

Abstract
Kv7.1 (KCNQ1) channels are regulators of several physiological processes including vasodilatation, repolarization of cardiomyocytes, and control of secretory processes. A number of Kv7.1 pore mutants are sensitive to extracellular potassium. We hypothesized that extracellular potassium also modulates wild-type Kv7.1 channels. The Kv7.1 currents were measured in Xenopus laevis oocytes at different concentrations of extracellular potassium (1–50 mM). As extracellular potassium was elevated, Kv7.1 currents were reduced significantly more than expected from theoretical calculations based on the Goldman-Hodgkin-Katz flux equation. Potassium inhibited the steady-state current with an IC50 of 6.0 ± 0.2 mM. Analysis of tail-currents showed that potassium increased the fraction of channels in the inactivated state. Similarly, the recovery from inactivation was slowed by potassium, suggesting that extracellular potassium stabilizes an inactivated state in Kv7.1 channels. The effect of extracellular potassium was absent in noninactivating Kv7.1/KCNE1 and Kv7.1/KCNE3 channels, further supporting a stabilized inactivated state as the underlying mechanism. Interestingly, coexpression of Kv7.1 with KCNE2 did not attenuate the inhibition by potassium. In a number of other Kv channels, including Kv1.5, Kv4.3, and Kv7.2–5 channels, currents were only minimally reduced by an increase in extracellular potassium as expected. These results show that extracellular potassium modulates Kv7.1 channels and suggests that physiological changes in potassium concentrations may directly control the function of Kv7.1 channels. This may represent a novel regulatory mechanism of excitability and of potassium transport in tissues expressing Kv7.1 channels.
Introduction
The voltage-gated potassium channel Kv7.1 (KCNQ1, KvLQT1) plays a physiological role in several tissues including the cardiovascular system (1–3), the pancreas (4), and the intestinal system (5–7). The physiological function and the regulation of Kv7.1 channels as well as their role in channelopathies have recently been reviewed (8,9).
Like other Kv channels, four Kv7.1 α-subunits assemble to form a potassium-conducting ion channel. Upon membrane depolarization, homomeric Kv7.1 channels activate fast and undergo a delayed inactivation process. The delayed onset of inactivation has been proposed to be the result of several open states that need to be occupied before the inactivated state can be reached (10). However, the degree of inactivation is limited and is appreciable only as a small hook on the tail currents representing recovery of channels from the inactivated state. When studied in isolation, the inactivation process is fast and independent of extracellular potassium (11,12). This makes it mechanistically different from traditional C-type inactivation observed in other Kv channels where inactivation is prevented by high extracellular potassium (13–15). The residues responsible for the delayed fast inactivation has been mapped to the S5 helix and the pore loop (16). Some Kv7.1 channels carrying mutations in the pore region have been shown to undergo additional slow inactivation. Interestingly, this slow inactivation process is modulated by extracellular potassium (elevation of extracellular potassium increases the rate of slow inactivation), suggesting a stabilization of the inactivated state in high potassium solutions (17), which again is different from C-type inactivation where high potassium generally prevents inactivation. The significant effect of extracellular potassium on the gating kinetics of the slow inactivating pore-mutants of Kv7.1 led us to hypothesize that also gating of wild-type Kv7.1 channels is modulated by extracellular potassium.
Here we demonstrate that Kv7.1 channels are inhibited by extracellular potassium. Our data strongly suggest that stabilization of an inactivated state is responsible for the reduction of Kv7.1 current in high extracellular potassium. Importantly, the modulation of Kv7.1 channels was observed at physiological relevant concentrations of extracellular potassium suggesting that extracellular potassium may indeed control the physiological function of Kv7.1 channels.
Materials and Methods
Unless otherwise mentioned all chemicals were obtained from Sigma-Aldrich (St. Louis, MO).
DNA constructs
cDNAs encoding human Kv1.5, Kv4.3, Kv11.1, Kv7.1, Kv7.2, Kv7.3, Kv7.4, Kv7.5, KCNE1, KCNE2, and KCNE3 were subcloned into pXOOM for expression in Xenopus laevis (X. laevis) oocytes (18). All plasmids were sequenced before use.
Expression in Xenopus laevis oocytes
cRNA for injection was prepared from the linearized DNA constructs using the T7 m-Message Machine kit (Ambion, Austin, TX) according to the manufacturer's instructions. RNA concentrations were measured using an ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE) and RNA quality was checked by gel electrophoresis. Xenopus laevis oocytes were either purchased from Ecocyte Bioscience (Castrop-Rauxel, Germany) or prepared in-house. In the latter case, X. laevis surgery and oocyte treatment were performed according to the guidelines of the Danish National Committee for Animal Studies as described previously (19). Oocytes were injected with cRNA (1–6 ng per oocyte; 50.6 nL injection volume) using a Nanoject microinjector (Drummond Scientific Company, Broomall, PA). For coexpression of Kv7.1/KCNE1, Kv7.1/KCNE2, Kv7.1/KCNE3, and Kv7.2/Kv7.3, cRNAs were mixed in a molar ratio of 1:1 before injection. Electrophysiological experiments were performed 2–3 days after injection.
Electrophysiological recordings
Measurements on X. laevis oocytes were performed with the two-electrode voltage-clamp technique using a Dagan CA-1B amplifier (Dagan, Minneapolis, MN). Experiments were performed at room temperature (22°C) under continuous superfusion with extracellular solution. The standard extracellular solution contained (in mM): 90 NaCl, 1 KCl, 1 MgCl2, 1 CaCl2, 5 HEPES, pH 7.4. In solutions containing higher concentration of potassium, NaCl was exchanged 1:1 for KCl. The order in which the different potassium solutions were applied was randomized to minimize any systematic effects of solution exchange in the experiments. The glass pipettes for the recording electrodes were filled with 2 M KCl and had tip resistances of 0.5–2.5 mOhm. Data were acquired with Pulse software (HEKA Elektronik, Lambrecht/Pfalz, Germany).
For all channel types, currents were recorded using a standard voltage-clamp step protocol: From a holding potential of −80 mV oocytes were clamped for 1–3 s at potentials between −80 and 40 mV (10 mV increments) followed by a step to −120 mV. The reversal potential for Kv7.1 channels was determined in each experiment by clamping the oocyte at 20 mV for 1 s before stepping to potentials between −120 and 20 mV (10 mV increments) and analyzing the resulting tail currents. The reversal potential for Kv7.1 channels was −93.1 ± 1.4 (n = 19) and −49.5 ± 0.6 (n = 19) mV in 1 and 10 mM [K+]o, respectively, which is close to the values reported by Sanguinetti et al. (1). To assess inactivation properties of Kv7.1, two different protocols, a triple-pulse and a tail-envelope protocol, were used. The triple pulse protocol consisted of a 2-s step to 40 mV, then a 20-ms step to −130 mV followed by a 500-ms step to potentials between 40 and −20 mV (in 10-mV decrements). The tail-envelope consisted of a step to 40 mV for a duration of 100–1600 ms (100-ms increments) followed by a step to −120 mV. All protocols used a −80 mV holding potential between test pulses.
In all current traces shown, the capacitive transients have been removed for clarity.
Data analysis
Data analysis was performed with Igor Pro (Wavemetrics, Lake Oswego, OR) and GraphPad Prism (GraphPad Software, San Diego, CA) software.
The ratio between the currents recorded in 1 and 10 mM [K+]o (I10 mM/I1 mM) was calculated for all channel types at a potential of 40 mV.
For Kv7.1 channels, steady-state current was converted to conductance by dividing with the reversal potential. The voltage dependence of activation was determined from normalized steady-state conductance-voltage (G/V) relationships. The G/V-relationship could be described by a standard Boltzmann function. The time constant of inactivation was estimated from the triple-pulse protocol: a single-exponential function was fitted to the current trace during the first 100 ms of the second depolarizing step to estimate the time constant of inactivation. The time constants of recovery from inactivation and deactivation as well as an estimate of the fraction of inactivated channels was obtained by fitting a four-state coupled ordinary differential equation (ODE) model to the tail currents recorded with the tail-envelope protocol. The ODE model is described in more detail in Results. The ODE model analysis was written in Python (www.python.org) using the SciPy (www.scipy.org) library. The model was fitted to the data using the Levenberg-Marquardt algorithm.
Statistical analyses were performed using the software “R” (V. 2.8.1; http://www.R-project.org). For all channel types, I10 mM/I1 mM-ratios were compared using a one-way ANOVA followed by Dunnett's test for multiple comparisons (using the predicted values—see Results—as the reference). The Wilcoxon signed-rank test was used to compare the delay and time constants for the onset of inactivation obtained from the ODE analysis of the tail-envelope data. The relationship between the fraction of inactivated channels (Finact) and the relative current amplitudes (Irel) obtained from the concentration-response experiment was fitted with a linear mixed-effects model (using the NLME package (20)). P values < 0.05 were considered statistically significant for all tests. Unless otherwise stated, data are represented as mean ± SE.
Results
In general, an increase in extracellular potassium reduces the driving force for potassium and results in reduced outward currents through potassium-selective ion channels. The theoretical reduction in current as a function of extracellular potassium concentration can be calculated from the GHK flux equation. The GHK flux equation for potassium can be written as (21)
| (1) |
where ΦK+ is the ionic flux of potassium, I is the current, A is the membrane area, PK+ is the permeability of potassium, Vm is the membrane potential, F is Faraday's number, R is the gas constant, T is the absolute temperature, and [K+]i and [K+]o are the intra- and extracellular concentrations of potassium, respectively. Assuming that all variables in the equation are independent of the extracellular potassium concentration (which would be the case for an ideal extracellular potassium-independent Kv channel), the theoretical ratio of the currents measured at two different concentrations, C1 and C2, of extracellular potassium can be calculated using Eq. 1:
| (2) |
Equation 2 is also known as the “independence relation” (21). Using Eq. 2 and the values for [K+]i in X. laevis oocytes given by Weber ((22), their Table 2), the predicted value for the ratio of currents recorded in 1 and 10 mM [K+]o, can be estimated to be I10 mM/I1 mM = 0.98 ± 0.002 (at a potential of 40 mV and a temperature of 22°C). In other words, an increase in [K+]o from 1 mM to 10 mM is expected to reduce the current by ∼2% (at a potential of 40 mV). Thus, under physiological conditions ([K+]o: 1–10 mM) outward potassium current amplitudes are expected to vary only little as a result of changes in driving force per se.
Extracellular potassium inhibits Kv7.1 steady-state currents
The effect of changing extracellular potassium in the physiological range from 1 mM to 10 mM was examined on heterologously expressed human Kv7.1 channels. In Fig. 1 A, representative current recordings obtained in 1 (solid) and 10 (shaded) mM [K+]o are shown. The steady-state currents measured at both 0 and 40 mV are markedly reduced by an elevation of extracellular potassium (I10 mM/I1 mM: 0.46 ± 0.06 and 0.57 ± 0.06 at 0 and 40 mV, respectively; n = 19). Summary data in the form of I/V-curves are shown in Fig. 1 B demonstrating that the steady-state current is reduced at all potentials above −50 mV where channels are open. To determine the voltage-dependency of activation, steady-state G/V-curves were constructed by converting the steady-state current to conductance. In Fig. 1 C, normalized steady-state G/V-curves for Kv7.1 channels in 1 (solid) and 10 (shaded) mM [K+]o are shown. The V50-values determined from Boltzmann fits to the normalized G/V-curves were not significantly different: −24.3 ± 1.1 (n = 13) and −25.0 ± 1.5 (n = 13) mV in 1 and 10 mM [K+]o, respectively.
Figure 1.
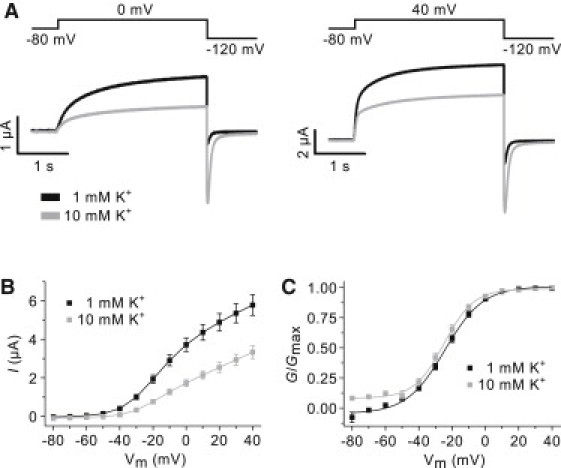
Kv7.1 current amplitude is reduced by extracellular potassium. (A) Representative currents from Kv7.1 channels recorded in 1 (solid) and 10 (shaded) mM [K+]o. The voltage-clamp protocols are shown above the corresponding recordings. (B) Steady-state current-voltage (I/V) relationship for Kv7.1 channels in 1 (solid) and 10 (shaded) mM [K+]o. (C) Normalized steady-state conductance-voltage (G-V) relationship for Kv7.1 channels in 1 and 10 mM [K+]o. Boltzmann functions (solid lines) were fitted to the data.
Effect of extracellular potassium on other Kv channels
To investigate whether other Kv channels were modulated by extracellular potassium to a similar extent, human Kv1.5, and Kv4.3 channels were examined. In addition, Kv7.1 was coexpressed with the ancillary subunit KCNE1. The Kv11.1 channels were included as a positive control of modulation by extracellular potassium. Elevation of extracellular potassium decreases inactivation of Kv11.1 channels resulting in increased current amplitude at steady-state conditions (23). Representative current traces for these channel types recorded during a voltage-clamp to 40 mV in 1 (solid) and 10 (shaded) mM [K+]o are shown in Fig. 2 A. The data show that Kv1.5, Kv4.3, and Kv7.1/KCNE1 steady-state currents are not significantly affected by extracellular potassium. As expected, steady-state Kv11.1 currents are increased in 10 mM extracellular potassium. Statistical comparison of the current ratios (I10 mM/I1 mM) measured at 40 mV (Fig. 2 B) demonstrate that only Kv7.1 (0.57 ± 0.01, n = 19 (data from Fig. 1), p < 0.05) and Kv11.1 (2.4 ± 0.2, n = 11, p < 0.05) current ratios are significantly different from the predicted value (0.98 ± 0.002). The I10 mM/I1 mM ratios for Kv1.5, Kv4.3, and Kv7.1/KCNE1 were 0.95 ± 0.006 (n = 18), 0.93 ± 0.02 (n = 15), and 0.95 ± 0.01 (n = 14), respectively.
Figure 2.
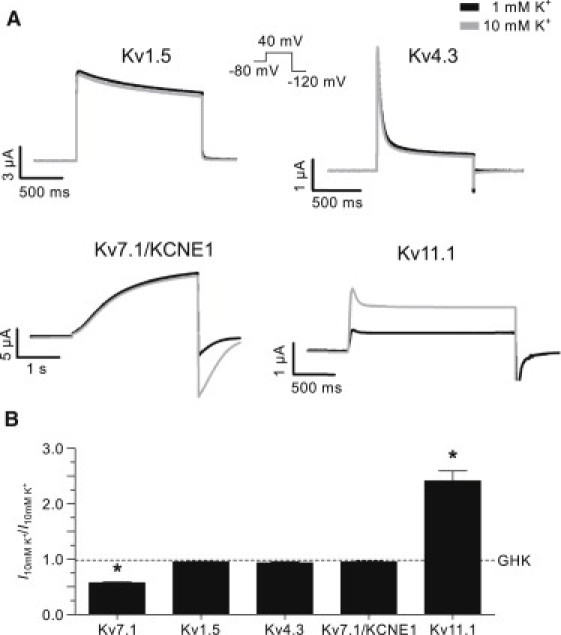
Response of other voltage-gated potassium channels to changes in extracellular potassium. (A) Representative currents from Kv1.5, Kv4.3, Kv7.1/KCNE1, and Kv11.1 channels recorded in 1 (solid) and 10 (shaded) mM [K+]o as indicated. The voltage-clamp protocol (inset) was used for all the recordings. Only the duration of the step to 40 mV was varied between channels. (B) Comparison of the ratio (I10 mM /I1 mM) of the current amplitude measured at 40 mV in 1 and 10 mM [K+]o to the theoretical ratio predicted by the GHK flux equation (dashed line). The asterisk (∗) indicates statistically significant difference compared to the predicted ratio (p < 0.05).
To further examine whether the effect of extracellular potassium was unique to Kv7.1 channels, all known Kv7 family members were studied in a separate experiment. None of these channels were significantly affected by extracellular potassium. Statistical comparison of the current ratios (I10 mM/I1 mM) measured at 40 mV demonstrate that only Kv7.1 (0.59 ± 0.03, n = 5, p < 0.05) current ratio is significantly different from the predicted value (0.98 ± 0.002). The current ratio for Kv7.2, Kv7.2/7.3, Kv7.4, and Kv7.5 channels were 1.0 ± 0.1 (n = 4), 0.95 ± 0.02 (n = 6), 0.94 ± 0.02 (n = 6), and 0.96 ± 0.05 (n = 6), respectively (representative and summary data are presented in Fig. S1 in the Supporting Material).
After observing that coexpression of Kv7.1 with KCNE1 attenuated the effect of extracellular potassium, KCNE2 and KCNE3 were also coexpressed with Kv7.1. For Kv7.1/KCNE2 channels, the inhibitory effect of extracellular potassium was preserved. Coexpression of Kv7.1 with KCNE3 attenuated the inhibition. The I10 mM/I1 mM ratio is significantly less than expected for Kv7.1/KCNE2 (0.72 ± 0.02; n = 6; p < 0.05) but not for Kv7.1/KCNE3 (0.91 ± 0.04; n = 11) (representative and summary data are presented in Fig. S2).
Concentration-dependence of potassium inhibition
To determine the IC50 of the potassium mediated inhibition of Kv7.1 steady-state current, the current at the end of a 3-s step to 40 mV was measured at concentrations of 1–50 mM extracellular potassium. Representative current traces obtained from one such experiment are shown in Fig. 3 A. The data show that Kv7.1 steady-state current is inhibited in a concentration-dependent fashion. In Fig. 3 B, the steady-state current, measured at 20-s intervals, is plotted as a function of time as the concentration of potassium was varied. The relative change in steady-state current (squares; normalized to current at 1 mM potassium) is summarized in Fig. 3 C. For comparison, the theoretical current ratios (calculated from the GHK flux equation) are also plotted (triangles). After correcting the experimental values for the expected theoretical reduction, the data was plotted as a function of the logarithm of the potassium concentration (Fig. 3 D). Fitting a standard Hill-function to the corrected data yielded an IC50 value of 6.0 ± 0.2 mM potassium (n = 6).
Figure 3.
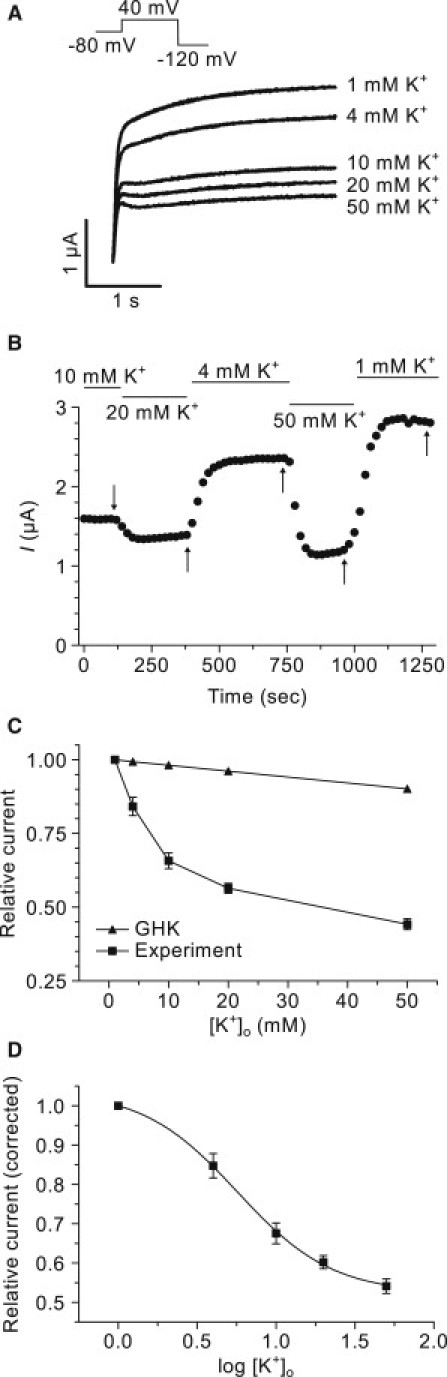
Concentration dependence of potassium inhibition on Kv7.1. (A) Representative currents recorded at 40 mV at various concentrations of extracellular potassium. (B) Steady-state current amplitudes measured at the end of the 3-s step to 40 mV. The current amplitude is plotted as a function of time (20-s intervals) as the potassium concentration was varied as indicated. The representative traces in panel A correspond to the points (indicated by the arrows). (C) Relative steady-state current amplitude as a function of potassium concentration as measured experimentally for Kv7.1 (squares) and theoretical values calculated from the GHK flux equation (triangles). The currents are normalized to the current measured in 1 mM [K+]o. (D) Concentration-response curve for extracellular potassium. The relative current was corrected for the theoretical reduction and plotted as a function of log [K+]o. A Hill-function was fitted to the data (solid line).
Effect of potassium on the inactivation properties of Kv7.1
Close examination of the shape of the current traces in Fig. 3 A, reveals that the apparent time-course of activation changes in a complex manner as the potassium concentration is increased. Notably, a small hook is appreciable in the beginning of the traces at higher concentrations of potassium. The presence of this hook may indicate that an increased number of channels are entering an inactivated state.
To investigate whether the apparent inhibition of steady-state currents could be explained by an effect of potassium on the inactivation properties of Kv7.1, two sets of experiments were performed.
First, a triple-pulse protocol was used to obtain a measure of the time constant of inactivation and to estimate the fraction of inactivated channels. With the triple-pulse protocol, channels are first activated by a prepulse to 40 mV. At the end of the 2-s prepulse, channels will primarily reside in open and inactivated states. By applying a short 20-ms pulse to −130 mV, a number of the inactivated channels will reenter the open state. Deactivation is minimized by the short pulse duration. Stepping back to depolarized potentials between −20 and 40 mV will cause a number of the channels in the open state to undergo reinactivation until equilibrium is again reached. During the third pulse, one can estimate the rate of inactivation by fitting an exponential function to the decrease in current. Furthermore, by using the fit to extrapolate back to the beginning of the third pulse, a measure of the fraction of channels recovered from the inactivated state during the 20 ms pulse to −130 mV can be obtained. In Fig. 4 A, a representative recording obtained with the triple-pulse protocol is shown. The currents on the left side are recorded at a concentration of 1 mM potassium. The enlargements on the right side show the onset of inactivation at 40 mV at three different concentrations of potassium. For comparison, the currents are normalized to the steady-state current before the hyperpolarizing step to −130 mV. Single-exponential functions described the current decay well. In the figure, the shaded lines represent the fitted functions. The time-course of reinactivation appears to be independent of potassium; however, the relative amplitudes are markedly increased at 10 and 50 mM [K+]o as compared to 1 mM [K+]o. The summary data show that the time constants of inactivation are independent of potassium concentration over a range of membrane potentials (Fig. 4 B, n = 7), whereas the fraction of channels recovered from inactivation is dependent on potassium concentration (Fig. 4 C, n = 7).
Figure 4.
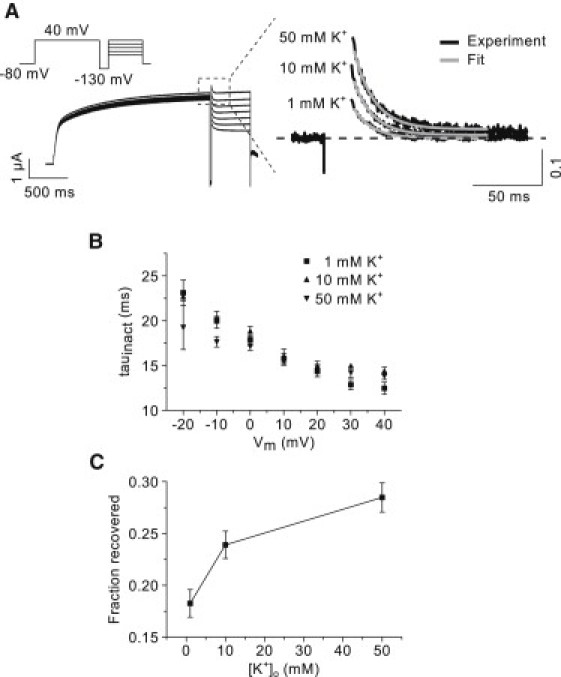
Inactivation of Kv7.1 measured by a triple-pulse protocol. (A) Representative currents recorded in 1 mM [K+]o using the triple-pulse protocol (shown on the left). The protocol is shown (inset). (Enlargement on the right) Three current traces measured at 40 mV at three different concentrations of potassium. The data are normalized to the current at the end of the prepulse to 40 mV (dashed line). (Shaded lines) Single-exponential functions fitted to the data. (B) Time constants of inactivation at three different concentrations of potassium. (C) Fraction of recovered channels as a function of [K+]o. See the text for details.
Second, a tail-envelope protocol was applied. The tail-envelope protocol consisted of depolarizing steps to 40 mV for various durations (100–1600 ms) before stepping to a potential of −120 mV. At −120 mV, only two processes occur to a significant extent for Kv7.1 channels: 1), recovery from inactivation; and 2), deactivation. By analyzing the tail-currents, estimates of the rate constants of these processes can be obtained as well as an estimate of the total fraction of inactivated channels at the end of the 40-mV prepulse. It is, however, nontrivial to estimate these parameters as the processes overlap significantly. In Fig. 5 A, representative tail currents obtained using the tail-envelope protocol are shown. A pronounced hook in the tail current indicates that a significant fraction of channels are recovering from an inactivated state. At 1 mM [K+]o, the hook is largely absent from the tail currents. In contrast, for both 10 and 50 mM [K+]o, the hook is visible already after a prepulse of 200–300 ms. As the prepulse duration increases, the hook becomes more pronounced. The enlarged tail current traces shown in Fig. 5 B compare the hook at 10 and 50 mM [K+]o. The hook is more pronounced at 50 than at 10 mM [K+]o. To estimate the time constants of recovery from inactivation and deactivation and to quantify the fraction of inactivated channels, a coupled ODE model was fit to the data. The tail current morphology suggested the presence of three distinct kinetic steps: 1), recovery from inactivation; 2), fast deactivation; and 3), slow deactivation.
Figure 5.
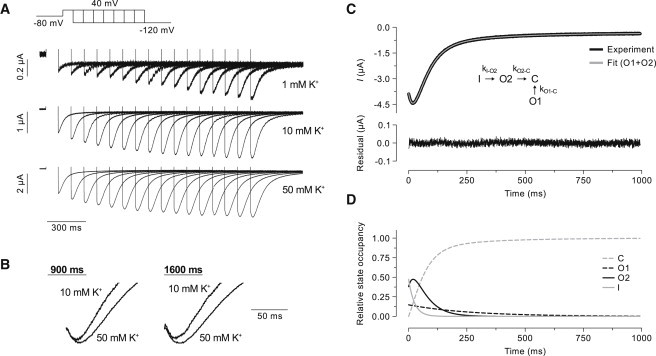
Tail-envelope protocol and ODE model fit. (A) Representative tail currents measured at −120 mV using a tail-envelope protocol at three different concentrations of potassium as indicated. The protocol is shown (Inset). The currents are scaled to visualize the development of the hook of the tail currents. (B) Enlarged tail currents at 10 and 50 mM [K+]o measured after pulse durations of 900 and 1600 ms as indicated. Currents are normalized to the instantaneous current at −120 mV to facilitate comparison of the tail hook. (C) Tail current recorded in 50 mM potassium after a 1600-ms step to 40 mV (solid). (Inset) The current was fitted to the coupled ODE model. See the text for more details on the model and fitting procedure. The fitted model is also shown (shaded). Below is shown the residuals of the fit. (D) The relative state occupancy of the closed (C), the two open (O1, O2), and the inactivated (I) states as predicted by the model fit.
Accordingly, the ODE model was designed with two open states (O1 and O2), one inactivated state (I) and a closed state (C). The transition from I to O2 represents the recovery from inactivation, the transition from O2 to C represents the fast deactivation, and the transition from O1 to C represents the slow deactivation. Only those transitions were allowed in the model. The kinetic scheme of the model and the corresponding transition rates are shown as an inset in Fig. 5 C. The model was only fit to the data obtained at 10 and 50 mM [K+]o because the low current amplitude at 1 mM [K+]o made a reliable fit difficult. In Fig. 5 C, a representative model fit is compared to the corresponding tail current measured after a 1600-ms prepulse in 50 mM [K+]o. The model fits the data very closely, as judged by the overlay and the residuals shown below. The corresponding channel state occupancies predicted by the model are shown in Fig. 5 D. In Fig. 6, summary data are shown for the fraction of inactivated channels (Fig. 6 A), the time constant of recovery (tauI-O2, Fig. 6 B), and the two time constants of deactivation (tauO1-C, Fig. 6 C and tauO2-C, Fig. 6 D) obtained from the model fits (n = 7). The ODE model analysis suggests that the fraction of inactivated channels is significantly greater in 10 than 50 mM [K+]o (Fig. 6 A).
Figure 6.
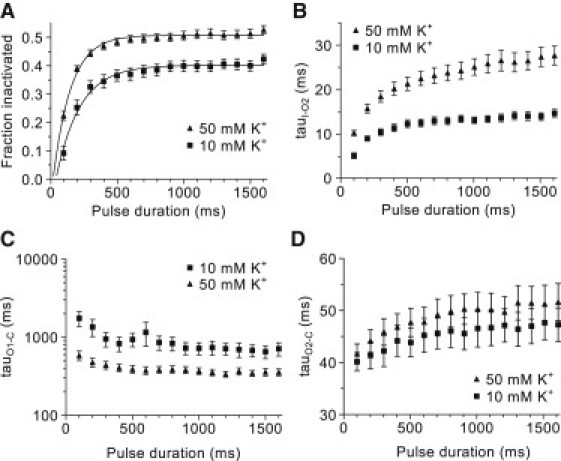
Summary of the ODE analysis of the tail-envelope protocol. (A) The fraction of inactivated channels plotted as a function of prepulse duration at 10 and 50 mM [K+]o. Also shown are single-exponential functions fitted to the fraction values (solid lines). (B–D) Time constants of recovery from inactivation (tauI-O2 (B)) and deactivation (tauO1-C (C) and tauO2-C (D)) plotted as a function of prepulse duration.
Furthermore, potassium slows the recovery from inactivation (Fig. 6 B). The time constant of recovery is time-dependent, possibly suggesting the presence of additional inactivated states. The time constant of slow deactivation (Fig. 6 C) is decreased by potassium, whereas the fast time constant of deactivation appears not to be affected by potassium (Fig. 6 D). Both the slow and the fast time constants of deactivation are not significantly time-dependent, suggesting that two open states are sufficient to model the deactivation process. The increase in the fraction of inactivated channels as a function of the prepulse duration could be described by a single-exponential function (solid lines in Fig. 6 A). From these fits we estimated the initial delay in onset of inactivation and the time constant of the increase in the fraction of inactivated channels. The delay in onset was not significantly different between 10 and 50 mM [K+]o (54 ± 11 ms and 31 ± 7 ms, respectively; n = 5). However, the time constant of the increase in the fraction of inactivated channels was slightly but significantly increased by potassium (135 ± 7 ms vs. 191 ± 22 ms in 10 and 50 mM [K+]o respectively; p < 0.05, n = 7).
The ODE model was also fitted to the tail currents recorded during the concentration-response experiment presented earlier. In Fig. 7 A, tail current traces from a representative experiment are shown together with the corresponding model fits for 1 and 50 mM [K+]o. The tail currents are recorded at −120 mV following a 3-s step to 40 mV (the protocol is shown in the inset in Fig. 3 A). In these experiments the tail currents in 1 mM [K+]o were large enough to obtain reliable fits. The fraction of inactivated channels was estimated from the fits and the summary data in Fig. 7 B clearly demonstrate that the fraction of inactivated channel increases as a function of extracellular potassium. Importantly, the fraction of inactivated channels and the corresponding relative current amplitudes were negatively correlated (r2 = 0.82, Fig. 7 C), suggesting that increased inactivation is the underlying mechanism for the reduction in steady-state current. The slope of the regression line was −0.45 ± 0.06 (p < 0.001).
Figure 7.
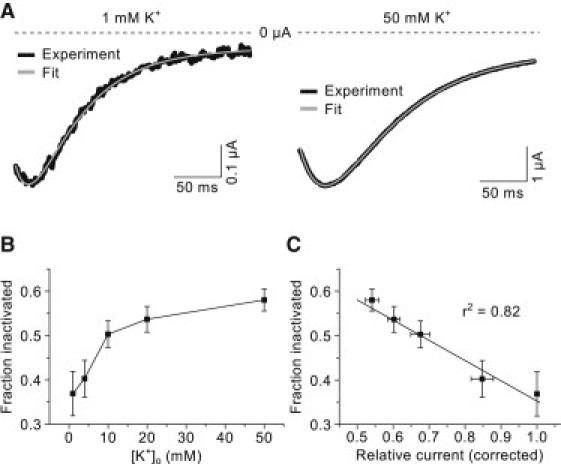
ODE analysis of tail currents at various concentrations of potassium. (A) Representative tail currents (solid) measured at −120 mV after a 3-s depolarizing step at 40 mV in 1 and 50 mM [K+]o as indicated. Only the initial 250 ms are shown. The voltage protocol is shown in the inset in Fig. 3A. (Dashed lines) Zero current level. The fits obtained with the ODE model are also shown (shaded). (B) Fraction of inactivated channels estimated from the model fits. (C) Correlation between fraction of inactivated channels and the corresponding relative current amplitudes (corrected for the theoretical reduction). The data are grouped by potassium concentration. The regression line (solid line) and the regression coefficient are also shown.
Discussion
In this study, we tested the hypothesis that Kv7.1 channels are modulated by extracellular potassium. We found that extracellular potassium inhibited Kv7.1 channels significantly more than expected based on theoretical calculations using the GHK flux equation. With the exception of Kv11.1 channels, which are known to be positively modulated by extracellular potassium (23), none of the other Kv channels we tested, including the other members of the Kv7 family, were modulated by extracellular potassium. Importantly, where Kv11.1 currents were increased by elevation of extracellular potassium, Kv7.1 currents were inhibited. Thus, inhibition by extracellular potassium seems to be a specific property of Kv7.1 channels. Our results suggest that potassium stabilizes an inactivated state of Kv7.1.
It has previously been shown that an inactivated state governed by fast kinetics is present in Kv7.1 channels (12). This fast inactivation process has been demonstrated to be independent of extracellular potassium concentration (12). Here, we confirm this finding in that the time constants of inactivation, obtained using the triple-pulse protocol, were independent of potassium concentration. However, we also show that the fraction of channels recovered during the short hyperpolarizing step in the protocol was increased at high concentrations of potassium. It is important to note that the triple-pulse protocol does not provide information about the number of channels residing in inactivated states. It only provides an estimate of the fraction of channels recovered during a certain period of time at a given voltage. Thus, a change in the fraction of channels recovered may be due to changes in the number of inactivated channels and/or in the kinetics of recovery from inactivation and deactivation. The analysis of tail currents from the tail-envelope protocol (see below for further discussion) shows that recovery from inactivation is slowed by potassium whereas the fast deactivation process is not affected. The slower recovery rate would result in fewer recovered channels at high concentrations of potassium in the triple-pulse protocol. Despite a slower rate of recovery, we observed that the fraction of recovered channels increased in high potassium—strongly suggesting that potassium increased the number of channels in an inactivated state.
Inactivation in Kv7.1 channels is visible as a hook in the tail currents (11,12). The hook represents channels recovering from inactivation and thus the fraction of inactivated channels can be estimated from tail current analysis. Here, we have used a coupled four-state ODE model fitted to Kv7.1 tail currents to estimate both the fraction of inactivated channels and the kinetics of recovery and deactivation. Previous studies have suggested the presence of at least two open states (11,12). The ODE model used to fit the tail currents in this study also incorporates two open states. In the model, channels may transition directly to the closed state from both open states. This choice was made for simplicity. However, in terms of the kinetics, the model is equivalent to a model with the two open states in sequence (O1-O2-C). Thus, the model we use is essentially similar to the gating models of Kv7.1 previously presented (11,12).
We used our model only to explain the behavior of the channel population during the deactivation process at −120 mV and to estimate the fraction of inactivated channels. The model is not intended to fully explain the gating behavior of Kv7.1. The tail current analysis demonstrates that the fraction of inactivated channels is significantly increased by potassium. The model was only fitted to the part of the tail currents that were not obstructed by the capacitive transients and it is therefore likely that the fit underestimates the fraction of inactivated channels especially at higher concentrations of extracellular potassium. Further, the time constant of recovery from inactivation was found to be significantly increased by potassium. The slower rate of recovery from inactivation likely indicates that potassium stabilizes an inactivated state in Kv7.1 and thereby shifts the equilibrium between open and inactivated channels resulting in fewer open channels at steady-state conditions. Moreover, the time-dependency of the time constant of recovery suggests the presence of more than one inactivated state.
There is further data to suggest that extracellular potassium may stabilize the gating of Kv7.1 channels.
First, barium has been shown to affect Kv7.1 channels via two discrete sites of action (24). One site is responsible for a direct pore blocking effect whereas the other modulates gating kinetics, including inhibition of channel inactivation. In high extracellular potassium only the pore blocking effect of barium is observed, suggesting that extracellular potassium stabilizes gating of Kv7.1 channels.
Second, in some pore mutants of Kv7.1 both a fast and a slow inactivation process can be detected (17). In these mutants, two distinct inactivated states have been shown to exist. One state is governed by slow kinetics, the other by fast kinetics. Importantly, the slow inactivation rate is significantly dependent on extracellular potassium. As extracellular potassium is increased, the slow inactivation rate also increases—indicating a stabilization of the inactivated state.
Based on these findings, we suggest that extracellular potassium modulates Kv7.1 channels by shifting the steady-state equilibrium between open and inactivated states toward the inactivated state. Furthermore, similar to the slow-inactivating pore mutants, it is possible that Kv7.1 channels may in fact be able to enter at least two distinct inactivated states. We cannot rule out a potential contributing effect of potassium on single channel conductance or trafficking/internalization effects, although the reversible nature of the inhibition suggests that trafficking/internalization is not a likely explanation. However, the negative correlation between the fraction of inactivated channels and relative current amplitude suggests that increased inactivation is the main mechanism underlying the inhibition of Kv7.1.
Importantly, we found that potassium inhibits Kv7.1 steady-state currents with an IC50 value of 6 mM, which is in the physiological range of potassium concentrations. This suggests that extracellular potassium may be an important regulator of Kv7.1 function under normal physiological conditions.
Inactivation of Kv7.1 is abolished upon coexpression with KCNE1 (1,2). Consistent with the hypothesis of a potassium-dependent inactivated state, Kv7.1/KCNE1 channels were not significantly affected by extracellular potassium in our experiments. Similarly, Kv7.1/KCNE3 channels were not affected by potassium. Interestingly, Kv7.1/KCNE2 channels were inhibited by potassium. Both KCNE1 and KCNE2 have been suggested to interact with residues in the pore region of Kv7.1 (25,26). It is likely that the site responsible for the modulation of Kv7.1 by extracellular potassium is located in the pore-region and that this site is altered in Kv7.1/KCNE1 and Kv7.1/KCNE3 channels but not in Kv7.1/KCNE2 channels. The presence of a potassium-sensitive site in the pore region would be consistent with the modulation of slow-inactivation observed in the pore-mutated Kv7.1 channels (17). The KCNE selective effect on potassium-mediated inhibition may help elucidate the molecular interactions between Kv7.1 and these subunits in future experiments.
In the heart, Kv7.1 channels are coexpressed with KCNE1, where they form the molecular basis for the repolarizing slow delayed rectifier current (IKs) (1,2). Consistent with our data, IKs has not been reported to be sensitive to extracellular potassium. Mutations in the genes encoding both Kv7.1 and KCNE1 leading to a reduction in IKs and corresponding prolongation of action potential duration have been associated with certain arrhythmic disorders, e.g., the long QT syndrome (27,28). It is possible that some disease-causing mutations in these genes may reduce the effect of KCNE1 on the modulation of Kv7.1 by extracellular potassium. Increased sensitivity of IKs to extracellular potassium may potentially lead to manifestation of the long QT syndrome. Future studies are needed to address the ability of potassium to inhibit Kv7.1/KCNE1 mutant channels associated with the long QT syndrome. Recently, studies have also suggested that Kv7.1 and KCNE2 interacts in the heart (29,30). Thus, under normal conditions, potassium may play a regulatory role in the heart by modulating Kv7.1/KCNE2 currents. Potentially, the selective effect of potassium on Kv7.1/KCNE2 versus Kv7.1/KCNE1 may be used to functionally discriminate between these channel complexes in the heart.
Kv7.1 channels are expressed in insulin-secreting β-cells in the pancreas (4). In the presence of KATP channel blockade, Kv7.1-specific blockers increased the secretion of insulin in INS-1 cells (4). Additionally, Kv7.1 has recently been associated with type 2 diabetes (31). Potentially, the modulation of Kv7.1 channels by extracellular potassium may be part of the mechanism underlying potassium-mediated insulin secretion.
Conclusion
In conclusion, we have found that Kv7.1 channels are significantly modulated by extracellular potassium. Specifically, Kv7.1 currents are inhibited by elevation of extracellular potassium. Extracellular potassium was found to increase the fraction of inactivated channels, indicating a stabilized inactivated state as the underlying mechanism. Importantly, inhibition by extracellular potassium was found to be unique to Kv7.1 compared to other voltage-gated potassium channels. Our results suggest that physiological changes in extracellular potassium may regulate the function of Kv7.1 channels. This may represent a novel regulatory mechanism of excitability in tissues expressing Kv7.1 channels.
Acknowledgments
This study was supported by a grant from the Niels Bohr Foundation (to A.P.L.) and the Danish National Research Foundation.
Supporting Material
References
- 1.Sanguinetti M.C., Curran M.E., Keating M.T. Coassembly of K(V)LQT1 and minK (IsK) proteins to form cardiac IKs potassium channel. Nature. 1996;384:80–83. doi: 10.1038/384080a0. [DOI] [PubMed] [Google Scholar]
- 2.Barhanin J., Lesage F., Romey G. K(V)LQT1 and IsK (minK) proteins associate to form the IKs cardiac potassium current. Nature. 1996;384:78–80. doi: 10.1038/384078a0. [DOI] [PubMed] [Google Scholar]
- 3.Zhong X.Z., Harhun M.I., Greenwood I.A. Participation of KCNQ (Kv7) potassium channels in myogenic control of cerebral arterial diameter. J. Physiol. 2010;588:3277–3293. doi: 10.1113/jphysiol.2010.192823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ullrich S., Su J., Lang F. Effects of IKs channel inhibitors in insulin-secreting INS-1 cells. Pflugers Arch. 2005;451:428–436. doi: 10.1007/s00424-005-1479-2. [DOI] [PubMed] [Google Scholar]
- 5.Dedek K., Waldegger S. Colocalization of KCNQ1/KCNE channel subunits in the mouse gastrointestinal tract. Pflugers Arch. 2001;442:896–902. doi: 10.1007/s004240100609. [DOI] [PubMed] [Google Scholar]
- 6.Grahammer F., Herling A.W., Warth R. The cardiac K+ channel KCNQ1 is essential for gastric acid secretion. Gastroenterology. 2001;120:1363–1371. doi: 10.1053/gast.2001.24053. [DOI] [PubMed] [Google Scholar]
- 7.Kunzelmann K., Hübner M., Greger R. Cloning and function of the rat colonic epithelial K+ channel KVLQT1. J. Membr. Biol. 2001;179:155–164. doi: 10.1007/s002320010045. [DOI] [PubMed] [Google Scholar]
- 8.Jespersen T., Grunnet M., Olesen S.P. The KCNQ1 potassium channel: from gene to physiological function. Physiology. (Bethesda) 2005;20:408–416. doi: 10.1152/physiol.00031.2005. [DOI] [PubMed] [Google Scholar]
- 9.Maljevic S., Wuttke T.V., Lerche H. KV7 channelopathies. Pflugers Arch. 2010;460:277–288. doi: 10.1007/s00424-010-0831-3. [DOI] [PubMed] [Google Scholar]
- 10.Silva J., Rudy Y. Subunit interaction determines IKs participation in cardiac repolarization and repolarization reserve. Circulation. 2005;112:1384–1391. doi: 10.1161/CIRCULATIONAHA.105.543306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pusch M., Magrassi R., Conti F. Activation and inactivation of homomeric KvLQT1 potassium channels. Biophys. J. 1998;75:785–792. doi: 10.1016/S0006-3495(98)77568-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tristani-Firouzi M., Sanguinetti M.C. Voltage-dependent inactivation of the human K+ channel KvLQT1 is eliminated by association with minimal K+ channel (minK) subunits. J. Physiol. 1998;510:37–45. doi: 10.1111/j.1469-7793.1998.037bz.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hoshi T., Zagotta W.N., Aldrich R.W. Two types of inactivation in Shaker K+ channels: effects of alterations in the carboxy-terminal region. Neuron. 1991;7:547–556. doi: 10.1016/0896-6273(91)90367-9. [DOI] [PubMed] [Google Scholar]
- 14.López-Barneo J., Hoshi T., Aldrich R.W. Effects of external cations and mutations in the pore region on C-type inactivation of Shaker potassium channels. Receptors Channels. 1993;1:61–71. [PubMed] [Google Scholar]
- 15.Baukrowitz T., Yellen G. Modulation of K+ current by frequency and external [K+]: a tale of two inactivation mechanisms. Neuron. 1995;15:951–960. doi: 10.1016/0896-6273(95)90185-x. [DOI] [PubMed] [Google Scholar]
- 16.Seebohm G., Scherer C.R., Lerche C. Identification of specific pore residues mediating KCNQ1 inactivation. A novel mechanism for long QT syndrome. J. Biol. Chem. 2001;276:13600–13605. doi: 10.1074/jbc.M008373200. [DOI] [PubMed] [Google Scholar]
- 17.Gibor G., Yakubovich D., Attali B. An inactivation gate in the selectivity filter of KCNQ1 potassium channels. Biophys. J. 2007;93:4159–4172. doi: 10.1529/biophysj.107.107987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jespersen T., Grunnet M., Olesen S.P. Dual-function vector for protein expression in both mammalian cells and Xenopus laevis oocytes. Biotechniques. 2002;32:536–538. doi: 10.2144/02323st05. 540. [DOI] [PubMed] [Google Scholar]
- 19.Grunnet M., Jensen B.S., Klaerke D.A. Apamin interacts with all subtypes of cloned small-conductance Ca2+-activated K+ channels. Pflugers Arch. 2001;441:544–550. doi: 10.1007/s004240000447. [DOI] [PubMed] [Google Scholar]
- 20.Pinheiro J.C., Bates D., Sarkar D., the R Core team NLME: linear and nonlinear mixed effects models. R Package Ver. 2009;3:1–90. [Google Scholar]
- 21.Hille B. Sinauer Associates; Sunderland, MA: 2001. Ion Channels of Excitable Membranes. [Google Scholar]
- 22.Weber W. Ion currents of Xenopus laevis oocytes: state of the art. Biochim. Biophys. Acta. 1999;1421:213–233. doi: 10.1016/s0005-2736(99)00135-2. [DOI] [PubMed] [Google Scholar]
- 23.Sanguinetti M.C., Jiang C., Keating M.T. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell. 1995;81:299–307. doi: 10.1016/0092-8674(95)90340-2. [DOI] [PubMed] [Google Scholar]
- 24.Gibor G., Yakubovich D., Attali B. External barium affects the gating of KCNQ1 potassium channels and produces a pore block via two discrete sites. J. Gen. Physiol. 2004;124:83–102. doi: 10.1085/jgp.200409068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Panaghie G., Tai K.K., Abbott G.W. Interaction of KCNE subunits with the KCNQ1 K+ channel pore. J. Physiol. 2006;570:455–467. doi: 10.1113/jphysiol.2005.100644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu X.S., Zhang M., Tseng G.N. Probing the interaction between KCNE2 and KCNQ1 in their transmembrane regions. J. Membr. Biol. 2007;216:117–127. doi: 10.1007/s00232-007-9047-7. [DOI] [PubMed] [Google Scholar]
- 27.Wang Q., Curran M.E., Keating M.T. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat. Genet. 1996;12:17–23. doi: 10.1038/ng0196-17. [DOI] [PubMed] [Google Scholar]
- 28.Splawski I., Tristani-Firouzi M., Keating M.T. Mutations in the hminK gene cause long QT syndrome and suppress IKs function. Nat. Genet. 1997;17:338–340. doi: 10.1038/ng1197-338. [DOI] [PubMed] [Google Scholar]
- 29.Lundby A., Ravn L.S., Schmitt N. KCNQ1 mutation Q147R is associated with atrial fibrillation and prolonged QT interval. Heart Rhythm. 2007;4:1532–1541. doi: 10.1016/j.hrthm.2007.07.022. [DOI] [PubMed] [Google Scholar]
- 30.Jiang M., Xu X., Tseng G.N. Dynamic partnership between KCNQ1 and KCNE1 and influence on cardiac IKs current amplitude by KCNE2. J. Biol. Chem. 2009;284:16452–16462. doi: 10.1074/jbc.M808262200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yasuda K., Miyake K., Kasuga M. Variants in KCNQ1 are associated with susceptibility to type 2 diabetes mellitus. Nat. Genet. 2008;40:1092–1097. doi: 10.1038/ng.207. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.