

Abstract
Pregnane derived steroids have agonistic and antagonistic actions at GABAA receptors. Putative binding sites for agonistic neurosteroids are located within the transmembrane (TM) regions. A mutation within the rat α1 TM3 region, S299C, caused the expressed receptors to have unusual and extreme sensitivity to agonistic neurosteroids. For mutant α1S299C receptors, with wild type β and γ subunits, expressed in Xenopus oocytes, steroids activated the GABAA receptors in the absence of GABA. Maximal steroid induced currents were about half of maximal GABA currents. The steroid activation was biphasic with EC50’s much lower than wild type, in subnanomolar and nanomolar concentrations, while the wild type had only one activation peak with near micromolar EC50. These currents could be blocked by both picrotoxin and an antagonist neurosteroid. The steroids did not seem to potentiate significantly submaximal GABA currents. The α1S299C mutation did not affect responses to the extracellularly acting partial agonist piperidine-4-sulfate. Substituted cysteine experiments indicate that this mutant can be modified by pCMBS− when the sulfhydryl reagent is added with the higher steroid concentration for activation but not the lower steroid concentration. The pCMBS− will also immediately block the high concentration steroid current. Taken together the data suggest that α1S299 is important in at least the in transduction of the steroid binding to the rest of the receptor.
Keywords: GABAA, neurosteroid, allopregnanolone, substituted cysteine, transmembrane, transduction
1. Introduction
Fast inhibitory neurotransmission in the central nervous system is generally signaled through GABAA receptors (Whiting 2003). This pentameric receptor of differing α, β and usually γ subunits can be positively or negatively modulated by many different drugs and ligands (Whiting 2003); some of these drugs include the clinically important benzodiazepines and anesthetics. An endogenous set of ligands with GABAA activity are the pregnan-derived steroids (Akk et al. 2007). The pregnane steroids have well established effects at GABAA receptors (Akk et al. 2007). 3α derivatives, such as 3α-hydroxy-5α-pregnan-20-one (allopregnanolone, ALLO), and 3α,21-hydroxy-5α-pregnan-20-one (tetrahydrodeoxycorticosterone, THDOC) are potent agonist steroids (Akk et al., 2007; Hosie et al., 2007), while 3β isoforms are antagonistic (Wang et al. 2002). Neurosteroids at GABAA receptors may play roles in some neurological such as depression, anxiety (Longone et al. 2008; Girdler and Klatzkin 2007), pain (Schlichter et al. 2006), schizophrenia, and bipolar disorder (Marx et al. 2006) These neurosteroid/GABAA effects are therefore an interesting puzzle, and of scientific and clinical importance.
For the agonistic pregnane steroids, their electrophysiological effects on GABAA receptors have been described well in many different model systems (Akk et al. 2007, Herd et al. 2007). Typically the agonistic neurosteroids enhance GABAA currents by increasing late channel openings (Zhu and Vicini 1997) or stabilizing an open state (Puia et al. 2003; Akk et al. 2004), and prolonging deactivation (Zhu and Vicini 1997). At higher concentrations, ALLO, THDOC and related agonistic steroids will open the receptor channel and pass current in the absence of GABA (Shu et al. 2004).
Less established is the location of the steroid binding site(s) on the GABAA receptor. Mutagenesis of transmembrane (TM) GABAA residues and modeling of the TMs using the nicotinic ACh receptor as a model suggest there are at least two steroid binding sites, one for potentiating effects and another for activating effects (Akk et al. 2004, Hosie et al. 2006, 2007); some reports indicate a separate antagonistic steroid binding site for 3β-OH or sulfated steroids may be distinct from the two agonistic ones (Akk et al. 2001; Wang et al. 2002; Li et al. 2006, Akk et al. 2007). The residues identified for the agonistic sites in these studies (Hosie et al. 2006, 2007, 2009) are transmembrane: the “potentiating” site consists of at least one α1TM1 residue and one α1TM4 residue, while the “activating” site contains at least one α1TM residue and one β2TM3 residue.
These regions, however, may not be the only residues or regions involved in the binding or actions of steroids (Ueno et al. 2004; Wardell et al. 2006; Twede et al. 2007; Akk et al. 2008). Closer looks reveal other areas that can influence the action of either agonistic or antagonistic steroids (Akk et al. 2001; Wang et al. 2002; Wardell et al. 2006; Li et al. 2006; Twede et al. 2007; Akk et al. 2007; Baker et al. 2010).
More problematic to the proposed sites (Hosie et al. 2006, 2007) is the modeling of the TM regions. Concerns about their orientation of the TMs in relation to each other now make it unclear if the α1TM - β2TM3 residues especially are within or near the actual binding site(s), or if they are residues that are important in the movement of the receptor upon steroid binding to cause the effect, or if they could be residues that allow steroid access to the TM regions. The observation that steroids do not compete with and in fact enhance azi-etomidate binding for a α1TM2-β2TM3 anesthetic site suggests a different orientation of the TM regions (Li et al. 2009). Further support is evidence that α1TM2 and β2TM3 do not cross link at the residues described as a binding pocket by Hosie et al. (2006, 2007), but instead cross link the α1TM2-β2TM3 anesthetic site residues (Bali et al. 2009). Therefore, at least the activating steroid binding modeled by Hosie et al. (2006,2007) would not be possible based on the newer, independent biochemical data (Li et al. 2009; Bali et al. 2009).
Nevertheless, the vast majority of evidence indicates that neurosteroids bind within TM regions with at least two distinct sites, one primarily responsible for potentiation and the other primarily responsible for activation (in the absence of GABA) (Akk et al. 2004; Hosie et al. 2006, 2007). As lipophilic compounds which may interact from the intracellular side of the membrane (Akk et al. 2009) a TM location for steroid binding sites is consistent with the biochemistry and evidence to date. The exact TM residues that bind steroids are still an open question. Though deeper residues in TM3, and residues in α1TM3 so far has not been shown to be involved in the actions of neurosteroids, the position of α1TM3 within the membrane and its known role in changing the conformation of the receptor upon ligand binding (Williams and Akabas 1999, 2000, 2002), make it an excellent target region for determining neurosteroid actions on GABAA receptors. Conformational movements or other residues that could be involved in steroid binding or transduction could be discovered by mutants in TM3. I already have a set of α1TM3 cysteine mutants from my previous work (Williams and Akabas 1999, 2000, 2002), and tested if any had unusual sensitivity to neurosteroids.
I have found a residue in TM3 of the α1 subunit, that when mutated to cysteine, exhibited extreme and unusual sensitivity to steroids; activation occurred at least 10× less steroid concentration, and instead of significant potentiation at lower concentrations, another set of activation occurs. Experimental evidence indicates that this residue is intimately involved in neurosteroid actions, most affecting the transduction of steroid binding, and/or by affecting steroid access to the TM regions. The homologous residue in glycine alpha subunits was found to affect ethanol and G protein modulation of those subunits (Yevenes et al. 201).
2. Materials and Methods
2.1 Preparation and electrophysiology
Oocytes were surgically isolated from Xenopus laevis (Xenopus Express, Brooksville, FL). Oocytes were incubated in 50 mg/ml collagenase for 30 min, separated, and washed 3× in OR-3 (70% Leibovitz’ L15/Gibco; with 1 mM HEPES to pH 7.5 and supplemented with 0.5 ml/ml tetracycline and 1% gentamicin). Mature, Stage IV-V isolated oocytes were injected with mRNA (50 ng/ml of each subunit) prepared from rat α1β1γ2S subunits cloned into pGEMHE as described (Williams and Akabas 1999, 2000, 2002). The synthesis and original GABA sensitivity of the α1S299C mutant have been described with no reported significant differences; therefore we assume it is unlikely that there are large conformational changes to the three dimensional structure (Williams and Akabas 1999). Steroids were from steraloids (Newport, RI) or Sigma (St. Louis, MO); pCMBS− was from Toronto Research Chemicals. All other chemicals were from Sigma or other commercial sources.
Electrophysiology was performed by whole-cell two-electrode voltage clamp, using a Warner TEV-700 amplifier, HAI118 interface and LabScribe software. After allowing 2–3 days (at 18ºC) for oocytes to express functional receptors, oocytes were clamped at −60 mV (−60 mV used to keep the oocytes stable longer). Electrodes were filled with 3M KCl and had a resistance about 2 mOhm. Expression was confirmed with an application of 1 mM GABA until peak current (20–30 sec). A 5 min wash period (to recover from desensitization) was next before adding any of the steroids. All chemicals were applied at a gravity driven perfusion of 5 ml/min; all chemicals were dissolved to their final concentrations in CFFR (115 mM NaCl, 2.5 mM KCl, 1.8 mM Mg2Cl, 10 mM HEPES, pH 7.5). Full details of the procedures and the components of CFFR are previously published (Williams and Akabas 1999; Williams 2008).
2.2 Dose response curves
2.2.1 Neurosteroid dose response curve
ALLO and THDOC were originally dissolved in DMSO at 1 mM concentrations. Final DMSO concentrations never exceeded 0.1% and had no significant effect on currents (not shown). Expression of GABAA receptors was confirmed by stable induced currents by 1 mM GABA; steroid induced currents are quantified as the percentage of the 1 mM GABA current. The steroid in question was added alone via the perfusion (in the absence of GABA) and the inward current measured. The range of steroid concentrations was from 10 pM to 5 μM for mutant containing receptors and from 50 nM to 10 μM for wt receptors (or from where there was no longer detectable steroid induced inward current to where the large concentration of steroid caused decreases in induced currents, as reported (Twyman and MacDonald 1992)). Steroids were typically added by perfusion 20–30 sec until current peaked. To look for ALLO or THDOC potentiation of GABA induced current, the steroid and 1 μM GABA (app EC30) were co-applied and the inward inflection measured. Concentrations of steroid ranged from 0.05 pM to 100 pM for S299C containing receptors and 100–500 nM for wild type receptors (or from about where there was clearly no steroid induced potentiation to where steroid alone induced current). For all doses, steroids were washed out 3–5 min between different concentrations (higher concentrations got longer wash). Oocytes received different steroid concentrations, and all steroid concentrations were tested in at least two different batches of oocytes for wt and mutant receptors. All potentiations are measured as percent increase of the 1 μM GABA current (approximate EC30). All dose responses were plotted using GraphPad Prism (San Diego, CA). Any significant differences in responses were sought using GraphPad Instat (ANOVA for within a single dose response; t-tests for the same concentration wild type v. mutant).
2.2.2 Piperidine-4-sulfonic acid dose response curve
Piperidine-4-sulfonic acid (P4S), a specific GABAA partial agonist used in basic electrophysiology experiments (Krossgaard-Larsen et al., 1980; Galvez-Ruano et al., 1995) was dissolved in CFFR as a 100 mM stock and stored at 4°C. Before the experiment, P4S was diluted further in CFFR to the necessary concentrations. Similar experiments were performed at wild type and S299C containing receptors. A maximal GABA current induced by 1 mM GABA at −60 mV was recorded. P4S at various concentrations (1 mM to 1 μM) was applied via perfusion with 5 minute washes with CFFR in between. The percent current induced by the concentrations of P4S was recorded. These percents were plotted into a dose response curve using GraphPad Prism.
2.2.3 Ethanol dose response
Control GABA currents were recorded at −60 mV for both wt or to S299C containing receptors. Ethanol in 1 mM to 100 mM was applied in the perfusion to wt or to S299C containing receptors with a concentration that was approximately the EC20 for that receptor isoform and the percent potentiation recorded.
2.3 Blocking ALLO-induced current
2.3.1 With picrotoxin
Picrotoxin was dissolved in CFFR with gentle heat. Control 5 nM or 100 nM ALLO-induced currents were established as in the dose responses above. Then, the ALLO was co-applied with 1 mM picrotoxin. Differences were calculated as percent change from control and compared by t-test.
2.3.2 With isoallopregnanolone
Isoallopregnanolone (3β-hydroxy-5α-pregnan-20-one: ISO) was dissolved in DMSO then CFFR as described for ALLO and THDOC. Stable control 5 nM or 100 nM ALLO currents were established. When the control was 5 nM ALLO, ISO at 5 nM or 100 nM were co-applied via the perfusion. When the control was 100 nM ALLO, ISO at 5 nM or 1μM were co-applied via the perfusion. Data are expressed as percent change from control; significant differences in currents were determined by t-test.
2.4 Substituted cysteine accessibility
Substituted cysteine accessibility (SCAM) experiments on the S299C mutant were performed by modifying the published procedure (Williams and Akabas 1999, 2000, 2002). Stable, control ALLO currents at 5 nM or 100 nM were obtained at −60 mV with appropriate (3–5 min) wash out times. 0.5 mM pCMBS− was perfusion applied in the presence of 5 nM or 100 nM for 1 min with 5 min wash out before the next application, then new 5 nM or 100 nM ALLO currents were found. Differences were calculated as the percent change of the after pCMBS− currents compared to the control currents. As a negative control and for comparison for statistical purposes, similar SCAM experiments were performed at S299C containing receptors with 5 nM or 100 nM ALLO inducing current, and the pCMBS− added in the presence of a high (100 nM -1 μM) or low (5 nM) concentration of ISO or 17-PA ((3α,5α)-17-phenylandrost-16-en-3-ol; Mennerick et al. 2004). Percent differences were analyzed by ANOVA with Student-Newman-Kuels post hoc tests, grouping by same control currents, and performed by Instat.
2.5 Extreme low dose assay
In the S299C containing receptors, 100 pM ALLO and 100 nM GABA (EC5) were perfusion applied alone and then simultaneously for 30 sec at −60 mV with 3 minutes between the different applications. Similar experiments were performed with 5 nM ALLO, and 100 nM GABA. The intent was to see if the currents were additive or synergistic to determine if any ALLO potentiation of currents still existed, or if activation only occurred. The currents induced by the low concentration of ALLO and the 100 nM GABA were added and compared to the current induced by co-application of the two. The average of the low ALLO/low GABA added currents was set as 1, and the rest of the added or co-applied obtained currents were scaled to that number. Statistics were done by Instat and described in the results.
3. Results
3.1 Neurosteroids have a double activation profile at the α1S299C mutant
ALLO induced current at wt α1β1γ2S receptors in a manner consistent with previous results (Liu et al. 2001; Shu et al. 2004), with one maximal about 43 ± 15% of 1 mM GABA and an EC50 of 0.87 μM (not shown). ALLO potentiated wild type receptors (not shown; 120 ± 28% at 0.5 μM ALLO; 1 μM GABA, about EC30 n =3). S299C containing receptors were responsive to 1 mM GABA similar to wt (not shown, and Williams and Akabas, 1999). At S299C α1β1γ2S receptors, ALLO when added in the absence of GABA, caused an inward current in concentrations ranging from 50 pM (6 ± 4% n = 4; not shown) to 5 μM, the concentration before inhibition of the currents (Fig 1A, 1B). The dose response was bimodal, with a peak at 5 nM and another at 100 nM, each peak calculated by the curve fit to be 58 ± 0.25% and the second to 42 ± 0.26% of maximal (1 mM GABA) current. Taking each plateau to valley as separate curves the EC50’s were fit as 0.59 nM and 70 nM (Fig. 2A). THDOC caused inward currents similarly to ALLO. In the absence of GABA, THDOC applications in the range of 5 μM to 10 pM resulted in inward currents (Fig 1C, 1D), again with a bimodal response. THDOC maximal responses were fit as 57 ± 5.3% and 48 ± 0.35% of maximal current. Taking each peak to valley as a separate curve the EC50’s were found to be 0.73 nM and 41 nM (Fig. 2B). Curves for both ALLO and THDOC plot best as two independent events. (S. Viscido, personal communication) These actions may not necessarily be two independent events, but fit best and are clearest that way.
Figure 1.

Low concentrations of agonistic neurosteroids cause current influx at α1S299C containing receptors. Tracings are examples of 3–31 samples. A: Representative tracings from the same oocyte expressing α1S299C containing receptors in the presence of 1 mM GABA, then 100 nM ALLO, then 5 nM ALLO. Each ALLO concentration passes approximately 50% of 1 mM GABA current. B: Representative trace of α1S299C containing receptors in the presence of 1 mM GABA, then 100 nM THDOC, with THDOC passing approximately 50% of 1 mM GABA current. C: In a different oocyte, representative trace of α1S299C containing receptors in the presence of 1 mM GABA, then 1 nM THDOC, with 1 nM THDOC passing more than 50% of 1 mM GABA current. For all panels, cells were from different experiments on different days with different injections into different oocytes; hence the variability in the amount of induced currents and the experiments in panel A taking longer to reach a clear maximum.
Figure 2.

Dose response curves of α1S299C containing receptors to ALLO or THDOC. Sample numbers for individual points (mean ± SEMs) were 3–31. A: α1S299C dose response to ALLO at a wide range of ALLO concentrations. The current peaks twice. The first peak was fit to 58 ± 0.25% and the second to 42 ± 0.26% of maximal (1 mM GABA) current. Taking each plateau to valley as separate curves the EC50’s were fit as 0.59 nM and 70 nM. B: α1S299C dose response to THDOC at a wide range of THDOC concentrations. THDOC maximal responses were fit as 57 ± 5.3% and 48 ± 0.35% of maximal current. Taking each peak to valley as a separate curve the EC50’s were found to be 0.73 nM and 41 nM. Curves are not connected as per Paul and Purdy (1992) as these neurosteroid curves fit best as two independent events (S. Viscido, personal communication), and not one smooth curve with two plateaus (Paul and Purdy, 1992, fig 4).
Hill numbers in all cases were greater than 1.8. There were no significant differences in the data between the two hormones.
3.2 Picrotoxin blocks ALLO activation at S299C containing mutants
To help ensure that the ALLO induced currents were from the heterologously expressed GABAA receptors and not from some kind of unknown receptor channel on the oocyte membrane, experiments attempting to block the ALLO induced current with the GABAA channel blocker picrotoxin were performed. Oocytes were exposed to 1 mM GABA to confirm expression of the receptors. Then control ALLO currents, 5 nM or 100 nM was added. After these currents were stable, 5 nM or 100 nM ALLO was co-applied with 1 mM PTN. Co-application of 1 mM PTN with the 5 nM ALLO blocked 80 ± 20% (n = 3) of induced currents induced by that ALLO concentration; 1 mM PTN co-applied with 100 nM ALLO blocked 82% ± 7% (n = 4) of currents induced by that ALLO concentration (Fig. 3). After wash out of PTN, the ALLO currents rebounded to near control.
Figure 3.

PTN inhibits on ALLO induced current at α1S299C containing receptors. A: 5 nM ALLO induces significant current at α1S299C containing receptors (first trace). That current is blocked by 1 mM PTN (second trace) but is recovered after the PTN is washed out and 5 nM ALLO reapplied (third trace). Representative of 3 experiments. B. 100 nM ALLO induces significant current at α1S299C containing receptors (first trace). That current is blocked by 1 mM PTN (second trace) but is recovered after the PTN is washed out and 100 nM ALLO reapplied (third trace). Representative of 4 experiments. C: Bar graph of blocks showing nearly 80% block of both 5 nM and 100 nM induced ALLO currents by 1 mM PTN.
3.3 Isoallopregnanolone blocks ALLO activation at S299C containing receptors
The 3β isoform of ALLO (ISO) does not have significant agonistic effects at GABAA receptors; in many ways it is an antagonist to the actions of ALLO (Wang et al. 2002); hence it was used to help determine the specificity of the strong agonistic actions of ALLO at S299C containing receptors. If these unusual agonistic properties of ALLO were due to specific binding of ALLO to the S299C containing receptors, then ISO should block at least one of the agonistic effects. In these experiments, ISO was used as an antagonist for activation effects of ALLO. Currents induced by 5 nM or 100 nM ALLO were attempted to be inhibited by a corresponding low or high concentration of ISO (Fig. 4). ISO significantly inhibited ALLO induced currents in all combinations. Current induced by 5 nM ALLO was blocked by both high and low concentrations of ISO. Current induced by 5 nM ALLO was blocked 73 ± 4% by 100 nM ISO (n = 3; Fig. 4D) and was blocked 90 ± 5 % by 5 nM ISO (n =3) (Fig. 4A). Current induced by 100 nM ALLO was also blocked 61 ± 20% by 1 μM ISO (n =3; Fig. 4D) and was blocked 86 ± 8% by 5 nM ISO (n = 3; Fig. 4B, 4C).
Figure 4.

ISO inhibits ALLO induced currents α1S299C containing receptors. A: 5 nM ALLO induces significant current at α1S299C containing receptors (first trace). That current is blocked by 5 nM ISO (second trace) but is mostly recovered after the ISO is washed out and 5 nM ALLO reapplied (third trace). Representative of 3 experiments. 100 nM ISO blocked current induced by 5 nM ALLO similarly (not shown). B: 100 nM ALLO induces significant current at α1S299C containing receptors (first trace). That current is blocked by 5 nM PTN (second trace) but is recovered after the ISO is washed out and 100 nM ALLO reapplied (third trace). Representative of 3 experiments. 1 μM ISO blocked current induced by 100 nM ALLO similarly (not shown). C: Bar graph of 5 nM ISO blocks on simultaneously added 100 nM or 5 nM ALLO on α1S299C containing receptors. 5 nM ISO inhibits both 100 nM and 5 nM ALLO induced currents 80–85%. All sample number of 3. D. Bar graph of 1 μM ISO block on simultaneously added 100 nM ALLO and 100 nM ISO block of 5 nM ALLO on α1S299C containing receptors. 1 μM ISO inhibits 100 nM ALLO currents about 60% and 100 nM ISO blocks 5 nM ALLO induced currents 90%. All sample number of 3.
3.4 Cysteine modification of S299C
Previous results indicated that S299C was not accessible to be modified by the water soluble sulfhydryl reagent pCMBS− when pCMBS− was added in the presence of GABA (Williams and Akabas 1999). Preliminary results also indicated no change in GABA induced current when pCMBS− was added in the presence of ALLO, with ALLO concentrations ranging from 1 μM to 5 nM (Williams 2009). To determine if S299C is important in the binding or action of steroids, as the ALLO activation and dose response suggest, a modification to the SCAM was performed. After confirming oocytes expressed GABAA receptors with a pulse of 1 mM GABA, currents were induced with either the “high” concentration (100 nM) or “low” concentration (5 nM) of activating ALLO. Then a concentration of ALLO was added in the presence of pCMBS−, so that there was a set of four different experiments: “low” ALLO control with “low” ALLO + pCMBS−; “low” ALLO control with “high” ALLO + pCMBS−; “high” ALLO control with “low” ALLO + pCMBS−; and “high” ALLO control with “high” ALLO + pCMBS−. Subsequent currents for the first concentration of ALLO were determined after the application of ALLO + pCMBS−. At wt receptors, pCMBS− when added in any concentration of ALLO, did not affect subsequent ALLO induced currents (not shown).
3.4.1 Effect of “high” ALLO + pCMBS- on ALLO induced currents
When 100 nM ALLO was added in the presence of pCMBS-, currents induced by both 100 nM ALLO and 5 nM ALLO were significantly inhibited. (ANOVA p<0.05 compared to negative controls) When added in the presence of 100 nM ALLO, pCMBS- inhibited subsequent currents induced by 100 nM ALLO by 63 ± 15% (n = 3; Fig. 5A, bottom bar). In addition, when co-applied with the pCMBS-, 100 nM ALLO currents were reduced 62 ± 17% (n =3; Fig. 5A, middle bar). When added in the presence of 100 nM ALLO, pCMBS− inhibited the subsequent 5 nM currents by 63 ± 15% (n = 3; Fig. 5A, top bar). Therefore high ALLO concentrations when added with pCMBS− caused significant decreases in subsequent ALLO induced currents at both “high” and “low” ALLO.
Figure 5.

ALLO induced currents affected by pCMBS− applied under different conditions. A: In a typical SCAM experiment, where 5 nM or 100 nM control currents were determined, 0.5 mM pCMBS−, when co-applied with 100 nM ALLO caused significant decreases in subsequent 5 nM (top) or 100 nM ALLO (bottom) induced currents. In addition, when 100 nM ALLO was the original control current, the co-application of 100 nM ALLO with 0.5 mM pCMBS− showed significantly less ALLO induced current (middle bar). Asterisks indicate a statistically significant effect. B: In a typical SCAM experiment with 5 nM ALLO (top) or 100 nM ALLO (bottom) control currents, 0.5 mM pCMBS− co-applied with 5 nM ALLO did not have as significant effects on subsequent currents; in addition the co-application of 5 nM ALLO with the pCMBS− had little effect on the induced 5 nM ALLO current (middle). C: In a series of control experiments for the 0.5 mM pCMBS− co-applied with 100 nM ALLO, 0.5 mM pCMBS− co-applied with 1 μM ISO showed significant effects (symbolized by the asterisk) on subsequent 100 nM ALLO induced current (middle bar) but not 5 nM ALLO induced current (top bar). However, an antagonist with absolutely no agonistic activity, 17-PA (Mennerick et al. 2004), when co-applied with pCMBS−, showed no significant effects on subsequent 100 nM ALLO currents (bottom bar.) (All n of 3).
3.4.2 Effect of “low” ALLO + pCMBS- on ALLO induced currents
However when pCMBS− was co-applied with only 5 nM ALLO, the effects were not as large and were not significant (ANOVA, p= 0.12 compared to the 100 nM SCAM’s and negative control). When 5 nM ALLO was added in the presence of pCMBS−, subsequent 5 nM ALLO induced currents were reduced 28 ± 8% (n = 6, Fig. 5B, top bar), while subsequent 100 nM ALLO induced currents were reduced only 22 ± 20% (n = 3, Fig. 5B, bottom bar). In addition, there was no significant decrease of 5 nM ALLO current when ALLO was simultaneously applied with pCMBS− (7 ± 29% decrease n = 4; Fig. 5B, middle bar). Based in my experience in many SCAM experiments (Williams and Akabas, 1999, 2000, 2001, 2002), I can conclude, despite the large error bars, that these numbers trend to near zero.
3.4.3 Effect of ISO + pCMBS on ALLO induced currents
To attempt to show that the significant effects of pCMBS− when co-applied with 100 nM ALLO on both subsequent 100 nM and 5 nM ALLO currents were due to modification of the introduced cysteine resulting from movement of TM3 induced by the agonistic ALLO, the 3β isoform, ISO, was used in a similar SCAM protocol. Control 100 nM ALLO or 5 nM ALLO currents were obtained, and then ISO added at 1 μM or 100 nM with pCMBS−. The percent difference in subsequent currents was then calculated. As expected, pCMBS− when co-applied with 100 nM ISO did not seem to affect subsequent 5 nM ALLO currents (3 ± 14% n= 4; Fig. 5C, top bar). 5 nM ISO plus pCMBS− did not affect subsequent 5 nM or 100 nM ALLO currents (not shown). Unexpectedly, pCMBS− when added with the 1 μM ISO did cause significant decreases in subsequent 100 nM ALLO currents (−71 ± 10%, n=4; ANOVA p< 0.03; Fig. 5C middle bar).
3.4.4 Effect of 17-PA + pCMBS on ALLO induced currents
Because of the unexpected significant effect of pCMBS− when co-applied with 1 μM ISO on subsequent 100 nM ALLO currents, another antagonistic steroid, 17-PA, that is more competitive for the agonistic steroids (Mennerick et al. 2004) was used in similar SCAM experiments. The 3β isoforms may have more general and noncompetitive responses (Wang et al. 2002; Mennerick et al. 2004), which may explain why “high” ISO caused a movement(s) that allowed S299C to become water accessible. 1 μM 17-PA when added with pCMBS−, did not have a significant effect on 100 nM ALLO induced currents, only a −4 ± 8% reduction (n = 3, Fig. 5C, bottom bar).
3.5 Low dose GABA and low dose ALLO are not synergistic
One question that remained from the low doses of ALLO activating the S299C containing receptors was if there was still some potentiation occurring, perhaps hidden by the activation. To look at this possibility, I used extremely low doses of GABA (approximate EC5, 100 nM) and ALLO (approximate EC5, 100 pM) separately, and then together. Separately, the currents were obtained, added together, and the average used as “1” to scale all the responses, as described in the methods. The co-application currents scaled to 1.21 ± 0.45, which was statistically insignificant to the added (1.0 ± 0.35) (n=4) (Fig 6A).
Figure 6.

S299C ALLO and GABA currents: additive or synergistic? Bar graphs show that the amount of current induced by 100 nM GABA and 100 pM ALLO (A) or 100 nM GABA and 5 nM ALLO (B) are not significantly different (20, 27% respectively) from when the two compounds at the same concentrations are co-applied at S299Cα1β1γ2S receptors. Numbers were scaled and analyzed as described in the methods.
In case that amount of ALLO was too low despite the extreme sensitivity of the S299C containing receptors, the same experiments were repeated with 5 nM ALLO. The co-application currents scaled to 1.27 ± 0.39, which was statistically insignificant to the added (1.0 ± 0.21) (n = 4) (Fig 6B).
3.6 P4S dose response
P4S concentrations ranging from 1 mM to 1 μM were added to either wild type receptors or S299C containing receptors. Currents induced by P4S were measured as the percentage of 1 mM GABA (saturating) current. There were no significant differences in the maximal amount of current induced (wt max 61 ± 3.7%; S299C max 58 ± 1.6% n of 3 for each) or in the Kd (wt 110 ± 37 μM; S299C 90 ± 18 μM) of P4S between wild type and S299C containing receptors (Fig. 7).
Figure 7.

Wild type v S299C P4S dose response curves. The amounts of current induced by various concentrations of P4S were measured at wild type α1β1γ2S receptors and S299Cα1β1γ2S receptors. Currents were calculated as the percentage of maximal (1 mM GABA) current. Error bars were removed for clarity. There is no significant difference in the dose response of the wild type and S299C containing receptors to P4S in the maximal amount of current induced (wt max 61 ± 3.7%; S299C max 58 ± 1.6% n of 3 for each) or in the Kd (wt 110 ± 37 μM; S299C 90 ± 18 μM).
3.7 Ethanol
Based in recently published work (Yevenes et al. 2010) where the homologous residue in TM3 of glycine α subunits was mutated causing significant effects on ethanol’s ability to potentiate the mutant glycine receptors, we decided to investigate if the α1S299C containing GABAA receptors showed significant differences in their ability to be potentiated by ethanol. A preliminary dose response (not shown) suggests a three-fold decrease in percent potentiation and a shift to a higher EC50, in general agreement with the glycine study (Yevenes et al. 2010). Future work will continue into the effect of this mutation on the effects of ethanol at GABAA receptors.
4. Discussion
The results indicate that agonistic neurosteroids such as ALLO and THDOC are significantly more potent and efficacious with a mutation at the S299 residue in the TM3 region of the rat α1 subunit for the GABAA receptor. This residue, when converted to a cysteine, most likely affects the transduction of the steroid binding. Alternatively, the accessibility, or perhaps one of the bindings of the steroid could be affected. Based in recent evidence of transduction differences for ethanol in homologous glycine α subunit mutations (Yevenes et al. 2010), and in the discussion below, changes in transduction are the most probable explanation. This research is the first report of a α1TM3 residue involved in the actions of neurosteroids at GABAA receptors. Previous studies indicated pre TM2, TM1, TM1-TM4, β2 TM3 (Rick et al. 1998, Akk et al. 2008, Wardell et al. 2006, Twede et al. 2007, Hosie et al. 2006, 2009) in the actions of agonistic neurosteroids at GABAA receptors.
The receptors containing the α1S299C did not show any significant potentiation of current induced by low concentrations of GABA with steroid doses as low as 100 pM as well as at 5 nM. However, receptors containing α1S299C mutant activated (passed current in the absence of GABA) in a distinct biphasic response with EC50’s that differed over 1500 fold and over 40 fold from wild-type receptors. The current at both these concentrations was inhibited by the antagonist ISO and by the more general GABAA pore blocker picrotoxin, strongly suggesting the currents at both of these steroid concentrations were induced at expressed GABAA receptors and specific for steroid binding into a specific site(s). The distinct biphasic activation of steroids at S299C containing receptors supports evidence of at least two sites for agonistic actions of steroids. The activation is distinct enough so that the curves fit best as separate plots with the percent activation increasing, severely dropping, and then increasing again. This pattern existed for both ALLO and THDOC, in multiple sets of oocytes, with different injection times, sometimes many months apart. Why such a phenomenon occurs cannot be explained by these experiments. Speculation includes maybe the mutation affects one part of steroid binding or induced movements more than another, or that the mutations, in receptors containing two mutant α subunits do not act in an equivalent fashion, causing extremely distinct effects. More studies are necessary to determine the exact mode of action of mutations in this locale. Based in mutations in a homologous residue of glycine α subunits, the most likely reason would be disrupted conformational changes (Yevenes et al. 2010). Both ALLO and THDOC had similar effects, with similar EC50’s and maximal responses and lack of significant potentiation (not shown for THDOC); evidence these steroids interact at same or similar sites with similar transduction of the binding on receptor structure.
Both ALLO and THDOC showed no significant potentiation of submaximal GABA current was detected, more clearly demonstrated when the co-application of ALLO and GABA added up to the amount of current induced by ALLO plus GABA applied separately (Fig. 6). This lack of synergy, in other words, the lack of a significant increase in EC5 GABA induced current with the co-application of 100 pM or 5 nM ALLO suggests that ALLO activates the S299C mutant but has little potentiating ability at this mutant.
Combining the activation and lack of potentiation results indicates that the α1S299C mutation either caused a higher affinity, or caused a change in the structure of α1TM3 that allowed transduction of the steroid binding to be more efficient, allowing binding in the potentiating (high affinity) site to cause activation, or allowed easier access of neurosteroids to the TM regions. Responses of α1S299C containing receptors to GABA, BZs, and propofol were also near wild type (Williams and Akabas 1999, 2000, 2002). The response to propofol being near normal for S299C containing receptors is particularly important as propofol binds in TM regions, like steroids, and has similar electrophysiological effects (Bai et al., 1999; Zhu and Vicini 1997; Puia et al. 2003; Akk et al. 2004). As well, α1S299C containing receptors had responses to P4S nearly identical to wild type (Fig. 7). However this mutant has decreased potentiation by ethanol (not shown); similar glycine receptor mutants show a similar decrease of potentiation by ethanol (Yevenes et al., 2010). Therefore, this mutation is not specific for the actions of neurosteroids, but could influence the actions of some TM binding ligands.
The SCAM experiments begin to reveal how α1S299C could be interacting with steroids. When added with 100 nM ALLO, pCMBS− significantly decreased subsequent currents induced by both 100 nM ALLO and 5 nM ALLO. In addition, the current induced by 100 nM ALLO when co-applied with pCMBS− was significantly reduced (Fig. 5). This immediate decrease in current markedly differs from pCMBS− added in the presence of GABA, where only changes were seen in subsequent currents, never simultaneously (Williams and Akabas, 1999). In this original case, currents are affected only afterward because GABA binds elsewhere (Amin and Weiss 1993); a conformation change is needed for reaction and effect later (Williams and Akabas 1999). That delay was due to the receptor needing to enter a desensitized state(s) for pCMBS− modification (Williams and Akabas 1999). At α1S299C, pCMBS− added in the presence of ALLO caused an immediate decrease in current. Therefore it is likely pCMBS− when added with 100 nM ALLO modifies α1S299C in an open state. The immediate actions of pCMBS− on 100 nM ALLO current could suggest a direct competition for lower affinity binding or accessibility. The lack of subsequent currents, whether induced by 5 nM ALLO or 100 nM ALLO suggest a binding site or accessibility to a site could have been blocked. The lack of significant effects on pCMBS− when added with 5 nM ALLO on currents, both simultaneous and subsequent suggest the α1S299C is more important in the binding or access to the lower affinity site; 5 nM ALLO was too low a concentration to significantly affect the α1S299C residue. The modification by pCMBS− in the presence of high concentration but not low concentration of ISO also suggests a lower affinity interaction is needed.
However, all the above data can also be explained if the α1S299C were part of a membrane steroid transduction site. Since hormones bind in the TMs (Hosie et al. 2006, 2007 2009; Rick et al. 1998; Akk et al. 2008; Wardell et al. 2006; Twede et al. 2007; Uneo et al. 2004), movements of TM residues are more likely to cause the actions of the neurosteroids, as opposed to extracellular acting ligands; extracellular ligands act more by moving ECM regions (Boileau and Czjakowski 1999; Jones-Davis et al. 2005; Keramidas et al. 2006). The change from S to C may subtly disrupt TM interactions, causing the steroid binding to cause greater movements, leading to increased apparent affinity and efficacy. Since steroids increase late channel openings (Zhu and Vicini 1997) or stabilize an open state (Puia et al. 2003; Akk et al. 2004), pCMBS− modification of 100 nM ALLO current in the open state should be expected and look like a direct competition block as a result, which looks like what happened. The rest of the SCAMs can also be explained if the modification occurs in an open state on a transduction residue rather than on a binding site residue. pCMBS− when added with 100 nM ALLO affected subsequent 5 nM ALLO currents; suggesting the residue was modified by the presence of the high concentration of ALLO, with the modification creating some kind of hindrance to not allow movements for activation by the 5 nM ALLO. The lack of significant effects on pCMBS− when added with 5 nM ALLO on currents, both simultaneous and subsequent suggest the α1S299C is more important in the movements from the lower affinity site; 5 nM ALLO was too low a concentration to significantly move the α1S299C residue. The modification by pCMBS− in the presence of high concentration but not low concentration of ISO suggests activating movements again are needed for the modification.
One major current hypothesis suggests that two steroid binding sites exist, a “potentiating site” between TM1-TM4 and the “activation” site between α1TM1 and β2 M3 (Hosie et al. 2006, 2009). My data is not inconsistent with the idea of two steroid binding sites; in fact, the mutation seems to better resolve two steroid binding sites by having the two distinct activation components. However, my data is not consistent with the presumed lack of involvement of α1TM3 in NS actions, as suggested by Hosie et al. (2006). This current data also does not suggest α1S299C as part of a binding site.
Conclusion
Using the orientation of the TMs as suggested by other biochemical methods (Li et al. 2009, Bali et al. 2009), using azi-etomidate cross-linking and disulfide trapping respectively (Fig. 8), the steroid actions described to a mutant α1S299 TM3 residue do not differentiate between those models and the TM orientation of Hosie et al. (2006,2009). This is because the α1TM3 S299 residue would face away from most of the other a subunit TMs, except for a small face of TM4. Otherwise α1S299C seems to orient to the membrane (Fig. 8). If α1S299 is part of a steroid binding site, it seems to be one not yet described. More likely, α1S299C is near a binding site and is involved in accessibility to the site and/or transduction of the steroid binding. The putative location of α1S299 (Fig 8) suggests it could be involved in the access of steroid from membrane lipid to the receptor complex (Akk et al. 2005, 2009). The recent data from glycine receptors with mutations in a homologous residue suggest a transduction role for α1S299C (Yevenes et al. 2010). More experiments will be needed to better differentiate between transduction and accessibility roles of this residue.
Figure 8.
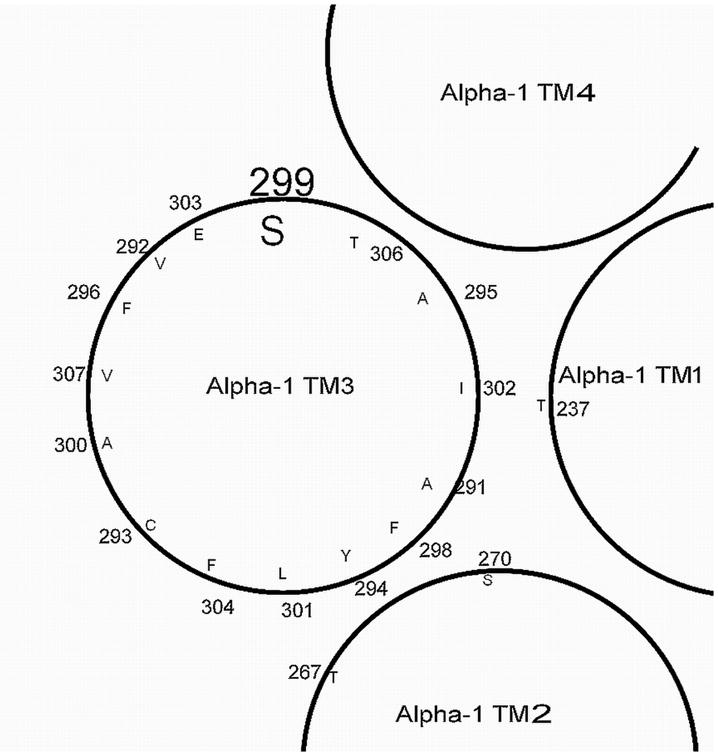
A GABAA receptor α1 subunit TM3 region helical plot. The orientation of the residues is as discussed in Williams and Akabas (1999). The residue of this paper is enlarged as compared to the others; S299 would be approximately halfway down the helix. Cysteines substituted for A291, Y294, F296, F298, A300, L301, and E303 react with pCMBS− in the presence of GABA (Williams and Akabas, 1999, 2000); S299 did not react to pCMBS− under that condition but is located on the same face as two others (E303 and F296) suggesting this face can become water accessible. The face that interacts with the α1 TM2 region has within it A291, F298, Y294, and L301; the face is defined according to the data of Mihic et al. (1997) and Bali and Akabas (2004) [using homolgous β residues] for S270-A291 and Jansen and Akabas (2006) for the T267 with L301, F304 and A305, with the approximate locations of T267 and S270 marked. The indicated TM1 (T237) residue is the one identified by Hosie et al. (2006) for steroid actions.
Acknowledgments
This work was supported by the NIH/NIMH grant 1R15MH076896-01.
I thank Alexandra E. Williams for help with the art for Figure 8.
Abbreviations
- TM
transmembrane
- ALLO
allopregnanolone, 3α derivatives, such as 3α-hydroxy-5α-pregnan-20-one
- THDOC
tetrahydroxycorticosterone, 3α,21-hydroxy-5α-pregnan-20-one
- CFFR
Calcium Free Frog Ringer’s
- pCMBS−
para-chloromercuribenzenesulfonic acid
- P4S
piperidine-4-sulfate
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akk G, Bracamontes J, Steinbach JH. Pregnenolone sulfate block of GABAA receptors: mechanism and involvement of a residue in the M2 region of the α subunit. J Physiol. 2001;546:641–646. doi: 10.1111/j.1469-7793.2001.0673e.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akk G, Bracamontes JR, Covey DF, Evers A, Dao T, Steinbach JH. Neuroactive steroids have multiple actions to potentiate GABAA receptors. J Physiol. 2004;558:59–74. doi: 10.1113/jphysiol.2004.066571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akk G, Shu H-J, Wang C, Steinbach JH, Zorumski CF, Covey DF, Mennerick S. Neurosteroid access to the GABAA receptor. J Neurosci. 2005;25:11605–11613. doi: 10.1523/JNEUROSCI.4173-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akk G, Covey DF, Evers AS, Steinbach JH, Zorumski CF, Mennerick S. Mechanisms of neurosteroid interactions with GABAA receptors. Pharmacol Ther. 2007;116:35–57. doi: 10.1016/j.pharmthera.2007.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akk G, Li P, Bracomontes J, Reichart DE, Covey DF, Steinbach JH. Mutations of the GABAA receptor of the α1 subunit M1 domain reveal unexpected complexity for modulation by neuroactive steroids. Mol Pharmacol. 2008;74:614–627. doi: 10.1124/mol.108.048520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akk G, Covey DF, Evers AS, Steinbach JH, Zorumski CF, Mennerick S. The influence of the membrane on neurosteroid actions at GABAA receptors. Psychoneuroendocrinology. 2009;345:559–566. doi: 10.1016/j.psyneuen.2009.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin J, Weiss DS. GABAA receptor needs two homologous domains of the beta subunit for activation by GABA but not by pentobarbital. Nature. 1993;366:565–569. doi: 10.1038/366565a0. [DOI] [PubMed] [Google Scholar]
- Bai D, Pennefather PS, MacDonald JF, Orser BA. The general anesthetic propofol slows deactivation and desensitization of GABAA receptors. J Neurosci. 1999;15:10635–10646. doi: 10.1523/JNEUROSCI.19-24-10635.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker C, Sturt BL, Bamber BA. Multiple roles for the first transmembrane domain of GABAA receptor subunits in neurosteroid modulation and spontaneous channel activity. Neurosci Lett. 2010;473:242–247. doi: 10.1016/j.neulet.2010.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bali M, Akabas MH. Defining the propofol binding site location on the GABAA receptor. Mol Pharmacol. 2004;65:68–76. doi: 10.1124/mol.65.1.68. [DOI] [PubMed] [Google Scholar]
- Bali M, Jansen M, Akabas MH. GABA-induced intersubunit conformational movement in the GABAA receptor α1M1-β2M3 transmembrane subunit interface: experimental basis for homology modeling of an intravenous anesthetic binding site. J Neurosci. 2009;29:3083–3092. doi: 10.1523/JNEUROSCI.6090-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boileau AJ, Czajkowski C. Identification of transduction for benzodiazepine regulation of the GABAA receptor: three residues are required for allosteric coupling. J Neurosci. 1999;19:10213–10220. doi: 10.1523/JNEUROSCI.19-23-10213.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvez-Ruano E, Aprison MH, Robertson DH, Lipkowitz KB. Identifying agonistic and antagonistic mechanisms operative at the GABA receptor. J Neurosci Res. 1995;42:666–673. doi: 10.1002/jnr.490420509. [DOI] [PubMed] [Google Scholar]
- Girdler SS, Klatzkin R. Neurosteroids in the context of stress: implications for depressive disorders. Pharamcol Ther. 2007;116:125–139. doi: 10.1016/j.pharmthera.2007.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herd MB, Belelli D, Lambert JJ. Neurosteroid modulation of synaptic and extrasynaptic GABAA receptors. Pharamcol Ther. 2007;116:20–34. doi: 10.1016/j.pharmthera.2007.03.007. [DOI] [PubMed] [Google Scholar]
- Hosie AM, Clarke L, da Silva H, Smart TG. Conserved site for neurosteroid modulation of GABAA receptors. Neuropharmacology. 2009;56:149–154. doi: 10.1016/j.neuropharm.2008.07.050. [DOI] [PubMed] [Google Scholar]
- Hosie AM, Wilkins ME, da Silva HMA, Smart TG. Endogenous neurosteroids regulate GABAA receptors through two discrete membrane sites. Nature. 2006;444:486–489. doi: 10.1038/nature05324. [DOI] [PubMed] [Google Scholar]
- Hosie AM, Wilkins ME, Smart TG. Neurosteroid binding sites on GABAA receptors. Pharmacol Ther. 2007;116:7–19. doi: 10.1016/j.pharmthera.2007.03.011. [DOI] [PubMed] [Google Scholar]
- Jansen M, Akabas MH. State-dependent cross-linking of the M2 and M2 segments: functional basis for the alignment of GABAA and acetylcholine receptor M3 segments. J Neurosci. 2006;26:4492–4499. doi: 10.1523/JNEUROSCI.0224-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones-Davis DM, Song L, Gallagher MJ, Macdonald RL. Structural determinants of benzodiazepine allosteric regulation of GABAA receptor currents. J Neurosci. 2005;25:8056–8065. doi: 10.1523/JNEUROSCI.0348-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogsgaard-Larsen P, Falch E, Schousboe A, Curtis DR, Lodge D. Piperidine-4-sulphonic acid, a new specific GABA agonist. J Neurochem. 1980;34:756–759. doi: 10.1111/j.1471-4159.1980.tb11211.x. [DOI] [PubMed] [Google Scholar]
- Keramidas A, Kash TL, Harrison NL. The pre-M1 segment of the alpha1 subunit is a transduction element in the activation of the GABAA receptor. J Biol Chem. 2006;275:11–22. doi: 10.1113/jphysiol.2005.102756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G-D, Chiara DC, Cohen JB, Olsen RW. Neurosteroids allosterically modulate binding of the anesthetic etomidate to γ-aminobutyric acid type A receptors. J Biol Chem. 2009;284:11771–11775. doi: 10.1074/jbc.C900016200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Covey DF, Steinbach JH, Akk G. Dual potentiating and inhibitory actions of a benz[e]indene neurosteroid analog on recombinant α1β2γ2 GABAA receptors. Mol Pharmacol. 2006;69:2015–2026. doi: 10.1124/mol.106.022590. [DOI] [PubMed] [Google Scholar]
- Liu Q-Y, Chang YH, Schaffner AE, Smith SV, Barker JL. Allopregnanolone activates GABAA receptor/Cl− channels in a multiphasic manner in embryonic rat hippocampal neurons. J Neurophysiol. 2001;88:1147–1158. doi: 10.1152/jn.00942.2001. [DOI] [PubMed] [Google Scholar]
- Longone P, Rupprecht R, Manieri GA, Bernardi G, Romeo E, Pasini A. The complex roles of neurosteroids in depression and anxiety disorders. Neurochem Int. 2008;52:596–601. doi: 10.1016/j.neuint.2007.10.001. [DOI] [PubMed] [Google Scholar]
- Marx CE, Stevens RD, Shampine LJ, et al. Neuroactive steroids are altered in schizophrenia and bipolar disorder: relevance to pathophysiology and therapeutics. Neuropsychopharmacology. 2006;31:1249–1263. doi: 10.1038/sj.npp.1300952. [DOI] [PubMed] [Google Scholar]
- Mennerick S, He Y, Jiang X, et al. Selective antagonism of 5α-reduced neurosteroid effects at GABAA receptors. Mol Pharmacol. 2004;65:1191–1197. doi: 10.1124/mol.65.5.1191. [DOI] [PubMed] [Google Scholar]
- Mihic SJ, Ye Q, Wick MJ, et al. Sites of alcohol and volatile anesthetic action on GABAA and glycine receptors. Nature. 1997;389:385–389. doi: 10.1038/38738. [DOI] [PubMed] [Google Scholar]
- Paul SM, Purdy RH. Neuroactive steroids. FASEB J. 1992;6:2311–2322. [PubMed] [Google Scholar]
- Puia G, Mienville J-M, Matsumoto K, Takahata H, Wantanabe H, Costa E, Guidotti A. On the putative physiological role of allopregnanolone on GABAA receptor function. Neuropharmacology. 2003;44:49–55. doi: 10.1016/s0028-3908(02)00341-6. [DOI] [PubMed] [Google Scholar]
- Rick CE, Ye Q, Finn SE, Harrison NL. Neurosteroids act on the GABAA receptor at sites on the N-terminal side of the middle of TM2. NeuroReport. 1998;9:373–383. doi: 10.1097/00001756-199802160-00004. [DOI] [PubMed] [Google Scholar]
- Schlichter R, Keller AF, De Roo M, Breton J-D, Inquimbert P, Poisbeau P. Fast genomic actions of steroids on synaptic transmission and role of endogenous neurosteroids in spinal pain pathways. J Mol Neurosci. 2006;28:33–51. doi: 10.1385/jmn:28:1:33. [DOI] [PubMed] [Google Scholar]
- Shu H-J, Eisenmann LN, Jinadasa D, Covey DF, Zorumski CF, Mennerick S. Slow actions of neuroactive steroids at GABAA receptors. J Neurosci. 2004;24:6667–6675. doi: 10.1523/JNEUROSCI.1399-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twede V, Tartaglia AL, Covey DF, Bamber BA. The neurosteroids dehydroepiandrosterone sulfate and pregnenlone sulfate inhibit the UNC-49 GABA receptor through a common set of residues. Mol Pharmacol. 2007;72:1322–1329. doi: 10.1124/mol.107.034058. [DOI] [PubMed] [Google Scholar]
- Twyman RE, Macdonald RL. Neurosteroid regulation of GABAA receptor single channel kinetic properties of mouse spinal cord neurons in culture. J Physiol. 1992;456:215–245. doi: 10.1113/jphysiol.1992.sp019334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno S, Tsutsui M, Toyohira Y, Minami K, Yanagihara N. Sites of allosteric modulation by neurosteroids on ionotropic γ-aminobutyric acid receptor subunits. FEBS Lett. 2004;566:213–217. doi: 10.1016/j.febslet.2004.04.030. [DOI] [PubMed] [Google Scholar]
- Wang M, He Y, Eisenman LN, et al. 3β-hydroxypregnane steroids are pregnenolone sulfate-like GABAA receptor antagonists. J Neurosci. 2002;22:3366–3375. doi: 10.1523/JNEUROSCI.22-09-03366.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wardell B, Marik PS, Piper D, et al. Residues in the first transmembrane domain of the Caenorhabditis elegans GABAA receptor confer sensitivity to the neurosteroid pregnenolone sulfate. Br J Pharmacol. 2006;148:162–172. doi: 10.1038/sj.bjp.0706719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiting PJ. GABAA receptor subtypes in the brain: a paradigm for CNS drug discovery? Drug Discovery Today. 2003;8:445–450. doi: 10.1016/s1359-6446(03)02703-x. [DOI] [PubMed] [Google Scholar]
- Williams DB. A novel, rapid inhibitory effect of insulin on α1β2γ2S γ-aminobutyric acid type A receptors. Neurosci Lett. 2008;443:27–31. doi: 10.1016/j.neulet.2008.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams DB. A partial comparison of the accessible cysteines induced by different neurosteroids in the M3 region of the α1 subunit of the GABAA receptor. Soc For Neuroscience. 2009;712.12 [Google Scholar]
- Williams DB, Akabas MH. Residues in the M3 membrane spanning segment of the γ-aminobutyric acid type A receptor are on the water accessible surface of the protein. Biophys J. 1999;77:2563–2574. doi: 10.1016/s0006-3495(99)77091-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams DB, Akabas MH. Benzodiazepines induce a conformational change in the region of the γ-aminobutyric acid type A receptor α1 subunit M3 membrane spanning segment. Mol Pharmacol. 2000;58:1129–1136. doi: 10.1124/mol.58.5.1129. [DOI] [PubMed] [Google Scholar]
- Williams DB, Akabas MH. Evidence for distinct conformations of the two α1 subunits in diazepam-bound GABAA receptors. Neuropharmacology. 2001;41:539–545. doi: 10.1016/s0028-3908(01)00099-5. [DOI] [PubMed] [Google Scholar]
- Williams DB, Akabas MH. Potentiating and activating concentrations of the intravenous anesthetic propofol induce distinct conformations of the γ-aminobutyric acid-A receptor α1 subunit M3 membrane segment. J Neurosci. 2002;22:7417–7424. doi: 10.1523/JNEUROSCI.22-17-07417.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yevenes GE, Mora-Cid G, Avila A, et al. Molecular requirements for ethanol differential allosteric modulation of glycine receptors based on selective Gβγ modulation. J Biol Chem. 2010;285:30203–30213. doi: 10.1074/jbc.M110.134676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu WJ, Vicini S. Neurosteroid prolongs GABAA channel deactivation by altering kinetics of desensitized states. J Neurosci. 1997;17:4022–4031. doi: 10.1523/JNEUROSCI.17-11-04022.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]