

Abstract
Objective
The examination of individuals who carry fully penetrant genetic alterations that result in familial Alzheimer’s disease (FAD) provides a unique model for studying the early presymptomatic disease stages. In AD, deficits in episodic and associative memory have been linked to structural and functional changes within the hippocampal system. This study used functional MRI (fMRI) to examine hippocampal function in a group of healthy, young, cognitively-intact presymptomatic individuals (average age 33.7 years) who carry the E280A presenilin-1 (PS1) genetic mutation for FAD. These PS1 subjects will go on to develop the first symptoms of the disease around the age of 45 years. Our objective was to examine hippocampal function years before the onset of clinical symptoms.
Methods
Twenty carriers of the Alzheimer’s-associated E280A PS1 mutation and 19 PS1-negative control subjects participated. Both groups were matched for age, sex, education level, and neuropsychological test performance. All participants performed a face-name associative encoding task while in a Philips 1.5T fMRI scanner. Analysis focused on the hippocampal system.
Results
Despite identical behavioral performance, presymptomatic PS1 mutation carriers exhibited increased activation of the right anterior hippocampus during encoding of novel face-name associations compared to matched controls.
Interpretation
Our results demonstrate that functional changes within the hippocampal memory system occur years before cognitive decline in FAD. These presymptomatic changes in hippocampal physiology in FAD suggest that hippocampal fMRI patterns during associative encoding may also provide a preclinical biomarker in sporadic AD.
As many as 5.3 million Americans are presently living with Alzheimer’s disease (AD).1 This progressive and fatal neurodegenerative disorder is the most common form of dementia, yet a cure remains elusive. Genetics research has identified 3 genetic mutations with a dominant inheritance structure linked to early-onset familial Alzheimer’s disease (FAD): the amyloid precursor protein mutation (APP), the presenilin-1 mutation (PS1), and the presenilin-2 mutation (PS2).2,3 The present study focuses specifically on a large Colombian family cohort of E280A PS1 mutation carriers that were first identified in 19954 and have been well characterized in terms of their genetic, neuropsychological, and neurophysiological profile.5-7 These PS1 mutation carriers develop the first signs and symptoms of AD at a younger age (45 years old), but in other respects their symptoms are indistinguishable from those of individuals with sporadic forms of the disease.5,8-10
Deterioration of episodic memory is one of the first cognitive symptoms of AD, and has been associated with functional and structural degeneration of the medial temporal lobe regions (MTL).11-13 In particular, the hippocampal formation and entorhinal cortex have been shown to exhibit pathological changes in early phases of AD, even before clinical symptoms are evident.14 There is current consensus that neuroimaging tools such as positron emission tomography (PET), structural MRI (sMRI), and functional MRI (fMRI) have the potential to identify subtle pathologic changes earlier in the disease’s progression and could be used as potential biomarkers of AD.15-17
A number of fMRI studies have examined brain activity in sporadic forms of AD and mild cognitive impairment (MCI) patients and have demonstrated functional changes during performance of episodic and associative memory tasks.15,16,18,19 In general, AD patients show decreased activation in MTL regions and increased activation in neocortical regions, including frontal cortices. Findings from studies comparing MCI patients to controls are more variable, with some reporting decreases in MTL activity during memory encoding,20 others reporting no differences during encoding and decreased activation in MTL regions during retrieval,21 and still others reporting increases in MTL activity during encoding of face-name pairs22 and objects23 in MCI patients. The functional variability within the MCI studies may reflect greater variability in the subject population being studied, both in terms of cognitive variability as well as variability in disease stage. Studies of individuals at genetic risk for AD are more consistent, with studies reporting increases in MTL activity during encoding of pictures24 and face-profession pairs.25 To our knowledge, Mondadori et al.’s study25 is the only fMRI study to examine memory encoding in presenilin mutation carriers, but the sample size was limited to 2 carriers, and both subjects were already cognitively symptomatic.
The present study was carried out in a cohort of Colombian families with the E-280A mutation in the PS1 gene. These families represent one of the world’s largest populations with a genetic form of AD caused by a single mutation.26 Our aim was to determine whether different brain activation patterns are observable during encoding of novel associations in individuals with the PS1 mutation years before the onset of the disease and in the absence of cognitive impairment. We hypothesized, based on the studies in MCI patients and earlier studies of subjects at risk for developing AD, that presymptomatic carriers would demonstrate hyperactivation within the hippocampal system compared to matched controls.
Patients and Methods
Participants
Subjects were recruited from the Familial Colombian Alzheimer’s disease population studied at the University of Antioquia, Medellin. A total of 39 young subjects were included in this study, 20 subjects were carriers of the E280A PS1 mutation (mean age ± SD = 33.70 ± 6.01 years) and 19 were PS1 mutation–negative and served as controls (mean age = 34.47 ± 6.46 years). Control participants were relatives of the PS1 carriers. Participants had a minimum of 5 years of education and lived in the city or suburbs of Medellin (where the scanner facility was located). Subject groups were matched for age, sex, education, and neuropsychological assessment performance (Table 1). Neuropsychological assessment consisted of the Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) battery, which has been adapted to this Colombian population.27 No evidence of cognitive impairments was found in all 39 subjects, as reported by their most recent neuropsychological assessment, which was done within 6 months prior to the time of the scanning session. All subjects were screened for MRI compatibility. Vision was normal or corrected-to-normal.
TABLE 1. Subject Demographic Information and CERAD Neuropsychological Test Battery.
| Controls (n = 19)a | PS1 Carriers (n = 20)a | t | p | |
|---|---|---|---|---|
| Gender | 16 females | 14 females | ||
| Age (yr) | 34.47 (6.46) | 33.70 (6.01) | 0.38 | 0.70 |
| Range | 24–49 | 25–44 | ||
| Education (yr) | 10.80 (3.28) | 10.70 (2.40) | −0.01 | 0.98 |
| Range | 5–16 | 6–16 | ||
| CERAD tests | ||||
| MMSE/30 | 29.5 (1.0) | 29.7 (0.5) | −0.65 | 0.52 |
| Verbal fluency | 19.26 (5.23) | 18.25 (4.37) | 0.65 | 0.51 |
| Naming/15 | 13.47 (1.50) | 13.80 (1.10) | −0.76 | 0.44 |
| Memory words | ||||
| Reading | 10 (0.00) | 10 (0.00) | ||
| Total correct/30 | 19.21 (3.53) | 19.85 (2.88) | −0.61 | 0.54 |
| Total intrusions | 1.68 (2.45) | 0.90 (1.02) | 1.29 | 0.20 |
| Recall of words | ||||
| Total correct/10 | 7.05 (1.47) | 6.75 (1.47) | 0.61 | 0.54 |
| Total intrusions | 0.32 (0.67) | 0.25 (0.44) | 0.35 | 0.72 |
| Recognition of words | ||||
| Correct “yes”/10 | 9.53 (0.84) | 9.80 (0.41) | −1.28 | 0.21 |
| Correct “no”/10 | 9.95 (0.22) | 10 (0.00) | −1.00 | 0.33 |
| Constructional praxis/11 | 9.84 (0.89) | 9.95 (0.99) | −0.35 | 0.72 |
| Recall of drawings/11 | 9.11 (1.96) | 8.40 (2.25) | 1.04 | 0.30 |
Values denoted mean (±standard deviation), except for ranges.
CERAD = Consortium to Establish a Registry for Alzheimer’s Disease; MMSE = Mini-Mental State Examination; PS1 = presenilin-1.
The study was approved by the Institutional Review Board (IRB) Committees of the University of Antioquia and of Boston University. All subjects gave signed informed consent before participating. Researchers were blind to the genetic status of the participants during data collection.
Study Design
We used a face-name paired-associate learning task based on the task developed by Sperling et al.15,28 We adapted the task for a Hispanic population by only including Hispanic faces and names. We used an fMRI mixed-design format with 3 conditions: novel face-name pairs, repeated face-name pairs, and fixation. Subjects performed a recognition test on a laptop computer after the scanning session was completed.
Face-Name Paired-Associate Learning Task
Subjects underwent fMRI scanning while viewing a face presented against a black background with a fictional name printed underneath. Each face-name pair was presented for 4.5 sec, followed by a brief variable duration white central fixation crosshair on a black background. A total of 86 (84 novel and 2 repeated) face-name pairs were presented during the experiment. The fMRI task consisted of 3 conditions that were alternated in blocks during each scanning run. (1) Novel face-name pairs: 7 novel face-name pairs were shown. (2) Repeated face-name pairs: 2 face-name pairs, 1 male and 1 female, were shown repeatedly. In order to familiarize the subject with the repeated face-name pairs, the 2 face-names were first shown during the practice run. (3) Fixation: a white cross in the center of the visual field on a black background was shown. Subjects were instructed to indicate whether they thought the name “fit” with the face and to remember the face-name pairs for later testing. Responses were collected with a response button-box that subjects used with their dominant hand. Each of 6 scanning runs consisted of the following blocks: fixation (10 sec), novel (40 sec), fixation (25 sec), repeated (40 sec), fixation (25 sec), novel (40 sec), fixation (25 sec), repeated (40 sec), and fixation (10 sec).
Postscan Recognition Test
Following the scanning session, subjects performed a memory recognition test in which the previously viewed face-name pairs were presented along with a set of 86 new face-name pairs. Subjects indicated by button-box whether each face-name pair was either old or new.
MRI Data Acquisition
Anatomical and functional data were acquired on a 1.5 Philips Achieva MR scanner at the Instituto de Alta Tecnología Médica (IATM) in Medellin, Colombia. For each subject, 2 high-resolution T1-weighted structural MRI scans were collected to examine cortical neuroanatomy (three-dimensional fast field echo [3D-FFE]; field of view = 256 × 256; voxel size = 1.0 × 1.0 × 1.0 mm; 176 slices). Automatic shimming procedures were performed. We ran 6 functional T2*-weighted gradient echo, echo-planar blood oxygenation level-dependent (BOLD) scans (repetition time [TR] = 2 sec; echo time = 40 msec; flip angle = 90 degrees; field of view = 200 × 200; 24 slices] and acquired 126 images during each scan (3.125 × 3.125 mm inplane resolution; 5mm slice thickness; 1mm skip between slices; resolution 64 × 64). Slices were aligned parallel to the anterior commissure (AC)– posterior commissure (PC)–.
Data Analysis
POSTSCAN BEHAVIORAL ANALYSIS
Within-group recognition accuracy was determined using a discrimination index Pr (percent hits – percent false alarms). Independent sample t-tests were implemented to compare the behavioral performance between groups (controls, PS1 carriers).
FMRI PREPROCESSING
fMRI data were preprocessed with SPM8 software (Wellcome Department of Cognitive Neurology, London, UK). Structural and functional image data were passed through a series of preprocessing steps: (1) Functional images were reoriented such that the origin (ie, coordinate xyz =[0 0 0]) was at the AC. (2) BOLD images were realigned to the first image within a run using INRIAlign toolbox29 to correct for variance due to susceptibility-by-movement interactions. (3) The high-resolution structural images were coregistered to the mean BOLD image created during the motion correction step. (4) The high-resolution structural images were segmented into white matter and gray matter images. A bias-corrected structural image was also created at this step using the default tissue probability maps as priors. (5) A group template was generated using the Diffeomorphic Anatomical Registration Through Exponentiated Lie Algebra (DARTEL) tool.30 (6) The bias-corrected structural images were spatially normalized to this template. (7) The coregistered BOLD images were spatially normalized into standard Montreal Neurological Institute (MNI) space using the DARTEL template and resampled to 3mm3 isotropic voxels. (8) Finally, all BOLD images were spatially smoothed using a 6mm3 full-width at half-maximum Gaussian filter.
FMRI ANALYSIS
The novel and repeated blocks were modeled using a boxcar function with a length of 40 sec convolved with the canonical hemodynamic response function. The movement parameters from the realignment procedure were additionally added as covariates to account for residual movement-related spurious activation. To assess the activation during learning of face-name pairs, linear contrasts of the novel stimuli blocks relative to repeated stimuli blocks and visual fixation were created for each subject.
A PRIORI REGION OF INTEREST ANALYSIS: RIGHT HIPPOCAMPUS AND LEFT HIPPOCAMPUS
Region of interest (ROI) analysis was performed on a priori defined ROIs. The coordinates for the ROIs were selected from a previously published study in young and elderly controls and subjects with mild AD using a face-name paired-associate task.15 Coordinates were taken from the within-group activations corresponding to the novel vs repeated contrast for young controls.15 Coordinates were converted from the original Talairach coordinates [left: −21, −27, −11; right: 24, −30, −11] to MNI coordinates −21, −27, −15; right: 24, −30, −15].
Parameter estimates (beta weights) were extracted separately for each ROI from a sphere with a radius of 5mm. Since the hippocampus is a small anatomical structure, a radius of 5mm (which equals 56 voxels at 3mm isotropic voxel resampling) allows us to confidently state that the ROI is within the hippocampus, and is consistent with the use of 5-mm ROIs in imaging studies of medial temporal structures.31,32 The spheres were centered at MNI coordinates in the left hippocampus [−21, −27, −15] and in the right hippocampus [24, −30, −15]. Within each subject only the voxels significantly active within the ROI at the threshold of 0.005 were included in the extractions. Parameter estimates were extracted using the SPM8 Volume toolbox. Parameter estimates were then averaged across subjects for each ROI. An omnibus mixed-factor analysis of variance (ANOVA) was constructed to estimate all main effects of condition (novel and repeated face-name pairs) and region (right and left hippocampus) on parameter estimates (beta weights), adjusting for age and years of education. Follow-up ANOVAs were performed within the right and left hippocampal ROIs to determine if laterality effects were present. Statistical analyses were performed using statistical software (SPSS version 16.0; SPSS Inc, Chicago, IL).
SPMs were generated using a combined right and left hippocampal Wake Forest University Pick Atlas33 anatomical mask. These maps were generated to illustrate where the between-group differences were localized to in the hippocampus.
WHOLE-BRAIN EXPLORATORY ANALYSIS
Both within-group and between-group comparisons were examined. Within-group and between-group SPMs were thresholded at a p < 0.005 (extent threshold = 20 voxels) uncorrected for the main linear contrasts of interest: novel face-name pairs vs. repeated face-name pairs and novel face-name pairs vs visual fixation. These brain activation maps were generated for exploratory purposes in order to identify peak regions of activation for future fMRI investigations of PS1 mutation carriers.
STRUCTURAL MRI ANALYSIS
The entire structure of the hippocampus was manually defined in structural T1 images after spatial normalization by a study investigator (R.N.), blind to the genetic status, using the ITK-snap software package34 (http://www.itksnap.org) and methods described in a previous study.35 Additionally, the Duvernoy atlas was used as a reference for anatomical details.36 The hippocampal tail was outlined from the first appearance of ovoid mass of gray matter inferiomedial to the trigone of the lateral ventricle, and boundary landmarks included the fasciolar gyrus and the crus of the fornix. The hippocampal head was evident by the emergence of the uncal recess in the superomedial region of the hippocampus. Important boundaries for identifying the hippocampal head included the uncal recess of the inferior horn of the lateral horn and the alveus.
To examine possible confounding effects of the right and left hippocampal volumes on their respective right and left hippocampal activation during both novel and repeated conditions (as measured by the beta weights), we implemented two separate partial correlations with the hippocampal activation as the dependent variable and the hippocampal volume as the independent variable. We controlled for age and years of education by conducting partial correlations and including them in the model. The partial correlations were also corrected for multiple comparisons using the Bonferroni method. All statistical analyses were performed using statistical software (SPSS version 16.0).
Results
Performance Data
The recognition accuracy on the discrimination index Pr was 0.43 (SD 0.10) for the control group and 0.42 (SD 0.11) for the PS1 carriers. The overall average of median reaction time for correct responses was 1376 ms (SD 238 ms) for the controls and 1468 ms (SD 256 ms) for the PS1 carriers. There were no significant differences in postscan recognition accuracy (t(37)= 0.53, p= 0.59) or reaction time (t(37)= −1.15, p= 0.25). Table 2 shows the behavioral data for both groups.
TABLE 2. Behavioral Performance for Controls and PS1 Mutation Carriers.
| Controlsa | PS1 Mutation Carriersa |
p | |
|---|---|---|---|
| Accuracy | |||
| Percent hits | 0.64 (0.08) | 0.68 (0.13) | 0.29 |
| Percent misses | 0.36 (0.08) | 0.32 (0.13) | 0.29 |
| Percent correct rejections | 0.79 (0.07) | 0.74 (0.14) | 0.12 |
| Percent false alarms | 0.21 (0.07) | 0.26 (0.14) | 0.12 |
| Index Pr | 0.43 (0.10) | 0.42 (0.11) | 0.59 |
| Reaction times (ms) | |||
| Old items | 1334 (248) | 1403 (256) | 0.40 |
| New items | 1406 (311) | 1620 (417) | 0.08 |
| Old and new items | 1376 (238) | 1468 (256) | 0.25 |
Values are mean (±standard deviation).
Index Pr = percent hits – percent false alarms; PS1 = presenilin-1.
A priori ROIs: Right Hippocampus and Left Hippocampus
For the novel condition, the mean parameter estimates were 0.19 (SD 0.33) for the right hippocampus and 0.06 (SD 0.76) for the left hippocampus for the controls, and 0.45 (SD 0.34) for the right hippocampus and 0.47 (SD 0.86) for the left hippocampus for PS1 carriers. For the repeated condition, the mean parameter estimates were −0.009 (SD 0.41) for the right hippocampus and 0.003 (SD 0.61) for the left hippocampus for the controls, and −0.008 (SD 0.36) for the right hippocampus and 0.18 (SD 0.67) for the left hippocampus for PS1 carriers. The omnibus mixed-factor ANOVA, which included group (PS1 carriers, controls), region (right hippocampus and left hippocampus), and condition (novel and repeated), as well as age and education covariates, revealed a significant interaction between group and condition [F(1, 35)= 5.230, p= 0.028]. This interaction indicates that although both controls and PS1 mutation carriers demonstrated greater hippocampal activity for novel face-name pairs than for repeated face-name pairs, this activity was significantly greater in the PS1 mutation carriers. Follow-up analyses within the right and left hippocampal ROIs revealed, within the right hippocampus, a significant main effect of condition [F(1,37) 33.441, p= 0.001] and a group by condition interaction [F(1,37)= 5.120, p= 0.030], such that activity in the right hippocampus was significantly greater in the carriers than in the controls during the novel condition but not during the repeated condition (Fig 1). Activity in the left hippocampus during both novel and repeated face-name conditions was not significantly different between groups.
FIGURE 1.
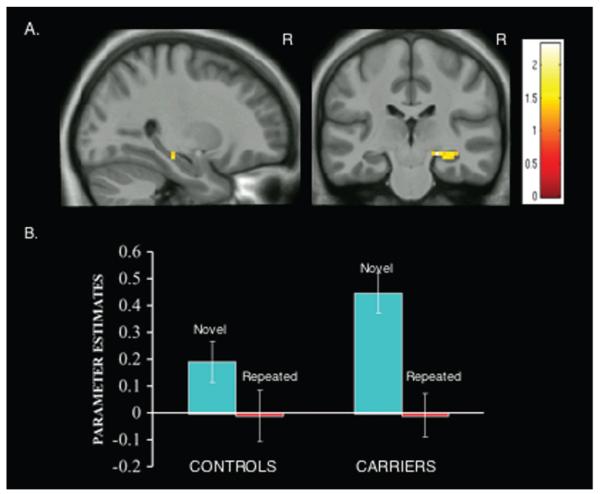
Carriers show greater right anterior hippocampus activity during encoding of face-name associations. (A) Statistical Parametric Maps (SPMs) for the comparison PS1 mutation carriers vs controls for the contrast novel face-name pairs vs repeated face-name pairs are displayed on anatomical images derived from an average obtained from the normalized structural images of all subjects using an anatomical mask of the combined right and left hippocampus with a statistical threshold of p < 0.005 uncorrected (extent threshold 5 voxels). PS1 mutation carriers (n = 20) demonstrate increased intensity changes in the right hippocampus when compared to controls (n = 19). MNI coordinates for peak activated voxel: [x,y,z][27, −24, −12]. Color bar represents t values for all activated voxels within the anatomical mask. (B) Parameter estimates extracted as a sphere with a radius of 5mm and centered at the coordinate [24, −30, −15; MNI] from the right hippocampus for the novel face pairs and the repeated face-name pairs for controls and PS1 mutation carriers. For the novel condition, the mean parameter estimates were 0.19 (SD 0.33) for the controls, and 0.45 (SD 0.34) for PS1 carriers. For the repeated condition, the mean parameter estimates were −0.009 (SD 0.41) for the controls, and −0.008 (SD 0.36) for PS1 carriers. The data in A and B suggest hyperactivation in the right hippocampus as an early functional change during associative memory encoding in presymptomatic FAD. MNI 5 Montreal Neurological Institute.
Whole-Brain Exploratory Analysis
The within-group analysis showed that for both groups there is an increase in the fMRI signal response of association areas in the frontal and parietal lobes and in the hippocampal formation to novel vs repeated stimuli (Table 3). The between-group analysis revealed differences in distinct regions of the superior parietal lobule, middle frontal gyrus, inferior frontal gyrus, parahippocampal gyrus, and hippocampal formation (Table 4). Controls showed greater activity in frontal and parietal regions whereas carriers exhibited greater activity in the right cingulate gyrus and the right anterior hippocampus (Fig 2; see Table 4). When examining novel face-name pairs vs fixation, both PS1 mutation carriers and controls showed activation in bilateral parietal regions, bilateral dorsolateral prefrontal cortices (DLPFC), and bilateral MTL regions (see Table 3). Between-group comparisons for this contrast revealed significant activations in the right cingulate gyrus, right hippocampal formation, right caudate nucleus, left precuneus, and right temporal gyrus for the PS1 carriers and significant activations in the superior parietal and bilateral DLPFC for the controls.
TABLE 3. Within-Group Activations, Whole-Brain Exploratory Analysis.
| Contrast | Region | MNI Coordinates | Z score | p (uncorrected) | ||
|---|---|---|---|---|---|---|
| x | y | z | ||||
| Controls | ||||||
| Novel pairs vs fixation | Left lingual gyrus | −21 | −90 | −18 | 6.23 | 0.0001 |
| Left superior medial gyrus | −3 | 21 | 42 | 5.57 | 0.0001 | |
| Right inferior frontal gyrus | 54 | 18 | 33 | 5.09 | 0.0001 | |
| Right hippocampal formation | 15 | −15 | −15 | 4.05 | 0.0001 | |
| Right middle frontal gyrus | 39 | 54 | 15 | 3.62 | 0.0001 | |
| Right cerebellum | 18 | −66 | −45 | 3.62 | 0.0001 | |
| Right middle frontal gyrus | 39 | 54 | 15 | 3.62 | 0.0001 | |
| Left middle temporal gyrus | −45 | −42 | 0 | 3.49 | 0.0001 | |
| Right post-central gyrus | 36 | −30 | 42 | 2.97 | 0.001 | |
| Novel vs repeated pairs | Left inferior frontal gyrus | −39 | 21 | 21 | 5.65 | 0.0001 |
| Left fusiform gyrus | −33 | −48 | −21 | 5.50 | 0.0001 | |
| Left superior medial gyrus | −6 | 21 | 42 | 4.87 | 0.0001 | |
| Left middle temporal gyrus | −60 | −30 | 6 | 4.35 | 0.0001 | |
| Left superior parietal lobule | −27 | −69 | 36 | 4.02 | 0.0001 | |
| Right parahippocampal gyrus | 24 | −3 | −24 | 3.70 | 0.0001 | |
| Left hippocampal formation | −27 | −30 | −9 | 3.76 | 0.0001 | |
| Right putamen | 21 | 6 | 6 | 3.74 | 0.0001 | |
| Right middle temporal gyrus | 57 | −39 | −9 | 3.27 | 0.001 | |
| Right superior temporal gyrus | 36 | 24 | −33 | 2.77 | 0.001 | |
| Carriers | ||||||
| Novel pairs vs fixation | Right calcarine gyrus | 18 | −96 | −3 | 6.72 | 0.0001 |
| Right middle frontal gyrus | 42 | 9 | 36 | 5.60 | 0.0001 | |
| Right superior medial gyrus | 6 | 30 | 42 | 5.23 | 0.0001 | |
| Left inferior frontal gyrus | −51 | 12 | 9 | 5.21 | 0.0001 | |
| Right inferior parietal lobule | 48 | −51 | 48 | 5.13 | 0.0001 | |
| Right middle frontal gyrus | 30 | 51 | 3 | 3.85 | 0.0001 | |
| Right superior temporal gyrus | 39 | −30 | −3 | 3.76 | 0.0001 | |
| Left middle orbital gyrus | −33 | 51 | −3 | 3.45 | 0.0001 | |
| Novel vs repeated pairs | Left inferior frontal gyrus | −45 | 21 | 21 | 4.42 | 0.0001 |
| Right parahippocampal gyrus | 21 | −27 | −15 | 4.13 | 0.0001 | |
| Left angular gyrus | −42 | −63 | 42 | 3.99 | 0.0001 | |
| Right angular gyrus | 45 | −60 | 48 | 3.67 | 0.0001 | |
| Left hippocampal formation | −15 | −6 | −21 | 3.50 | 0.0001 | |
| Left fusiform gyrus | −36 | −9 | −27 | 3.28 | 0.0001 | |
| Right hippocampal formation | 27 | −18 | −18 | 3.25 | 0.001 | |
MNI = Montreal Neurological Institute.
TABLE 4. Between-Group Differences, Whole-Brain Exploratory Analysis.
| Contrast | Region | MNI Coordinates | Z score | p (uncorrected) | ||
|---|---|---|---|---|---|---|
| x | y | z | ||||
| Controls > carriers | ||||||
| Novel pairs vs fixation |
Right inferior occipital gyrus | 36 | −81 | −15 | 4.33 | 0.0001 |
| Left inferior frontal gyrus | −33 | 36 | 15 | 4.24 | 0.0001 | |
| Left caudate nucleus | −6 | 6 | −3 | 4.19 | 0.0001 | |
| Left middle temporal gyrus | −57 | −60 | 18 | 3.69 | 0.0001 | |
| Right temporal pole | 48 | 21 | −21 | 3.69 | 0.0001 | |
| Right superior parietal lobule | 21 | −48 | 51 | 3.46 | 0.0001 | |
| Right middle frontal gyrus | 27 | 54 | 21 | 2.63 | 0.0001 | |
| Novel vs repeated pairs |
Left postcentral gyrus | −60 | −6 | 24 | 4.70 | 0.0001 |
| Left middle occipital gyrus | −42 | −75 | 24 | 4.19 | 0.0001 | |
| Right postcentral gyrus | 63 | 0 | 18 | 4.14 | 0.0001 | |
| Left insular lobe | −30 | 24 | 3 | 4.02 | 0.0001 | |
| Left middle temporal gyrus | −45 | 6 | 27 | 3.59 | 0.0001 | |
| Left lingual gyrus | −18 | −87 | −18 | 3.57 | 0.0001 | |
| Left parahippocampal gyrus | −27 | −21 | −27 | 3.19 | 0.0001 | |
| Right superior medial gyrus | 9 | 45 | 0 | 3.49 | 0.0001 | |
| Right insular lobe | 39 | −18 | 0 | 3.49 | 0.0001 | |
| Left precuneus | −3 | −60 | 39 | 3.44 | 0.0001 | |
| Right postcentral gyrus | 48 | −9 | 36 | 3.17 | 0.0001 | |
| Carriers > controls | ||||||
| Novel pairs vs fixation |
Right extrastriate/lingual gyrus | 18 | −90 | −3 | 4.82 | 0.0001 |
| Right middle frontal gyrus | 30 | 27 | 30 | 3.47 | 0.0001 | |
| Right caudate nucleus | 15 | 12 | 12 | 3.05 | 0.001 | |
| Left precuneus | −15 | −66 | 33 | 3.00 | 0.001 | |
| Right middle temporal gyrus | 39 | −9 | −18 | 2.75 | 0.001 | |
| Right cingulate gyrus | 15 | −21 | 51 | 2.74 | 0.001 | |
| Right superior temporal gyrus | 39 | −33 | −15 | 2.19 | 0.001 | |
| Right hippocampal formation | 21 | −24 | −15 | 2.16 | 0.001 | |
| Novel vs repeated pairs |
Right middle frontal gyrus | 24 | −3 | 39 | 3.09 | 0.001 |
| Right cingulate gyrus | 15 | −39 | 39 | 2.95 | 0.001 | |
| Right superior parietal lobule | 33 | −42 | 57 | 2.81 | 0.001 | |
| Right inferior parietal lobule | 51 | −36 | 54 | 2.71 | 0.001 | |
| Right hippocampal formation | 21 | −24 | −15 | 2.67 | 0.003 | |
| Right inferior frontal gyrus | 33 | 27 | 24 | 2.56 | 0.003 | |
| Right middle frontal gyrus | 42 | 12 | 42 | 2.47 | 0.005 | |
MNI = Montreal Neurological Institute.
FIGURE 2.

Whole brain exploratory analyses. Statistical parametric maps (SPMs) for both groups, controls and PS1 mutation carriers, for the contrast novel face-name pairs vs repeated face-name pairs are displayed on anatomical images derived from an average obtained from the normalized structural images of all subjects with a statistical threshold of p < 0.005 uncorrected (extent threshold = 20 voxels). (A) Within-group analysis: both groups showed an increase in the fMRI signal response of association areas in the frontal and parietal lobes and in the hippocampal formation to novel vs repeated stimuli. (B) Between-group analysis: controls showed greater activity in frontal and parietal regions whereas carriers exhibited greater activity in the right cingulate gyrus and the right anterior hippocampus to novel vs repeated stimuli. Color bar represents t values for all activated voxels in the whole brain.
Volumetric Measurements: Right Hippocampus and Left Hippocampus
A repeated measures ANOVA revealed no statistically significant differences between the volumes of the left and right hippocampus in both the controls and the PS1 mutation carriers [F(1, 37)= 2.31, p= 0.13]. The partial correlation analysis between left and right hippocampal volumes and their respective novel and repeated parameter estimates also did not reveal significant findings, suggesting that hippocampal volume did not influence the parameter estimates of the BOLD signal response.
Discussion
The examination of individuals who carry fully-penetrant genetic alterations that result in FAD provides a unique model for studying the early presymptomatic disease stages. We used fMRI to examine the functional integrity of the hippocampal system during the encoding of novel associations in young, cognitively-intact, presymptomatic individuals who carry the E280A presenilin-1 (PS1) genetic mutation for AD. The average age of our subjects was 33.7 years, and the average age of disease onset in this cohort is 45 years.5 The cognitive and neuropsychological profile of our PS1 subjects matched our controls (see Table 1). The fMRI data demonstrate that functional changes occur in presymptomatic PS1 individuals well in advance of the average age of onset of clinical symptoms. Presymptomatic carriers showed greater right anterior hippocampus activation compared to matched controls. This difference in brain activity was observed despite the fact that the groups did not differ in performance on the postscan recognition test or in hippocampal volume.
Neuropathological studies of AD have demonstrated changes in medial temporal lobe and parietal regions. In the medial temporal lobe, neurofibrillary tangles appear first in the transition zone between the entorhinal and perirhinal cortex and progress to superficial layers of the lateral entorhinal cortex. The pathology appears to spread progressively back from anterior medial temporal areas into more posterior medial temporal areas,37 and from more limbic cortical areas into neocortical regions. Encoding memory paradigms, such as the face-name paired-association task that we used here, primarily result in activity in the anterior MTL regions.15,28 Anterior MTL activity is also present during long-term encoding of novel stimuli in working memory tasks.38,39 Encoding of novel associations has been investigated in MCI, populations at genetic risk for AD, and AD patients.15,19,22,25 These studies found that hippocampal activation was significantly greater when comparing individuals at preclinical stages (MCI or genetic risk) to AD patients. Dickerson et al.22 demonstrated during a face-name associative encoding task that MCI patients had greater hippocampal activation whereas AD patients had hippocampal hypoactivation compared to controls. They proposed that in early preclinical stages of AD, there is a phase of hyperactivation that is followed by a decrease in activation that accompanies clinical AD progression. Celone et al.16 showed that this change in hippocampal regions from hyperactivation to hypoactivation could also be observed when comparing individuals at different stages of MCI. They demonstrated that hypoactivation was present in more impaired MCI patients compared to controls, whereas hyperactivation was observed in the early, less impaired stages of MCI.
In the present study, we found that cognitively-intact PS1 mutation carriers demonstrate hyperactivation within the right anterior hippocampus during the encoding of novel associations. This result is notable for several reasons. First, this result demonstrates that changes in hippocampal physiology are present several years prior to the onset of detectable neuropsychological changes and subsequent clinical symptoms. Second, this result demonstrates that fMRI is capable of detecting these presymptomatic changes and suggests that hippocampal fMRI patterns during associative encoding may also provide a preclinical biomarker in sporadic AD.
Two possible interpretations could explain why cognitively-intact PS1 mutation carriers demonstrate hyperactivation within the right anterior hippocampus during the encoding of novel associations. First, as has been suggested in MCI populations, the increase in activity may reflect compensatory recruitment of resources needed to maintain normal cognition at presymptomatic stages of AD. For example, previous studies in healthy young subjects have shown that effective memory encoding and recognition may be associated with decreases in neuronal activity in the hippocampal formation.40,41 The hyperactivation observed in the carriers compared to controls may reflect more efficient encoding processes used by the controls, so that over time they recruit fewer “resources,” while the carriers exhibit less efficient encoding processes which require greater recruitment of the hippocampus during the entire memory task in order to perform equally well.
A second interpretation of the results is that the hippocampal hyperactivation noted in our presymptomatic subjects and also in earlier MCI studies reflects an early excitotoxic disease process. There is evidence of calcium dysregulation in AD from human and mouse models,42-44 and it may be that calcium homeostasis is perturbed in our PS1 carriers and that early neurotoxicity is already present in hippocampal neurons as early as a decade before the onset of clinical symptoms. Excitotoxicity could also result from the PS1 mutation altering the amyloid precursor protein regulation of axonal pruning,45 resulting in excess formation of synapses within cortical circuits.46 Excitotoxic synaptic changes could result in the fMRI hyperactivation within the right anterior hippocampus seen in our study, and would fit well with both the known pathological progression of AD from anterior medial temporal areas into more caudal medial temporal areas37 and with data demonstrating a progression of fMRI activity in MCI patients and AD patients from early stages of hyperactivation to later stages of hypoactivation.16,22
In conclusion, the present study extends and increases our understanding of functional changes in the brain during the presymptomatic stages of AD. Using fMRI, we demonstrated that increased hippocampal activation can be detected years before cognitive impairment and clinical diagnosis.
Neuroimaging studies with PS1 mutation carriers represent a unique opportunity to understand the trajectory of AD. Our data show how relevant it could be to start examining individuals at risk for AD as early as 20 years before possible clinical onset of the disease. A logical next step is to carry out longitudinal fMRI studies in PS1 mutation populations in order to fully understand the progression of the disease and to determine whether changes in fMRI patterns have the potential both as an early marker of AD and as a marker of treatment efficacy.
Acknowledgments
This study was supported by the Boston University Center for Neuroscience (to CES and AEB), Department of Psychology (to CES and YTQ), a Boston University Alzheimer’s Disease Center pilot award to CES and AEB (P30 AG13846), and COLCIENCIAS-Colombia to FL (Project: 1115-343-19127).
We thank Karin Schon and Robert Ross for helpful discussions about data analysis, and Shelly Cooper for assistance with task development and preprocessing of data. We thank the Instituto de Alta Tecnología Médica (IATM) staff for their help with data acquisition, especially Simon Rascovsky and Carlos Vasquez. We thank Thomas Benner at the Martinos Center for his help with developing the imaging sequences. We thank the PS1 Colombian families for contributing their valuable time and effort, without whom this study would not have been possible.
Footnotes
Potential Conflict of Interest:
Nothing to report.
References
- 1.Alzheimer’s Association . Early onset dementia: a national challenge, a future crisis. Alzheimer’s Association; [Accessed July 18, 2010]. 2006. Available at: http://www.alz.org/national/documents/report_earlyonset_summary.pdf. [Google Scholar]
- 2.Goate A, Chartier-Harlin MC, Mullan M, et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature. 1991;349:704–706. doi: 10.1038/349704a0. [DOI] [PubMed] [Google Scholar]
- 3.Levy-Lahad E, Wasco W, Poorkaj P, et al. Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science. 1995;18:973–977. doi: 10.1126/science.7638622. [DOI] [PubMed] [Google Scholar]
- 4.Alzheimer’s Disease Collaborative Group The structure of the presenilin 1 (S182) gene and identification of six novel mutations in early onset AD families. Nat Genet. 1995;11:219–222. doi: 10.1038/ng1095-219. [DOI] [PubMed] [Google Scholar]
- 5.Lopera F, Ardilla A, Martínez A, et al. Clinical features of early-onset Alzheimer disease in a large kindred with an E280A presenilin-1 mutation. JAMA. 1997;277:793–799. [PubMed] [Google Scholar]
- 6.Arango-Lasprilla JC, Iglesias J, Lopera F. Neuropsychological study of familial Alzheimer’s disease caused by mutation E280A in the presenilin 1 gene. Am J Alzheimers Dis Other Demen. 2003;18:137–146. doi: 10.1177/153331750301800306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bobes MA, García YF, Lopera F, et al. ERP generator anomalies in presymptomatic carriers of the Alzheimer’s disease E280A PS1 mutation. Hum Brain Mapp. 2010;31:247–265. doi: 10.1002/hbm.20861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gómez-Isla T, Wasco W, Pettingell WP, et al. A novel presenilin-1 mutation: increased beta-amyloid and neurofibrillary changes. Ann Neurol. 1997;41:809–813. doi: 10.1002/ana.410410618. [DOI] [PubMed] [Google Scholar]
- 9.Ardila A, Lopera F, Rosselli M, et al. Neuropsychological profile of a large kindred with familial Alzheimer’s disease caused by the E280A single presenilin-1 mutation. Arch Clin Neuropsychol. 2000;15:515–528. [PubMed] [Google Scholar]
- 10.Rosselli MC, Ardila AC, Moreno SC, et al. Cognitive decline in patients with familial Alzheimer’s disease associated with E280A presenilin-1 mutation: a longitudinal study. J Clin Exp Neuropsychol. 2000;22:483–495. doi: 10.1076/1380-3395(200008)22:4;1-0;FT483. [DOI] [PubMed] [Google Scholar]
- 11.Petersen RC, Smith GE, Ivnik RJ, et al. Memory function in very early Alzheimer’s disease. Neurology. 1994;44:867–872. doi: 10.1212/wnl.44.5.867. [DOI] [PubMed] [Google Scholar]
- 12.Gainotti G, Marra C, Villa G, et al. Sensitivity and specificity of some neuropsychological markers of Alzheimer dementia. Alzheimer Dis Assoc Disord. 1998;12:152–162. doi: 10.1097/00002093-199809000-00006. [DOI] [PubMed] [Google Scholar]
- 13.Remy F, Mirrashed F, Campbell B, Richter W. Verbal episodic memory impairment in Alzheimer’s disease: a combined structural and functional MRI study. Neuroimage. 2005;25:253–266. doi: 10.1016/j.neuroimage.2004.10.045. [DOI] [PubMed] [Google Scholar]
- 14.Kordower JH, Chu YP, Stebbins GT, et al. Loss and atrophy of layer II entorhinal cortex neurons in elderly people with mild cognitive impairment. Ann Neurol. 2001;49:202–213. [PubMed] [Google Scholar]
- 15.Sperling RA, Bates JF, Chua EF, et al. fMRI studies of associative encoding in young and elderly controls and mild Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 2003;74:44–50. doi: 10.1136/jnnp.74.1.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Celone K, Calhoun VD, Dickerson BC, et al. Alterations in memory networks in mild cognitive impairment and Alzheimer’s disease: an independent component analysis. J Neurosci. 2006;26:10222–10231. doi: 10.1523/JNEUROSCI.2250-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dickerson BC, Sperling RA. Large-scale functional brain network abnormalities in Alzheimer’s disease: insights from functional neuroimaging. Behav Neurol. 2009;21:63–75. doi: 10.3233/BEN-2009-0227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dickerson BC, Salat D, Bates J, et al. Medial temporal lobe function and structure in mild cognitive impairment. Ann Neurol. 2004;56:27–35. doi: 10.1002/ana.20163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nestor PJ, Scheltens P, Hodges JR. Advances in the early detection of Alzheimer’s disease (Review) Nat Med. 2004;10:S34–S41. doi: 10.1038/nrn1433. [DOI] [PubMed] [Google Scholar]
- 20.Machulda MM, Ward HA, Borowski B, et al. Comparison of memory fMRI response among normal, MCI, and Alzheimer’s patients. Neurology. 2003;61:500–506. doi: 10.1212/01.wnl.0000079052.01016.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Petrella JR, Krishnan S, Slavin MJ, et al. Mild cognitive impairment: evaluation with 4-T functional MR imaging. Radiology. 2006;240:177–186. doi: 10.1148/radiol.2401050739. [DOI] [PubMed] [Google Scholar]
- 22.Dickerson BC, Salat DH, Greve DN, et al. Increased hippocampal activation in mild cognitive impairment compared to normal aging and AD. Neurology. 2005;65:404–411. doi: 10.1212/01.wnl.0000171450.97464.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hämäläinen A, Pihlajamäki M, Tanila H, et al. Increased fMRI responses during encoding in mild cognitive impairment. Neurobiol Aging. 2007;28:1889–1903. doi: 10.1016/j.neurobiolaging.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 24.Bookheimer SY, Strojwas MH, Cohen MS, et al. Patterns of brain activation in people at risk for Alzheimer’s disease. N Engl J Med. 2000;343:450–456. doi: 10.1056/NEJM200008173430701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mondadori CR, Buchmann A, Mustovic H, et al. Enhanced brain activity may precede the diagnosis of Alzheimer’s disease by 30 years. Brain. 2006;129:2908–2922. doi: 10.1093/brain/awl266. [DOI] [PubMed] [Google Scholar]
- 26.Lendon CL, Martinez A, Behrens IM, et al. E280A PS1 mutation causes Alzheimer’s disease but age of onset is not modified by ApoE alleles. Hum Mutat. 1997;10:186–195. doi: 10.1002/(SICI)1098-1004(1997)10:3<186::AID-HUMU2>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 27.Aguirre-Acevedo DC, Gómez RD, Moreno S, et al. [Validity and reliability of the CERAD-Col neuropsychological battery] Rev Neurol. 2007;45:655–660. Spanish. [PubMed] [Google Scholar]
- 28.Sperling RA, Bates JF, Cocchiarella AJ, et al. Encoding novel face name associations: a functional MRI study. Hum Brain Mapp. 2001;14:129–139. doi: 10.1002/hbm.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Freire L, Roche A, Mangin J-Fr. What is the best similarity measure for motion correction in fMRI? IEEE Trans Med Imaging. 2002;21:470–484. doi: 10.1109/TMI.2002.1009383. [DOI] [PubMed] [Google Scholar]
- 30.Ashburner J. A fast diffeomorphic image registration algorithm. Neuroimage. 2007;38:95–113. doi: 10.1016/j.neuroimage.2007.07.007. [DOI] [PubMed] [Google Scholar]
- 31.Trivedi MA, Schmitz TW, Ries ML, et al. Reduced hippocampal activation during episodic encoding in middle-aged individuals at genetic risk of Alzheimer’s disease: a cross-sectional study. BMC Med. 2006;4:1. doi: 10.1186/1741-7015-4-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dickerson BC, Miller SL, Greve DN, et al. Prefrontal-hippocampal-fusiform activity during encoding predicts intraindividual differences in free recall ability: an event-related functional-anatomic MRI study. Hippocampus. 2007;17:1060–1070. doi: 10.1002/hipo.20338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maldjian JA, Laurienti PJ, Kraft RA, Burdette JH. An automated method for neuroanatomic and cytoarchitectonic atlas-based interrogation of fMRI data sets. Neuroimage. 2003;19:1233–1239. doi: 10.1016/s1053-8119(03)00169-1. [DOI] [PubMed] [Google Scholar]
- 34.Yushkevich PA, Piven J, Hazlett HC, et al. User-guided 3D active contour segmentation of anatomical structures: significantly improved efficiency and reliability. Neuroimage. 2006;31:1116–1128. doi: 10.1016/j.neuroimage.2006.01.015. [DOI] [PubMed] [Google Scholar]
- 35.Pruessner JC, Li LM, Serles W, et al. Volumetry of hippocampus and amygdala with high-resolution MRI and three dimensional analysis software: minimizing the discrepancies between laboratories. Cerebral Cortex. 2000;10:433–442. doi: 10.1093/cercor/10.4.433. [DOI] [PubMed] [Google Scholar]
- 36.Duvernoy HM. The human hippocampus, functional anatomy, vascularization and serial sections with MRI. 3rd ed. Springer-Verlag; New York: 2005. [Google Scholar]
- 37.Braak H, Braak E. Staging of Alzheimer’s disease-related neurofibrillary changes. Neurobiol Aging. 1995;16:271–278. doi: 10.1016/0197-4580(95)00021-6. [DOI] [PubMed] [Google Scholar]
- 38.Stern CE, Sherman SJ, Kirchhoff BA, Hasselmo ME. Medial temporal and prefrontal contributions to working memory tasks with novel and familiar stimuli. Hippocampus. 2001;11:337–346. doi: 10.1002/hipo.1048. [DOI] [PubMed] [Google Scholar]
- 39.Schon K, Hasselmo ME, LoPresti ML, et al. Persistence of parahippocampal representation in the absence of stimulus input enhances long-term encoding: a functional magnetic resonance imaging study of subsequent memory after a delayed match-to-sample task. J Neurosci. 2004;24:11088–11097. doi: 10.1523/JNEUROSCI.3807-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gonsalves BD, Kahn I, Curran T, et al. Memory strength and repetition suppression: multimodal imaging of medial temporal cortical contributions to recognition. Neuron. 2005;47:751–761. doi: 10.1016/j.neuron.2005.07.013. [DOI] [PubMed] [Google Scholar]
- 41.Otten LJ, Henson RN, Rugg MD. Depth of processing effects on neural correlates of memory encoding: relationship between findings from across- and within-task comparisons. Brain. 2001;124:399–412. doi: 10.1093/brain/124.2.399. [DOI] [PubMed] [Google Scholar]
- 42.Yu JT, Chang RC, Tan L. Calcium dysregulation in Alzheimer’s disease: From mechanisms to therapeutic opportunities. (Review) Prog Neurobiol. 2009;89:240–255. doi: 10.1016/j.pneurobio.2009.07.009. [DOI] [PubMed] [Google Scholar]
- 43.Smith IF, Green KN, LaFerla FM. Calcium dysregulation in Alzheimer’s disease: recent advances gained from genetically modified animals. Cell Calcium. 2005;38:427–437. doi: 10.1016/j.ceca.2005.06.021. [DOI] [PubMed] [Google Scholar]
- 44.Saitoh T, Horsburgh K, Masliah E. Hyperactivation of signal transduction systems in Alzheimer’s disease. Ann N Y Acad Sci. 1993;695:34–41. doi: 10.1111/j.1749-6632.1993.tb23023.x. [DOI] [PubMed] [Google Scholar]
- 45.Nikolaev A, McLaughlin T, O’Leary DD, Tessier-Lavigne M. APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature. 2009;457:981–989. doi: 10.1038/nature07767. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 46.Hasselmo ME. A computational model of the progression of Alzheimer’s disease. MD Comput. 1997;14:181–189. [PubMed] [Google Scholar]