

Abstract
Hippocampal volume reductions and functional impairments are reliable findings in post-traumatic stress disorder (PTSD) imaging studies. However, it is not clear if and how hippocampal dysfunction contributes to the etiology and maintenance of PTSD. Individuals with PTSD are often described as showing fear responses to trauma reminders outside of contexts in which these cues would reasonably predict danger. Animal studies suggest that the hippocampus is required to form and recall associations between contextual stimuli and aversive events. For example, the hippocampus is critical for encoding memories in which a complex configuration of multiple cues is associated with the aversive event. Conversely, the hippocampus is not required for associations with discrete cues. In animal studies, if configural memory is disrupted, learning strategies using discrete cue associations predominate. These data suggest poor hippocampal function could bias the organism towards forming multiple simple cue associations during trauma, thus increasing the chances of fear responses in multiple environments (or contexts) in which these cues may be present. Here we will examine clinical and animal literature to support a theory of hippocampal dysfunction as a primary contributory factor to the etiology of PTSD, and discuss future research required to test these hypotheses.
1. Introduction
Some of the most robust and significant neural markers associated with post-traumatic stress disorder (PTSD) are altered hippocampal volume and activation differences. The hippocampal complex is critical for encoding emotional memory and modulating appropriate emotional responses to fear relevant stimuli. Here we will review the findings of neural circuit abnormalties in PTSD patients with focus on hippocampal alterations, discuss if hippocampal differences are markers of vulnerability traits or, rather, are linked to alterations post trauma, and describe how these abnormalities might disrupt fear learning processes during trauma exposure and recall. We will also propose future research avenues to test the hypothesis that hippocampal abnormalities contribute to the etiology of this disorder.
1a. PTSD prevalence and symptoms
PTSD is unique among the mental health disorders in that symptoms are induced by exposure to a known environmental stimulus, i.e. a life-threatening or traumatic event. This external stimulus has been incorporated into the DSM diagnostic criteria as Criterion A, defined as having experienced both a traumatic stress (“experienced or witnessed actual or threatened death, injury or threat to the physical integrity of self or others”) and reacted to that experience with “intense fear, helplessness or horror”. Lifetime prevalence rates for experiencing Criterion A events vary widely depending on the country studied (~80% in the U.S. vs. ~20–30% in Germany and Switzerland; Breslau, 2009). However, of those that experience this type of trauma in their lifetime, only a subset will go on to develop chronic PTSD symptoms (~10% in the US; Breslau, 2009; Kessler et al, 2005) suggesting individual vulnerability or risk factors play a role in development of PTSD (Vogt et al, 2007). Also, the conditional risk for developing PTSD varies depending upon factors such as gender and type of trauma, with females and individuals exposed to assaultive violence at highest risk (Breslau, 2009; Galea, Nandi, & Vlahov, 2005; Breslau et al, 1998).
The symptoms that result from exposure to a Criterion A event in vulnerable individuals are intrusive recollections of the traumatic event (Criterion B), avoidance of trauma reminders and emotional numbing (Criterion C), and hyperarousal, e.g. irritability, difficulty sleeping, and increased startle reactivity (Criterion D). If these symptoms persist for longer than 3 months, the condition is referred to as “chronic” (APA, 2000).
These symptoms invoke a significant toll on the individual suffering from the disorder, as well as their families and their place of employment (Schnurr et al., 2009). PTSD is associated with high levels of impairment across social and occupational domains (Alonso, 2004; Druss, 2008) with a work loss index and disability rivaling that of neurological disorders and exceeding that of diabetes (Alonso, 2004). Annual productivity loss from PTSD in the United States is estimated to be over 3 billion US dollars (Brunello, 2001). In contrast to other anxiety disorders, PTSD is independently and significantly associated with both suicidal ideation and suicide attempts (Sareen, 2005; Sareen, 2007). The lifetime prevalence rate for PTSD has been estimated at ~ 7% (Kessler, et al, 2005), and the current treatments are mainly psychotherapy based treatments (e.g. exposure therapy and cognitive therapy), with modest efficacy of pharmacological treatments (Institute of Medicine, 2007). There is a clear need for development of novel therapeutic approaches, which undoubtedly will emerge with better understanding of the etiology of this disorder (Baker et al, 2009, Steckler & Risbrough, this issue).
For a contemporary example of trauma and symptoms associated with PTSD, consider a soldier in Iraq or Afghanistan who experiences attack by an improvised explosive device (IED). Exposure to IED or mortar blasts are a common experience for soldiers deployed to these theatres. Approximately 15% of infantry soldiers experience a significant blast, with ~33% of those going on to develop PTSD (Hoge et al., 2008). IEDs are often hidden in walls, garbage bags, or piles of rubbish along side the road. Following exposure to an IED attack, the soldier may develop a constellation of symptoms consistent with PTSD. For instance, the soldier may experience distressing dreams or recollections of the explosion, intense psychological and physiological reactions when exposed to cues that resemble important aspects of the event (e.g., driving on roads littered with trash), avoidance of such cues or conversations that may trigger memories of the event, a diminished interest in activities or neglect of relationships, hypervigilance for signs of danger, irritability/anger, insomnia, and increased startle reactivity. While we have provided a current, Western-centric example of combat-related PTSD, it is important to note that this example holds across various trauma types (i.e., a female raped by a bald man may experience intense physiological and psychological reactions when later confronted by a different bald man, etc.).
1b. Vulnerability factors and biomarkers associated with PTSD
As stated above, only ~10% of individuals exposed to traumatic stress develop chronic PTSD (Breslau, 2009; Kessler, 2005). Thus, there is considerable variability in trauma stimuli and their perception, as well as differential individual vulnerability to trauma effects. Therapeutic interventions have been limited in part because the pathways that mediate vulnerability to trauma, and hence that contribute to the etiology of PTSD, are not understood.
Factors associated with increased risk for development of PTSD include psychosocial factors and past trauma exposures (Vogt et al, 2007), as well as genetic and epigenetic factors (Afifi et al., 2010; Yehuda & Bierer, 2009; Charney 2004; Crow 2004; Kent et al. 2002; Meaney et al. 2000; Merikangas & Pine 2004; Nemeroff 2004; Rasmusson et al. 2001; Saigh & Bremner 1999). Epigenetic factors that contribute to increased stress responses in adulthood are particularly interesting as these can both be non heritable (e.g. methylation occurring due to early life stress; McGowan et al., 2009) and heritable depending upon the developmental period of epigenetic programming (e.g. in utero methylation occurring in primordial germ cells that is passed on to subsequent offspring; Dunn et al., in press). These factors are beyond the scope of this review but are reviewed in this extensively elsewhere (Kremen et al, this issue; Mehta & Binder, this issue; Skelten et al., this issue).
PTSD has also been associated with abnormal hypothalamic-pituitary-adrenal axis markers (e.g. Bauer et al. 2010, Yehuda 2009) as well as neural circuit abnormalities (see below). In the case of biological markers associated with PTSD however, there is debate over which of these markers are related to a vulnerability traits vs. those that are related to biological adaptations specifically after trauma exposure (e.g. state).
2. Neural Circuit Abnormalities Reported in PTSD
2a. Cortical and subcortical findings
Structural MRI and functional MRI studies in PTSD report differences in volume and activation of limbic circuitry mediating stress responding, fear memory, and emotional modulation (Garfinkel & Liberzon, 2009; Rauch, Shin, & Phelps, 2006). Given its central role in the acquisition of the conditioned fear response, the amygdala is a region of interest in imaging studies with hypotheses of increased function based on the increased anxiety/fear responding in these patients (Shin et al., 2006). Due to its central role in the inhibition of fear responses (Quirk & Mueller, 2008), the medial prefrontal cortex (particularly the ventral region) has also been intensely investigated in this population where it is hypothesized to have reduced function. Importantly, the medial prefrontal cortex is thought to exert a top-down regulatory influence on the amygdala, and thus dysfunction in these areas may be linked. A recent meta-analysis of functional neuroimaging studies in the anxiety disorders supports the notion of dysfunction in prefrontal-limbic areas in PTSD, showing modest amygdala hyperactivation along with more pronounced hypoactivation in the medial prefrontal cortex as well as the rostral and dorsal anterior cingulate cortex (ACC; Etkin & Wager, 2007). Ventromedial PFC activation however has been shown to be increased in PTSD during a working memory task with combat imagery distractors (e.g. Morey, Dolcos, Petty, Cooper, Hayes, LaBar & McCarthy, 2009). For a more in-depth review of frontal dysfunction findings in PTSD see (Aupperle et al., this issue).
Another robust finding in neuroimaging research is reduced gray matter volume in the ACC in patients with PTSD (Yamasue et al., 2003; Corbo et al, 2005; Kitayama et al., 2006). Kasai and colleagues (2008) found that combat-exposed twins with PTSD had lower gray matter volumes in the subgenual ACC compared to their trauma-exposed non-PTSD co-twins. This pattern of finding suggests that subgenual ACC volume reductions are an acquired feature of PTSD, rather than a vulnerability factor. However, in another recent study, Shin and colleagues (2009) used positron emission tomography (PET) to assess resting metabolic activity in the dorsal ACC in veterans with PTSD and their non-combat exposed co-twins. They found that veterans with PTSD and their co-twin both exhibited higher resting metabolic rates than veterans without PTSD and their co-twins. This pattern of results suggests that metabolic abnormalities in the dorsal ACC represents a pre-existing vulnerability factor for the development of PTSD subsequent to trauma. More research is needed to clarify the nature of prefrontal/ACC abnormalities in PTSD, and how they may contribute to the development or maintenance of the disorder.
2b. Hippocampal abnormalities reported in PTSD
i. Structural MRI findings
The suggestion that PTSD may be associated with reduced hippocampal volume has generated a large body of human structural MRI research and been the subject of three recent meta-analyses (Kitayama et al., 2005; Smith, 2005; Karl et al., 2006). Smith (2005) completed a meta-analysis of 13 structural MRI studies comparing adult PTSD patients to matched controls (215 total patients and 325 total controls). Pooled effect size estimates suggested significant bilateral hippocampal volume reduction in PTSD patients compared to controls. Specifically, PTSD patients had, on average, 7.2% lower right hippocampal volume and 7.0% lower left hippocampal volume. These reductions were somewhat attenuated (Right: 4.3%; Left: 4.5%) when comparing PTSD patients to trauma-exposed controls rather than control subjects without trauma exposure. A meta-analysis by Kitayama and colleagues (2005) examined 9 studies comparing adult patients with chronic PTSD (133 total) to healthy controls (148 total), and trauma-exposed controls (53 total). They also confirmed bilateral hippocampal volume reductions in the PTSD patients compared to healthy controls and trauma-exposed individuals without PTSD.
Using an expanded sample of structural MRI studies investigating hippocampal volume abnormalities in PTSD, the meta-analysis by Karl and colleagues (2006) included several moderators including MRI methodology, age, gender, PTSD severity and duration, and medication use. Their results also supported findings of smaller hippocampal volumes in PTSD patients relative to healthy controls. Bilateral hippocampal volumes were also reduced in PTSD patients relative to trauma-exposed non-PTSD subjects, though this was evident only in studies using samples with severe PTSD, suggesting a relationship between hippocampus volume and PTSD severity. This relationship has been reported in some previous studies (e.g., Gilbertson et al., 2002) though not in others (e.g., Pederson et al., 2004). However, a positive correlation between effect sizes and PTSD severity in this meta-analysis supports the relationship (Karl et al., 2006). Interestingly, hippocampal volume differences were not observed in studies utilizing pediatric samples (Karl et al. 2006), suggesting detectable volumetric abnormalities may be more specific to adult PTSD.
Thus, three recent meta-analyses have confirmed the presence of volumetric reductions in PTSD subjects relative to healthy controls as well as trauma-exposed non-PTSD subjects. There is some suggestion that these differences are more pronounced with more chronic, severe PTSD, and may also be present in trauma exposed individuals without PTSD, suggesting it may be a result of trauma as opposed to a pre-existing vulnerability factor. Recent research has begun to investigate the structure of individual hippocampal subfields in PTSD. Wang et al. (2010), using high-resolution MRI, found that the disorder was associated with specific volume reductions in the cornu ammonis 3 (CA3)/dentate gyrus (DG) subfields. These subfields are specifically involved in the encoding and retrieval of spatial memory, as well as the encoding of geometric representations of the environment (Colgin et al., 2008; Kesner, 2007), a finding of importance to our later discussion of fear learning and PTSD etiology.
ii. Magnetic Resonance Spectroscopy findings
Magnetic Resonance Spectroscopy (MRS) is an MRI-associated method of determining metabolite concentrations in predefined areas of the brain. Specifically, MRS collects information on concentrations of N-Acetylaspartate (NAA), creatine (Cr), and choline (Cho), indicators of neuronal integrity. Specifically, NAA is found localized to mature neurons, and is not found in glia (Karl & Werner, 2010; Urenjak et al, 1993). Reductions in NAA have been interpreted as evidence of neuronal death, glial proliferation, and metabolic dysfunction, and are thought to be a general marker of altered neuronal structure/function (Karl & Werner, 2010; Martin et al, 2001a; Schuff et al, 2001). Though these interpretations are not without controversy (see Sullivan et al., 2001; Arnold et al., 2001; Marting et al., 2001b), NAA alterations have been reported in a number of pathologic and psychiatric conditions, including multiple sclerosis, ischemia, schizophrenia, depression, bipolar disorder, and cognitive decline, strongly suggesting an association with neuropathology (Glodzik-Sobanska et al, 2007; Olvera et al, 2007; Szulc et al, 2007 Sijens et al, 2006; Ross et al, 2005; Gruber et al, 2003).
In PTSD patients, studies have shown hippocampal dysfunction in the form of decreased NAA even in the absence of structural abnormalities (Freeman et al., 2006; Schuff et al., 2001). Karl and Werner (2010) conducted a recent meta-analysis of 16 total MRS studies, 12 of which provided data specific to the hippocampus. They found consistent bilateral hippocampal NAA reductions in PTSD as compared to trauma-exposed non-PTSD subjects, but not when compared to healthy controls. The authors suggested that these findings might indicate resilience, indexed by higher pre-trauma NAA levels, in the trauma-exposed non-PTSD controls, relative to a mixed sample of vulnerable and non-vulnerable individuals in the healthy control groups. This explanation assumes that NAA levels are a pre-existing vulnerability or protective factor, and not related to stress-induced changes in hippocampal function.
In support of this notion, Siegmund and colleagues (2009) measured NAA levels and fear behaviors pre and post-“trauma” (i.e. footshock exposure) in a mouse model of PTSD. Hippocampal NAA levels were assessed before the mice underwent a procedure in which they were placed in a conditioning chamber and received an unsignaled foot-shock. The mice were then re-exposed to the conditioning chamber and a neutral chamber on weeks 4, 5, 18, and 32 post-shock and their fear behavior (freezing) in each chamber was assessed (see Box 1 for a brief review of methods for studying context fear in animals). NAA levels in the left dorsal hippocampus were negatively correlated with fear behaviors exhibited in the conditioning chamber (i.e. contextual fear), the durability of this behavior over time, as well as freezing in the neutral chamber (interpreted as generalized fear). These findings support the hypothesis that pre-existing variation in hippocampal function (neuronal integrity) is a vulnerability factor for the development of enduring anxiety-like behaviors following exposure to stress. This conclusion is somewhat contradictory to the structural MRI findings discussed above, and suggests that subtle neuronal abnormalities may be present in the absence of detectable structural abnormalities. Whether these abnormalities represent state vs. trait (or vulnerability) factors will be discussed below.
Box 1. Methods for Studying Fear Learning in Animals.
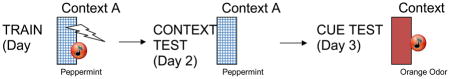
Animal (and human) models of aversive learning such as context and cued fear conditioning have played a large role in identifying potential mechanisms in the etiology of PTSD and other fear learning disorders (see Siegmund & Wotjak, 2007; Rau, DeCola & Fanselow, 2005, Johnson et al. this issue). These tasks attempt to model cardinal symptoms of PTSD - the intrusive recollection of the trauma as well as fear responses to trauma-related cues.
Contextual learning in animals is typically studied via fear conditioning paradigms (which are highly relevant to memory processes in PTSD). Fear conditioning is a simple form of Pavlovian or classical associative conditioning in which an animal learns that one or more stimuli in the environment reliably predict an aversive event. This association results in robust, long-lasting memory (Sweatt, 2003) as measured by fear responses when presented with the training environment or cues. Typically, a fear conditioning protocol in rodents involves the following sequence of events. A rodent is first placed in a novel context (e.g., a conditioning chamber with a shock grid floor) and allowed to explore for a set period of time (e.g., 2 min). The rodent is then presented with a salient, novel stimulus (e.g., a tone) for several seconds. During the last second of the tone, the rodent receives a mild foot-shock. Foot-shock, the unconditioned stimulus (US), elicits numerous reflex fear responses, including defecation, elevated blood pressure, ultrasonic vocalizations, and freezing (Fanselow, 2000). Freezing is defined as the absence of all movements except that which is required for respiration (Fanselow, 2000) and is a common defensive behavior of prey animals under threat. These tone-shock pairings can be repeated several more times within the conditioning session, with a brief interval (approximately 1 min) between pairings. Rodents tested in this conditioning protocol learn two associations. First is the association between the training environment (regardless of whether it has been encoded elementally or conjunctively) and shock (US). Once this association is formed, subsequent visits to the chamber itself will invoke conditioned fear responses (such as freezing), even in the absence of a shock. This environment-shock association is called contextual fear conditioning. The second learned association is that the tone also predicts shock. Thus, even when the tone is presented in a novel environment, the rodent will freeze. This tone-shock association is referred to as auditory, or cued, fear conditioning. In both context and cued fear conditioning, retention of the learned associations is assessed by measuring fear responses, such as freezing, following conditioning. It is important to use a novel environment when measuring retention of the tone-shock association in order to dissociate cued conditioning from context conditioning. Importantly, this simple tone-US pairing is able to elicit conditioned fear in many types of environments, and is thus context independent. That is, the animal learns the association between tone and shock, not that the tone predicts shock only in a certain time or place. This is a form of elemental learning. The presence of the fear response during retention tests reflects the extent to which context-US associations have been consolidated. Once formed, these memories can persist for days, weeks, or years (Goelet et al., 1986).
iii. Functional Imaging Findings
To date, few functional neuroimaging studies have examined hippocampal function in PTSD (Shin et al., 2006). Shin and colleagues (2004) used positron emission tomography (PET) to measure hippocampal regional cerebral blood flow (rCBF) in both PTSD patients and controls during performance of an explicit memory task. Relative to controls, PTSD patients displayed higher rCBF in the bilateral hippocampus, which was positively associated with symptom severity, though the groups did not differ on recall accuracy. Additionally, Osuch and colleagues (2001) found that hippocampal rCBF measured in PTSD patients experiencing script-induced flashbacks while undergoing fMRI was positively correlated with severity. In a recent study employing functional MRI measures of hippocampal blood oxygen level-dependent response (BOLD), PTSD patients appear to show greater hippocampal activation when successfully encoding negative imagery compared to trauma matched controls, as well as greater coupling of hippocampal and amygdala activiation when viewing negative images (Brohawn, Offringa, Pfaff, Hughes & Shin, 2010). However when successfully encoding images that are trauma-related, PTSD subjects have been reported to exhibit reduced hippocampal BOLD responses (Hayes, LaBar, McCarthy, Selgrade, Nasser et al, in press). This study also found that reduced hippocampal activation was associated with increased arousal symptoms. These findings suggest an interesting possibility that hippocampal activation is high when presented with novel aversive stimuli but low when presented with trauma-related stimuli in PTSD patients. Finally, hippocampal activation was reduced in PTSD subjects compared to controls when completing a spatial memory task (virtual-reality Morris water maze), and was negatively correlated with PTSD severity (Astur et al., 2006). These subjects performed as well as controls in the task, suggesting that they may have been utilizing non-hippocampal strategies to complete the task.
A note of caution is that in fMRI studies can be difficult to interpret what the rCBF/BOLD response differences means in terms of task performance or stimulus processing. Increases in rCBF/BOLD response can be interpreted as a lack of efficiency and that this region is “working harder” than the same region in the comparison group or simply that it is more engaged by the task. Low levels can be interpreted that the region is either not functionally engaged in the task or the opposite, that it is engaged and working with more “efficiency”. rCBF/BOLD response to the various tasks in the above studies showed inconsistent directional changes compared to controls. However these tasks engage different hippocampal processes (response to trauma cues, spatial navigation learning, declarative memory recall) and thus are likely engaging different hippocampal neural circuits (e.g. hippocampus-cortex vs. hippocampus-amygdala). Nonetheless, these findings do suggest that PTSD is characterized by abnormalities in hippocampal rCBF/BOLD response. More functional imaging research combined with behavioral performance assessments are needed to further clarify the nature of these differences.
3. Abnormalities in Hippocampus-dependent processes
A substantial body of research in both animals and humans has implicated the hippocampus and parahippocampus in declarative memory (Squire, 1992; Eichenbaum, 2000). Declarative memory refers to the ability to recall facts and events. It is commonly divided into semantic memory (memory for specific facts) and episodic memory (memory for specific events). The observation of hippocampal volumetric and neurochemical abnormalities in PTSD has led to the suggestion that declarative memory may be affected in this disorder (Woodward et al., 2009). Indeed, patients with PTSD experience problems with declarative memory including loss of specific narrative memory for the traumatic event as well as intrusive, inappropriate, emotion-laden memories (Bremner, 2006; Brewin, 2010).
Many neuropsychological studies have found deficits in verbal declarative memory in patients with PTSD (Gilbertson et al., 2006; Buckley et al., 2000; Elzinga & Bremner, 2002; Brewin, 2001; Golier & Yehuda, 1998; Bremner et al., 1993; Bermner et al., 1995; Gilbertson et al., 2001; Jenkins et al, 1998; Moradi et al., 1999; Roca & Freeman, 2001; Uddo et al., 1993; Vasterling et al., 1998; Vasterling et al., 2002; Yehuda et al., 1995; Barrett e al, 1996; Gil et al., 1990; Sachinvala et al., 2000; Golier et al, 1997), though some conflicting findings have been reported (Stein et al., 1999; Zalewski et al., 1994). Brewin and colleagues (2007) reviewed 27 studies on verbal and/or visual memory differences for emotionally neutral material between PTSD and healthy controls. The results consistently supported a small to moderate effect for poorer performance on verbal as opposed to visual memory in PTSD subjects. This pattern of results suggests that declarative memory is an important link between hippocampal abnormalities and PTSD symptomatology. However, Woodward et al. (2009), while observing verbal declarative memory deficits in PTSD patients, failed to find any correlation between these deficits and hippocampal volume. This is surprising given that PTSD patients in this sample showed significant volume reductions relative to controls in the hippocampus, parahippocampus, and total cerebral cortex (Woodward et al., 2009). Further research is needed to clarify the relationship between declarative memory deficits in PTSD and specific brain volumes.
4. Genetic Risk Factors Related to Hippocampal Function in PTSD
To date, there are few research studies linking candidate genetic risk factors with hippocampal abnormalities in PTSD. Animal and human studies suggest that mutations in the BDNF (brain-derived neurotrophic factor, a member of the neurotrophin family of polypeptide growth factors) can influence acquisition of fear learning and extinction (for a recent review see Frielingsdorf et al., 2010). A methionine in place of a valine in position 66 of the prodomaine of BDNF (Val66Met) has been linked with poor episodic memory and low hippocampal activity in healthy controls (Dempster et al., 2005; Hariri et al., 2003), and is also linked to reduced hippocampal gray matter in subjects exposed to early life stress (Gatt et al. 2009). Although this variant has been linked to depression in some studies, there is not suffient evidience to indicate it is linked to PTSD (Frustaci et al., 2008; Zhang et al., 2006). Animal models suggest that stress alters BDNF expression levels in the hippocampus via DNA methylation (Roth et al., in press; Kozlovsky et al., 2007), thus it is possible that stressors that induce epigenetic modificaiton of BDNF expression could contribute to either risk for development or maintenance of PTSD symptoms.
Recently a mutation in the estrogen response binding element of the pituitary adenylate cyclase-activating polypeptide receptor (PAC1) gene was associated with PTSD in women (Ressler et al. 2011). Interestingly this receptor is involved in hippocampal plasticity (Yang et al. 2010), but it is unknown if this mutation is linked to hippocampal function or volume in humans. Finally, there has been recent evidence that low expression of FKBP5, a cochaperone peptide that inhibits nuclear translocation of bound glucocorticoid receptors, is linked to risk for PTSD in subjects with early life stress (see Mehta and Binder review in this issue). There are no studies to our knowledge examining the relationship between FKBP5 and hippocampal function or structure in humans, however studies in animals suggest that low FKBP5 expression in hippocampus is linked to reduced short term memory (Soontornniymokij et al., 2010). Thus some candidate genes that alter hippocampal function are linked to PTSD, however a mechanistic link between these genes, hippocampal structure/function and PTSD has not been established. For a more extensive review of genetic risk factors for PTSD see Cornelius et al. (2010).
5. Hippocampus abnormalities: state or trait?
In order to understand how hippocampal abnormalities contribute to development and/or maintenance of PTSD, an important question is whether this reduced volume is a trait in vulnerable individuals or, rather, a manifestation of the disease state after experiencing trauma. Recent research is beginning to support a conceptualization of these abnormalities as a pre-existing vulnerability factor for development of the disorder. Gilbertson and colleagues (2002; 2007) examined hippocampal volume and function in a sample of monozygotic twins discordant for trauma exposure. In the first study, the authors compared hippocampal volume, as assessed by structural MRI, across twins with and without PTSD, and also correlated volume with PTSD severity in the exposed twin (Gilbertson et al., 2002). Findings supported smaller, predominately right, hippocampal volume in severe PTSD vs. trauma-exposed non-PTSD subjects. Interestingly, twins of severe PTSD subjects showed hippocampal volumes comparable to their brothers and significantly lower than both trauma-exposed non-PTSD patients as well as their non-trauma-exposed twins. Disorder severity in PTSD subjects was negatively correlated with both their own hippocampal volumes and with those of their trauma-discordant twin. Thus, hippocampal volume may be a vulnerability trait that contributes to the development of PTSD upon trauma exposure.
In a follow up study, Gilbertson and colleagues (2007) tested the same subjects in a measure of configural cue processing, a hippocampus-dependent function related to processing of spatial cues (described in detail below). PTSD subjects showed significantly impaired performance on the task relative to trauma-exposed non-PTSD subjects. This impaired performance was also reflected in the trauma-unexposed twins of the PTSD subjects. These deficits were significantly related to hippocampal volume in both twins and PTSD severity in the PTSD twin.
Further support for hippocampal abnormalities as a vulnerability factor for disordered fear processes comes from the study by Siegmund and colleagues (2009) mentioned above. While human studies must infer from post-hoc twin studies, animal research allows for the direct manipulation of a traumatic event following pre-trauma hippocampal measurements. Indeed, in Siegmund et al. (2009), pre-trauma differences in hippocampal integrity, as measured by MRS, predicted the development of PTSD-like symptoms.
A recent human structural MRI study, while replicating the finding of smaller hippocampal volume in chronic PTSD patients, found no differences between Gulf War veterans without PTSD and those who had successfully recovered from the disorder (Apfel et al., 2011). The authors suggest that this pattern of findings may indicate that low hippocampal volume is a state factor. However, the cross-sectional design of the study precludes any causal inference in this regard. It is also possible, as the authors mention, that measurable structural abnormalities are a trait factor predisposing individuals toward a more severe, treatment resistant form of the disorder, or that hippocampal neurogenesis is a mediating factor in successful recovery. Longitudinal imaging studies are necessary to disentangle these competing hypotheses. Ongoing large prospective studies, such as the Marine Resiliency Study (Veteran’s Affairs) and Army STARRS (National Institute of Mental Health) will be critical for indentifying vulnerability factors such as hippocampal function in the etiology of PTSD.
Support for the alternative idea that hippocampal reductions manifest only after trauma (i.e. “state”) is predominantly based on evidence for stress- and glucocorticoid signaling induced reductions of hippocampal volume (for review see Conrad, 2008; Bremner, 1999). Stress induces acute release of glucocorticoids via activation of the Hypothalamic-Pituitary-Adrenal (HPA) axis. The hippocampus has a large number of glucocorticoid receptors and plays an important role in negative feedback to shut off HPA axis activation (Sapolsky, 2003). Severe and chronic stress, with concurrent elevations in circulating glucocorticoids has been shown to induce hippocampal atrophy in rodents and primates (Magarinos et al. 1996; Sapolsky et al. 1990; McEwen et al. 1992; Sunada et al. 1995). These effects of stress on hippocampal volume have also been shown to result in poor hippocampal-dependent memory performance (Luine et al. 1994). In humans, high cortisol levels (e.g. Cushing’s Disease) are associated with low hippocampal volume and cognitive deficits, which are reversed with successful treatment (Starkman et al., 2003). Based on these studies it has been hypothesized that that trauma exposure induces HPA axis dysregulation, resulting in hippocampal atrophy in vulnerable individuals (Elzinga and Bremner, 2002). Measures of circulating cortisol levels in PTSD patients have been inconsistent however, predominantly with reports of normal or low levels of cortisol and increased negative feedback (for review see Yehuda, 2009). Thus there is not a clear link between dysregulation of cortisol and the hippocampal abnormalities observed in PTSD. Recent findings of alterations in second messenger systems involved in glucocorticoid signaling (Binder, 2009) suggest that, although glucocorticoid levels may be relatively normal, patient’s ability to regulate signaling at the cellular level may be impaired. It remains to be determined if these downstream modifiers of glucocorticoid signaling are linked to the hippocampal abnormalities reported in PTSD patients. Taken together evidence supports both the state and trait hypothesis of hippocampal abnormalities in PTSD. Nevertheless, it is reasonable to ask how these clear circuit abnormalities support development and/or maintenance of PTSD symptoms.
5. Hippocampal abnormalities can explain PTSD symptomatology
5a. Brewin’s Model of Intrusive Memory/Flashbacks
Brewin and colleagues (1996, 2001, 2010) have developed a dual representation model of intrusive memories in PTSD involving two complementary forms of memory, one of which is dependent upon the hippocampus. The hippocampal-dependent memory is termed “verbally-accessible memory” or “context representation” (C-Rep, Brewin et al., 2010). Under ordinary circumstances, C-reps are comprised of the spatial context for a given event, which is encoded as allocentric (view-independent) representations of space in the hippocampal and parahippocampal regions of the medial temporal lobe (MTL). During recall, this allocentric representation is translated to an egocentric (view-dependent) frame of reference in the posterior parietal cortex with head-direction input from Papez’s circuit.
The second form of memory is a sensation-based representation of the same event termed S-reps (also termed situational-accessible memory). S-reps include autonomic markers of affective valence, essentially amygdala and insula-mediated conditioned fear responses to trauma stimuli. Theoretically, S-reps may be activated via a bottom-up pathway triggered by environmental or internal cues reminiscent of the event, or via top-down activation of higher-level mental representations of the event.
The crux of dual representation theory is the integration of C-reps and S-reps in the precuneus (located in the medial longitudinal fissure of the superior parietal lobe) to provide an accurate mental representation/imagery for the event. When an S-rep is activated, perhaps by an environmental cue, its corresponding C-rep is also activated. The integration of the S-rep with its corresponding C-rep allows for the event to be placed in the appropriate semantic and autobiographical context and prevents it from being re-experienced in the present, as well as allowing for top-down control of the S-rep through connections between the prefrontal cortex and MTL. Thus, reminders (environmental cues) of an aversive event trigger an appropriate memory of a past event, and not the perception of immediate threat in the present context. Interestingly, there is some functional neuroimaging evidence that the precuneus is less active during memory encoding in PTSD patients relative to controls (Gueze, Vermetten, de Kloet, & Westenberg, 2008) and that precuneal activation is positively related with a tendency to produce false recall of memory for trauma information (Hayes et al, in press). More research is needed however to clarify the role of precuneal dysfunction in PTSD. For example it is not clear if there is an abnormality in this region per se, or if reduced activity is due to abnormalities in upstream neural circuits (e.g. hippocampus).
According to Brewin and colleagues (2010), intrusive memories associated with PTSD are associated with strong S-reps for a traumatic event in conjunction with relatively impoverished or absent C-reps, due to decreases in hippocampal function. When an S-rep for an aversive event is activated, perhaps by an external cue, an adequate C-rep is not concurrently activated to place the S-rep in its appropriate context. This reduction in C-rep strength results in both the event/threat being experienced as occurring in the present, and in the failure to activate appropriate top-down control over autonomic arousal associated with the event. In this situation, the individual will respond to trauma-related cues as if the threat is imminent, regardless of the actual spatiotemporal context in which these cues are placed. Brewin and colleagues did not speculate if deficiencies in C-rep are a trait of vulnerable individuals or are due to post-trauma pathology to the hippocampus and related structures.
Research supporting the dual representation model of intrusive memory is still in its early stages. Bisby and colleagues (2009, 2010) have conducted 2 studies using alcohol (0.4 and 0.8 g/kg) to putatively perturb hippocampal functions while the subjects viewed a video of horrific scenes (e.g. traffic accidents). Alcohol consumption did increase subsequent self reports of intrusive memories of the horrific scenes in the following days (interpreted as an increase in S-reps), as well as impair subjects in task of allocentric memory (a task of requiring hippocampal function). The authors argued that these findings supported the idea that spontaneous aversive memories (e.g. of the traffic accident scenes) are supported by an imbalance between intact S-reps and disrupted C-reps. However, these studies contain important limitations. First, frequency of spontaneous memories could only be measured by self-report. Second, alcohol consumption is not a selective manipulation of hippocampal function, thus leaves the question of a hippocampal specific role in C-reps vs. S-reps largely untested. Additionally, as the authors note, it is unclear to what extent viewing the video is an appropriate analog for real-life trauma (Bisby et al., 2010). Future studies seeking to test this theory would benefit from employing fear learning paradigms that directly measure fear (e.g. skin conductance changes, startle reactivity) as opposed to self-report. Such paradigms are validated probes of the neurocircuitry underlying emotional memory, and may be more representative of aversive experience (e.g. shock) and hence aversive association learning. These paradigms could be employed across subjects with high and low hippocampal volume (and/or high and low performance on hippocampal mediated tasks), to directly ask the question of how hippocampal volume may affect C-rep vs. S-rep measures. A complementary approach would be the employment of animal models in which specific hippocampal manipulations can be employed (see below).
5b. Rudy Model of Context Representations
As discussed above, one of the hallmark symptoms of PTSD patients is inappropriate fear responses to reminders of trauma in environments (or contexts) in which these cues lack predictive meaning (e.g. the soldier who exhibits fearful and avoidant responses to litter on the street after he/she returns to the US, a context in which IEDs are no longer predicted). This deficient contextual control of fear to internal or external trauma cues could contribute to both physiological symptoms of fear/anxiety (and possibly intrusive memories) and be an important functional consequence of the reported hippocampal abnormalities in PTSD patients. Work in animals by Rudy and colleagues (2004 Rudy and colleagues (2009) supports the notion that the hippocampus is critical for contextual control of conditioned fear responding.
The extent to which a memory for a traumatic event is recalled depends upon a number of factors including 1) the form in which the features associated with the aversive event are encoded and 2) the relative strength of the association between these features and events. Rudy and colleagues suggest that a given memory for an event can occur as a result one of two ways (forms) in which the features are initially represented in the brain (for review see Rudy et al., 2004; Rudy, 2009). In the first form, features (or elements, such as the tactile, visual, odor, spatial, temporal stimuli) present at the same time as the aversive event are encoded individually. In this view, referred to from here on as elemental strategy, each element of the environment or “context” in which the trauma occurred subsequently becomes independently associated with the aversive event, and thus presentation of each specific element alone has the potential to induce a conditioned (fear) response. In the second form, a conjunctive representation comprised of many individual elements present in the environment is encoded as a whole. In this case the individual elements comprising the environment are bound into one representation that defines a place (i.e. context) where the event occurs (Rudy et al., 2004), and a conditioned response would only occur in the presence of an environment with stimuli that matches the full representation, with no single cue sufficient to induce a conditioned response. Conjunctive representation of the context is also referred to as a cognitive map (O’Keefe and Nadel, 1978), hierarchical representation (Nadel and Willner, 1980), unitary representation (Fanselow, et al., 1993), or configural representation (Sutherland & Rudy, 1989; Rudy & O’Reilly, 1999), although in some of these views the combined singular representation of multiple cues are also bound in a particular spatial and temporal construct, whereas conjunctive strategies stresses that the original spatial configuration is not essential, only that a majority of the original salient contextual elements are present (see figure 1). For a brief review of context fear conditioning procedures used in rodents, see Box 1.
Figure 1.

Schematic representation of the role of hippocampal function in context fear memory. Here we suggest that such a shift to a predominantly elemental strategy would thus allow elemental cues to have a much larger role in behavioral responses to the environment, with each discrete cue encoded during trauma able to induce conditioned fear responses across multiple contexts. The left side of the figure demonstrates how normal hippocampal function allows for the formation of a conjunctive context representation consisting of a combination of individual elements. This conjunctive representation is then associated with the traumatic event (in this case an exploding grenade). Upon later exposure to a single element of the original context (in this case the garbage bag), no fear response is triggered. The right side of the figure demonstrates how impaired hippocampal function precludes formation of a conjunctive representation. Instead, each individual element of the context is independently associated with the traumatic event. Due to this single-element association, later exposure to only the garbage bag (independent of other contextual elements) is then sufficient to trigger a fear response.
i. Evidence for hippocampal involvement in context representations
Evidence suggests that conjunctive and elemental forms of memory rely upon different neural substrates. Whereas elemental associations rely upon the neocortex, the hippocampus is required for conjunctive associations (discussed further in 5b.ii; Fanselow, 2000; Rudy et al., 2002; 2004). Although both associative strategies can result in measurable contextual fear conditioning due to the either individual elements in the environment or the conjunctive representation associated with an aversive event, it has been suggested that the rate of learning when the neocortex is involved (elemental) is slower than when the hippocampus is involved (conjunctive) (O’Reilly and Rudy, 2001; Wiltgen et al. 2006). There is also evidence that these different representational strategies compete with one another, with the conjunctive strategy typically dominating the formation of elemental representations (Rudy, 2004; 2009). This last point is particularly relevant to our and other’s suggestion that hippocampal dysfunction could thus bias learning strategies towards discrete (elemental) associations between environmental features (stimuli) and the aversive event, and in relation to PTSD, bias fear learning and recall towards simple associations over conjunctive or contextual associations.
The clearest evidence of hippocampal involvement in the formation of a conjunctive representation for an event comes from examination of the phenomena known as the context preexposure facilitation effect (CPFE) (Rudy et al., 2002). CPFE occurs when pre-exposure to a fear conditioning chamber is combined with an immediate shock (Rudy et al., 2002). As the name suggests, in the immediate shock protocol a rat is presented with a single shock immediately upon placement in the conditioning chamber. It is well established that a single, immediate shock results in minimal contextual fear learning, a finding referred to as the immediate shock deficit (Fanselow, 1986; Fanselow, 1990). If, however, a rat is placed in the conditioning chamber for several minutes one day prior to the immediate shock session (context pre-exposure), then the rat will exhibit substantial amounts of context fear memory (as measured by the amount of freezing behavior) and the immediate shock deficit will be eliminated. This occurrence is known as CPFE (Fanselow, 2000; Kiernan and Westbrook, 1993; Rudy et al., 2002; Rudy and O’Reilly, 2001). Importantly, the CPFE depends upon the acquisition of a conjunctive, rather than an elemental, representation of the context (Rudy and O’Reilly, 1999). That is, whereas preexposure to the context in which the shock occurs (i.e. an illuminated rectangular chamber with grid flooring) facilitates fear learning, preexposure to the individual features of the context (i.e. a rectangular chamber, or a grid floor or an illuminated, non-rectangular, chamber) does not (Rudy and O’Reilly, 1999). Findings from hippocampal lesion and pharmacological studies indicate that the hippocampus is critically involved in the conjunctive-dependent CPFE. For example, neurotoxic dorsal hippocampal lesions prior to pre-exposure (Rudy et al., 2002) or infusion of anisomycin (a protein synthesis inhibitor) into the hippocampus following context pre-exposure (Barrientos et al., 2002; Rudy and Matus-Amat, 2005) significantly attenuate the facilitation effect when long-term retention is tested. Transient dorsal hippocampal inactivation by muscimol before context preexposure has also been shown to disrupt CPFE (Matus-Amat et al., 2004). Similar results have been reported with transient inactivation prior to the immediate shock and prior to the test for context fear expression (Matus-Amat et al., 2004; Matus-Amat et al. 2007). Taken together, the CPFE studies indicate that conjunctive representations of the context rely upon the hippocampus.
Recent findings using a novel paradigm also support the notion that the hippocampus plays a critical role in the conjunctive encoding of a context and, furthermore, when the hippocampus is damaged via lesion or temporary pharmacological inactivation, encoding can only occur via an elemental strategy (Iordanova et al., 2009). Specifically, Iordanova and colleagues first demonstrated that a “what-where-when” paradigm (a rodent model of episodic memory in humans) in which rats learn that an auditory stimulus is played in a “spotted” chamber in the morning (as opposed to a clicker played in a checkered chamber in the afternoon)), requires conjunctive processing (i.e. the association of multiple elements, in this case chamber type, time and auditory stimulus into one predictive cue, Iordanova et al., 2009). Either excitoxic lesions of the hippocampus prior to learning of this task or muscimol inactivation of the hippocampus prior to retrieval significantly impaired performance (Iordanova et al., 2009). Rats with hippocampal damage performed normally in tasks which only required elemental associations (e.g. learning simple associations between “what-where” or “what-when”) (Iordanova et al., 2009). The hippocampal findings are consistent with the findings from the CPFE described above and strongly suggest that the hippocampus is necessary for retrieval of conjunctive, but not elemental, associations (memories). Thus, these findings suggest that elemental strategies remain intact in the presence of hippocampal dysfunction, and it is reasonable to assume that elemental learning will thus predominate as the associative learning strategy.
ii. Conjunctive representation deficits and the etiology of PTSD: a hypothesis
Given that individuals who develop PTSD subsequent to trauma may have predisposing hippocampal abnormalities and possibly deficits in hippocampal-dependent memory (Gilbertson et al., 2002; 2007; Siegmund et al., 2009), it is logical to consider that difficulty forming conjunctive representations of context may play a functional role in PTSD development and/or maintenance. This difficulty would leave individuals potentially more dependent upon an elemental strategy when encoding and recalling the separate features of the context and subsequently forming associations between the features and threat. That is, rather than associating threat with the context as a whole, these individuals would associate threat with individual elements of the context, independent upon their relation to one another. An individual with intact conjunctive/configural abilities might encode a representation of a roadside bomb explosion in association with multiple elements of the context as a whole (trash bag on the ground next to the humvee in the Iraqi marketplace in the sweltering desert), and a certain number of individual elements must be present on subsequent occasions to reach threshold of similarity to the conjunctive memory and activate a fear response. In contrast, a vulnerable individual might encode the same representation in association with individual elements rather than the whole, and thus one highly salient individual element (i.e., the trash bag) may be able to subsequently trigger a conditioned fear response in and of itself without the presence of any other contextual cues (humvee, marketplace, desert). Thus, these individuals may respond with fear to stimuli outside of the threat context (i.e., trash bag/litter on the roadside in the United States). Interestingly, as indicated in section 2b.1., PTSD patients exhibit the most pronounced volume reduction in the CA3/DG subfield of the hippocampus (Wang et al., 2010). This region of the hippocampus has been implicated in particular in context memory (e.g. McHugh & Tonegawa, 2009). These data suggest that the hippocampal abnormalities in PTSD could specifically impinge on this form of conjunctive memory function.
As we indicated above, there are two schools of thought on whether hippocampal volume differences are a trait or a state effect in PTSD individuals. This distinction is important to consider when developing a learning model of PTSD etiology. That is, could such a model account for only one effect or can it account for both? Animal studies suggest that the hippocampus contributes to both the encoding and the retrieval of context fear memory, thus in PTSD, either trait dysfunction before trauma or subsequent hippocampal dysfunction after trauma can disrupt use of context/configural fear memory. Early lesion studies suggest that both pre- and post-training hippocampal lesions severely impaired contextual fear memory, while leaving cued fear memory intact (Kim and Fanselow, 1992; Phillips and LeDoux, 1992; Young et al., 1994). Pharmacological and genetic disruption of hippocampal signaling also produces selective effects on contextual fear memory (Young et al., 1994; Bast et al., 2001; Wallenstein & Vago, 2001; Quinn et al., 2005; Nakashiba et al., 2008). These data suggest that hippocampal disruption either before or after fear learning will disrupt contextual fear, hence both trait hippocampal dysfunction pre-trauma and post-trauma hippocampal dysfunction could result in poor contextual fear memory.
More recent studies however have suggested that pretraining lesions of the hippocampus do not always result in context fear memory deficits. These studies have found that in the absence of the hippocampus, alternative regions (neocortex) can subserve context memory (Frankland et al., 1998; Wiltgen et al., 2006; Maren et al., 1997). Along these lines, a recent report suggests that while both hippocampal and extrahippocampal regions can subserve context memory, the hippocampal-dependent memory usually predominates (Biedenkapp and Rudy, 2008). For example, when alternative non-hippocampal regions are used, they may be less efficient at forming context memory (perhaps due to use of an elemental strategy) – indeed in instances where pretraining hippocampal lesions do not result in context fear memory deficits, more training trials (i.e. more time in the context) are needed in order to form context-event (shock) association. At first glance, these data contradict our argument that hippocampus is necessary for contextual fear memory using a configural strategy. However, a critical limitation to these studies is that it was not tested if hippocampal lesioned animals were relying on conjunctive representation of the context or if they were simply learning a simple (elemental) association with a few discrete elements within the context (e.g. the shock grid). Hence, future studies should determine if the context as a whole or just discrete elements of the environment are responsible for intact contextual fear in lesioned animals. This distinction is critical to test our hypothesis that when conjunctive memory formation is disrupted during encoding (e.g. trait hippocampal dysfunction in PTSD vulnerable individuals), biases towards elemental learning will predominate. Indeed, in both cases of pre and post hippocampal lesions, elemental associations, which are context independent (like the tone-US pairing), are left intact. We suggest that such a shift to a predominantly elemental strategy would thus allow elemental cues to have a much larger role in behavioral responses to the environment, with each discrete cue encoded during trauma able to induce conditioned fear responses across multiple contexts.
6. Future research
The theory outlined above suggests a testable hypothesis. Namely, that hippocampus volume, identified through structural MRI, will be correlated with performance in a fear conditioning paradigm which requires conjunctive processing in order to predict presence or absence of an aversive event. In order to test this hypothesis, tasks need to be developed that allow the researcher to demonstrate the use of either a conjunctive or elemental encoding strategy by forcing the use of conjunctive memory recall for successful task performance during some test phase. In addition, an ideal task would have a rodent analogue to allow for cross-species translational research, and thus the possibility of direct manipulation of hippocampal function. One potential task may be an object/cue array task, modified for fear conditioning.
This task would involve presenting an array of objects or cues, which may be naturalistic or arbitrary, that are repeatedly paired with an aversive event. This cue array represents the context in which the aversive event occurs, and allows for the prediction of the event using either a conjunctive or elemental representational strategy. More specifically, when this cue array is paired with an aversive event, a conditioned response should develop regardless of individual hippocampal volume. This conditioning may reflect the use of either a conjunctive or elemental strategy for representing context. Additionally, when presented with an array that contains no cues presented in the original array, all individuals should show an absence of fear.
However, when presented with an array in which only a limited number of original cues are present, differences among individuals should become apparent. Individuals with hippocampal function in the normal range should show limited fear, as their original fear response was conditioned to a representation of the conjunctive whole. That is, they will require all of the cues of the array to be present in the original conjunctive arrangement in order to display a conditioned response. Conversely, individuals with low hippocampal function should display a fear response when presented with a limited number of cues scattered among irrelevant objects. This result would reflect their reliance on an elemental strategy due to weakened hippocampal function. Thus, the original fear response can be demonstrated to have been conditioned to individual elements of the context rather than the conjunctive whole, allowing these individual cues to trigger a fear response on their own. In this example, the individuals with low hippocampal volumes are responding inappropriately to a context that contains only a few elements of the original context, but nevertheless is distinct. In order to recognize this context as safe, the individual must have employed a conjunctive strategy during encoding.
Various parameters of the object array can be altered, including number of objects present and the relationship of objects to one another, which provides additional flexibility in testing this and related hypotheses of configural fear learning. In addition, the ability to use arbitrary cues allows for a cross-species analogue that is basically identical to the human version of the task. This procedure may allow for considerable versatility in conducting translational research, as well as increased confidence in the generalizability of the results. Together, these aspects of the task would make it a potentially powerful tool for investigating the role of hippocampal function and conjunctive processing in the development of PTSD following a traumatic experience, including testing the initial hypothesis that hippocampal dysfunction can affect successful processing of contextual fear cues.
7. Conclusions
The presence of hippocampal abnormality is a robust finding in neurobiological investigations of PTSD, with some evidence that this may be a pre-existing vulnerability factor relevant to the etiology of the condition. Moreover, a large body of animal research documents the contribution of the hippocampal formation to the encoding and retrieval of memory for context. The present article proposes a mechanism through which hippocampal dysfunction may interact with traumatic experience to influence the etiology and maintenance of PTSD. Namely, an inability to adequately form conjunctive context representations may leave an individual dependent upon an elemental context representation strategy, and thus prone to respond with fear in the future presence of single elements encoded during the trauma. This proposition suggests a potential avenue for future research attempting to increase understanding of the pathophysiology of this disorder, and, if supported, has potential implications for treatment and prevention efforts. For example, the identification of hippocampal biomarkers may aid in more accurately targeting at risk individuals for preventative interventions, such as resiliency training targeted specifically at improving hippocampus-dependent functions. Further, elucidating the nature of hippocampal dysfunction and its relationship to maintenance of fear responding in PTSD may point toward targets for novel pharmaceutical or cognitive remediational interventions, or as adjuncts to existing clinical treatments.
Acknowledgments
Dr. Acheson is supported by a Veteran’s Affairs Mental Illness Research, Education, and Clinical Center (MIRECC) fellowship and the Department of Defense, Dr. Gresack is supported by a NARSAD Young Investigator award, and Dr. Risbrough is supported by NIMH MH074697, the Veteran’s Affairs Center of Excellence for Stress and Mental Health and a VA Merit Grant. We would also like to acknowledge the helpful comments of Dr. Robert Clark.
References
- Afifi TO, Asmundson GJ, Taylor S, Jang KL. The role of genes and environment on trauma exposure and posttraumatic stress disorder symptoms: A review of twin studies. Clinical Psychology Review. 2010;30:101–112. doi: 10.1016/j.cpr.2009.10.002. [DOI] [PubMed] [Google Scholar]
- Alonso J, Angermeyer MC, Bernert S, Bruffaerts R, Brugha TS, Bryson H, Girolamo G, Graaf R, Demyttenaere K, et al. Disability and quality of life impact of mental disorders in Europe: results from the European Study of the Epidemiology of Mental Disorders (ESEMeD) project. Acta Psychiatrica Scandanavica Supplement. 2004;109:21–27. doi: 10.1111/j.1600-0047.2004.00329.x. [DOI] [PubMed] [Google Scholar]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (DSM-IV-TR) Washington, D.C: Author; 2000. [Google Scholar]
- Apfel BA, Ross J, Hlavin J, Meyerhoff DJ, Metzler TJ, Marmar CR, Weiner MW, Schuff N, Neylan TC. Hippocampal volume differences in Gulf War veterans with current versus lifetime posttraumatic stress disorder symptoms. Biological Psychiatry. 2011;69:541–548. doi: 10.1016/j.biopsych.2010.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold DL, de Stefano N, Matthews PM, Trapp BD. N-acetylaspartate: Usefulness as an indicator of viable neuronal tissue. Annals of Neurology. 2001;50:823. doi: 10.1002/ana.10013. [DOI] [PubMed] [Google Scholar]
- Astur RS, St Germain SA, Tolin D, Ford J, Russell D, Stevens M. Hippocampus function predicts severity of post-traumatic stress disorder. CyberPsychology and Behavior. 2006;9:234–240. doi: 10.1089/cpb.2006.9.234. [DOI] [PubMed] [Google Scholar]
- Baker DG, Nievergelt CM, Risbrough VB. Post-traumatic stress disorder: Emerging concepts of pharmacotherapy. Expert Opinion on Emerging Drugs. 2009;14:251–272. doi: 10.1517/14728210902972494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett DH, Green ML, Morris R, Giles WH, Croft JB. Cognitive functioning and posttraumatic stress disorder. American Journal of Psychiatry. 1996;153:1492–1494. doi: 10.1176/ajp.153.11.1492. [DOI] [PubMed] [Google Scholar]
- Barrientos RM, O’Reilly RC, Rudy JW. Memory for context is impaired by injecting anisomycin into dorsal hippocampus following context exploration. Behavioral Brain Research. 2002;134:299–306. doi: 10.1016/s0166-4328(02)00045-1. [DOI] [PubMed] [Google Scholar]
- Bast T, Zhang W, Feldon J. The ventral hippocampus and fear conditioning in rats: Different anterograde amnesias of fear after tetrodotoxin inactivation and infusion of the GABAA agonist muscimol. Experimental Brain Research. 2001;139:39–52. doi: 10.1007/s002210100746. [DOI] [PubMed] [Google Scholar]
- Bauer ME, Wiek A, Lopes RP, Teixeira AL, Grassi-Oliveira R. Interplay between neuroimmunoendocrine systems during post-traumatic stress disorder: A minireview. Neuroimmunomodulatoin. 2010;17:192–195. doi: 10.1159/000258721. [DOI] [PubMed] [Google Scholar]
- Biedenkapp JC, Rudy JW. Hippocampal and extrahippocampal systems compete for control of contextual fear: Role of ventral subiculum and amygdala. Learning and Memory. 2008;16:38–45. doi: 10.1101/lm.1099109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder EB. The role of FKBP5, a co-chaperone of the glucocorticoid receptor in the pathogenesis and therapy of affective and anxiety disorders. Psychoneuroendocrinology. 2009;34:S186–S195. doi: 10.1016/j.psyneuen.2009.05.021. [DOI] [PubMed] [Google Scholar]
- Bisby JA, Brewin CR, Leitz JR, Curran HV. Acute effects of alcohol on the development of intrusive memories. Psychopharmacology. 2009;204:655–666. doi: 10.1007/s00213-009-1496-5. [DOI] [PubMed] [Google Scholar]
- Bisby JA, King JA, Brewin CR, Burgess N, Curran HV. Acute effects of alcohol on intrusive memory development and viewpoint dependence in spatial memory support a dual representational model. Biological Psychiatry. 2010;68:280–286. doi: 10.1016/j.biopsych.2010.01.010. [DOI] [PubMed] [Google Scholar]
- Bremner JD, Scott TM, Delany RC, Southwick SM, Mason JW, Johnson DR, Innis RB, McCarthy G, Charney DS. Deficits in short-term memory in posttraumatic stress disorder. American Journal of Psychiatry. 1993;150:1015–1019. doi: 10.1176/ajp.150.7.1015. [DOI] [PubMed] [Google Scholar]
- Bremner JD, Randall P, Scott TM, Capelli S, Delany R, McCarthy G, Charney DS. Deficits in short-term memory in adult survivors of childhood abuse. Psychiatry Research. 1995;59:97–107. doi: 10.1016/0165-1781(95)02800-5. [DOI] [PubMed] [Google Scholar]
- Bremner JD. Does stress damage the brain? Biological Psychiatry. 1999;45:797–805. doi: 10.1016/s0006-3223(99)00009-8. [DOI] [PubMed] [Google Scholar]
- Bremner JD. The relationship between cognitive and brain changes in posttraumatic stress disorder. Annals of the New York Academy of Sciences. 2006;1071:80–86. doi: 10.1196/annals.1364.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breslau N. The epidemiology of trauma, PTSD, and other posttrauma disorders. Trauma, Violence, and Abuse. 2009;10:198–210. doi: 10.1177/1524838009334448. [DOI] [PubMed] [Google Scholar]
- Breslau N, Kessler RC, Chilcoat HD, Schultz LR, Davis CG, Andreski P. Trauma and posttraumatic stress disorder in the community; the 1996 Detroit Area Survey of Trauma. Archives of General Psychiatry. 1998;55:626–632. doi: 10.1001/archpsyc.55.7.626. [DOI] [PubMed] [Google Scholar]
- Brewin CR, Dalgleish T, Joseph S. A dual representation theory of posttraumatic stress disorder. Psychological Review. 1996;103:670–686. doi: 10.1037/0033-295x.103.4.670. [DOI] [PubMed] [Google Scholar]
- Brewin CR. A cognitive neuroscience account of post-traumatic stress disorder and its treatment. Behaviour Research and Therapy. 2001;39:373–393. doi: 10.1016/s0005-7967(00)00087-5. [DOI] [PubMed] [Google Scholar]
- Brewin CR, Kleiner JS, Vasterling JJ, Field AP. Memory for emotionally neutral information in posttraumatic stress disorder: A meta-analytic investigation. Journal of Abnormal Psychology. 2007;116:448–463. doi: 10.1037/0021-843X.116.3.448. [DOI] [PubMed] [Google Scholar]
- Brewin CR, Gregory JD, Lipton M, Burgess N. Intrusive images in psychological disorders: Characteristics, neural mechanisms, and treatment implications. Psychological Review. 2010;117:210–232. doi: 10.1037/a0018113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brohawn KH, Offringa R, Pfaff DL, Hughes KC, Shin LM. The neural correlates of emotional memory in posttraumatic stress disorder. Biological Psychiatry. 2010;68:1023–1030. doi: 10.1016/j.biopsych.2010.07.018. [DOI] [PubMed] [Google Scholar]
- Brunello N, Davidson JR, Deahl M, Kessler RC, Mendlewicz J, Racagni G, Shalev AY, Zohar J. Posttraumatic stress disorder: diagnosis and epidemiology, comorbidity and social consequences, biology and treatment. Neuropsychobiology. 2001;43:150–162. doi: 10.1159/000054884. [DOI] [PubMed] [Google Scholar]
- Buckley TC, Blanchard EB, Neill WT. Information processing and PTSD: A review of the empirical literature. Clinical Psychology Review. 2000;28:1041–1065. doi: 10.1016/s0272-7358(99)00030-6. [DOI] [PubMed] [Google Scholar]
- Charney DS. Psychobiological mechanisms of resilience and vulnerability: Implications for successful adaptation to extreme stress. American Journal of Psychiatry. 2004;161:195–216. doi: 10.1176/appi.ajp.161.2.195. [DOI] [PubMed] [Google Scholar]
- Colgin LL, Moser EI, Moser M. Understanding memory through hippocampal remapping. Trends in Neurosciences. 2008;31:469–477. doi: 10.1016/j.tins.2008.06.008. [DOI] [PubMed] [Google Scholar]
- Conrad CD. Chronic stress-induced hippocampal vulnerability: The glucocorticoid vulnerability hypothesis. Reviews in the Neurosciences. 2008;19:395–411. doi: 10.1515/revneuro.2008.19.6.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornelius MC, Nugent NR, Amstadter AB, Koenen KC. Genetics of post-traumatic stress disorder: A review and recommendations for genome-wide association studies. Current Psychiatry Reports. 2010;12:313–326. doi: 10.1007/s11920-010-0126-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempster E, Toulopoulou T, McDonald C, Bramon E, Walshe M, Filbey F, Wickham PCS, et al. Association between BDNF Val66Met genotype and episodic memory. American Journal of Medical Genetics Neuropsychiatric Genetics. 2005;134:73–75. doi: 10.1002/ajmg.b.30150. [DOI] [PubMed] [Google Scholar]
- Corbo V, Clement MH, Armony JL, Pruessner JC, Brunet A. Size versus shape differences: Contrasting voxel-based and volumetric analyses of the anterior cigulate cortex in individuals with acute posttraumatic stress disorder. Biological Psychiatry. 2005;58:119–124. doi: 10.1016/j.biopsych.2005.02.032. [DOI] [PubMed] [Google Scholar]
- Crow RR. Molecular Genetics of Anxiety. In: Stein DJ, Hollander E, editors. Neurobiology of Mental Illness. New York: Oxford Press; 2004. pp. 535–545. [Google Scholar]
- Druss BG, Hwang I, Petukhova M, Sampson NA, Wang PS, Kessler RC. Impairment in role functioning in mental and chronic medical disorders in the United States: Results from the National Comorbidity Survey Replication. Molecular Psychiatry. 2008;14:728–737. doi: 10.1038/mp.2008.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn GA, Morgan CP, Bale TL. Sex-specificity in transgenerational epigenetic programming. Hormones and Behavior. doi: 10.1016/j.yhbeh.2010.05.004. (in press) [DOI] [PubMed] [Google Scholar]
- Eichenbaum H. A cortical-hippocampal system for declarative memory. Nature Reviews Neuroscience. 2000;1:41–50. doi: 10.1038/35036213. [DOI] [PubMed] [Google Scholar]
- Elzinga BM, Bremner JD. Are the neural substrates of memory the final common pathway in PTSD? Journal of Affective Disorders. 2002;70:1–17. doi: 10.1016/s0165-0327(01)00351-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etkin A, Wager TD. Functional neuroimaging of anxiety: A meta-analysis of emotional processing in PTSD, social anxiety disorder, and specific phobia. American Journal of Psychiatry. 2007;164:1476–1488. doi: 10.1176/appi.ajp.2007.07030504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanselow MS. Associative vs topographical accounts of the immediate shock-freezing deficit in rats: Implications for the response selection rules governing species-specific defensive reactions. Learning and Motivation. 1986;17:16–39. [Google Scholar]
- Fanselow MS. Factors governing one-trial contextual conditioning. Animal Learning & Behavior. 1990;18:264–270. [Google Scholar]
- Fanselow MS, DeCola JP, Young S. Mechanisms responsible for reduced contextual conditioning with massed unsignaled unconditional stimuli. Journal of Experimental Psychology: Animal Behavior Process. 1993;19:121–137. [PubMed] [Google Scholar]
- Fanselow MS. Contextual fear, gestalt memories, and the hippocampus. Behavioral Brain Research. 2000;110:73–81. doi: 10.1016/s0166-4328(99)00186-2. [DOI] [PubMed] [Google Scholar]
- Frankland PW, Cestari V, Filipkowski RK, McDonald RJ, Silva AJ. The dorsal hippocampus is essential for context discrimination but not for contextual conditioning. Behavioral Neuroscience. 1998;112:863–874. doi: 10.1037//0735-7044.112.4.863. [DOI] [PubMed] [Google Scholar]
- Freeman T, Kimbrell T, Booe L, Myers M, Cardwell D, Lindquist DM, Hart J, Komoroski RA. Evidence of resilience: Neuroimaging in former prisoners of war. Psychiatry Research: Neuroimaging. 2006;146:59–64. doi: 10.1016/j.pscychresns.2005.07.007. [DOI] [PubMed] [Google Scholar]
- Frielingsdorf H, Bath KG, Soliman F, DiFede J, Casey BJ, Lee FS. Variant brain-derived neurotrophic factor Val66Met endophenotypes: Implications for posttraumatic stress disorder. Annals of the New York Academy of Sciences. 2010;1208:150–157. doi: 10.1111/j.1749-6632.2010.05722.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frustaci A, Pozzi G, Gianfagna F, Manzoli L, Boccia S. Meta-analysis of the brain-derived neurotrophic factor gene (BDNF) Val66Met polymorphism in anxiety disorders and anxiety-related personality traits. Neuropsychobiology. 2008;58:163–170. doi: 10.1159/000182892. [DOI] [PubMed] [Google Scholar]
- Galea S, Nandi A, Vlahov D. The epidemiology of post-traumatic stress disorder after disasters. Epidemiologic Reviews. 2005;27:78–91. doi: 10.1093/epirev/mxi003. [DOI] [PubMed] [Google Scholar]
- Garfinkel SN, Liberzon I. Neurobiology of PTSD: A review of neuroimaging findings. Psychiatric Annals. 2009;39:370–381. [Google Scholar]
- Gatt JM, Nemeroff CB, Dobson-Stone C, Paul RH, Bryant RA, Schofield PR, et al. Interactions between BDNF Val66Met polymorphism and early life stress predict brain and arousal pathways to syndromal depression and anxiety. Molecular Psychiatry. 2009;14:681–695. doi: 10.1038/mp.2008.143. [DOI] [PubMed] [Google Scholar]
- Geuze E, Vermetten E, de Kloet CS, Westenberg HGM. Precuneal activity during encoding in veterans with posttraumatic stress disorder. Progress in Brain Research. 2008;167:293–297. doi: 10.1016/S0079-6123(07)67026-5. [DOI] [PubMed] [Google Scholar]
- Gil T, Calev A, Greenberg D, Kugelmass S, Lerer B. Cognitive functioning in post-traumatic stress disorder. Journal of Traumatic Stress. 1990;3:29–45. [Google Scholar]
- Gilbertson MW, Gurvits TV, Lasko NB, Orr SP, Pitman RK. Multivariate assessment of explicit memory function in combat veterans with posttraumatic stress disorder. Journal of Traumatic Stress. 2001;14:413–432. doi: 10.1023/A:1011181305501. [DOI] [PubMed] [Google Scholar]
- Gilbertson MW, Shenton ME, Ciszewski A, Kiyoto K, Lasko NB, Orr SP, Pitman RK. Smaller hippocampal volume predicts pathologic vulnerability to psychological trauma. Nature Neuroscience. 2002;5:1242–1247. doi: 10.1038/nn958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbertson MW, Paulus LA, Williston SK, Gurvits TV, Lasko NB, Pitman RK, Orr SP. Neurocognitive function in monozygotic twins discordant for combat exposure: Relationship to posttraumatic stress disorder. Journal of Abnormal Psychology. 2006;115:484–495. doi: 10.1037/0021-843X.115.3.484. [DOI] [PubMed] [Google Scholar]
- Gilbertson MW, Williston SK, Paulus LA, Lasko NB, Gurvits TV, Shenton ME, Pitman RK, Orr SP. Configural cue performance in identical twins discordant for posttraumatic stress disorder: Theoretical implications for the role of hippocampal function. Biological Psychiatry. 2007;62:513–520. doi: 10.1016/j.biopsych.2006.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glodzik-Sobanska L, Li J, Mosconi L, Slowik A, Walecki J, Szczudlik A, Sobiecka B, de Leon MJ. Prefrontal N-acetylaspartate and poststroke recovery: A longitudinal proton spectroscopy study. American Journal of Neuroradiology. 2007;28:470–474. [PMC free article] [PubMed] [Google Scholar]
- Golier J, Yehuda R, Cornblatt B, Harvey P, Gerber D, Levengood R. Sustained attention in combat-related posttraumatic stress disorder. Integrative Psychological and Behavioral Science. 1997;32:52–61. doi: 10.1007/BF02688613. [DOI] [PubMed] [Google Scholar]
- Goelet P, Castelucci V, Schacter S, Kandel E. The long and the short of long-term memory – a molecular framework. Nature. 1986;332:419–422. doi: 10.1038/322419a0. [DOI] [PubMed] [Google Scholar]
- Golier J, Yehuda R. Neuroendocrine activity and memory-related impairments in posttraumatic stress disorder. Developmental Psychopathology. 1998;10:857–869. doi: 10.1017/s0954579498001904. [DOI] [PubMed] [Google Scholar]
- Gruber S, Frey R, Mlynarik V, Stadlbauer A, Heiden A, Kasper S, Kemp G, Graham J, Moser E. Quantification of metabolic differences in the frontal brain of depressive patients and controls obtained by 1H-MRS at 3 Tesla. Investigative Radiology. 2003;38:403–408. doi: 10.1097/01.rli.0000073446.43445.20. [DOI] [PubMed] [Google Scholar]
- Hariri AR, Goldberg TE, Mattay VS, Kolachana BS, Callicott JH, Egan MF, Weinberger DR. Brain-derived neurotrophic factor Val66Met polymorphism affects human memory-related hippocampal activity and predicts memory performance. Journal of Neuroscience. 2003;23:6690–6694. doi: 10.1523/JNEUROSCI.23-17-06690.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes JP, LaBar KS, McCarthy G, Selgrade E, Nasser J, Dolcos F, et al. Reduced hippocampal and amygdala activity predicts memory distortions for trauma reminders in combat-related PTSD. Journal of Psychiatric Research. doi: 10.1016/j.jpsychires.2010.10.007. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoge CW, McGurk D, Thomas JL, Cox AL, Engel CC, Castro CA. Mild traumatic brain injury in U.S. soldiers returning from Iraq. New England Journal of Medicine. 2008;358:453–463. doi: 10.1056/NEJMoa072972. [DOI] [PubMed] [Google Scholar]
- Iordanova MD, Burnett DJ, Aggleton JP, Good M, Honey RC. The role of the hippocampus in mnemonic integration and retrieval: Complementary evidence from lesion and inactivation studies. European Journal of Neuroscience. 2009;30:2177–2189. doi: 10.1111/j.1460-9568.2009.07010.x. [DOI] [PubMed] [Google Scholar]
- Jenkins MA, Langlais PJ, Delis D, Cohen R. Learning and memory in rape victims with posttraumatic stress disorder. American Journal of Psychiatry. 1998;155:278–279. doi: 10.1176/ajp.155.2.278. [DOI] [PubMed] [Google Scholar]
- Karl A, Schaefer M, Malta LS, Dorfel D, Rohleder N, Werner A. A meta-analysis of structural brain abnormalities in PTSD. Neuroscience and Biobehavioral Reviews. 2006;30:1004–1031. doi: 10.1016/j.neubiorev.2006.03.004. [DOI] [PubMed] [Google Scholar]
- Karl A, Werner A. The use of proton magnetic resonance spectroscopy in PTSD research – meta-analyses of findings and methodological review. Neuroscience and Biobehavioral Reviews. 2010;34:7–22. doi: 10.1016/j.neubiorev.2009.06.008. [DOI] [PubMed] [Google Scholar]
- Kasai K, Yamasue H, Gilbertson MW, Shenton ME, Rauch SL, Pitman RK. Evidence for acquired pregenual anterior cignulate gray matter loss from a twin study of combat-related posttraumatic stress disorder. Biological Psychiatry. 2008;63:539–541. doi: 10.1016/j.biopsych.2007.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent JM, Mathew SJ, Gorman JM. Molecular targets in the treatment of anxiety. Biological Psychiatry. 2002;52:1008–1030. doi: 10.1016/s0006-3223(02)01672-4. [DOI] [PubMed] [Google Scholar]
- Kesner RP. Behavioral functions of the CA3 subregion of the hippocampus. Learning and Memory. 2007;14:771–781. doi: 10.1101/lm.688207. [DOI] [PubMed] [Google Scholar]
- Kessler RC, Chiu WT, Demler O, Merikangas KR, Walters EE. Prevalence, severity, and comorbidity of 12-month DSM-IV disorders in the National Comorbidity Survey Replication. Archives of General Psychiatry. 2005;62:617–627. doi: 10.1001/archpsyc.62.6.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiernan MJ, Westbrook RF. Effects of exposure to a to-be-shocked environment upon the rat’s freezing response: Evidence for facilitation, latent inhibition, and perceptual learning. Quarterly Journal of Experimental Psychology: Comparative and Physiological Psychology. 1993;46:271–288. doi: 10.1080/14640749308401089. [DOI] [PubMed] [Google Scholar]
- Kim JJ, Fanselow MS. Modality-specific retrograde amnesia of fear. Science. 1992;256:675–677. doi: 10.1126/science.1585183. [DOI] [PubMed] [Google Scholar]
- Kitayama N, Vaccarino V, Kutner M, Weiss P, Bremner JD. Magnetic resonance imaging (MRI) measurement of hippocampal volume in posttraumatic stress disorder: A meta-analysis. Journal of Affective Disorders. 2005;88:79–86. doi: 10.1016/j.jad.2005.05.014. [DOI] [PubMed] [Google Scholar]
- Kitayama N, Quinn S, Bremner JD. Smaller volume of anterior cigulate cortex in abuse-related posttraumatic stress disorder. Journal of Affective Disorders. 2006;90:171–174. doi: 10.1016/j.jad.2005.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlovsky N, Matar MA, Kaplan Z, Kotler M, Zohar J, Cohen H. Long-term down-regulation of BDNF mRNA in rat hippocampal CA1 subregion correlates with PTSD-like behavioural stress response. International Journal of Neurophychopharmacology. 2007;10:741–758. doi: 10.1017/S1461145707007560. [DOI] [PubMed] [Google Scholar]
- Luine VN, Villegas M, Martinez C, McEwan BS. Repeated stress causes impairments of spatial memory performance. Brain Research. 1994;639:167–170. doi: 10.1016/0006-8993(94)91778-7. [DOI] [PubMed] [Google Scholar]
- Maren S, Aharonov G, Fanselow MS. Neurotoxic lesions of the dorsal hippocampus and Pavlovian fear conditioning in rats. Behavioral Brain Research. 1997;88:261–274. doi: 10.1016/s0166-4328(97)00088-0. [DOI] [PubMed] [Google Scholar]
- Margarinos AM, McEwan BS, Flugge G, Fluchs E. Chronic psychosocial stress causes apical dendritic atrophy of hippocampal CA3 pyramidal neurons in subordinate tree shrews. Journal of Neuroscience. 1996;16:3534–3540. doi: 10.1523/JNEUROSCI.16-10-03534.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin E, Capone A, Schneider J, Henning J, Thiel T. Absence of N-acetylaspartate in the human brain: Impact on neurospectroscopy. Annals of Neurology. 2001a;49:518–521. [PubMed] [Google Scholar]
- Martin E, Thiel T, Capone A, Henning J, Schneider J. Reply. Annals of Neurology. 2001b;50:824. [PubMed] [Google Scholar]
- Matus-Amat P, Higgins EA, Barrientos RM, Rudy JW. The role of the dorsal hippocampus in the acquisition and retrieval of context memory representations. The Journal of Neuroscience. 2004;24:2431–2439. doi: 10.1523/JNEUROSCI.1598-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matus-Amat P, Higgins EA, Sprunger D, Wright-Hardesty K, Rudy JW. The role of the dorsal hippocampus and basolateral amygdale NMDA receptors in the acquisition and retrieval of context and contextual fear memories. Behavioral Neuroscience. 2007;121:721–731. doi: 10.1037/0735-7044.121.4.721. [DOI] [PubMed] [Google Scholar]
- McEwan BS, Angulo J, Cameron H, Chao HM, Daniels D, Gannon MN, Gould E, Mendelson S, Sakai R, Spencer R, et al. Paradoxical effects of adrenal steroids on the brain: Protection versus degeneration. Biological Psychiatry. 1992;31:177–199. doi: 10.1016/0006-3223(92)90204-d. [DOI] [PubMed] [Google Scholar]
- McGowan PO, Sasaki A, D’Alessio AC, Dymov S, Labonte B, Szyf M, et al. Epigenetic regulation of the gluccocorticoid receptor in human brain associates with childhood abuse. Nature Neuroscience. 2009;12:342–348. doi: 10.1038/nn.2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHugh TJ, Tonegawa S. CA3 NMDA receptors are required for the rapid formation of a salient contextual representation. Hippocampus. 2009;19:1153–1158. doi: 10.1002/hipo.20684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meaney MJ, Diorio J, Francis D, Weaver S, Yau J, Chapman K, Seckl JR. Postnatal handling increases the expression of cAMP-inducible transcription factors in the rat hippocampus: The effects of thyroid hormones and serotonin. Journal of Neuroscience. 2000;20:3926–3935. doi: 10.1523/JNEUROSCI.20-10-03926.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merikangas K, Pine D. Genetic and other vulnerability factors for anxiety and stress disorders. In: Davis KL, Charney DS, Coyle JT, Nemeroff C, editors. Neuropsychopharmacology: The Fifth Generation of Progress. Philadelphia: Lippincott Williams & Wilkins; 2004. pp. 867–882. [Google Scholar]
- Moradi AR, Neshat-Doost HT, Taghavi MR, Yule W, Dalgleish T. Everyday memory deficits in children and adolescents with PTSD: Performance on the Rivermead Behavioural Memory Test. Journal of Child Psychology and Psychiatry. 1999;40:357–361. [PubMed] [Google Scholar]
- Morey RA, Dolcos F, Petty CM, Cooper DA, Hayes JP, LaBar KS, McCarthy The role of trauma-related distractors on neural systems for working memory and emotion processing in posttraumatic stress disorder. Journal of Psychiatric Research. 2009;43:809–817. doi: 10.1016/j.jpsychires.2008.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadel L, Willner J. Context and conditioning: A place for space. Physiology and Behavior. 1980;8:218–228. [Google Scholar]
- Nakashiba T, Young JZ, McHugh TJ, Buhl DL, Tonegawa S. Transgenic inhibition of synaptic transmission reveals role of CA3 output in hippocampal learning. Science. 2008;319:1260–1264. doi: 10.1126/science.1151120. [DOI] [PubMed] [Google Scholar]
- Nemeroff CB. Neurobiological consequences of childhood trauma. Journal of Clinical Psychiatry. 2004;65:18–28. [PubMed] [Google Scholar]
- O’Keefe J, Nadel L. The Hippocampus as a Cognitive Map. Oxford University Press; 1978. [Google Scholar]
- Olvera RL, Caetano SC, Fonseca M, Nicoletti M, Stanley JA, Chen HH, et al. Low levels of N-acetyl aspartate in the left dorsolateral prefrontal cortex of pediatric bipolar patients. Journal of Child and Adolescent Psychopharmacology. 2007;17:461–473. doi: 10.1089/cap.2007.0102. [DOI] [PubMed] [Google Scholar]
- O’Reilly RC, Rudy JW. Conjunctive representations in learning and memory: Principles of cortical and hippocampal function. Psychological Review. 2001;108:311–345. doi: 10.1037/0033-295x.108.2.311. [DOI] [PubMed] [Google Scholar]
- Ousch EA, Benson B, Geraci M, Podell D, Herscovitch P, McCann UD, Post RM. Regional cerebral blood flow correlated with flashback intensity in patients with posttraumatic stress disorder. Biological Psychiatry. 2001;50:246–253. doi: 10.1016/s0006-3223(01)01107-6. [DOI] [PubMed] [Google Scholar]
- Pederson CL, Maurer SH, Kaminski PL, Zander KA, Peters CM, Stokes-Crowe LA, Osborn RE. Hippocampal volume and memory performance in a community-based sample of women with posttraumatic stress disorder secondary to child abuse. Journal of Traumatic Stress. 2004;17:37–40. doi: 10.1023/B:JOTS.0000014674.84517.46. [DOI] [PubMed] [Google Scholar]
- Phillips RG, LeDoux JE. Differential contribution of amygdale and hippocampus to cued and contextual fear conditioning. Behavioral Neuroscience. 1992;106:274–285. doi: 10.1037//0735-7044.106.2.274. [DOI] [PubMed] [Google Scholar]
- Quinn JJ, Loya F, Ma QD, Fanselow MS. Dorsal hippocampus NMDA receptors differentially mediate trace and contextual fear conditioning. Hippocampus. 2005;15:665–674. doi: 10.1002/hipo.20088. [DOI] [PubMed] [Google Scholar]
- Quirk GJ, Mueller D. Neural mechanisms of extinction learning and retrieval. Neuropsychopharmocology. 2008;33:56–72. doi: 10.1038/sj.npp.1301555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmusson AM, Lipschitz DS, Wang S, Hu S, Vojvoda D, Bremner JD, Southwick SM, Charney DS. Increased pituitary and adrenal reactivity in premenopausal women with posttraumatic stress disorder. Biological Psychiatry. 2001;50:965–977. doi: 10.1016/s0006-3223(01)01264-1. [DOI] [PubMed] [Google Scholar]
- Rauch SL, Shin LM, Phelps EA. Neurocircuitry models of posttraumatic stress disorder and extinction: Human neuroimaging research – past, present, and future. Biological Psychiatry. 2006;60:376–382. doi: 10.1016/j.biopsych.2006.06.004. [DOI] [PubMed] [Google Scholar]
- Ressler KJ, Mercer KB, Bradley B, Jovanovic T, Mahan A, Kerley K, et al. Post-traumatic stress disorder is associated with PACAP and the PAC1 receptor. Nature. 2011;470:492–497. doi: 10.1038/nature09856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roca V, Freeman TW. Complaints of impaired memory in veterans with PTSD. American Journal of Psychiatry. 2001;158:17–38. doi: 10.1176/appi.ajp.158.10.1738-a. [DOI] [PubMed] [Google Scholar]
- Ross AJ, Sachdey PS, Wen W, Valenzuela MJ, Brodaty H. Cognitive correlates of 1H MRS measures in the healthy elderly brain. Brain Research Bulletin. 2005;66:9–16. doi: 10.1016/j.brainresbull.2005.01.015. [DOI] [PubMed] [Google Scholar]
- Roth TL, Zoldz PR, Sweatt JD, Diamond DM. Epigenetic modification of hippocampal BDNF in adult rats in an animal model of post-traumatic stress disorder. Journal of Psychiatric Research. doi: 10.1016/j.jpsychires.2011.01.013. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudy JW, O’Reilly RC. Contextual fear conditioning, conjunctive representations, pattern completion, and the hippocampus. Behavioral Neuroscience. 1999;113:867–880. doi: 10.1037//0735-7044.113.5.867. [DOI] [PubMed] [Google Scholar]
- Rudy JW, O’Reilly RC. Conjunctive representations, the hippocampus, and contextual fear conditioning. Cognitive, Affective, & Behavioral Neuroscience. 2001;1:66–82. doi: 10.3758/cabn.1.1.66. [DOI] [PubMed] [Google Scholar]
- Rudy JW, Barrientos RM, O’Reilly RC. Hippocampal formation supports conditioning to memory of a context. Behavioral Neuroscience. 2002;116:530–538. doi: 10.1037//0735-7044.116.4.530. [DOI] [PubMed] [Google Scholar]
- Rudy JW, Huff NC, Matus-Amat P. Understanding contextual fear conditioning: Insights from a two-process model. Neuroscience and Biobehavioral Reviews. 2004;7:675–685. doi: 10.1016/j.neubiorev.2004.09.004. [DOI] [PubMed] [Google Scholar]
- Rudy JW, Matus-Amat P. The ventral hippocampus supports a memory representation of context and contextual fear conditioning: Implications for a unitary function of the hippocampus. Behavioral Neuroscience. 2005;119:154–163. doi: 10.1037/0735-7044.119.1.154. [DOI] [PubMed] [Google Scholar]
- Rudy JW. Context representations, context functions, and the parahippocampal-hippocampal system. Learning and Memory. 2009;16:573–585. doi: 10.1101/lm.1494409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachinvala N. Memory, attention, function, and mood among patients with chronic posttraumatic stress disorder. Journal of Nervous and Mental Disease. 2000;12:818–823. doi: 10.1097/00005053-200012000-00005. [DOI] [PubMed] [Google Scholar]
- Saigh PA, Bremner JD. Posttraumatic Stress Disorder. Needham Heights: Allyn and Bacon; 1999. [Google Scholar]
- Sapolsky RM, Uno H, Rebert CS, Finch CE. Hippocampal damage associated with prolonged glucocorticoid exposure in primates. Journal of Neuroscience. 1990;10:2897–2902. doi: 10.1523/JNEUROSCI.10-09-02897.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapolsky RM. Stress and plasticity in the limbic system. Neurochemical Research. 2003;28:1735–42. doi: 10.1023/a:1026021307833. [DOI] [PubMed] [Google Scholar]
- Sareen J, Houlahan T, Cox BJ, Asmundson GJ. Anxiety disorders associated with suicidal ideation and suicide attempts in the National Comorbidity Survey. Journal of Nervous and Mental Disease. 2005;193:450–454. doi: 10.1097/01.nmd.0000168263.89652.6b. [DOI] [PubMed] [Google Scholar]
- Sareen J, Cox BJ, Stein MB, Afifi TO, Fleet C, Asmundson GJ. Physical and mental comorbidity, disability, and suicidal behavior associated with posttraumatic stress disorder in a large community sample. Psychosomatic Medicine. 2007;69:242–248. doi: 10.1097/PSY.0b013e31803146d8. [DOI] [PubMed] [Google Scholar]
- Schnurr PP, Lunney CA, Bovin MJ, Marx BP. Post-traumatic stress disorder and quality of life: Extension of findings to veterans of the wars in Iraq and Afghanistan. Clinical Psychology Review. 2009;29:727–735. doi: 10.1016/j.cpr.2009.08.006. [DOI] [PubMed] [Google Scholar]
- Schuff N, Neylan TC, Lenoci MA, Du A, Weiss DS, Marmar CR, Weiner MW. Decreased hippocampal N-Acetylaspartate in the absence of atrophy in posttraumatic stress disorder. Biological Psychiatry. 2001;50:952–959. doi: 10.1016/s0006-3223(01)01245-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin LM, Shin PS, Heckers S, Krangel TS, Macklin ML, Orr SP, Lasko N, Segal E, Makris N, Richert K, Levering J, Schacter DL, Alpert NM, Fischman AJ, Pitman RK, Rauch SL. Hippocampal function in posttraumatic stress disorder. Hippocampus. 2004;14:292–300. doi: 10.1002/hipo.10183. [DOI] [PubMed] [Google Scholar]
- Shin LM, Rauch SL, Pitman RK. Amygdala, medial prefrontal cortex, and hippocampal function in PTSD. Annals of the New York Academy of Sciences. 2006;1071:67–79. doi: 10.1196/annals.1364.007. [DOI] [PubMed] [Google Scholar]
- Shin LM, Lasko NB, Macklin ML, Karpf RD, Milad MR, Orr SP, Goetz JM, Fischman AJ, Rauch SL, Pitman RK. Resting metabolic activity in the cigulat cortex and vulnerability to posttraumatic stress disorder. Archives of General Psychiatry. 2009;66:1099–1107. doi: 10.1001/archgenpsychiatry.2009.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegmund A, Kaltwasser SF, Holsboer F, Czisch M, Wotjak CT. Hippocampal N-acetylaspartate levels before trauma predict the development of long-lasting posttraumatic stress disorder-like symptoms in mice. Biological Psychiatry. 2009;65:258–262. doi: 10.1016/j.biopsych.2008.08.023. [DOI] [PubMed] [Google Scholar]
- Sijens PE, Mostert JP, Oudkerk M, De Keyser J. 1H MR spectroscopy of the brain in multiple sclerosis subtypes with analysis of the metabolite concentrations in gray and white matter: Initial findings. European Radiology. 2006;16:489–495. doi: 10.1007/s00330-005-2839-1. [DOI] [PubMed] [Google Scholar]
- Smith ME. Bilateral hippocampal volume reduction in adults with post-traumatic stress disorder: A meta-analysis of structural MRI studies. Hippocampus. 2005;15:798–807. doi: 10.1002/hipo.20102. [DOI] [PubMed] [Google Scholar]
- Soontornniyomkij V, Risbrough VB, Young JW, Wallace CK, Soontornniyomkij B, Jeste DV, et al. Short-term recognition memory impairment is associated with decreased expression of FK506 binding protein 51 in the aged mouse brain. Age. 2010;32:309–322. doi: 10.1007/s11357-010-9145-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squire LR. Memory and the hippocampus: A synthesis from finding with rats, monkeys, and humans. Psychological Review. 1992;99:195–231. doi: 10.1037/0033-295x.99.2.195. [DOI] [PubMed] [Google Scholar]
- Starkman MN, Giordani B, Gebarski SS, Schteingart DE. Improvement in learning associated with increase in hippocampal formation volume. Biological Psychiatry. 2003;53:233–238. doi: 10.1016/s0006-3223(02)01750-x. [DOI] [PubMed] [Google Scholar]
- Stein MB, Hanna C, Vaerum V, Koverola C. Memory functioning in adult women traumatized by childhood sexual abuse. Journal of Traumatic Stress. 2005;12:527–534. doi: 10.1023/A:1024775222098. [DOI] [PubMed] [Google Scholar]
- Sullivan EV, Adalsteinsson E, Spielman DM, Hurd RE, Pfefferbaum A. N-acetylaspartate – A marker of neuronal integrity. Annals of Neurology. 2001;50:823. doi: 10.1002/ana.1279. [DOI] [PubMed] [Google Scholar]
- Sunada M, Rao MS, Raju TR. Effect of chronic restraint stress on dendritic spines and excrescenes of hippocampal CA3 pyramidal neurons – a quantitative study. Brain Research. 1995;694:312–317. doi: 10.1016/0006-8993(95)00822-8. [DOI] [PubMed] [Google Scholar]
- Sutherland RJ, Rudy JW. Configural association theory: The role of the hippocampal formation in learning, memory, and amnesia. Psychobiology. 1989;17:129–144. [Google Scholar]
- Sweatt JD. Mechanisms of Memory. New York: Elsevier Academic Press; 2003. [Google Scholar]
- Szulc A, Galinska B, Tarasow E, Kubas B, Dzienis W, Konarzewska B, Poplawska R, et al. N-acetylaspartate (NAA) levels in selected areas of the brain in patients with chronic schizophrenia treated with typical and atypical neuroleptics: A proton magnetic resonance spectroscopy (1H MRS) study. Medical Science Monitor. 2007;13:17–22. [PubMed] [Google Scholar]
- Uddo M, Vasterling JJ, Brailey K, Sutker PB. Memory and attention in combat-related post-traumatic stress disorder (PTSD) Journal of Psychopathology and Behavioral Assessment. 1993;15:43–52. [Google Scholar]
- Urenjak J, Williams SR, Gadian DG, Noble M. Proton nuclear magnetic resonance spectroscopy unambiguously identifies different neural cell types. Journal of Neuroscience. 1993;13:981–989. doi: 10.1523/JNEUROSCI.13-03-00981.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasterling JJ, Brailey K, Constans JI, Sutker PB. Attention and memory dysfunction in posttraumatic stress disorder. Neuropsychology. 1998;12:125–133. doi: 10.1037//0894-4105.12.1.125. [DOI] [PubMed] [Google Scholar]
- Vasterling JJ, Duke LM, Brailey K, Constans JI, Allain AN, Sutker PB. Attention, learning, and memory performances and intellectual resources in Vietnam veterans: PTSD and no disorder comparisons. Neuropsychology. 2002;1:5–14. doi: 10.1037//0894-4105.16.1.5. [DOI] [PubMed] [Google Scholar]
- Vogt DS, King DW, King LA. Risk pathways for PTSD: Making sense of the literature. In: Friedman MJ, Keane TM, Resick PA, editors. Handbook of PTSD: Science and Practice. New York: The Guilford Press; 2007. [Google Scholar]
- Wallenstein GV, Vago DR. Intrahippocampal scopolamine impairs both acquisition and consolidation of contextual fear conditioning. Neurobiology of Learning and Memory. 2001;75:245–252. doi: 10.1006/nlme.2001.4005. [DOI] [PubMed] [Google Scholar]
- Wang Z, Neylan TC, Mueller SG, Lenoci M, Truran D, Marmar CR, Weiner MW, Schuff N. Magnetic resonance imaging of hippocampal subfields in posttraumatic stress disorder. Archives of General Psychiatry. 2010;67:296–303. doi: 10.1001/archgenpsychiatry.2009.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiltgen BJ, Sanders MJ, Anagnostaras SG, Sage JR, Fanselow MS. Context fear learning in the absence of the hippocampus. The Journal of Neuroscience. 2006;26:5484–5491. doi: 10.1523/JNEUROSCI.2685-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodward SH, Schaer M, Kaloupek DG, Cediel L, Eliez S. Smaller global and regional cortical volume in combat-related posttraumatic stress disorder. Atchives of General Psychiatry. 2009;66:1373–1382. doi: 10.1001/archgenpsychiatry.2009.160. [DOI] [PubMed] [Google Scholar]
- Woodward SH, Kaloupek DG, Grande LJ, Stegman WK, Kutter CJ, Leskin L, Prestel R, Schaer M, Reiss AL, Eliez S. Hippocampal volume and declarative memory function in combat-related PTSD. Journal of the International Neuropsychological Society. 2009;15:830–839. doi: 10.1017/S1355617709990476. [DOI] [PubMed] [Google Scholar]
- Yamasue H, Kasai K, Iwanami A, Ohtani T, Yamada H, Abe O, Kuroki N, Fukuda R, Tochigi M, Furukawa S, Sadamatsu M, Sasaki T, Aoki S, Ohtomo K, Asukai N, Kato N. Voxel-based analysis of MRI reveals anterior cingulate gray-matter volume reduction in posttraumatic stress disorder due to terrorism. Proceedings of the National Academy of Sciences. 2003;100:9039–9043. doi: 10.1073/pnas.1530467100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K, Lei G, Jackson M, MacDonald J. The involvement of PACAP/VIP system in synaptic transmission in the hippocampus. Journal of Molecular Neuroscience. 2010;42:319–326. doi: 10.1007/s12031-010-9372-7. [DOI] [PubMed] [Google Scholar]
- Yehuda R. Status of glucocorticoid alterations in post-traumatic stress disorder. Annals of the New York Academy of Sciences. 2009;1179:56–69. doi: 10.1111/j.1749-6632.2009.04979.x. [DOI] [PubMed] [Google Scholar]
- Yehuda R, Bierer LM. The relevance of epigenetics to PTSD: Implications for the DSM-IV. Journal of Traumatic Stress. 2009;22:427–434. doi: 10.1002/jts.20448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yehuda R, Keefe RS, Harvey PD, Levengood DK, Gerber JG, Siever LJ. Learning and memory in combat veterans with posttraumatic stress disorder. American Journal of Psychiatry. 1995;152:137–139. doi: 10.1176/ajp.152.1.137. [DOI] [PubMed] [Google Scholar]
- Young SL, Bohenek DL, Fanselow MS. NMDA processes mediate anterograde amnesia of contextual fear conditioning induced by hippocampal damage: Immunization against amnesia by context preexposure. Behavioral Neuroscience. 1994;108:19–29. doi: 10.1037//0735-7044.108.1.19. [DOI] [PubMed] [Google Scholar]
- Zalewski C, Thompson W, Gottesman I. Comparison of neuropsychological test performance in PTSD, generalized anxiety disorder, and control Vietnam veterans. Assessment. 1994;1:133–142. doi: 10.1177/1073191194001002003. [DOI] [PubMed] [Google Scholar]
- Zhang H, Ozbay F, Lappalainen J, Kranzler HR, van Dyck CH, Charney DS, et al. Brain derived neurotrophic factor (BDNF) gene variants and Alzheimer’s disease, affective disorders, posttraumatic stress disorder, schizophrenia, and substance dependence. American Journal of Medical Genetics Neuropsychiatric Genetics. 2006;141:387–393. doi: 10.1002/ajmg.b.30332. [DOI] [PMC free article] [PubMed] [Google Scholar]