

Abstract
Rationale
Pressure overload cardiac hypertrophy, a risk factor for heart failure, is associated with reduced mitochondrial fatty acid oxidation (FAO) and oxidative phosphorylation (OXPHOS) proteins that correlate in rodents with reduced PGC-1α expression.
Objective
To determine the role of PGC-1β in maintaining mitochondrial energy metabolism and contractile function in pressure overload hypertrophy.
Methods and Results
PGC-1β deficient (KO) mice and wildtype controls (WT) were subjected to transverse aortic constriction (TAC). Although LV function was modestly reduced in young KO hearts, there was no further decline with age so that LV function was similar between KO and WT when TAC was performed. WT – TAC mice developed relatively compensated LVH, despite reduced mitochondrial function and repression of OXPHOS and FAO genes. In non-stressed KO hearts, OXPHOS gene expression and palmitoyl-carnitine supported mitochondrial function were reduced to the same extent as banded WT, but FAO gene expression was normal. Following TAC, KO mice progressed more rapidly to heart failure and developed more severe mitochondrial dysfunction, despite a similar overall pattern of repression of OXPHOS and FAO genes as WT – TAC. However, relative to WT TAC, PGC-1β deficient mice exhibited greater degrees of oxidative stress, decreased cardiac efficiency, lower rates of glucose metabolism and repression of hexokinase II protein.
Conclusions
PGC-1β plays an important role in maintaining baseline mitochondrial function and cardiac contractile function following pressure overload hypertrophy by preserving glucose metabolism and preventing oxidative stress.
Keywords: Mitochondria, Cardiac Hypertrophy, Heart Failure, Gene Expression
PGC-1α and PGC-1β are primarily expressed in oxidative tissues including heart and skeletal muscle and regulate mitochondrial biogenesis and genes encoding for enzymes of mitochondrial metabolism and proteins that comprise the electron transport chain 1–3. In non-stressed hearts from PGC-1α-KO mice, impaired fatty acid oxidation (FAO) and oxidative phosphorylation (OXPHOS) gene expression was observed, but cardiac function was preserved 4. Repression of PGC-1α and its transcriptional partners has been suggested as a potential mechanism responsible for the shift away from FAO towards glucose oxidation and impaired ATP production in pressure overload hypertrophy (POH) and heart failure 5–7. Mice lacking PGC-1α show accelerated cardiac dysfunction and heart failure following POH 8. Interestingly, loss of PGC-1α expression in non-stressed hearts reduced expression of FAO and OXPHOS genes to the same extent as aortic banding in control mice and in both instances without overt cardiac dysfunction 8. However, in banded PGC-1α deficient hearts there was further repression of OXPHOS and FAO gene expression that coincided with the onset of heart failure. PGC-1α deficient hearts also exhibit increased oxidative stress in response to POH, which contributes to accelerated heart failure 9. These observations suggest that a component of the mitochondrial dysfunction that occurs in the transition from compensated hypertrophy to heart failure might be PGC-1α independent and that a threshold expression of PGC-1α might be required to sustain mitochondrial function in compensated cardiac hypertrophy.
The role of PGC-1β and its contribution to cardiac mitochondrial and metabolic adaptations in non-stressed hearts and in response to POH is incompletely understood. We previously reported that PGC-1β deficiency reduces OXPHOS gene expression in the heart and reduced mitochondrial size. In isolated soleus muscle, absence of PGC-1β reduced electron transport chain gene expression, decreased mitochondrial volume density and impaired mitochondrial oxygen consumption and ATP-synthesis 10. We therefore sought to determine if PGC-1β contributed to the maintenance of mitochondrial and contractile function in response to POH induced by transverse aortic constriction (TAC). We observed that PGC-1β deficiency represses OXPHOS but not FAO gene expression in non-stressed hearts. PGC-1β deficient hearts develop accelerated heart failure and greater mitochondrial dysfunction following POH. This is associated with increased oxidative stress and a more rapid decline in myocardial glucose utilization. A subset of OXPHOS genes whose expression levels are further repressed during POH in hearts that lack PGC-1β was also identified. Thus, PGC-1α and PGC-1β play complementary but partially non-overlapping roles in maintaining mitochondrial function in the hypertrophied and failing heart.
Methods
Animals
PGC-1β−/− germ-line KO mice (βKO) and wildtype littermate controls (WT) were generated as previously described 10. The animals were housed at 22°C with a 12h light/12h dark cycle with free access to water and standard chow (see Online Supplement for composition of chow). Studies were performed in mice of both genders as indicated in the figure legends. All animal experiments were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee of the University of Utah.
Surgeries
Transverse aortic constriction (TAC) was performed at the age of 8 to 10 weeks or at 23–30 weeks using methods previously described 11. Animals were sacrificed either three or eight weeks after surgery. See online supplement for surgical details.
Transthoracic echocardiography
Mouse echocardiography was performed as previously described 12. Efficacy of the aortic ligation was determined by comparing flow velocity in right and left carotid arteries using pulsed wave Doppler. Full details are provided in the online supplement.
Hemodynamic studies
Heart rate, left ventricular systolic pressure (LVSP), left ventricular end-diastolic pressure (LVEDP) and the maximal rate of pressure change (+dP/dt and −dP/dt) were recorded and analyzed as previously described 13, following insertion of a micromanometer tipped pressure catheter (Millar Instruments, Houston, TX) that was retrogradely introduced into the left ventricle (LV) via the right carotid artery. Studies were performed in non-stressed mice before and after dobutamine 10 and following 3 or 8-weeks of TAC or sham surgery. Detailed methods are summarized in the online supplement.
Isolated Working Hearts
Cardiac substrate metabolism, left ventricular developed pressure, cardiac output, cardiac power, oxygen consumption and cardiac efficiency were measured in isolated working hearts obtained after 3-weeks of banding as previously described 14. Hearts were perfused with 5 mM glucose and 0.4 mM palmitate without insulin.
Mitochondrial function
Mitochondrial oxygen consumption and ATP production rates were measured in ventricular muscle fibers as described before, with palmitoyl carnitine or pyruvate as substrate 15. See online supplement for details.
RNA extraction and quantitative RT-PCR
Total RNA was isolated, reverse transcribed to cDNA, which was quantified by Real-Time PCR as previously described and transcript levels normalized to Cyclophilin A16. Primer sequences and accession numbers are listed in Online Table I. See online supplement for detailed methods.
Western Blot analysis
Immunoblotting was performed with heart homogenates as described in the online supplement.
Electron Microscopy
Left ventricular samples were prepared for electron microscopy as previously described 17. Mitochondrial number and volume density were assessed in a blinded fashion using point quantification counting 18. For volume density, 2 pictures per sample were analyzed using 2 grids per picture. For mitochondrial number, 3 pictures per sample were analyzed.
Measurement of ROS levels
Reactive oxygen species levels were measured by 2′-7′-dichlorofluorescein-diacetate (DCFDA) fluorescence as described in the online supplement.
Histology and Stereology
Myocardial fragments were stained by hematoxylin-eosin, Masson’s trichrome, TUNEL, and DAPI stains. Stereological analysis was performed as described in the online supplement 19.
Statistical analysis
Statistical analysis is described in the online supplement. Data are presented as means ± SEM. RT-PCR results are presented as fold change vs. WT Sham. A p-value of < 0.05 was considered significantly different.
Results
Inotropic reserves in PGC-1β−/− hearts
LV hemodynamics was determined in PGC-1β−/− (βKO) and wildtype (WT) hearts as a function of age. As shown in Figure 1A, peak rates of ventricular contraction (+dP/dt) were significantly higher in 7-week-old WT hearts, after which an age-dependent decline was observed. By contrast, despite an initial reduction, no age-related decline in +dP/dt was observed in βKO hearts so that by 17 and 27-weeks of age respectively, there were no significant differences between the genotypes. Similar patterns were observed for LV developed pressure (LVDevP) and peak rates of ventricular relaxation (− dP/dt) (data not shown). To determine if the reduction in contractility in 7-week-old βKO hearts was associated with decreased contractile reserve, hemodynamic measurements were performed following administration of a graded infusion of dobutamine. Heart rates were similar at baseline, but βKO mice developed a blunted chronotropic response (Figure 1B). By contrast, the fold change (from respective baselines) in LVDevP and ±dP/dt was similar in βKO and WT hearts (Figure 1C, D). Because baseline values were lower in βKO than WT, absolute values remained lower in βKO throughout the dobutamine infusion.
Figure 1.
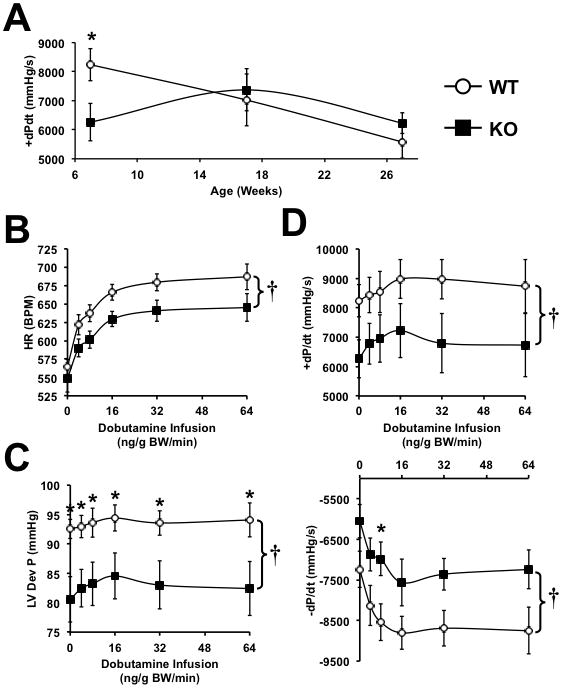
(A) Age-dependent changes in peak rate of ventricular contraction (+dP/dt) in non-stressed WT and PGC-1β−/− hearts (n=5–9). (B–D) In vivo hemodynamic parameters in 7-week-old WT and PGC-1β−/− hearts following graded dobutamine infusion as indicated. * = p<0.05 vs. WT at equivalent dobutamine dose, † = p<0.05 for analysis of covariance comparing genotypes (n=5–7). Data were obtained in male and female mice and are combined.
Increased left ventricular dysfunction after TAC in PGC-1β −/− hearts
TAC was performed in mice at two ages (8–10 weeks of age and 23–30 weeks of age). The first cohort was studied after 8-weeks of TAC (age 16–18 weeks) and the second cohort after 3-weeks of TAC (age 26–33-weeks of age). At these ages, there were no differences in in vivo hemodynamics between KO and WT sham operated mice (Figure 1A). In sham-operated hearts, heart weights of βKO mice were similar to WT and exhibited no histological abnormalities. After eight weeks of TAC, WT and βKO hearts developed comparable degrees of hypertrophy (Figure 2A–C and Online Table II), and similar increases in mean cardiomyocyte cross-sectional surface area (Figure 2D,F). However, βKO hearts developed increased fibrosis, independent of any increase in TUNEL positive nuclei (Figure 2E,G–I)). We also analyzed left ventricular function at three and eight weeks following TAC. Three weeks following TAC, WT and βKO hearts exhibited modest degrees of left ventricular (LV) dysfunction as evidenced by LV dilation. However, these changes were not statistically different between banded WT and banded βKO (Figure 3B). Moreover, fractional shortening was not different from sham-operated animals (Figure 3B and Online Table III). LV function progressively declined as evidenced by decreased ejection fraction (EF) and fractional shortening (FS) in βKO and WT mice between 3 and 8-weeks post TAC (Figure 3B, C). Thus after eight weeks of TAC, the degree of reduction of EF and FS was greater in PGC-1β KO hearts, which developed overt heart failure as evidenced by decreased left ventricle systolic pressure (LVSP), impaired peak rates of ventricular contraction and relaxation (±dP/dt) and increased left ventricular cavity dimensions (Figure 3A, C, D).
Figure 2.
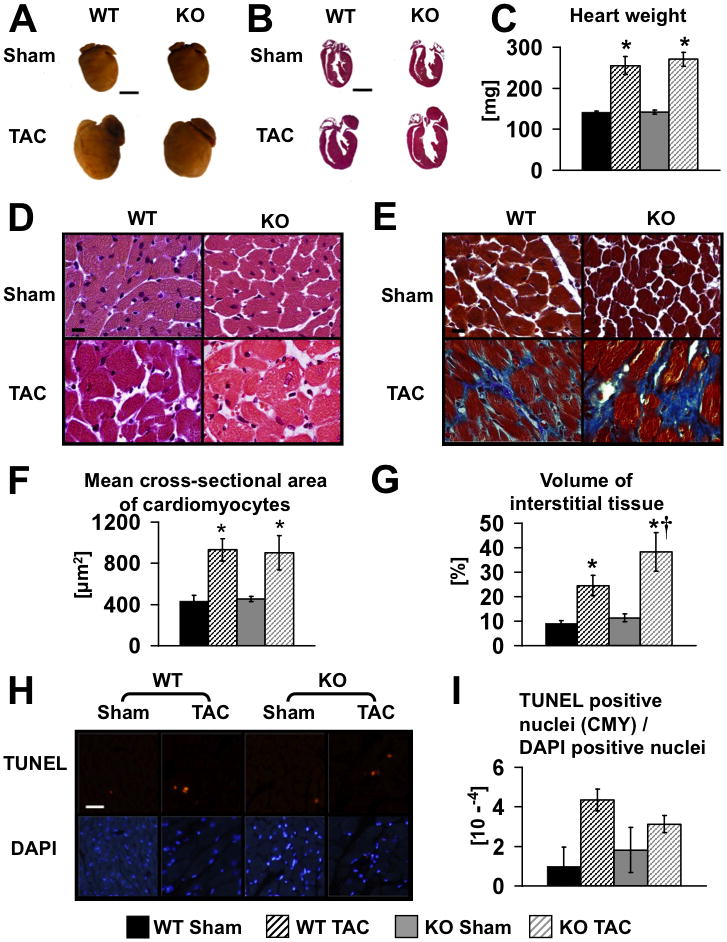
(A) Representative photographs of male WT and KO hearts following eight weeks of TAC or Sham surgery (age 16–18 weeks). Scale bars, 3 mm. (B) Hematoxylin-eosin stains of longitudinal sections of the same hearts as in A. Scale bars, 3 mm. (C) Mean heart weights eight weeks after surgery, n=12. Hematoxylin-eosin (D) and trichrome stains (E) (scale bars, 10 μm) and quantification of mean cross-sectional area of cardiomyocytes (F) and volume of interstitial fibrosis (G). Representative TUNEL (upper) and DAPI (lower) staining (H) and quantification (I); Scale bar, 50 μm; CMY – cardiomyocytes. Data were obtained from 3 to 7 hearts per group. * = p<0.05 vs. Sham same genotype, † = p<0.05 vs. WT same treatment.
Figure 3.
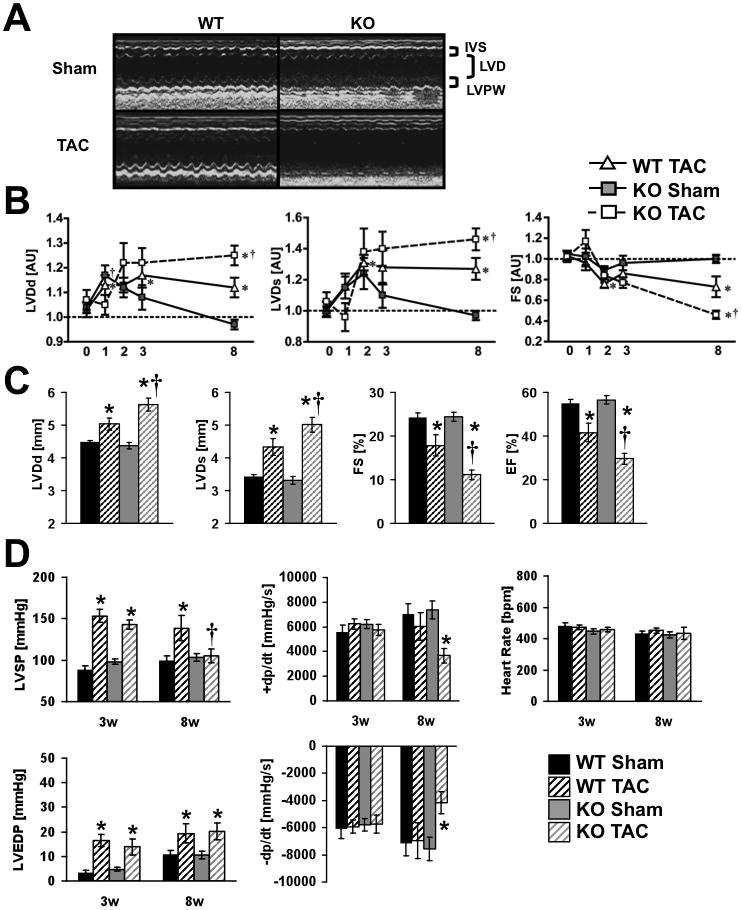
(A) Representative M-Mode echocardiographs from Sham (upper) or TAC (lower) male WT and KO mice illustrating cardiac contractile dysfunction in PGC-1β hearts after eight weeks of pressure overload (same mice as figure 2). IVS=interventricular septum, LVD=left ventricular diameter, LVPW=LV posterior wall; (B) Time course for LVDd - Left ventricular cavity diameter at diastole, LVDs - Left ventricular cavity diameter at systole, and FS - Fractional shortening presented as fold change vs. WT Sham (= 1.0). The X-axes show the time in weeks after surgery. (C) LVDd, LVDs, FS, and EF - Ejection fraction 8-weeks after Sham or TAC. (D) In vivo, left ventricular hemodynamic parameters, three and eight weeks after Sham or TAC surgery. * = p<0.05 vs. Sham same genotype, † = p<0.05 vs. WT same treatment (n = 6–13). Week 1–3 mice were banded at 23–30 weeks of age and week 8 data are from 16–18-week-old mice
Mitochondrial dysfunction following TAC in WT and PGC-1β KO hearts
Mitochondrial function was determined after 8-weeks of TAC (Figure 4A, B). With pyruvate as a substrate, maximal ADP-stimulated mitochondrial oxygen consumption (VADP) and ATP-synthesis rates were unchanged in sham-operated βKO and were decreased equivalently following TAC in both WT and βKO mice. Palmitoyl-carnitine (PC)-supported VADP and ATP synthesis were decreased in sham-operated β KO mice and TAC reduced PC-supported VADP and ATP synthesis in WT hearts to levels that were similar to sham βKO. TAC modestly reduced VADP in βKO further so that relative to WT-TAC, VADP was significantly reduced. ATP synthesis rates followed a similar pattern, but the decline in ATP synthesis in βKO after TAC was more pronounced, being lowest in βKO-TAC mice relative to all other groups (Figure 4B). Mitochondrial dysfunction was not associated with any noticeable change in mitochondrial ultrastructure, number or volume density (Online Figure I). To determine if the decline in mitochondrial function induced by TAC was associated with independent evidence of energetic stress, we determined AMPK phosphorylation, which was significantly increased in the banded βKO after 8-weeks of TAC, but not in banded WT (Figure 4C).
Figure 4.
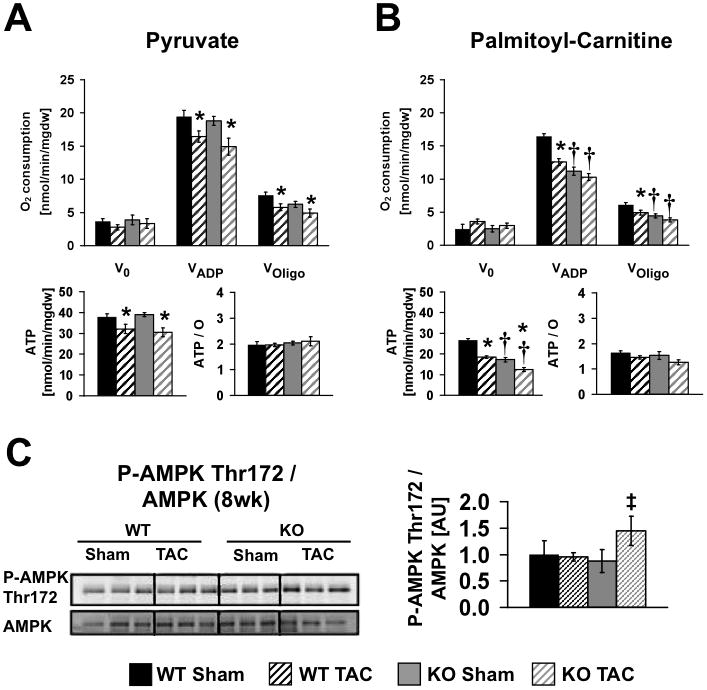
Mitochondrial function of male and female WT and KO hearts (age 16–18 weeks) following eight weeks of TAC or Sham surgery (4 to 7 hearts per group). Mitochondrial respiration, ATP synthesis rates and ATP/O ratios were measured in saponin-permeabilized cardiac fibers. Pyruvate-malate (A) or palmitoyl-carnitine-malate (B) were used as substrates. (C) Western blot analysis and densitometric ratios of AMPK phosphorylation eight weeks post surgery (n=5–6). Data are presented as fold change vs. WT Sham (=1.0). * = p<0.05 vs. Sham same genotype, † = p<0.05 vs. WT same treatment ‡ = p<0.05 vs. KO Sham (Wilcoxon test)
Oxidative stress is increased in banded PGC-1β−/− hearts
To gain additional insight into the mechanisms that increased heart failure progression in banded βKO mice, we evaluated the potential contribution of oxidative stress. Total levels of tissue ROS were increased in banded βKO hearts 3 and 8 weeks after TAC respectively (Figure 5A, B). Pressure overload was associated with repression of MnSOD protein after 3-weeks of banding in βKO but not in banded WT hearts. After 8-weeks of banding, MnSOD was equivalently repressed in banded mice of both genotypes (Figure 5C). Thus loss of antioxidant capacity occurs at an earlier stage after pressure overload in βKO mice. Uncoupling proteins such as UCP3, mediate reduction of membrane potential under conditions of increased ROS 20. UCP3 protein levels were increased in sham βKO mouse hearts relative to sham WT. Banding equivalently repressed UCP3 in both genotypes at 3 and 8 weeks following TAC, which paralleled changes in UCP3 mRNA (Figure 7 and Online Figure II).
Figure 5.
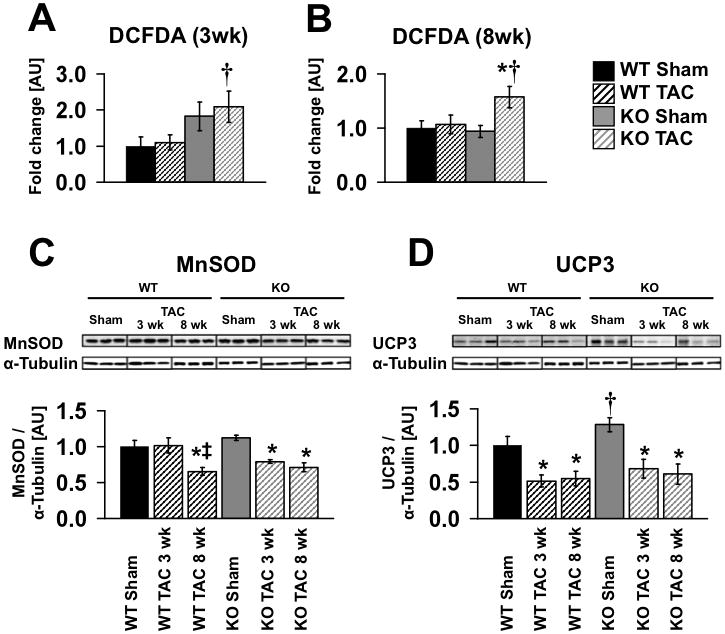
Reactive oxygen species levels were measured by 2′-7′-dichlorofluorescein diacetate (DCFDA) fluorescence in whole tissue extract three (A) and eight (B) weeks after Sham or TAC surgery, n=4–7. (C, D) Representative western blots showing protein levels of MnSOD and UCP3 three and eight weeks after surgery and densitometric analysis normalized to α-tubulin (n = 5–11). Data are presented as fold change vs. WT Sham. * = p<0.05 vs. Sham same genotype, † = p<0.05 vs. WT same treatment, ‡ = p<0.05 vs. same genotype three weeks post surgery. TAC 3wk and TAC 8 wk were 8 and 13-weeks of age respectively at the time of sacrifice.
Figure 7.
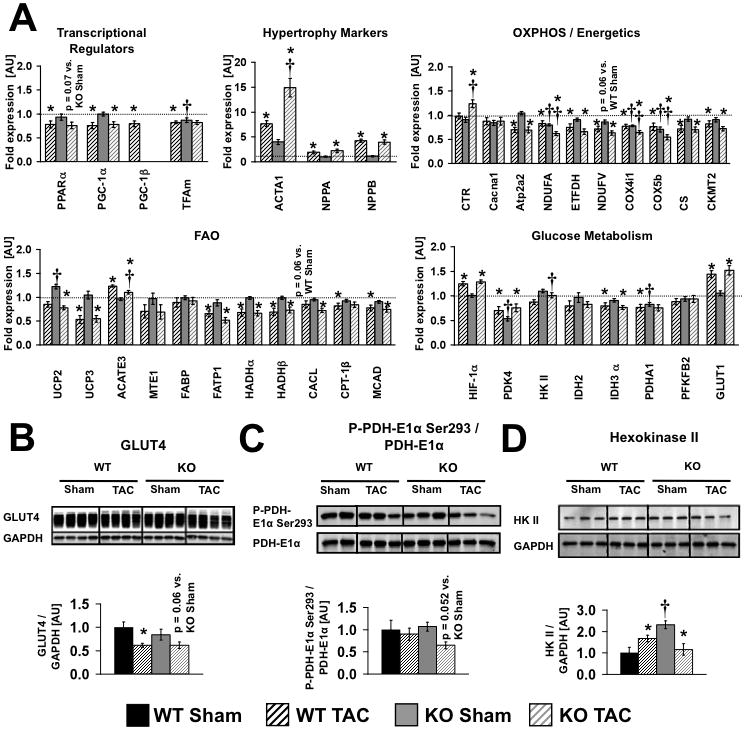
(A) Gene expression eight weeks after surgery (n = 8, same mice as Figure 2). (B, C, D) Western blot analysis and densitometric ratios of GLUT4 protein expression, PDH-E1α phosphorylation and Hexokinase II protein expression in WT and KO hearts eight weeks post surgery (n = 5–6, same mice as Figure 5). * = p<0.05 vs. Sham same genotype, † = p<0.05 vs. WT same treatment. Gene names are shown in Online Table III.
Substrate metabolism and cardiac efficiency in PGC-1β−/− hearts following TAC
Substrate metabolism was determined in isolated working hearts, 3-weeks after sham or banding surgery. Relative to WT shams, TAC increased glucose oxidation and glycolysis rates in WT hearts. Rates of glucose oxidation and glycolysis were also increased in non-stressed βKO hearts. However, in response to banding there was a significant reduction both in glucose oxidation and glycolysis to levels that were lower than WT TAC. Palmitate oxidation rates were not altered by banding in WT hearts. By contrast FAO was increased in βKO hearts and tended to fall with banding (p=0.08), but did not fall below WT levels (Figure 6). Left ventricular developed pressure, cardiac output, cardiac power, oxygen consumption and cardiac efficiency were also determined in isolated working hearts. In sham-operated hearts, there were no differences in cardiac function, but myocardial oxygen consumption rates (MVO2) were increased in βKO hearts resulting in decreased cardiac efficiency. LV developed pressure declined with banding in both genotypes but to a greater extent in βKO. Cardiac output and cardiac power declined equivalently in banded mice of both genotypes. MVO2 remained persistently increased in banded βKO hearts leading to a greater reduction in cardiac efficiency in banded βKO relative to banded WT. Thus, PGC-1β deficiency leads to diminished glucose utilization and decreased cardiac efficiency following TAC.
Figure 6.
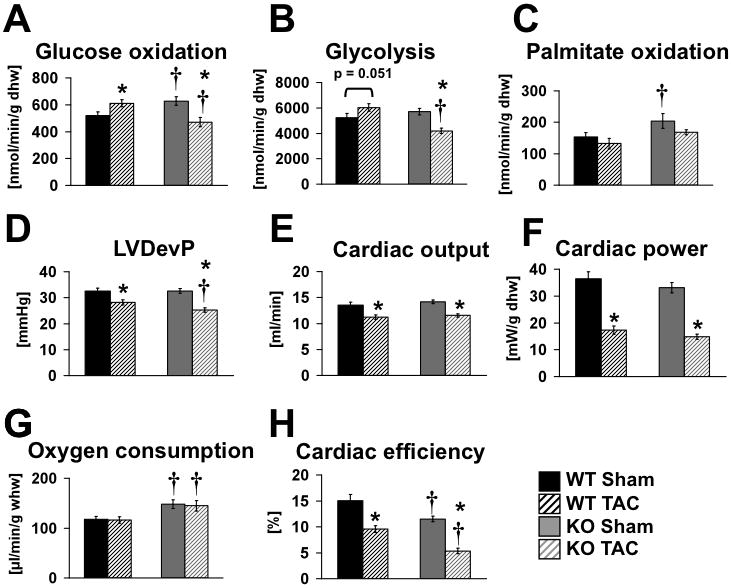
(A–H) Cardiac substrate metabolism and function in isolated working hearts. Glucose oxidation, glycolysis, palmitate oxidation, aortic developed pressure (DevP), cardiac output, cardiac power, oxygen consumption and cardiac efficiency in isolated working hearts isolated from wildtype (WT) and PGC-1β KO (KO) mice three weeks after TAC or Sham surgery, performed in mice between the ages of 23–30 weeks. Hearts were perfused with 5mM glucose and 0.4mM palmitate. n=4 hearts per group for metabolism, MVO2 and efficiency and 8/group for cardiac function (pooled data from glucose and FA perfusions). * = p<0.05 vs. Sham same genotype, † = p<0.05 vs. WT same treatment.
Changes in expression of metabolic regulators following banding in PGC-1β−/− hearts
Targeted gene expression analysis was performed to evaluate the impact of TAC vs. loss of PGC-1β on mitochondrial and metabolic genes (Figure 7 and Online Figure II). In WT mice, TAC leads to reduced expression of PGC-1α, OXPHOS and FAO genes as early as 3–weeks after banding and reduced expression of PGC-1β by 8–weeks. Similar reductions of PGC-1α and PGC-1β mRNA expression, and their downstream targets involved in OXPHOS and FAO was also observed in rat hearts following two weeks of TAC (Online Table IV and V), indicating a conserved transcriptional response of mitochondrial regulatory genes to POH in two rodent species. After three weeks of TAC, expression levels of PGC-1β were increased in WT and expression of PPARα and PGC-1α were equivalently reduced by 30% and 60% respectively in βKO and WT. OXPHOS gene expression was globally reduced in sham-operated βKO, which mirrored the pattern observed in WT-TAC, suggesting redundant roles for PGC-1α and PGC-1β in the regulation of these genes. For the majority of the OXPHOS genes examined, there was no further reduction in expression levels following TAC in βKO mice despite additional reductions in PGC-1α expression. By contrast, three OXPHOS genes (ndfua9, cox4i and cox5b) were additionally repressed in βKO after TAC suggesting synergistic interactions of PGC-1α and PGC-1β in the regulation of these genes. KO of PGC-1β had no impact on FAO gene expression in non-stressed hearts. In contrast, FAO gene expression was reduced equivalently in WT and βKO hearts after three weeks of TAC. Thus, impaired FAO gene expression following TAC is most likely the consequence of reduced levels of PGC-1α and PPARα. Similar patterns of gene expression were noted following eight weeks of TAC, but in addition, reduced expression of PGC-1β occurred in WT-TAC. In non-stressed βKO hearts, expression levels of most glucose regulatory genes were unchanged with the exception of the alpha subunit of PDH (PDHA1), which was reduced in sham βKO hearts. With the exception of HIF-1α mRNA which was induced by banding in WT and βKO, most genes involved in glucose metabolism were equivalently repressed by banding in WT and βKO. GLUT1 exhibited a biphasic response, initially being repressed by banding at 3-weeks, but induced by 8-weeks in both WT and βKO. We also examined the protein levels of GLUT4 and Hexokinase II, and assessed the phosphorylation state of PDH. Banding repressed GLUT4 content in WT and βKO hearts (Figure 7B). The phosphorylation of the PDH-E1α subunit was unchanged in all groups with the exception of banded βKO hearts at 3 and 8 weeks respectively, where the phosphorylation of this subunit was reduced (Figure 7C and Online Figure II). Hexokinase II (HKII) protein was induced in WT hearts after 8-weeks of banding and was increased in sham βKO hearts in the 8-week cohort. However, after 8-weeks there was a significant reduction in HKII protein in βKO following TAC (Figure 7D).
Discussion
The present study identified important roles for PGC-1β in the maintenance of cardiac OXPHOS gene expression and mitochondrial function in non-stressed hearts and cardiac function following pressure overload hypertrophy (POH). PGC-1β plays an important role in maintaining cardiac function in compensated POH, as evidenced by the increased susceptibility of PGC-1β deficient hearts to heart failure. The mechanisms by which PGC-1β contributes to the maintenance of cardiac function in response to pressure overload are complex and cannot be completely accounted for by changes in the expression of nuclear genes that regulate mitochondrial OXPHOS, FAO or glucose metabolism, given the similarity in the transcriptional responses of banded WT and KO mice. Moreover, we identify some circumstances where changes in gene expression do not necessarily correlate with changes at the protein levels as exemplified by hexokinase II in banded PGC-1β KO hearts. Rather, a number of defects became apparent with banding in PGC-1β deficient hearts that include increased oxidative stress that may be precipitated in part by an early loss of anti-oxidant defenses, a precipitous reduction in glucose utilization and reduced cardiac efficiency that precede the subsequent decline in cardiac function. By the time that heart failure ensues, additional defects were noted in β KO hearts such as decreased palmitoyl carnitine ATP generation, increased AMPK phosphorylation and decreased HKII content. It is however challenging to discern if these additional molecular defects are a cause or a consequence of the more severe heart failure in β KO hearts.
In non-stressed hearts, no differences in heart weights were observed between WT and βKO mice and expression of natriuretic peptide precursor type A and B were unchanged. At the ages at which the banded hearts were analyzed, there were no differences in contractile function between sham operated βKO and WT, which is consistent with previous reports 10, 21. Interestingly however, in young β KO mice a reduction in basal contractility was observed, suggesting that deficiency of PGC-1β led to a constitutive defect in contractile function that did not progress with age. Importantly, inotropic reserve in response to dobutamine was relatively preserved in young β KO mouse hearts, which supports observations by Lai et al. who reported that LV contractile function following exercise was not impaired in β KO mice of similar age 21. We confirmed a defect in chronotropic reserve in young β KO mice that we previously observed in an older cohort of mice 10. However, there were no differences in heart rate following banding in β KO mice. Taken together, these data suggest that PGC-1β plays an important role in preserving LV function in response to pressure overload. However, we cannot completely rule out the possibility that this constitutive defect could contribute in part to the more rapid decline in cardiac function when pressure overload was imposed. Cardiac phenotypes have been described for two independently generated lines of PGC-1α KO mice. One model showed normal contractile function under basal conditions 4, 22 and the other exhibited age-dependent contractile dysfunction 23. Deletion of PGC-1α, results in impaired expression of both FAO and OXPHOS genes in non-stressed hearts 4. In contrast, we observed that whereas PGC-1β plays an important role in the regulation of OXPHOS gene expression, it does not appear to be necessary for regulating FAO genes, suggesting divergent roles for PGC-1 isoforms in regulating FAO gene expression. Similar changes in mitochondrial gene expression were noted in the hearts of 2 other independently generated PGC-1β deficient mouse models21, 24. Importantly, repression of OXPHOS and/or FAO gene expression in these models is not sufficient to consistently impair cardiac function in the non-stressed heart.
Deficiency of PGC-1α in the heart reduces maximal palmitoyl-carnitine (PC) supported (VADP) respiration and increases pyruvate supported VADP respiration 4. In the present study, we observed impaired PC-supported VADP respiration and ATP-synthesis in non-stressed hearts lacking PGC-1β. But in contrast to PGC-1α-KO (α KO) hearts, KO of PGC-1β had no effect on pyruvate-supported mitochondrial function. The basis for the differences in pyruvate-supported respiration in α vs. β KO mice is not fully understood. We speculate that because PGC-1α deficiency significantly represses OXPHOS and FAO gene expression, there is a greater reliance of α KO mitochondria on glucose carbons, which drives adaptive responses that increase pyruvate utilization. In this regard, we observed a reduction in expression levels of the PDHA1 subunit in sham β KO hearts, which could also limit the induction of pyruvate utilization. In contrast, there was no repression of FAO genes in β KO hearts, nor was there any defect in FA utilization in isolated working hearts. PC-supported respiration and ATP synthesis rates were reduced in β KO mitochondria. However, the reductions were not as striking as we previously reported in α KO mitochondria where PC-supported ATP generation was reduced by 65% 4, which contrasts with the 30% reduction in β KO mitochondrial ATP synthesis, observed in the present study. These findings are consistent with the hypothesis that the greater decline in mitochondrial function with FA substrates in α KO mice reflects the combined effects of reduced OXPHOS and FAO capacity. Electron microscopy revealed normal mitochondrial volume density and number for non-stressed PGC-1β deficient hearts, which is consistent with previous reports that examined younger PGC-1β KO hearts 21. It is important to note though that mitochondrial volume density declines with age in PGC-1β KO hearts 10. Thus analyses of non-stressed βKO hearts reveal divergent effects for PGC-1α vs. PGC-1β on the expression of mitochondrial metabolic genes and mitochondrial function. However, both of these models suggest that mitochondrial dysfunction that develops on the basis of loss of either PGC-1α or PGC-1β is not sufficient to impair cardiac function throughout life in the absence of a superimposed stress. They also support the existence of overlapping or redundant roles for both of these transcriptional regulators in the regulation of nuclear-encoded genes that regulate mitochondrial metabolism and suggest that loss of PGC-1α may compensate for the loss of PGC-1β in maintaining contractile function under non-stressed conditions and vice versa. Support for redundant roles for PGC-1α and PGC-1β in maintaining basal mitochondrial function in the heart was also provided by analysis of mice that lack both PGC-1α and PGC-1β, which die of heart failure shortly after birth with bradycardia, small hearts and reduced cardiac output 21.
We examined the impact of PGC-1β deletion on pressure overload cardiac hypertrophy (POH). PGC-1β KO mice progressed more rapidly to heart failure than banded WT mice, and this was clearly associated with evidence of energetic stress, namely increased phosphorylation of AMPK. An earlier study demonstrated that mice lacking PGC-1α developed heart failure eight weeks following TAC. This was associated with additional repression of FAO and OXPHOS genes in PGC-1α-KO mice in response to TAC, versus WT-TAC or sham-operated PGC-1α-KO mice 8. It is important to note that the additional repression of OXPHOS and FAO gene expression in banded PGC-1α-KO mice occurred despite normal expression of PGC-1β. These data suggest that PGC-independent mechanisms must exist that account for reduced expression of mitochondrial target genes in pressure overload hypertrophy. They also suggest that there is a critical threshold of mitochondrial activity that is required to cope with increased workload, which cannot be substituted for or sustained by PGC-1β. The transition to heart failure was not associated with striking changes in mitochondrial morphology in β KO hearts. Although unexpected, it is probable that ultrastructural studies were performed at a time point shortly after the transition to heart failure and that a longer period of observation might be needed before changes in mitochondrial morphology become apparent.
It is also likely that energetic limitations might not be the only mechanism that contributed to heart failure in PGC-1α and PGC-1β KO hearts following TAC. A recent study in banded PGC-1α-KO mice suggested that increased oxidative stress partially drove the LV dysfunction that was observed 9. In the present study we also obtained evidence of significant oxidative stress that developed in PGC-1β KO hearts and was evident as early as three weeks post-TAC, prior to any differences in LV function. In this regard, it is interesting to note that protein levels of MnSOD were significantly repressed as early as three weeks in banded PGC-1β KO hearts but not in banded WT hearts in which repression of MnSOD was only evident after 8-weeks. It is also noteworthy that preserved MnSOD content, 3-weeks post-TAC correlated with increased PGC-1β expression, despite reduced PGC-1α expression in WT hearts, whereas at 8-weeks, the expression of PGC-1α and β were both reduced in banded WT hearts. Taken together, these data indicate that PGC-1β plays an important role in supporting anti-oxidant mechanisms that may limit oxidative stress in the compensated state of pressure overload cardiac hypertrophy and represents another function that overlaps with that of PGC-1α.
Interesting changes were observed in the expression levels and content of uncoupling proteins, which could also contribute to the cardiac responses observed. UCP3 protein and mRNA were increased in sham β KO hearts, and were repressed by TAC in KO and WT hearts. UCP2 mRNA also exhibited a similar pattern. Uncoupling proteins have been proposed to play a role in reducing mitochondrial membrane potential under conditions where ROS is increased 20. Thus the increase in UCP2 and UCP3 in the PGC-1β deficient hearts could reflect an adaptation to low levels of oxidative stress that might also be present in non-stressed β KO hearts. The increase in UCPs could also contribute to the increase in MVO2 and reduced cardiac efficiency that characterized these hearts. ROS are potent activators of uncoupling proteins, thus the persistently elevated MVO2 in banded β KO hearts could represent ROS-mediated activation of UCPs in the face of progressive oxidative stress, despite falling levels of UCPs following TAC. Isolated working hearts revealed significant differences in substrate utilization between PGC-1β deficient hearts and WT controls. As reported by others, compensated LVH in wildtype hearts was associated with increased glucose utilization. Unexpectedly, glucose and FA utilization was increased in non-stressed β KO hearts. The molecular mechanisms for these adaptations are not immediately apparent, and will require additional studies in the future. Importantly, these adaptations were not sustained in β KO hearts following TAC. The global reduction in glucose utilization is not accounted for by differences in GLUT4 protein or by increased phosphorylation of PDH. There is the suggestion that hexokinase II (HKII) levels might fall more rapidly in banded β KO hearts and could contribute to decreased glucose utilization, although it is likely that additional mechanisms are also involved. An association between HKII and VDAC in mitochondria plays an important role in cardioprotection 25. We did not measure mitochondrial localization of HKII in response to banding in β KO hearts. If the decline in HKII in whole heart homogenates parallels a decline in mitochondrial HKII, this could potentially contribute to exacerbated mitochondrial dysfunction.
Our results support a model that PGC-1α and PGC-1β play partially overlapping but somewhat distinct roles in maintaining cardiac mitochondrial energetics in the unstressed heart with both pathways regulating the expression of OXPHOS genes, while PGC-1α predominantly regulates FAO capacity. Both PGC-1 isoforms likely contribute to the maintenance of cardiac function in the context of pressure overload. While it is clear that PGC-1α expression is repressed much earlier in the course of POH than is PGC-1β, it seems that the heart can at least in the short term tolerate a 50% reduction of PGC-1α and complete absence of PGC-1β, with relative preservation of LV function as was observed at 3-weeks. However changes in patterns of substrate utilization and evidence of oxidative stress were already present at this stage, implying that maladaptation as a result of reduced expression of these co-activators likely contributed to the subsequent development of heart failure. The time course for the development of overt heart failure in PGC-1α and PGC-1β KO hearts after TAC are remarkably similar, implying that there is reserve mitochondrial capacity that is capable of maintaining myocardial energetics and contractile function in the face of reduced expression or activity of these transcriptional co-activators. Moreover, our study underscores the potential role of additional mechanisms such as oxidative stress that is exacerbated by deficiency of these transcriptional co-activators that likely contributes to the accelerated transition to decompensated heart failure.
In conclusion, the present study describes the contribution of PGC–1β to mitochondrial function and gene expression in non-stressed hearts and identifies its contribution to maintaining contractile function under pathological increased workload. Thus, modulation of PGC-1 activity may represent a promising target for limiting the transition from pressure overload cardiac hypertrophy to heart failure.
Supplementary Material
Novelty and Significance.
What Is Known?
PGC-1α and PGC-1β are transcriptional co-activators that regulate mitochondrial biogenesis and oxidative capacity.
Pressure overload cardiac hypertrophy and heart failure is associated with impaired mitochondrial function that correlates with repression of PGC-1α expression.
PGC-1α deficient hearts have reduced oxidative phosphorylation (OXPHOS) and fatty acid (FAO) capacity and develop heart failure more rapidly in response to pressure overload.
What New Information Does This Article Contribute?
PGC-1β regulates OXPHOS capacity but not FAO capacity of cardiac mitochondria
PGC-1β deficient hearts rapidly decompensate following pressure overload
PGC-1β deficiency promotes oxidative stress and reduces glucose metabolism in response to pressure overload hypertrophy
PGC-1α and β are critical mediators of mitochondrial biogenesis and oxidative capacity in the heart. Although repression of PGC-1α may contribute to mitochondrial dysfunction in pressure overload hypertrophy (POH) and heart failure, the role of PGC-1β was unknown. Using PGC-1β deficient mice we show that PGC-1β regulates mitochondrial OXPHOS but not FAO capacity, but LV function is maintained. In contrast to PGC-1α deficiency, there is no compensatory increase in mitochondrial metabolism of glucose-derived carbons. Following transverse aortic constriction (TAC) PGC-1β KO hearts more rapidly decompensate to heart failure as a result of increased oxidative stress, decreased cardiac efficiency and failure to upregulate glycolysis and glucose oxidation, despite only modest additional reductions in mitochondrial function. In control animals, TAC initially is associated with induction of PGC-1β that correlates with increased glucose utilization, despite mitochondrial dysfunction. Thus we show for the first time PGC-1β plays a critical role in maintaining antioxidant mechanisms and in activating cardiac glucose utilization in POH, both of which are believed to be important adaptations that prevent the transition from compensated POH to heart failure. Our study underscores that modulation of PGC-1β may represent a novel approach for retarding the transition from compensated POH to heart failure.
Acknowledgments
We thank Deborah Jones, Chase Andrizzi, Alfred P McQueen, Heather A Theobald, Jamie Soto, Joseph Tuinei and Ping Hu, for important technical help during the course of these studies. PGC1β−/− mice were generated and provided by the AstraZeneca Transgenics & Comparative Genomics Department, Mölndal, Sweden.
Sources of Funding
These studies were supported by NIH grant RO1HL73167 to EDA who is an established investigator of the American Heart Association, the British Heart Foundation, MRC Programme Grant and FP7-European Commission (MITIN, HEALTH-F4-2008-223450) to AV-P and a VA Merit award to SEL. TD was a Heisenberg Professor of the German Research Foundation (DFG) and was supported by grants from the DFG (Do602/4-1, 6-1, and 8-1). CR was supported by a student fellowship from the German Academic Exchange Service (DAAD). VZ was supported by a post-doctoral fellowship from the American Heart Association (AHA) Western Affiliates. ARW was supported by NIH Grant 5T32 HL007576 and a post-doctoral fellowship from the AHA Western Affiliates, KP and ABM were supported by scholarships from the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES, Brazil), HB was supported by a post-doctoral fellowship grant from the German Research Foundation (DFG).
Abbreviations
- FAO
Fatty Acid Oxidation
- GLUT1
Glucose Transporter-isoform 1
- GLUT4
Glucose Transporter-isoform 4
- KO
Knockout
- LV
Left Ventricle
- LVSP
LV Systolic Pressure
- LVEDP
LV End Diastolic Pressure
- MVO2
Myocardial Oxygen Consumption Rate
- OXPHOS
Oxidative Phosphorylation
- PC
Palmitoyl Carnitine
- PGC-1α
PPAR Gamma Coactivator 1 alpha
- PGC-1β
PPAR Gamma Coactivator 1 beta
- PPARα
Peroxisome Proliferator Activated Receptor Alpha
- POH
Pressure Overload Hypertrophy
- Pyr
Pyruvate
- RT-PCR
Reverse Transcriptase-Polymerase Chain Reaction
- TAC
Transverse Aortic Constriction
- WT
Wildtype
Footnotes
Disclosures
C. Lelliott is employed by Astra Zeneca. The other authors have no conflicts of interest to disclose.
References
- 1.Meirhaeghe A, Crowley V, Lenaghan C, Lelliott C, Green K, Stewart A, Hart K, Schinner S, Sethi JK, Yeo G, Brand MD, Cortright RN, O’Rahilly S, Montague C, Vidal-Puig AJ. Characterization of the human, mouse and rat pgc1 beta (peroxisome-proliferator-activated receptor-gamma co-activator 1 beta) gene in vitro and in vivo. Biochem J. 2003;373:155–165. doi: 10.1042/BJ20030200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lin J, Tarr PT, Yang R, Rhee J, Puigserver P, Newgard CB, Spiegelman BM. Pgc-1beta in the regulation of hepatic glucose and energy metabolism. J Biol Chem. 2003;278:30843–30848. doi: 10.1074/jbc.M303643200. [DOI] [PubMed] [Google Scholar]
- 3.Fan M, Rhee J, St-Pierre J, Handschin C, Puigserver P, Lin J, Jaeger S, Erdjument-Bromage H, Tempst P, Spiegelman BM. Suppression of mitochondrial respiration through recruitment of p160 myb binding protein to pgc-1alpha: Modulation by p38 mapk. Genes Dev. 2004;18:278–289. doi: 10.1101/gad.1152204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lehman JJ, Boudina S, Banke NH, Sambandam N, Han X, Young DM, Leone TC, Gross RW, Lewandowski ED, Abel ED, Kelly DP. The transcriptional coactivator pgc-1alpha is essential for maximal and efficient cardiac mitochondrial fatty acid oxidation and lipid homeostasis. Am J Physiol Heart Circ Physiol. 2008;295:H185–196. doi: 10.1152/ajpheart.00081.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Garnier A, Fortin D, Delomenie C, Momken I, Veksler V, Ventura-Clapier R. Depressed mitochondrial transcription factors and oxidative capacity in rat failing cardiac and skeletal muscles. J Physiol. 2003;551:491–501. doi: 10.1113/jphysiol.2003.045104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lehman JJ, Kelly DP. Transcriptional activation of energy metabolic switches in the developing and hypertrophied heart. Clin Exp Pharmacol Physiol. 2002;29:339–345. doi: 10.1046/j.1440-1681.2002.03655.x. [DOI] [PubMed] [Google Scholar]
- 7.Sambandam N, Lopaschuk GD, Brownsey RW, Allard MF. Energy metabolism in the hypertrophied heart. Heart Fail Rev. 2002;7:161–173. doi: 10.1023/a:1015380609464. [DOI] [PubMed] [Google Scholar]
- 8.Arany Z, Novikov M, Chin S, Ma Y, Rosenzweig A, Spiegelman BM. Transverse aortic constriction leads to accelerated heart failure in mice lacking ppar-gamma coactivator 1alpha. Proc Natl Acad Sci U S A. 2006;103:10086–10091. doi: 10.1073/pnas.0603615103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lu Z, Xu X, Hu X, Fassett J, Zhu G, Tao Y, Li J, Huang Y, Zhang P, Zhao B, Chen Y. Pgc-1 alpha regulates expression of myocardial mitochondrial antioxidants and myocardial oxidative stress after chronic systolic overload. Antioxid Redox Signal. 2010;13:1011–1022. doi: 10.1089/ars.2009.2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lelliott CJ, Medina-Gomez G, Petrovic N, Kis A, Feldmann HM, Bjursell M, Parker N, Curtis K, Campbell M, Hu P, Zhang D, Litwin SE, Zaha VG, Fountain KT, Boudina S, Jimenez-Linan M, Blount M, Lopez M, Meirhaeghe A, Bohlooly YM, Storlien L, Stromstedt M, Snaith M, Oresic M, Abel ED, Cannon B, Vidal-Puig A. Ablation of pgc-1beta results in defective mitochondrial activity, thermogenesis, hepatic function, and cardiac performance. PLoS Biol. 2006;4:e369. doi: 10.1371/journal.pbio.0040369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hu P, Zhang D, Swenson L, Chakrabarti G, Abel ED, Litwin SE. Minimally invasive aortic banding in mice: Effects of altered cardiomyocyte insulin signaling during pressure overload. Am J Physiol Heart Circ Physiol. 2003;285:H1261–1269. doi: 10.1152/ajpheart.00108.2003. [DOI] [PubMed] [Google Scholar]
- 12.Belke DD, Betuing S, Tuttle MJ, Graveleau C, Young ME, Pham M, Zhang D, Cooksey RC, McClain DA, Litwin SE, Taegtmeyer H, Severson D, Kahn CR, Abel ED. Insulin signaling coordinately regulates cardiac size, metabolism, and contractile protein isoform expression. J Clin Invest. 2002;109:629–639. doi: 10.1172/JCI13946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sena S, Rasmussen IR, Wende AR, McQueen AP, Theobald HA, Wilde N, Pereira RO, Litwin SE, Berger JP, Abel ED. Cardiac hypertrophy caused by peroxisome proliferator- activated receptor-gamma agonist treatment occurs independently of changes in myocardial insulin signaling. Endocrinology. 2007;148:6047–6053. doi: 10.1210/en.2006-1559. [DOI] [PubMed] [Google Scholar]
- 14.Mazumder PK, O’Neill BT, Roberts MW, Buchanan J, Yun UJ, Cooksey RC, Boudina S, Abel ED. Impaired cardiac efficiency and increased fatty acid oxidation in insulin-resistant ob/ob mouse hearts. Diabetes. 2004;53:2366–2374. doi: 10.2337/diabetes.53.9.2366. [DOI] [PubMed] [Google Scholar]
- 15.Boudina S, Sena S, O’Neill BT, Tathireddy P, Young ME, Abel ED. Reduced mitochondrial oxidative capacity and increased mitochondrial uncoupling impair myocardial energetics in obesity. Circulation. 2005;112:2686–2695. doi: 10.1161/CIRCULATIONAHA.105.554360. [DOI] [PubMed] [Google Scholar]
- 16.Boudina S, Sena S, Theobald H, Sheng X, Wright JJ, Hu XX, Aziz S, Johnson JI, Bugger H, Zaha VG, Abel ED. Mitochondrial energetics in the heart in obesity-related diabetes: Direct evidence for increased uncoupled respiration and activation of uncoupling proteins. Diabetes. 2007;56:2457–2466. doi: 10.2337/db07-0481. [DOI] [PubMed] [Google Scholar]
- 17.O’Neill BT, Kim J, Wende AR, Theobald HA, Tuinei J, Buchanan J, Guo A, Zaha VG, Davis DK, Schell JC, Boudina S, Wayment B, Litwin SE, Shioi T, Izumo S, Birnbaum MJ, Abel ED. A conserved role for phosphatidylinositol 3-kinase but not akt signaling in mitochondrial adaptations that accompany physiological cardiac hypertrophy. Cell Metab. 2007;6:294–306. doi: 10.1016/j.cmet.2007.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weibel E. Steriological principles for morphometry in electron microscopic cytology. Int Rev Cytol. 1979;26:235–302. doi: 10.1016/s0074-7696(08)61637-x. [DOI] [PubMed] [Google Scholar]
- 19.Mandarim-de-Lacerda CA. Stereological tools in biomedical research. An Acad Bras Cienc. 2003;75:469–486. doi: 10.1590/s0001-37652003000400006. [DOI] [PubMed] [Google Scholar]
- 20.Boudina S, Abel ED. Mitochondrial uncoupling: A key contributor to reduced cardiac efficiency in diabetes. Physiology (Bethesda) 2006;21:250–258. doi: 10.1152/physiol.00008.2006. [DOI] [PubMed] [Google Scholar]
- 21.Lai L, Leone TC, Zechner C, Schaeffer PJ, Kelly SM, Flanagan DP, Medeiros DM, Kovacs A, Kelly DP. Transcriptional coactivators pgc-1{alpha} and pgc-l{beta} control overlapping programs required for perinatal maturation of the heart. Genes Dev. 2008;22:1948–1961. doi: 10.1101/gad.1661708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leone TC, Lehman JJ, Finck BN, Schaeffer PJ, Wende AR, Boudina S, Courtois M, Wozniak DF, Sambandam N, Bernal-Mizrachi C, Chen Z, Holloszy JO, Medeiros DM, Schmidt RE, Saffitz JE, Abel ED, Semenkovich CF, Kelly DP. Pgc-1alpha deficiency causes multi-system energy metabolic derangements: Muscle dysfunction, abnormal weight control and hepatic steatosis. PLoS Biol. 2005;3:e101. doi: 10.1371/journal.pbio.0030101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arany Z, He H, Lin J, Hoyer K, Handschin C, Toka O, Ahmad F, Matsui T, Chin S, Wu PH, Rybkin, Shelton JM, Manieri M, Cinti S, Schoen FJ, Bassel-Duby R, Rosenzweig A, Ingwall JS, Spiegelman BM. Transcriptional coactivator pgc-1 alpha controls the energy state and contractile function of cardiac muscle. Cell Metab. 2005;1:259–271. doi: 10.1016/j.cmet.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 24.Sonoda J, Mehl IR, Chong LW, Nofsinger RR, Evans RM. Pgc-1beta controls mitochondrial metabolism to modulate circadian activity, adaptive thermogenesis, and hepatic steatosis. Proc Natl Acad Sci U S A. 2007;104:5223–5228. doi: 10.1073/pnas.0611623104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zuurbier CJ, Smeele KM, Eerbeek O. Mitochondrial hexokinase and cardioprotection of the intact heart. J Bioenerg Biomembr. 2009;41:181–185. doi: 10.1007/s10863-009-9209-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.