually accepted when the system has reached equilibrium.
Potential of mean force
The potential of mean force for ions (see Fig. 4 B) was calculated by taking the logarithm of the density of each ion type i along the channel axis (z axis), according to G(z) = −kBT ln [ni(z)], where kB is the Boltzmann constant, T is the temperature, and ni(z) is the ion density as a function of the channel coordinate. Ion coordinates were analyzed from a trajectory of 100 ns length, using bins of 1 Å width and by normalizing to the channel cross-sectional area. The PMF value of the bulk z system boundary was set to G = 0 (41,42). Errors were estimated by subdividing the trajectory into four parts of equal length and calculating the standard deviation of the average value within each bin.
Figure 4.
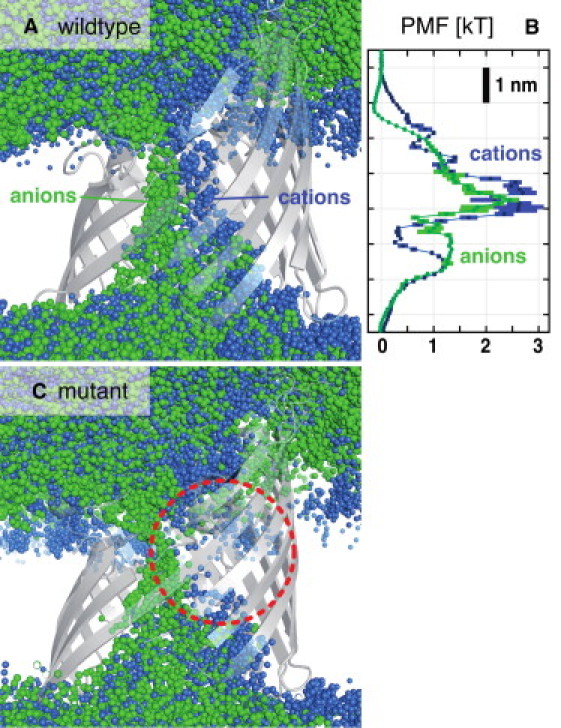
(A) Overlay of ion positions from 500 snapshots of a 100-ns PorB simulation at a charge imbalance of Δq = 8 e. (B) Potential of mean force for Cl− (green) and Na+ ions (blue) for wild-type PorB. (C) As A, but for PorB mutant G103K. The mutant shows a disrupted cation pathway (red circle).
Calculation of ion selectivity and conductance
Fig. 3 A shows the net ion flux accumulated until the end of the simulation as a function of the set transmembrane charge imbalance Δq. For charge imbalance levels of up to 12 e, the ion flux can be described by a linear function. Conductance calculations Λ = I/ΔU were based on ion current I as a function of the potential difference ΔU between the compartments. To obtain an error estimate, and because potential and flux are time-dependent, ΔU(t) and I(t) were determined within 20-ns time windows, with 10-ns overlap among consecutive slices. For each window, ΔU(t) was determined as shown in Fig. 1 C, whereas I(t) was derived by fitting a straight line to the ion flux in that time window. This way, each data point from Fig. 3 A results in a cluster of data points in Fig. 3 B, separated by shading. Combining the results from the three different Δq values and then fitting linear functions to those data gives the straight lines shown in Fig. 3 B for wild-type PorB (solid) and the G103K mutant (dashed). The dotted lines provide 95% confidence intervals for the slope estimate. Note that separate fits have been carried out for positive and negative U. The selectivity ratio (Cl−/Na+) is provided by the slope ratios.
Results and Discussion
As shown in Fig. 1, a double-bilayer system was used to separate two compartments in the periodic box. Particle exchanges occur according to one of two different protocols: In the deterministic protocol, each permeation event is followed by an exchange after a slight delay. In the nonequilibrium switching protocol, similar to a grand canonical ensemble (43), the sum of ions in each compartment fluctuates around a mean value, whereas frequent Monte Carlo attempts ensure a preserved charge gradient over time.
Because the MC protocol is computationally more expensive, due to the large number of rejected exchange attempts, we adhered to the deterministic protocol for the majority of the simulations.
Conduction pathway and ionic conductance of PorB
The ion transfer mechanism inside PorB has been suggested to be unusual, as on the basis of its crystal structure, individual, almost noninteracting pathways for the diffusion of anions and cations were postulated (12). Our method enabled us to study the molecular mechanism, ionic selectivity, and energetics of ion flux through PorB by applying electrochemical potential gradients on PorB trimers inserted into lipid bilayers.
As shown by an overlay of ionic positions from 500 simulation snapshots that cover 100 ns simulated time (Fig. 4 A), we find that there is indeed virtually no overlap of the pathway that translocating cations and anions take along nearly the entire length of the PorB pore. A similar but somewhat less pronounced split between anionic and cationic pathways had previously been found in the general diffusion porin OmpF from Escherichia coli (44). In PorB, anions almost exclusively pass along a cluster of basic residues on one side of the eyelet, whereas cations move along acidic residues lining the opposite wall, and this effect continues throughout the channel.
The flux of anions and cations, IA and IC, through wild-type PorB trimers during the simulations is plotted in Fig. 2. Transmembrane charge imbalances of Δq = 4, 8, and were probed and are indicated by shading. These data readily enable extraction of the principal experimental observables of electrophysiological channel measurements, conductance Λ = I/ΔU and ion selectivity IA/IC. The potential difference ΔU needed to calculate the conductance, and arising from a given electrochemical gradient Δq, was calculated by solving the Poisson equation (Fig. 1 C, see Methods). We determined the conductance in time windows of 20 ns. Fig. 3 displays the ionic currents through PorB in 1 M salt, dependent on the actual transmembrane potential, taking into account fluctuations around the expected values according to Δq. The currents show a slight dependence on channel orientation. The fluctuations occur as a result of differing transient ion configurations in the system, and reflect ionic fluctuations in cells and experiment. At a salt concentration of 1 M, the wild-type conductance at positive polarizations is predicted to be 2.5 ± 0.1 nS, that at negative polarizations 2.0 ± 0.1 nS. The anion selectivities are calculated to be 2.3 ± 0.1 and 2.7 ± 0.2, respectively. This demonstrates an influence of PorB orientation on the electrophysiological observables. The mutant channel has a conductance of 2.4 ± 0.1 nS and 2.0 ± 0.1 nS at positive and negative membrane polarization, respectively. Its selectivity for anions is markedly raised, reaching values of 5.9 ± 0.6 and 12.3 ± 1.7 for a positive or negative potential gradient.
Experimentally, values at a NaCl concentration of 200 mM are available. At a NaCl concentration of 200 mM, our protocol predicts an average conductance of 0.8 ± 0.1 nS and an ion selectivity of 3.0 ± 0.6 (Cl−/Na+) for PorB. These values are in excellent agreement with the respective experimentally determined quantities for recombinant Nme class 3 PorB (single trimer conductance: ∼1.0 nS, preference for Cl− over Na+: 2.8) (45), and also correspond to the consensus values across Neisserial PorB channels (19).
PorB antibiotic resistance mutation
Porins are the major gateways for entry of small molecules into the cells of Gram-negative bacteria. Thus, for successful antibacterial therapy, porin channels must be passable for antibiotic molecules (18). However, single-point mutations in PorB suffice to contribute to the development of resistance toward antibiotics, often in conjunction with efflux mechanisms (19). It is expected that Neisserial infections are becoming a very difficult infection to treat within the next five years, so efforts to develop new antibiotic therapies are highly desirable (20).
We investigated the effect of a functionally important single-point mutation in the channel interior on ionic current. The mutation G120K on loop 3 of Ngo PorB is known to contribute to resistance toward antibiotic molecules such as tetracycline and penicillin (19). Fig. 2 demonstrates that the corresponding mutant, G103K Nme PorB (dashed), exhibits a fourfold raised anion selectivity. As highlighted by Fig. 4 C, this can be ascribed to an almost complete block of the independent cationic pathway by the additional single charge in the eyelet region, whereas the anionic conduction pathway remains unaffected. Thus, the electrostatic barrier inside the channel eyelet becomes almost insurmountable for cations. We suggest that the drastic alteration of the dipolar properties of the pore contributes to reduced permeability for molecules with a polar core such as the β-lactam antibiotic penicillin.
Impact of particle exchanges in the deterministic protocol
To quantify the extent of the unphysical discontinuity in the forces on nearby particles, caused by ion exchanges in the deterministic protocol, we examined the effect of a single ion/water exchange on all atoms by monitoring the (absolute) forces acting on the atoms before and after the exchange. The wild-type PorB system at 1 M was used at a representative particle exchange event.
Fig. 5 shows the absolute value of the atomic forces (one value per atom) at the time step before the exchange (thin dots) as well as their average (straight black line). The forces are shown as a function of their distance r to the nearest of the two exchanged molecules. Immediately after the exchange, forces were recalculated. Thick dots represent the absolute difference of the forces before and after the exchange.
Figure 5.
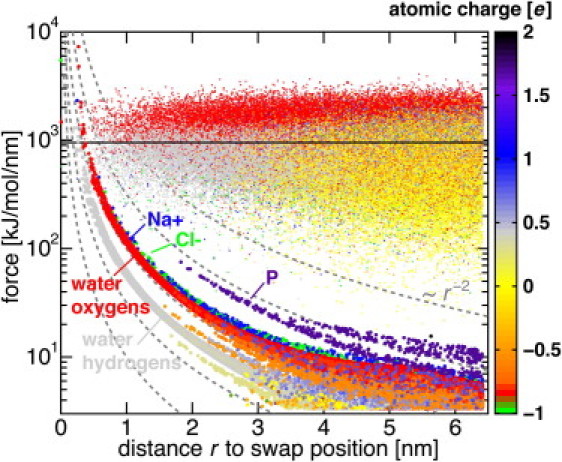
Perturbation of the atomic forces caused by a single H2O/Cl− exchange. Thin dots mark all interatomic forces in an unperturbed system at a representative time step. Thick dots display the absolute difference of the interatomic forces before and after the water/ion exchange. The atomic charge is color-coded. Dashed lines show c × r−2 with 10 ≤ c ≤ 1000.
The force difference is expectably largest near the exchanged molecules (at small r). Within 0.5 nm distance to the exchanged molecules, the force change exceeds the average MD force by almost an order of magnitude (Fig. 5). This, however, only affects about a dozen atoms, which are located far from the membrane channels. The affected atoms are largely comprised of the exchanged atoms themselves and their first solvation shell. Overall, the average force change amounts to only 0.55% of the mean force. For distances r > 1 nm, the force change is at least an order of magnitude smaller than the mean force.
Because of this low level of perturbation, energy minimization steps are unnecessary after exchanges. In practice, the impact of the swaps on the membranes and channels can be further reduced by extending the compartments in z direction.
Nonequilibrium switching
We applied the nonequilibrium switching MC protocol as described in Methods to the double-membrane wild-type PorB system at 1M NaCl, and compared it to the deterministic protocol. As can be seen from Fig. 6 A, starting from initially neutral compartments, the system responds to the selected energy offset W0 of 175 kJ/mol by adjusting the ratio of anions (green) to cations (blue) in the compartments within 1 ns, whereas the total number of ions (black) only slightly fluctuates around its mean. As a result, a charge imbalance of Δq ≈ 10 (for W0 = 125 kJ/mol, cyan) and Δq ≈ 20 (for W0 = 175 kJ/mol, red) rapidly evolves between the compartments, and is then sustained against continuous dissipation by the channel currents by accepted MC moves (black stars) (Fig. 6 B).
In contrast to the deterministic protocol, in which the desired ion counts per compartment are restored precisely every 0.2 ps, the MC protocol yields larger fluctuations of the total charge imbalance between the compartments, which occur on timescales of a nanosecond. Fig. 6 C shows the ionic current that is evoked as a result of these charge imbalances and, ultimately, of the energy offset W0.
For comparison with the results from the deterministic protocol, potential differences ΔU(W0) and ionic currents I(U) for both channels were calculated as described; however, a single time slice of t = 1–3.5 ns was used here. The values are displayed as gray diamonds in Fig. 3 B; note that the values for W0 = 175 kJ/mol result in |ΔU |> 500 mV and are thus only shown in the inset. The values agree surprisingly well with the data recorded with the deterministic protocol. Note, however, that the cationic currents are based on a small number of channel permeations, and hence may suffer from poorer statistics.
Comparison to alternative approaches and conclusion
For comparison to other approaches, and using a similar computational effort as in the deterministic protocol, we computed the one-dimensional potential of mean force (PMF), which is commonly used to address questions around ion permeation and selectivity of ion channels. We performed equilibrium simulations of ∼100 ns length, from which the PMF profile was calculated according to GPMF(z) = −kBT ln n(z), using the residence frequencies n(z) within bins of 0.1 nm width along the pore axis z as measure of the free energy a particular ion type experiences along the channel axis. Fig. 4 B demonstrates that, in contrast to our approach, the interpretation of PMF landscapes to extract information on ion flux is not straightforward. As they stand, the main energy barriers in the PMF curve would suggest an anion selectivity of PorB at 200 mM salt of 1.3 ± 0.7 (rather than 3.0 as observed from computational electrophysiology and 2.8 from experiment).
Based on the PMF, we estimated the conductance of PorB by determining local diffusion rates, calculated as the transition rates of ions between bins of 1 Å width along the pore axis (42). Extrapolation of these rates to the nonequilibrium case of applied voltages yields a conductance of 3.4 ± 1.6 nS at 200 mM (in contrast to ∼1 nS from experiment and computational electrophysiology). This value suggests a substantially larger channel, whereas its uncertainty is in the range of the experimental conductance. Possible reasons for this discrepancy are the assumption of a low-dimensional reaction coordinate and a local diffusion model.
In the computational electrophysiology approach presented here, no such assumptions are required, thus allowing a direct and accurate estimation of the conductance and selectivity. Simultaneously, it provides detailed mechanistic insight into channel function and channel modulation by mutation and molecular interactions, not as readily obtainable through BD or continuum methods (3,46).
Compared to the application of external electric fields, computational electrophysiology is computationally only slightly more expensive, considering the fact that two channels and polarizations can be probed simultaneously. In addition, the usually applied periodic boundary conditions complicate a direct and quantitative comparison to experiment using an external electric field. As noted by Neumann (47), the relation between the applied electric field and the locally acting field is not a priori known, and depends on the used electrostatics scheme (cut-off, reaction field, Ewald summation, and boundary conditions) as well as on the dielectric distribution across the simulation box. The latter effect is particularly noticeable in systems in which the dielectric distribution is variable in time, for example during membrane pore formation (8). Moreover, some applications such as the calculation of reversal potentials require two well-separated compartments.
Computational electrophysiology is applicable to channels of small and large conductance and is able to control both concentration and potential gradients. Thus we expect it to be useful for studies of the molecular mechanisms of ion passage, e.g., for the improvement of drugs to overcome resistance mutations in bacterial porins, and in drug design on ion channel targets. Its implementation into the freely available, efficient MD software GROMACS (21) allows simulations of several microseconds and thus enables direct comparison to experiment and biology.
Acknowledgments
We thank Bernhard Egwolf and Dirk Matthes for helpful discussions and critical reading of the manuscript.
The work was partially funded by the Dutch TI Pharma project D2-101 (B.dG. and U.Z.).
Footnotes
This is an Open Access article distributed under the terms of the Creative Commons-Attribution Noncommercial License (http://creativecommons.org/licenses/by-nc/2.0/), which permits unrestricted noncommercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
References
- 1.Wulff H., Castle N.A., Pardo L.A. Voltage-gated potassium channels as therapeutic targets. Nat. Rev. Drug Discov. 2009;8:982–1001. doi: 10.1038/nrd2983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kaczorowski G.J., McManus O.B., Garcia M.L. Ion channels as drug targets: the next GPCRs. J. Gen. Physiol. 2008;131:399–405. doi: 10.1085/jgp.200709946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roux B., Allen T., Im W. Theoretical and computational models of biological ion channels. Q. Rev. Biophys. 2004;37:15–103. doi: 10.1017/s0033583504003968. [DOI] [PubMed] [Google Scholar]
- 4.Roux B. The membrane potential and its representation by a constant electric field in computer simulations. Biophys. J. 2008;95:4205–4216. doi: 10.1529/biophysj.108.136499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jensen M.Ø., Borhani D.W., Shaw D.E. Principles of conduction and hydrophobic gating in K+ channels. Proc. Natl. Acad. Sci. USA. 2010;107:5833–5838. doi: 10.1073/pnas.0911691107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tieleman D.P., Leontiadou H., Marrink S.J. Simulation of pore formation in lipid bilayers by mechanical stress and electric fields. J. Am. Chem. Soc. 2003;125:6382–6383. doi: 10.1021/ja029504i. [DOI] [PubMed] [Google Scholar]
- 7.Sachs J.N., Crozier P.S., Woolf T.B. Atomistic simulations of biologically realistic transmembrane potential gradients. J. Chem. Phys. 2004;121:10847–10851. doi: 10.1063/1.1826056. [DOI] [PubMed] [Google Scholar]
- 8.Böckmann R.A., de Groot B.L., Grubmüller H. Kinetics, statistics, and energetics of lipid membrane electroporation studied by molecular dynamics simulations. Biophys. J. 2008;95:1837–1850. doi: 10.1529/biophysj.108.129437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dzubiella J., Allen R.J., Hansen J.P. Electric field-controlled water permeation coupled to ion transport through a nanopore. J. Chem. Phys. 2004;120:5001–5004. doi: 10.1063/1.1665656. [DOI] [PubMed] [Google Scholar]
- 10.Murad S., G. Powles J. A computer simulation of the classic experiment on osmosis and osmotic pressure. J. Chem. Phys. 1993;99:7271–7272. [Google Scholar]
- 11.Kalra A., Garde S., Hummer G. Osmotic water transport through carbon nanotube membranes. Proc. Natl. Acad. Sci. USA. 2003;100:10175–10180. doi: 10.1073/pnas.1633354100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tanabe M., Nimigean C.M., Iverson T.M. Structural basis for solute transport, nucleotide regulation, and immunological recognition of Neisseria meningitidis PorB. Proc. Natl. Acad. Sci. USA. 2010;107:6811–6816. doi: 10.1073/pnas.0912115107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cowan S.W., Schirmer T., Rosenbusch J.P. Crystal structures explain functional properties of two E. coli porins. Nature. 1992;358:727–733. doi: 10.1038/358727a0. [DOI] [PubMed] [Google Scholar]
- 14.Zachariae U., Helms V., Engelhardt H. Multistep mechanism of chloride translocation in a strongly anion-selective porin channel. Biophys. J. 2003;85:954–962. doi: 10.1016/S0006-3495(03)74534-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zachariae U., Klühspies T., Zeth K. High resolution crystal structures and molecular dynamics studies reveal substrate binding in the porin Omp32. J. Biol. Chem. 2006;281:7413–7420. doi: 10.1074/jbc.M510939200. [DOI] [PubMed] [Google Scholar]
- 16.Massari P., Ram S., Wetzler L.M. The role of porins in Neisserial pathogenesis and immunity. Trends Microbiol. 2003;11:87–93. doi: 10.1016/s0966-842x(02)00037-9. [DOI] [PubMed] [Google Scholar]
- 17.Kozjak-Pavlovic V., Dian-Lothrop E.A., Rudel T. Bacterial porin disrupts mitochondrial membrane potential and sensitizes host cells to apoptosis. PLoS Pathog. 2009;5:e1000629. doi: 10.1371/journal.ppat.1000629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pagès J.M., James C.E., Winterhalter M. The porin and the permeating antibiotic: a selective diffusion barrier in Gram-negative bacteria. Nat. Rev. Microbiol. 2008;6:893–903. doi: 10.1038/nrmicro1994. [DOI] [PubMed] [Google Scholar]
- 19.Olesky M., Zhao S., Nicholas R.A. Porin-mediated antibiotic resistance in Neisseria gonorrhoeae: ion, solute, and antibiotic permeation through PIB proteins with penB mutations. J. Bacteriol. 2006;188:2300–2308. doi: 10.1128/JB.188.7.2300-2308.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.GRASP Steering Group . Health Protection Agency; London, UK: 2008. The Gonococcal Resistance to Antimicrobials Surveillance Programme (GRASP) Year 2007 Report. [Google Scholar]
- 21.Hess B., Kutzner C., Lindahl E. GROMACS 4.0: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 22.Hallermann S., Heckmann S., Heckmann M. Short openings in high resolution single channel recordings of mouse nicotinic receptors. J. Physiol. 2005;563:645–662. doi: 10.1113/jphysiol.2004.080606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Corry B., Hoyles M., Chung S.H. Reservoir boundaries in Brownian dynamics simulations of ion channels. Biophys. J. 2002;82:1975–1984. doi: 10.1016/S0006-3495(02)75546-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ballard A.J., Jarzynski C. Replica exchange with nonequilibrium switches. Proc. Natl. Acad. Sci. USA. 2009;106:12224–12229. doi: 10.1073/pnas.0900406106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jarzynski C. Nonequilibrium equality for free energy differences. Phys. Rev. Lett. 1997;78:2690–2693. [Google Scholar]
- 26.Kirkwood J. Statistical mechanics of fluid mixtures. J. Chem. Phys. 1935;3:300–313. [Google Scholar]
- 27.Tieleman D., Berendsen H. Molecular dynamics simulations of a fully hydrated dipalmitoylphosphatidylcholine bilayer with different macroscopic boundary conditions and parameters. J. Chem. Phys. 1996;105:4871–4880. [Google Scholar]
- 28.Gurtovenko A.A., Vattulainen I. Calculation of the electrostatic potential of lipid bilayers from molecular dynamics simulations: methodological issues. J. Chem. Phys. 2009;130:215107. doi: 10.1063/1.3148885. [DOI] [PubMed] [Google Scholar]
- 29.Wolf M.G., Hoefling M., Groenhof G. g_membed: Efficient insertion of a membrane protein into an equilibrated lipid bilayer with minimal perturbation. J. Comput. Chem. 2010;31:2169–2174. doi: 10.1002/jcc.21507. [DOI] [PubMed] [Google Scholar]
- 30.Hornak V., Abel R., Simmerling C. Comparison of multiple AMBER force fields and development of improved protein backbone parameters. Proteins. 2006;65:712–725. doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Berger O., Edholm O., Jähnig F. Molecular dynamics simulations of a fluid bilayer of dipalmitoylphosphatidylcholine at full hydration, constant pressure, and constant temperature. Biophys. J. 1997;72:2002–2013. doi: 10.1016/S0006-3495(97)78845-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Berendsen H., Grigera J., Straatsma T. The missing term in effective pair potentials. J. Phys. Chem. 1987;91:6269–6271. [Google Scholar]
- 33.Miyamoto S., Kollman P. SETTLE—an analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992;13:952–962. [Google Scholar]
- 34.Hess B., Bekker H., Fraaije J. LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 1997;18:1463–1472. doi: 10.1021/ct700200b. [DOI] [PubMed] [Google Scholar]
- 35.Hess B. P-LINCS: a parallel linear constraint solver for molecular simulation. J. Chem. Theory Comput. 2008;4:116–122. doi: 10.1021/ct700200b. [DOI] [PubMed] [Google Scholar]
- 36.Bussi G., Donadio D., Parrinello M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007;126:014101. doi: 10.1063/1.2408420. [DOI] [PubMed] [Google Scholar]
- 37.Feenstra K., Hess B., Berendsen H. Improving efficiency of large time-scale molecular dynamics simulations of hydrogen-rich systems. J. Comput. Chem. 1999;20:786–798. doi: 10.1002/(SICI)1096-987X(199906)20:8<786::AID-JCC5>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 38.Darden T., York D., Pedersen L. Particle mesh Ewald: an N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993;98:10089–10092. [Google Scholar]
- 39.Essmann U., Perera L., Lee H. A smooth particle mesh Ewald method. J. Chem. Phys. 1995;103:8577–8593. [Google Scholar]
- 40.Berendsen H., Postma J., Haak J. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984;81:3684–3690. [Google Scholar]
- 41.Fischer G., Kosinska-Eriksson U., Lindkvist-Petersson K. Crystal structure of a yeast aquaporin at 1.15 Ångstrom reveals a novel gating mechanism. PLoS Biol. 2009;7:e1000130. doi: 10.1371/journal.pbio.1000130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.de Groot B.L., Frigato T., Grubmüller H. The mechanism of proton exclusion in the aquaporin-1 water channel. J. Mol. Biol. 2003;333:279–293. doi: 10.1016/j.jmb.2003.08.003. [DOI] [PubMed] [Google Scholar]
- 43.Im W., Seefeld S., Roux B. A grand canonical Monte Carlo-Brownian dynamics algorithm for simulating ion channels. Biophys. J. 2000;79:788–801. doi: 10.1016/S0006-3495(00)76336-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Im W., Roux B. Ion permeation and selectivity of OmpF porin: a theoretical study based on molecular dynamics, Brownian dynamics, and continuum electrodiffusion theory. J. Mol. Biol. 2002;322:851–869. doi: 10.1016/s0022-2836(02)00778-7. [DOI] [PubMed] [Google Scholar]
- 45.Song J., Minetti C.A., Colombini M. Successful recovery of the normal electrophysiological properties of PorB (class 3) porin from Neisseria meningitidis after expression in Escherichia coli and renaturation. Biochim. Biophys. Acta. 1998;1370:289–298. doi: 10.1016/s0005-2736(97)00279-4. [DOI] [PubMed] [Google Scholar]
- 46.Koumanov A., Zachariae U., Karshikoff A. Improved 3D continuum calculations of ion flux through membrane channels. Eur. Biophys. J. 2003;32:689–702. doi: 10.1007/s00249-003-0330-y. [DOI] [PubMed] [Google Scholar]
- 47.Neumann M. Dipole moment fluctuation formulas in computer simulations of polar systems. Mol. Phys. 1983;50:841–858. [Google Scholar]