

Abstract
Airway diseases such as asthma involve increased airway smooth muscle (ASM) contractility and remodelling via enhanced proliferation. Neurotrophins (NTs) such as brain-derived neurotrophic factor (BDNF), well-known in the nervous system, can regulate Ca2+ signalling, and interact with cytokines in contributing to airway hyperreactivity. In this study, we determined whether and how BDNF regulates human ASM cell proliferation in the presence of inflammation, thus testing its potential role in airway remodelling. Cells were treated with 10 nM BDNF, 25 ng/ml tumour necrosis factor (TNF-α) or interleukin-13 (IL-13), or 10 ng/ml platelet-derived growth factor (PDGF). Proliferation was measured using CyQuant dye, with immunoblotting of cell cycle proteins predicted to change with proliferation. Forty-eight hours of BDNF enhanced ASM proliferation to ∼50% of that by PDGF or cytokines. Transfection with small interfering RNAs (siRNAs) targeting high-affinity tropomyosin-related kinase B receptor abolished BDNF effects on proliferation, whereas low-affinity 75 kD neurotrophin receptor (p75NTR) siRNA had no effect. Systematic pharmacologic inhibition of different components of ERK1/2 and PI3K/Akt1 pathways blunted BDNF or TNF-α–induced proliferation. BDNF also induced IκB phosphorylation and nuclear translocation of p50 and p65 NF-κB subunits, with electron mobility shift assay confirmation of NF-κB binding to consensus DNA sequence. These results demonstrate that NTs such as BDNF can enhance human ASM cell proliferation by activating proliferation-specific signalling pathways and a versatile transcription factor such as NF-κB, which are common to cytokines and growth factors involved in asthma.
Keywords: brain-derived neurotrophic factor, tropomyosin related kinase, lung, cytokine, asthma, growth factor
Introduction
Exaggerated airway narrowing, airflow limitation and accompanying dyspnoea are hallmark symptoms of diseases such as asthma, chronic bronchitis and emphysema [1,2]. In this regard, it is now clear that in addition to passively responding to bronchoconstrictor stimuli (i.e. hyperreactivity), the diseased airway also undergoes ‘remodelling’ via increased airway smooth muscle (ASM) mass (hypertrophy and hyperplasia). There is also increasing recognition that ASM itself produces several mediators of airways disease (e.g. growth factors, cytokines) that may have both negative and positive effects on airway narrowing and remodelling [3]. In this setting, the recent discovery of NTs in normal and diseased lung (recently reviewed in Refs. 4–6) raises the exciting possibility that these growth factors contribute not only to normal ASM function, but also to airway diseases.
The NT family of growth factors includes nerve growth factor (NGF), BDNF, neurotrophin 3 (NT3) and neurotrophin 4 (NT4). NTs are well-known to regulate differentiation, morphology, function and gene expression of neuronal cells [7-9]. BDNF, NT4 and NT3 exert their effects via receptors [the low-affinity 75 kD NT receptor p75NTR and the high-affinity tropomyosin-related (tyrosine) kinases, TrkB and TrkC], initiating receptor autophosphorylation and activating several intracellular signalling cascades (e.g. PLC, phospholipase C; PI3K, phosphatidylinositol 3 kinase; MAPK, mitogen-activated protein kinases). NTs have also been found to affect intracellular Ca2+ ([Ca2+]i) and neurotransmitter release in neurons on more rapid time scales [10,11]. However, an important aspect of NT signalling is that by virtue of being growth factors, they can potentially influence a myriad of signalling mechanisms, which are species, cell type and context specific.
NTs and their receptors have recently been found in different lung components [4,12–16]. For example, BDNF is produced by epithelium, sensory neurons, ASM itself and a range of immune cells [17] (also see Refs. 4–6, for review). Altered NT (BDNF as well as NGF) and receptor expression have been observed in asthma, allergy and even lung cancer [4,6]. A role for BDNF has been suggested in airway inflammation, remodelling and hyperreactivity [18,19], but the mechanisms by which NTs such as BDNF can affect ASM are still under investigation. We recently demonstrated that NTs such as BDNF can enhance [Ca2+]i regulation in human ASM, and potentiate the enhancing effects of the pro-inflammatory cytokine TNF-α on [Ca2+]i [15].
Considering the fact that diseases such as asthma involve both altered contractility as well as remodelling where ASM hyperplasia is a key component, and the recent evidence for BDNF signalling within ASM, we hypothesized that BDNF activates ASM cell proliferative pathways, thus contributing to ASM hyperplasia observed in airway diseases. We tested this hypothesis using primary human ASM cells as a paradigm, and determined the role of signalling mechanisms most commonly associated with BDNF as well as cell proliferation. We compared BDNF effects and signalling mechanisms with those of two cytokines thought to be key in asthma: the early Th1 cytokine TNF-α, which functions via a plasma membrane receptor TNFR1 and activates the transcription factor NF-κB [20] as well as the Th2 cytokine IL-13 [21-23]. Platelet-derived growth factor (PDGF), a potent mitogen was also used for comparison [24,25].
Materials and methods
Human ASM cells
The techniques for isolating human ASM cells have been previously described [15,16]. Briefly, human bronchi from lung specimens incidental to patient surgery were used to obtain ASM cells. Specimens were usually derived from lobectomies or pneumenectomies for localized tumour or transplant, whereas specimens removed for metastatic tumour or infection were not used. Bronchial samples were immersed in ice-cold Hanks’ balanced salt solution (HBSS; 2.5 mM Ca2+), the epithelial layer removed by blunt dissection, and the ASM layer excised and finely minced in ice-cold Ca2+-free HBSS. Cells were isolated using collagenase digestion, followed by ovomucoid–albumin separation. Subsequent cell pellets were resuspended in DMEM-F12 supplemented with 10% foetal bovine serum (DMEM Complete), centrifuged, resuspended, seeded in culture flasks and were passaged to a maximum of two subcultures. Cells were serum-deprived at least for 24 hrs prior to all experiments.
Western analyses
ASM cell lysates were prepared using previously described standard techniques [15,16]. Briefly, cells were rinsed twice with ice-cold phosphate-buffered saline (PBS), harvested and subjected to sonication in cell lysis buffer (Cell Signaling Technologies, Beverly, MA, USA) containing protease inhibitors, and the resultant supernatants were assayed for total protein content using the DC protein Assay kit (BioRad, Hercules, CA, USA). Approximately, 30-μg total protein was loaded onto a denaturing SDS-PAGE and electrophoresed according to standard protocols. Proteins were transferred onto a PVDF membrane, blocked with 5% non-fat milk and probed with antibodies of interest. Protein detection was performed with the Supersignal Pico substrate kit from Pierce Biotechnology (Rockford, IL, USA). Blots were exposed to BioMax-XAR film (Eastman Kodak Co., Rochester, NY, USA), and scanned for densitometric analyses using Kodak ImageStation 4000 (Carestream Health, Rochester, NY, USA).
BDNF antibodies were obtained from Abcam; Akt1, pAkt1 and GAPDH antibodies from Cell Signaling; ERK1/2, pERK1/2, IκB, pIκB, NF-κB-p50, NF-κB-p65, and PCNA, Cyclin E and p27Kip1 antibodies from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-rabbit or antimouse secondary antibodies were obtained from Sigma-Aldrich (St. Louis, MO, USA).
Cytokine and BDNF treatment
ASM cells were exposed to serum-free medium for at least 24 hrs before treatment. Cells were then exposed for 48 hrs to 10 nM human recombinant BDNF (R&D Systems Minneapolis, MN, USA), 25 ng/ml TNF-α (Calbiochem), 25 ng/ml IL-13 (Calbiochem, Gibbstown, NJ, USA) or 10 ng/ml PDGF (Sigma-Aldrich).
Pharmacological inhibition
A range of inhibitors for different components of signalling cascades were used to determine their involvement in BDNF effects on cell proliferation. Specifics of inhibitor use are provided in the results section in individual protocols. We recognize that most inhibitors are specific only within certain concentrations, and may differ in their inhibitory potency between cell types. Accordingly, only one fixed concentration of each inhibitor was selected based on the published or vendor-provided IC50 in vitro. In general, the approach was to expose ASM cells to an inhibitor for at least 30 min. prior to subsequent exposure to BDNF (or other stimuli) for 48 hrs in the continued presence of inhibitor. Control studies involved exposure to the inhibitor alone for 48 hrs. The list of inhibitors is as follows: Akt Inhibitor XIII, ERK inhibitor peptide, IKK inhibitor III, PD98059, Raf Kinase inhibitor, RSK inhibitor, SN-50 and wortmannin (Calbiochem).
siRNA transfection
ASM cells were grown to approximately 50% confluence in 96-well culture plates, and were exposed to serum- and antibiotic-free medium for 24 hrs before transfection with 200 pM non-specific (scrambled) RNA or siRNA against TrkB, TrkC, TNFR1 or P75NTR (Ambion-Applied Biosystems, Austin, TX, USA). The siRNA was allowed to complex with Lipofectamine reagent (Invitrogen, Carlsbad, CA, USA) and added to cells. After 24 hrs, transfection medium was replaced with serum-free medium for 24 hrs and the cells were then treated with BDNF for 24 hrs (thus, maintaining the total experimental period at 48 hrs).
CyQuant cell proliferation assay
Proliferation of ASM cells grown in 96-well culture plates was assayed using the CyQuant NF kit (Invitrogen). For all experiments, 48 hrs after the treatments (stimulus, with or without inhibitor or siRNA as above), cells were washed with PBS and exposed to the CyQuant dye for 1 hr at room temperature. Dye binding to DNA (fluorescence) was measured on a Flex Station3 microplate reader (Molecular Devices, Sunnyvale, CA, USA). Dye calibrations were performed empirically using different cell counts and used to obtain estimates of baseline proliferation and with drug exposures over the 48 hrs period. Extent of proliferation was normalized to baseline (i.e. time zero).
Flow cytometry
ASM cells from different individuals were grown in 100 mm culture plates and were treated with 10 nM BDNF or 25 ng/ml TNF for 0 or 24 hrs before ethanol fixation. Cells in medium alone for the same time periods were used as controls. Cells were then stained with propidium iodide, and analysed using either an FACSAria or FACSVantage SE Flow Cytometer (Mayo Flow Cytometry Core Facility). Approximately 20,000 events were captured per sample and cell populations in G1, S and G2/M phases were analysed using the ModFit LT software (Verity Software House, Topsham, ME, USA).
Electrophoresis mobility shift assay (EMSA)
ASM cells were grown on 10-cm plates to ∼80% confluence, and exposed to serum-free medium for 24 hrs prior to treatment with BDNF, TNF-α, IL-13 or PDGF. After 2 hrs, cells were washed and fractionated to obtain cytoplasmic and nuclear extracts using the NE-PER kit (Pierce Biotechnology). Biotinylated or non-biotinylated forward and reverse oligonucleotides corresponding to the NF-κB binding site consensus (5′-AGTTGAGGGGACTTTCCCAGGC-3′) were obtained from Mayo Clinic DNA Synthesis Core Facility, and used with the nuclear extracts and non-radioactive EMSA kit from Pierce Biotechnology to analyse NF-κB binding in the EMSA assay. Nuclear extracts allowed to bind to the NF-κB oligonucleotide, and the complex was run on a non-denaturing 5% polyacrylamide gel buffered with 0.5% Tris-Borate-EDTA (TBE). Nucleoprotein–DNA complexes were then transferred to a nylon membrane, and cross-linked with ultraviolet light. Membranes were treated with streptavidin–horseradish peroxidase conjugate and the shift in mobility was detected via treatment with a chemiluminescence substrate and exposure to BioMax-XAR film.
Statistical analyses
All proliferation experiments were performed in quadruplicate using different sets of cells isolated from four different individuals. For CyQuant proliferation assay, data from at least six wells per protocol were collected for cells from each individual and averaged. Data for graphs were generated using these averages. Controls represent unstimulated samples or those not exposed to drugs (and thus a baseline measurement). For immunoblotting, extracts were obtained from three to five different individuals. Net band intensity was normalized to that of the housekeeping gene product glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Comparisons were performed across groups with independent Student’s t-test or two-way ANOVA as appropriate. Repeated comparisons were tested via Bonferroni correction. Statistical significance was tested at the P < 0.05 level. Values are reported as means ± S.E. ‘n’ values representing numbers of individuals are provided in the legends to the figures.
Results
ASM phenotype
Before initiating proliferation studies, we verified that the ASM cells used maintained a smooth muscle phenotype during the passaging process. Western analysis for smooth muscle myosin and actin, as well as muscarinic receptor and vimentin, showed sustained expression of these proteins in ASM cells up to the third passage, even with 48 hrs serum deprivation (Fig. 1; representative data from n = 6 individuals). Such sustained expression of smooth muscle markers was observed even at passage 5; however, to rule out any possibility of de-differentiation, earlier passages were used. Furthermore, we found no reduction in expression of smooth muscle markers between untreated cells, and those exposed to BDNF, TNF-α, IL-13 or PDGF after 48 hrs (not shown).
Fig 1.
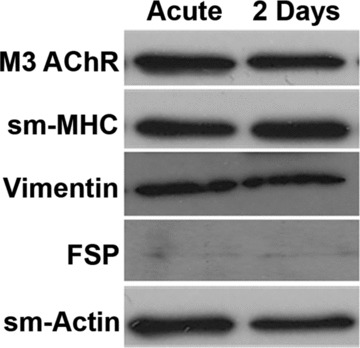
Phenotype of human airway smooth muscle (ASM) cells. Isolated ASM cells maintained a smooth muscle phenotype in culture (up to third passage of subculture, serum deprivation for 48 hrs), demonstrating continued expression of muscarinic receptors (M3 subtype; M3/AChR in figure), smooth muscle (SM) myosin heavy chain (MHC) and actin, as well as vimentin. Fibroblast-specific protein expression was negligible verifying that lack of contamination by fibroblasts. Representative sample from ASM derived from n = 6 individuals.
BDNF effect on cell proliferation
Cell proliferation was measured using fluorescence changes of the CyQuant dye. We first verified that the dye has a fairly linear detection range from as low as 50 to 25,000 ASM cells in a fixed volume (as claimed by the vendor). This dilution curve was used to generate an in situ empirical calibration for human ASM cells. Because we used the same dye concentration and the initial cell density (i.e. before drug or other exposures) of ∼5000 (mid-range for the dye), we converted the fluorescence values to changes in cell numbers following treatments. Based on this analysis, serum-deprived human ASM cells showed a baseline proliferation of ∼5.5% over a 48-hr period, compared to time zero (Fig. 2).
Fig 2.
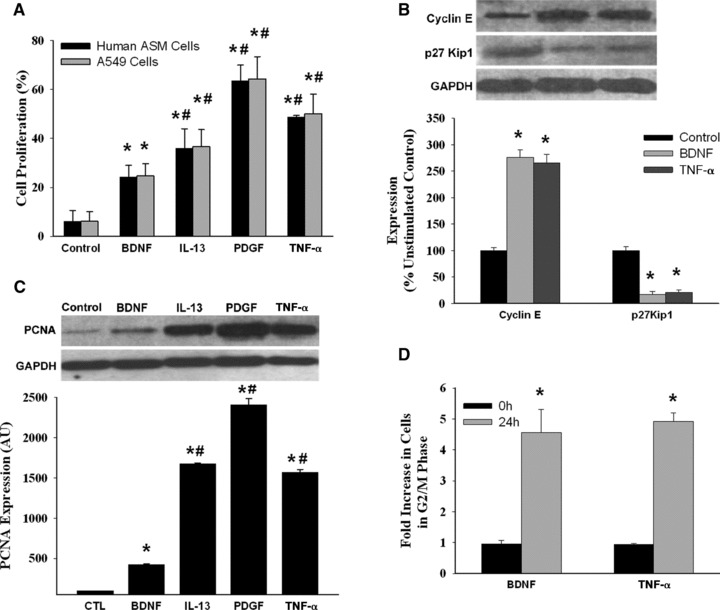
Effect of brain-derived neurotrophic factor (BDNF) on human ASM cell proliferation. (A) Compared to unstimulated controls (which showed a small baseline proliferation over a 48-hr period measured using a fluorescent CyQuant assay), exposure of ASM cells to 10 nM BDNF induced significant increase in cell proliferation, albeit less than that induced by the pro-inflammatory cytokines tumour necrosis factor (TNF-α) and interleukin-13 (IL-13) or by platelet-derived growth factor (PDGF) (n = 4 individuals for each set, quadruplicate measurements per individual). The proliferative effect of these stimuli on ASM cells was comparable to that seen in a lung alveolar cancer cell line (A459). Enhanced proliferation by BDNF or other stimuli was further demonstrated by increased Cyclin E and decreased p27Kip1 expression (B) and increased proliferating cell nuclear antigen (PCNA) levels (C) (n = 3 individuals for each set; quadruplicate measurements per individual). Changes in ASM proliferation profile were also measured by propidium iodide staining followed by flow cytometry at 0 and 24 hrs after BDNF or TNF-α treatment, which caused a marked increase in the fraction of cells in S and G2/M phases (D) (n = 3 individuals for each set). Values are mean ± S.E. * indicates significant difference from control. #indicates significant effect of cytokine or PDGF (P < 0.05).
Compared to baseline proliferation, exposure of serum-deprived human ASM cells to 48 hrs BDNF significantly increased proliferation (P < 0.05; Fig. 2A; n = 4 individuals, quadruplicate measurements per individual and condition). BDNF effects on cell proliferation were smaller than that of TNF-α, IL-13 or PDGF (with PDGF having considerably greater effect compared to the other compounds; P < 0.05). In addition, we tested the effect of different concentrations of BDNF, TNF-α or PDGF on cell proliferation (Table 1). We found that proliferation rates were not increased beyond 10 nM BDNF, 25 ng/ml TNF-α or 10 ng/ml PDGF. Accordingly, these concentrations were used in further analyses. We also explored whether at a fixed BDNF concentration, duration of exposure (12, 24, 48 and 72 hrs) was important, and found that >48 hrs did not substantially influence proliferation (data not shown): hence our selection of 48 hrs for subsequent studies.
Table 1.
ASM cell proliferation dose responses
| BDNF (nM) | Proliferation | TNF-α (ng/ml) | Proliferation | PDGF (ng/ml) | Proliferation |
|---|---|---|---|---|---|
| 0.1 | 6.1 ± 0.5% | 0.25 | 6.4 ± 1.1% | 0.1 | 10.0 ± 1.5% |
| 1 | 18.3 ± 1.1% | 2.5 | 16.3 ± 2.5% | 1 | 18.9 ± 3.7% |
| 10 | 24.3 ± 2.5% | 25 | 45.0 ± 4.5% | 10 | 59.6 ± 8.1% |
| 100 | 27.3 ± 4.5% | 250 | 52.7 ± 6.8% | 100 | 67.3 ± 10.4% |
Values are means ± S.E. BDNF: brain-derived neurotrophic factor; TNF-α: tumour necrosis factor α; PDGF: platelet-derived growth factor.
To verify that BDNF, TNF-α, IL-13 or PDGF effects on ASM cell proliferation were not artefact, we performed additional experiments where the same stimuli (48 hrs, same concentrations) were given to the unrelated alveolar epithelial tumour cell line A549 (ATCC), and found comparable increases in A549 proliferation (Fig. 2).
Proliferation was further verified by Western analysis of ASM cell fractions which showed increased proliferating cell nuclear antigen (PCNA) levels following exposure to BDNF as well as cytokines or PDGF (Fig. 2; P < 0.05 compared to controls; P < 0.05 for BDNF being less than other stimuli which had comparable effects). In addition, we examined the expression of Cyclin E (a G1/S Cyclin which, along with CDK2, enables cells to enter the synthesis phase of the cell cycle), and p27Kip1, a CDK inhibitor and a target for the phosphorylating activity of the Cyclin E–CDK2 complex. Increased Cyclin E and decreased p27Kip1 abundance are thus indicative of cell cycle progression from G1 or quiescence phase [26,27]. We observed that Cyclin E expression was increased and p27Kip1 expression concurrently decreased when ASM cells were exposed for 24 hrs to BDNF or TNF-α (Fig. 2; P < 0.05 for either agent), suggesting that both stimulate pathways that signal a shift from quiescence to division phase in ASM cells. Consistent with this scenario, flow cytometric analysis of ASM cells treated with BDNF or TNF-α for 24 hrs showed a substantial increase in the fraction of cells entering G2/M phase (Fig. 2; compared to untreated cells; P < 0.05), indicating that the downstream effect of BDNF and TNF-α signalling is cell cycle activation, directing ASM cells to undergo mitosis.
An alternative mechanism for BDNF action was the possibility that BDNF might simply enhance the survival of ASM cells rather than actively promote their proliferation. Accordingly, we examined viability of ASM cells treated with BDNF or TNF-α. Trypan Blue staining followed by cell counting using a haemocytometer revealed that the BDNF- or TNF-α–treated samples had an increased number of cells compared to the untreated cells. More importantly, the ratio of live to dead cells (Trypan Blue excluded versus stained cells) was unaltered irrespective of whether the cells had been exposed to BDNF (or TNF-α) or not (data not shown). These data supported the idea that BDNF actually increases cellular proliferation instead of just enhancing cell survival.
Mechanisms of BDNF effect
Role of receptors
BDNF can activate the high-affinity receptor TrkB as well as the low-affinity pan-NT receptor p75NTR, both which have been previously shown to be expressed by human ASM cells [15]. Accordingly, we used siRNAs against specific receptors to determine their role in cell proliferation. We have previously used this set of siRNAs to demonstrate specificity for human TrkB with absence of effect by nonsense siRNA or Lipofectamine [15].
In cells transfected with Lipofectamine alone, there was no significant change in baseline proliferation (demonstrating lack of vehicle toxicity). In cells not exposed to BDNF, inhibition of TrkB expression decreased proliferation to below baseline (Fig. 3; P < 0.05). A similar effect on baseline proliferation was observed with siRNAs against TrkC (receptor for the NT3 with no known activation by BDNF) and p75NTR (Fig. 3). Transfection with scrambled (nonsense) siRNA did not inhibit BDNF- or TNF-α–induced cell proliferation. Constitutive expression of NTs has been reported in ASM [4,13]. Accordingly, these siRNA effects may reflect baseline NT expression (including BDNF and NT3). Indeed, treatment of ASM cells with 1 μg/ml TrkB-Fc chimeric molecule (BDNF chelator; Sigma-Aldrich) inhibited basal cell proliferation to extents similar to that induced by TrkB siRNA (Fig. 3; P < 0.05 compared to control). Chelation of other NTs was not examined.
Fig 3.
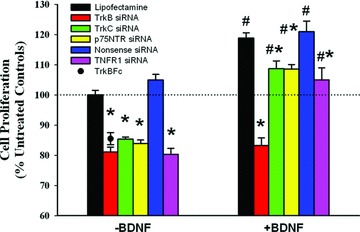
Role of neurotrophin (NT) receptors in ASM cell proliferation. Suppression the high-affinity BDNF receptor tropomyosin-related kinase B (TrkB) using siRNA blunted baseline cellular proliferation (cells not exposed to exogenous BDNF), suggesting endogenous BDNF production by ASM cells. This was confirmed when chelation of extracellular BDNF by the chimeric TrkBFc protein blunted proliferation to levels comparable to that by TrkB siRNA. Blunting effects on proliferation were also observed with the neurotrophin-3 receptor TrkC (not activated by BDNF), and the pan-NT low-affinity receptor p75NTR, suggesting baseline release of other NTs. The increased proliferation induced by 10 nM BDNF was significantly reduced by TrkB siRNA, but to a much smaller extent by p75NTR siRNA, suggesting a major role for the high-affinity receptor. Furthermore, suppression of the receptor for TNF-α (TNFR1) also reduced BDNF effects. Scramble (nonsense) siRNAs did not significantly alter the effects of BDNF on proliferation. Values are mean ± S.E. * indicates significant siRNA effect compared to vehicle (Lipofectamine) control; # indicates significant BDNF effect (P < 0.05). n = 4 individuals for each set and condition, with summary of quadruplicate measurements within each set/conditions.
Inhibition of TrkB expression completely abolished BDNF-induced increase in human ASM cell proliferation, compared to Lipofectamine control (Fig. 3; P < 0.05 for siRNA effect). Inhibiting TrkC had only a small effect on proliferation in the presence of BDNF (likely reflecting continued baseline NT3 expression). siRNA inhibition of p75NTR expression also had only slightly (but significantly) blunted BDNF effects (Fig. 3; P < 0.05). Chelation with TrkBFc was not an appropriate experiment in the presence of exogenous BDNF. Furthermore, BDNF and TNF-α appear to interact in terms of their effects on ASM cell proliferation. The effects of BDNF on cell proliferation were significantly reduced by siRNA suppression of TNFR1 (TNF-α receptor).
Signalling cascades
As mentioned earlier, NTs such as BDNF can potentially activate a number of intracellular signalling cascades. Given that NT-activated cascades are species and cell-type specific, it was first necessary to establish which mechanisms were relevant to human ASM. Previous studies have implicated the MAP kinase and PI3/Akt pathways in airway remodelling [25,28–30]. These pathways are also known to be activated by cytokines [31,32]. Accordingly, we initiated our studies with these cascades.
Western analysis of lysates of cells treated with BDNF, TNF-α, IL-13 or PDGF showed substantial (P < 0.05 compared to control) enhancement of Akt1 phosphorylation (with no change in total Akt1 levels) with these stimuli (Fig. 4). The effect of BDNF, IL-13 and PDGF were comparable, but less than that of TNF-α (Fig. 4; P < 0.05). Expression of total ERK1/2 was not substantially altered by any of the four stimuli. However, phosphorylation of ERK1/2 was significantly (P < 0.05) increased by all stimuli, albeit to different extents: BDNF had significantly less effect compared to TNF-α, IL-13 or PDGF (Fig. 4; P < 0.05).
Fig 4.
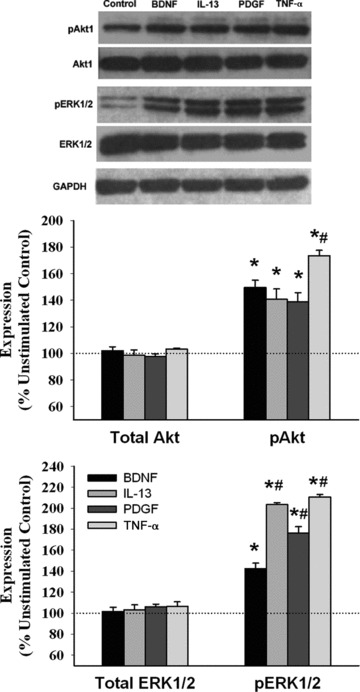
Effect of BDNF on Akt1 and ERK1/2. Western analysis of human ASM cell lysates exposed to BDNF, IL-13, PDGF or TNF-α demonstrated significant phosphorylation of both Akt1 and ERK1/2 by all four stimuli (compared to untreated controls), with no change in total Akt1 or ERK1/2 expression. The effect of BDNF on Akt1 phosphorylation was comparable to that of IL-13 and PDGF, but less than that of TNF-α. Effect of BDNF on ERK1/2 phosphorylation was substantially lesser than the other three stimuli. Values are mean ± S.E. *indicates significant stimulus effect compared to control; #indicates significant difference from BDNF (P < 0.05). n = 3 individuals for each set and condition.
Based on enhanced pERK1/2 and pAkt1, we explored putative upstream and downstream signalling intermediates that could be involved in BDNF enhanced proliferation. Inhibition of ERK (2.5 μM ERK inhibitor peptide), the upstream Raf kinase (10 nM Raf kinase inhibitor peptide) and MEK (2.0 μM PD98059), or the downstream p90 ribosomal S6 kinase (RSK; 100 nM of inhibitor SL0101) resulted in substantial blunting of proliferation induced by BDNF, TNF-α, IL-13 or PDGF (Fig. 5; P < 0.05 for inhibitor effect). Similarly, inhibiting PI3 kinase with 50 nM wortmannin or Akt with 500 nM inhibitor peptide XIII also resulted in decreased proliferation of ASM cells (Fig. 5; P < 0.05).
Fig 5.
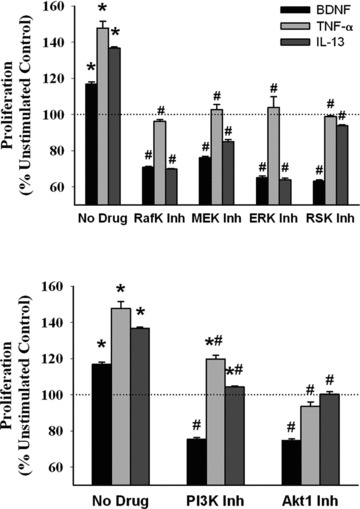
Effect of inhibiting MAP kinase and PI3/Akt pathways on BDNF effects. Details of individual inhibitors is provided in Materials and Methods section and in specific protocols of Results section. Inhibition of the upstream (RAF kinase, MEK) and downstream (RSK) intermediates as well as ERK1/2 itself resulted in substantial reduction in BDNF-induced cellular proliferation. As expected, TNF-α and IL-13 effects were also affected by these inhibitors, albeit to different extents. Similarly, inhibition of PI3K and Akt suppressed BDNF-induced cellular proliferation to a greater extent than for TNF-α or IL-13. * indicates significant stimulus effect compared to control; # indicates significant inhibitor effect (P < 0.05). n = 4 individuals for each set and condition. Quadruplicate measurements of proliferation and triplicate Western analyses per individual/set/condition.
Signalling via PI3K and Akt1 can either proceed towards a β-catenin-mediated cell cycle stimulation or activate the IκB kinase IKK, which in turn promotes phosphorylation and degradation of IκB, thus enabling the subunits of the transcription factor NF-κB to localize to the nucleus [33,34]. Previous studies have suggested that NTs such as NGF can activate the NF-κB pathway [35-37]. In human ASM cells, inhibition of IKK or NF-κB resulted in significantly reduced proliferation by BDNF, TNF-α and IL-13, whereas PDGF-induced proliferation was largely unaffected (Fig. 6).
Fig 6.
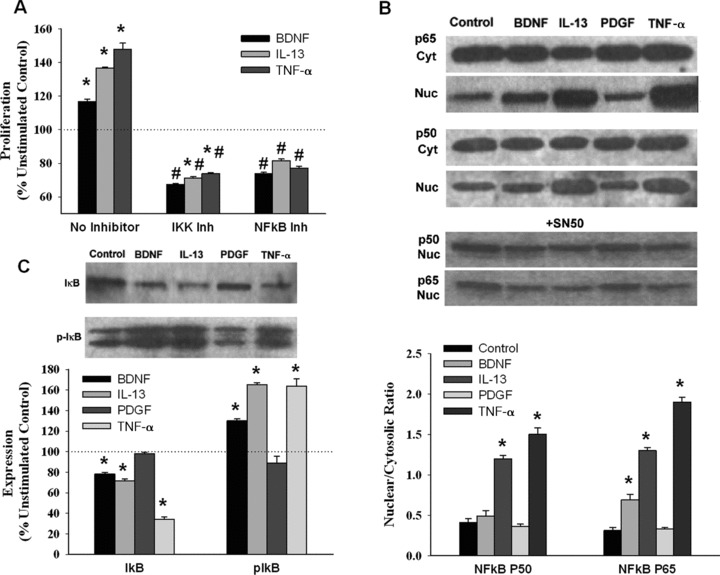
Role of NF-κB in BDNF effects on ASM cells. Inhibition of IκB as well as of NF-κB resulted in substantial reduction in BDNF-induced ASM cell proliferation, comparable to inhibitor effect when cells were exposed to TNF-α or IL-13 (A). BDNF exposure resulted in nuclear translocation especially of the p65 subunit of NF-κB, albeit not the same extent as with the p50 and p65 subunits in TNF-α–exposed cells, effects suppressed by the NF-κB inhibitor SN50 (B). PDGF did not appear to work through this mechanism. Similarly, exposure to BDNF was associated with increased IκB phosphorylation to extents comparable to that by cytokines (while PDGF did not have much effect; C). * indicates significant stimulus effect compared to control; # indicates significant inhibitor effect (P < 0.05). n = 4 individuals for each set and condition. Quadruplicate measurements of proliferation and triplicate Western analyses per individual/set/condition.
To fully understand the role of NF-κB in BDNF-induced proliferation, we immunoblotted cytoplasmic and nuclear extracts from BDNF-, TNF-α– or IL-13–treated cells with antibodies against NF-κB subunits p65 and p50. When not activated, NF-κB subunits are sequestered in the cytoplasm by IκB; activation signals enhance the phosphorylation and the subsequent proteasomal degradation of IκB and the release of NF-κB subunits p65 and p50. In lysates from cells treated with BDNF, TNF-α or IL-13, IκB expression was attenuated whereas levels of phospho-IκB were augmented (Fig. 6), bolstering the idea that proliferative signals issued by stimuli such as BDNF induce NF-κB nuclear translocation. Cytoplasmic levels of p65 and p50 were not altered by BDNF, TNF-α or IL-13; however, BDNF caused a moderate increase in nuclear p65 (less so for p50), albeit less than that induced by TNF-α (Fig. 6; P < 0.05 compared to control, and for difference between BDNF and TNF-α). Nuclear localization of NF-κB subunits by BDNF or TNF-α or IL-13 was blocked by pre-treatment with 20 μM SN-50, a known NF-κB inhibitor (Fig. 6; P < 0.05 for inhibitor effect).
Finally, applying gel mobility shift assay (EMSA) to ASM nuclear extracts, we confirmed that NFκB is involved in ASM proliferation in response to BDNF, TNF-α or IL-13 by acting as a transcriptional regulator. NF-κB consensus oligonucleotide sequence exhibited a shift in mobility (Fig. 7, arrow) when in association with nuclear extracts from BDNF, IL-13 or TNF-α–treated cells, but not when mixed with the extract from PDGF-treated cells. The specificity of this DNA–protein binding was evident when a competing unlabeled oligonucleotide included in the reaction mix caused significant decrease in the mobility shift.
Fig 7.
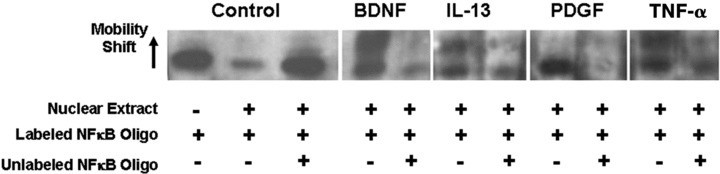
BDNF induces NF-κB activation that binds to target genes. Gel mobility shift assay showed NF-κB consensus binding to DNA in human ASM cells stimulated with BDNF, at levels comparable to that achieved by TNF-α. Representative sample of data from n = 3 individuals.
BDNF interactions with cytokines
Based on the above data suggesting common mechanisms of action of BDNF versus the other agents tested here, we determined whether BDNF has additive or potentiating effects when the latter are present. ASM cells were exposed first to TNF-α, IL-13 or PDGF, with BDNF added midway at 24 hrs. The presence of BDNF resulted in additive effects for cell proliferation induced by all three agents to comparable extents, but no potentiation was observed (Fig. 8; P < 0.05 for BDNF effects compared to no BDNF being present when any of TNF-α, IL-13 or PDGF are present).
Fig 8.
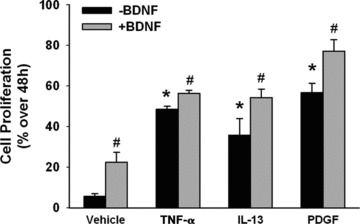
BDNF interactions with cytokines. When ASM cells were exposed first to TNF-α, IL-13 or PDGF, with BDNF added midway at 24 hrs, cell proliferation was even greater compared to that when BDNF was absent. This additive (but not potentiating) effect of BDNF was comparable for all three agents. * indicates significant stimulus effect compared to control; # indicates significant BDNF effect (P < 0.05). n = 4 individuals for each set and condition. Quadruplicate measurements of proliferation per individual/set/condition.
Discussion
Airway remodelling is an important aspect of several diseases including asthma and COPD. Regulation of ASM cell population (hyperplasia or hypertrophy) can occur via a number of mechanisms including cytokines derived from neighbouring cells [38,39], as well as extracellular matrix–derived growth factors [40,41]. In this regard, the presence of NTs (which are well-known to be neuronal growth factors) within the lung [4,5] raises the exciting possibility that airway remodelling involves NTs. The results of this study are the first to demonstrate that the NT BDNF (previously shown to be present in human airway [15,16]) enhances human ASM cell proliferation to levels comparable to that induced by potent growth factors such as PDGF and pro-inflammatory cytokines such as TNF-α and IL-13 relevant to airway disease. Furthermore, our study demonstrates that BDNF activates specific cell growth and survival signalling pathways such as ERK1/2, PI3/Akt and NF-κB that overlap with cytokine and growth factor signalling. Accordingly, we believe that BDNF, derived from potentially multiple sources in the lung (including ASM itself) can serve to work with cytokines to influence ASM proliferation and function, thus contributing to airway hyperresponsiveness in diseases such as asthma.
NTs such as BDNF have been extensively studied in the nervous system [7-9]. When cleaved extracellularly, secreted BDNF specifically binds to high-affinity TrkB receptors (especially the full-length form that is key to biological activity) [37,42], as well as the low-affinity pan-NT p75NTR receptor. Both receptors activate multiple, distinct signalling pathways including ERK, PI3K/Akt, phospholipase C, and in the case of p75NTR (a member of the TNF receptor family) NF-κB [37,42].
There is now considerable evidence (including our own studies) that NTs and their receptors are present in the lung. Specifically, BDNF is produced by airway epithelium, sensory innervation to the airway, ASM itself, and immune cells known to be involved in airway diseases (see Refs. 4–6 for review). BDNF and receptor expression have been found to be increased in blood, tissue and lung lavages of asthma, allergy and even lung cancer [4,6]. Based on these observations, a role for BDNF has been suggested in airway inflammation, remodelling and hyperreactivity [18,19], but the mechanisms of action are still under investigation. Here, ASM cells themselves can be both a potential source [13,16], as well as a target [15,16], with effects being mediated via TrkB as well as p75NTR [13,15]. From a functional standpoint, we recently reported that that BDNF acutely (within 30 min.) enhances [Ca2+]i and force responses of human ASM to agonists such as acetylcholine [16]. Other studies have also demonstrated that NTs (potentially derived from nerves) can influence airway tone [43,44]. Whether NTs also modulate ASM proliferation was not known, and this study is the first to demonstrate such an effect.
This study found that BDNF induces significant cellular proliferation, as demonstrated by the proliferation marker PCNA, measurement of DNA content and cell cycle phase analysis. We used ASM cells isolated from human airways and maintained in culture, albeit limiting the subcultures. Furthermore, initial ASM maturity (in the absence of serum) was confirmed by low proliferation (quiescent state) with increased expression of contractile proteins such as smooth muscle α-actin (sm-α-actin), calponin and smooth muscle myosin heavy chain (sm-MHC) [31,41]. The switch to a proliferative state involves ERK activation, biasing cells toward a less contractile phenotype and PI3K pathway associated with decreased contractility [45]. Such events usually occur in the presence of serum, which contains growth factors including (perhaps) NTs. Accordingly, we felt it important to determine the effect of exogenous BDNF on cell proliferation in the absence of serum. Although this likely decreased the overall extent of ASM proliferation, it is still remarkable that BDNF produced significant increases in proliferation, comparable to that induced by mitogens such as PDGF or TNF-α. Accordingly, it is likely that the effect of BDNF on ASM cell proliferation in vivo is even greater.
Enhanced ASM proliferation by BDNF builds on our previous report on enhanced [Ca2+]i in ASM by BDNF [15]. Modulation of [Ca2+] has long been known to persuade cells to enter a proliferative state, and is thought to be relevant to ASM as well [46-48], and may involve several [Ca2+]i regulatory mechanisms. Here, store-operated Ca2+ influx appears important [47,49], a mechanism we have previously demonstrated to be enhanced by BDNF in human ASM [16]. Whether this is achieved by increased expression of Ca2+ regulatory proteins remains to be determined.
BDNF effects on ASM cell proliferation appear to involve TrkB, but not p75NTR. Here, an interesting observation was that even though p75NTR siRNA did not significantly affect BDNF-induced proliferation, in cells not stimulated with BDNF (i.e. controls), all of TrkB, TrkC and p75NTR siRNAs reduced proliferation to below baseline. We interpret this data as there being a basal secretion of NTs (BDNF and NT3) that have autocrine/paracrine effects via their respective receptors to produce a background level of proliferation. NTs other than BDNF (e.g. NT3) may be activating p75NTR, and thus p75NTR siRNA reduces proliferation. However, in the presence of exogenous BDNF (which is acting only via TrkB), other NTs may still be present, and may account for the small reduction in proliferation in the presence of TrkC or p75NTR siRNA. More detailed examination (beyond the scope of this study) is required to determine the role of (and interaction between) different NTs in basal ASM proliferation, and the signalling mechanisms involved.
The downstream effectors of TrkB signalling have been previously established in cell types other than ASM [37] and involve PI3/Akt (enhancing cell survival) and ERK1/2 via upstream kinases. Both of these major cascades appear to be important in BDNF-induced ASM proliferation, demonstrated by the effects of pharmacological inhibitors for the various signalling intermediates. Activation of ERK1/2 suggests involvement of CREB-mediated gene regulation, an important aspect of ASM proliferation [50,51]. Although this study stopped short of examining CREB phosphorylation per se, studies in other cell types have demonstrated the importance of this transcription factor in mediating NT effects [52].
Activation of NF-κB also appears to be important in mediating BDNF effects on cellular proliferation. NF-κB activation is typically associated with p75NTR signalling. Accordingly, this result was somewhat surprising because p75NTR siRNA did not substantially influence BDNF-induced cell proliferation. However, the strong involvement of PI3/Akt in BDNF effects suggests an alternative route for NF-κB activation. Thus, the PI3/Akt pathway may be particularly important in mediating BDNF effects on cellular proliferation.
In this study, we chose to compare BDNF effects with a routinely used mitogen (PDGF) and two cytokines of importance in the airway. Although the list of pro- and anti-inflammatory mediators in the airway is ever expanding, one well-studied cytokine is TNF-α [53-55]. TNF-α is known to activate MAP kinases, NF-κB and other transcription factors that are involved in cell proliferation [20,32,56–58]. Finally, TNF-α itself stimulates ASM cells to release other cytokines which can further ASM proliferation [59,60]. Th2 cytokines such as IL-13 are also recognized as key to asthma [61], activating MAP kinases, PI3K/Akt and NFκB, and enhancing proliferation of different airway cell types. We previously showed that TNF-α increases expression of BDNF and TrkB in human ASM [15], with BDNF and TNF-α having synergistic enhancing effects on [Ca2+]i and force responses to agonists. Based on this idea, we examined interactions between cytokines and BDNF in ASM proliferation, but found only additive (and not synergistic) effects. Although more detailed studies are required, these data do suggest that in the presence of inflammation, BDNF, working via several signalling mechanisms (even those common to cytokines), may additionally contribute to ASM proliferation, as represented in our model (Fig. 9). Such effects could contribute to altered airway structure and function, and thus airway responsiveness.
Fig 9.
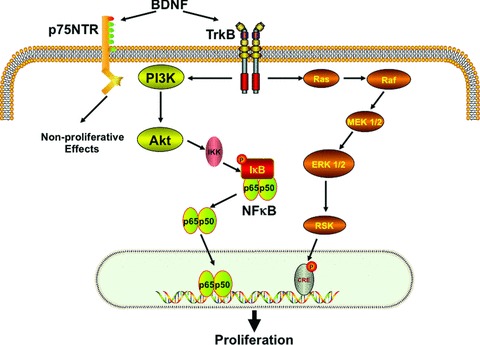
Schematic of the role of BDNF in ASM cell proliferation. In human ASM cells, exposure to BDNF results in binding to the high-affinity TrkB receptor that activates the PI3/Akt and ERK1/2 pathways. PI3/Akt further stimulates nuclear translocation of NF-κB, which binds to promoters of genes involved in cellular proliferation. BDNF activation of the low-affinity p75NTR receptor may lead to other, non-proliferative effects in ASM.
In conclusion, this study is the first to report that physiologically relevant levels of BDNF induce substantial proliferation of human ASM cells, comparable to that by pro-inflammatory cytokines such as TNF-α and IL-13 which are important in the pathogenesis of asthma. Here, BDNF works via signalling mechanisms common to these cytokines, and may thus by itself (or in combination with other mediators), contribute to the pro-inflammatory environment of the airway.
Acknowledgments
This work is supported by grants HL088029 and HL56470 (YSP) from the National Institutes of Health, USA, and by a Clinical Innovator Grant from the Flight Attendants Medical Research Institute (FAMRI; YSP).
Authors’ contributions
B.A., C.M.P. and Y.S.P. designed the research study, and wrote the manuscript. B.A. and M.A.T. performed the research and analysed the data.
Conflict of interest
The authors declare no conflict of interest.
References
- 1.Hirota S, Helli PB, Catalli A, et al. Airway smooth muscle excitation-contraction coupling and airway hyperresponsiveness. Can J Physiol Pharmacol. 2005;83:725–32. doi: 10.1139/y05-070. [DOI] [PubMed] [Google Scholar]
- 2.Joubert P, Hamid Q. Role of airway smooth muscle in airway remodeling. J Allergy Clin Immunol. 2005;116:713–6. doi: 10.1016/j.jaci.2005.05.042. [DOI] [PubMed] [Google Scholar]
- 3.Doherty T, Broide D. Cytokines and growth factors in airway remodeling in asthma. Curr Opin Immunol. 2007;19:676–80. doi: 10.1016/j.coi.2007.07.017. [DOI] [PubMed] [Google Scholar]
- 4.Prakash Y, Thompson MA, Meuchel L, et al. Neurotrophins in lung health and disease. Expert Rev Respir Med. 2010;4:395–411. doi: 10.1586/ers.10.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Scuri M, Samsell L, Piedimonte G. The role of neurotrophins in inflammation and allergy. Inflamm Allergy Drug Targets. 2010;9:173–80. doi: 10.2174/187152810792231913. [DOI] [PubMed] [Google Scholar]
- 6.Hoyle GW. Neurotrophins and lung disease. Cytokine Growth Factor Rev. 2003;14:551–8. doi: 10.1016/s1359-6101(03)00061-3. [DOI] [PubMed] [Google Scholar]
- 7.Kaplan DR, Miller FD. Neurotrophin signal transduction in the nervous system. Curr Opin Neurobiol. 2000;10:381–91. doi: 10.1016/s0959-4388(00)00092-1. [DOI] [PubMed] [Google Scholar]
- 8.Friedman WJ, Greene LA. Neurotrophin signaling via Trks and p75. Exp Cell Res. 1999;253:131–42. doi: 10.1006/excr.1999.4705. [DOI] [PubMed] [Google Scholar]
- 9.Zweifel LS, Kuruvilla R, Ginty DD. Functions and mechanisms of retrograde neurotrophin signalling. Nat Rev Neurosci. 2005;6:615–25. doi: 10.1038/nrn1727. [DOI] [PubMed] [Google Scholar]
- 10.Kovalchuk Y, Holthoff K, Konnerth A. Neurotrophin action on a rapid timescale. Curr Opin Neurobiol. 2004;14:558–63. doi: 10.1016/j.conb.2004.08.014. [DOI] [PubMed] [Google Scholar]
- 11.Rose CR, Blum R, Pichler B, et al. Truncated TrkB-T1 mediates neurotrophin-evoked calcium signalling in glia cells. Nature. 2003;426:74–8. doi: 10.1038/nature01983. [DOI] [PubMed] [Google Scholar]
- 12.Lommatzsch M, Braun A, Renz H. Neurotrophins in allergic airway dysfunction: what the mouse model is teaching us. Ann N Y Acad Sci. 2003;992:241–9. doi: 10.1111/j.1749-6632.2003.tb03154.x. [DOI] [PubMed] [Google Scholar]
- 13.Ricci A, Felici L, Mariotta S, et al. Neurotrophin and neurotrophin receptor protein expression in the human lung. Am J Respir Cell Mol Biol. 2004;30:12–9. doi: 10.1165/rcmb.2002-0110OC. [DOI] [PubMed] [Google Scholar]
- 14.Ricci A, Greco S, Amenta F, et al. Neurotrophins and neurotrophin receptors in human pulmonary arteries. J Vasc Res. 2000;37:355–63. doi: 10.1159/000025751. [DOI] [PubMed] [Google Scholar]
- 15.Prakash YS, Thompson MA, Pabelick CM. Brain-derived neurotrophic factor in TNF-alpha modulation of Ca2+ in human airway smooth muscle. Am J Respir Cell Mol Biol. 2009;41:603–11. doi: 10.1165/rcmb.2008-0151OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Prakash YS, Iyanoye A, Ay B, et al. Neurotrophin effects on intracellular Ca2+ and force in airway smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2006;291 doi: 10.1152/ajplung.00501.2005. : L447–56. [DOI] [PubMed] [Google Scholar]
- 17.Rochlitzer S, Nassenstein C, Braun A. The contribution of neurotrophins to the pathogenesis of allergic asthma. Biochem Soc Trans. 2006;34:594–9. doi: 10.1042/BST0340594. [DOI] [PubMed] [Google Scholar]
- 18.Braun A, Lommatzsch M, Neuhaus-Steinmetz U, et al. Brain-derived neurotrophic factor (BDNF) contributes to neuronal dysfunction in a model of allergic airway inflammation. Br J Pharmacol. 2004;141:431–40. doi: 10.1038/sj.bjp.0705638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lommatzsch M, Quarcoo D, Schulte-Herbruggen O, et al. Neurotrophins in murine viscera: a dynamic pattern from birth to adulthood. Int J Dev Neurosci. 2005;23:495–500. doi: 10.1016/j.ijdevneu.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 20.Pimentel-Muinos FX, Seed B. Regulated commitment of TNF receptor signaling: a molecular switch for death or activation. Immunity. 1999;11:783–93. doi: 10.1016/s1074-7613(00)80152-1. [DOI] [PubMed] [Google Scholar]
- 21.Gao YD, Zou JJ, Zheng JW, et al. Promoting effects of IL-13 on Ca2+ release and store-operated Ca2+ entry in airway smooth muscle cells. Pulm Pharmacol Ther. 2010;23:182–9. doi: 10.1016/j.pupt.2009.12.005. [DOI] [PubMed] [Google Scholar]
- 22.Kellner J, Gamarra F, Welsch U, et al. IL-13Ralpha2 reverses the effects of IL-13 and IL-4 on bronchial reactivity and acetylcholine-induced Ca+ signaling. Int Arch Allergy Immunol. 2007;142:199–210. doi: 10.1159/000097022. [DOI] [PubMed] [Google Scholar]
- 23.Zhu Z, Homer RJ, Wang Z, et al. Pulmonary expression of interleukin-13 causes inflammation, mucus hypersecretion, subepithelial fibrosis, physiologic abnormalities, and eotaxin production. J Clin Invest. 1999;103:779–88. doi: 10.1172/JCI5909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nunes RO, Schmidt M, Dueck G, et al. GSK-3/beta-catenin signaling axis in airway smooth muscle: role in mitogenic signaling. Am J Physiol Lung Cell Mol Physiol. 2008;294:L1110–8. doi: 10.1152/ajplung.00500.2007. [DOI] [PubMed] [Google Scholar]
- 25.Walker TR, Moore SM, Lawson MF, et al. Platelet-derived growth factor-BB and thrombin activate phosphoinositide 3-kinase and protein kinase B: role in mediating airway smooth muscle proliferation. Mol Pharmacol. 1998;54:1007–15. doi: 10.1124/mol.54.6.1007. [DOI] [PubMed] [Google Scholar]
- 26.Moroy T, Geisen C. Cyclin E. Int J Biochem Cell Biol. 2004;36:1424–39. doi: 10.1016/j.biocel.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 27.Sherr CJ, Roberts JM. Living with or without cyclins and cyclin-dependent kinases. Genes Dev. 2004;18:2699–711. doi: 10.1101/gad.1256504. [DOI] [PubMed] [Google Scholar]
- 28.Hayashi K, Saga H, Chimori Y, et al. Differentiated phenotype of smooth muscle cells depends on signaling pathways through insulin-like growth factors and phosphatidylinositol 3-kinase. J Biol Chem. 1998;273:28860–7. doi: 10.1074/jbc.273.44.28860. [DOI] [PubMed] [Google Scholar]
- 29.Krymskaya VP, Penn RB, Orsini MJ, et al. Phosphatidylinositol 3-kinase mediates mitogen-induced human airway smooth muscle cell proliferation. Am J Physiol. 1999;277:L65–78. doi: 10.1152/ajplung.1999.277.1.L65. [DOI] [PubMed] [Google Scholar]
- 30.Zhou L, Baumgartner BJ, Hill-Felberg SJ, et al. Neurotrophin-3 expressed in situ induces axonal plasticity in the adult injured spinal cord. J Neurosci. 2003;23:1424–31. doi: 10.1523/JNEUROSCI.23-04-01424.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hirst SJ, Martin JG, Bonacci JV, et al. Proliferative aspects of airway smooth muscle. J Allergy Clin Immunol. 2004;114 doi: 10.1016/j.jaci.2004.04.039. : S2–17. [DOI] [PubMed] [Google Scholar]
- 32.Tirumurugaan KG, Jude JA, Kang BN, et al. TNF-alpha induced CD38 expression in human airway smooth muscle cells: role of MAP kinases and transcription factors NF-kappaB and AP-1. Am J Physiol Lung Cell Mol Physiol. 2007;292 doi: 10.1152/ajplung.00472.2006. [DOI] [PubMed] [Google Scholar]
- 33.Oeckinghaus A, Ghosh S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb Perspect Biol. 2009;1:a000034. doi: 10.1101/cshperspect.a000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yamamoto Y, Gaynor RB. IkappaB kinases: key regulators of the NF-kappaB pathway. Trends Biochem Sci. 2004;29:72–9. doi: 10.1016/j.tibs.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 35.Freund-Michel V, Frossard N. The nerve growth factor and its receptors in airway inflammatory diseases. Pharmacol Ther. 2008;117:52–76. doi: 10.1016/j.pharmthera.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 36.Furuno T, Nakanishi M. Neurotrophic factors increase tumour necrosis factor-alpha-induced nuclear translocation of NF-kappaB in rat PC12 cells. Neurosci Lett. 2006;392:240–4. doi: 10.1016/j.neulet.2005.09.031. [DOI] [PubMed] [Google Scholar]
- 37.Reichardt LF. Neurotrophin-regulated signalling pathways. Philos Trans R Soc Lond B Biol Sci. 2006;361:1545–64. doi: 10.1098/rstb.2006.1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Howell JE, McAnulty RJ. TGF-beta: its role in asthma and therapeutic potential. Curr Drug Targets. 2006;7:547–65. doi: 10.2174/138945006776818692. [DOI] [PubMed] [Google Scholar]
- 39.Peng Q, Lai D, Nguyen TT, et al. Multiple beta 1 integrins mediate enhancement of human airway smooth muscle cytokine secretion by fibronectin and type I collagen. J Immunol. 2005;174:2258–64. doi: 10.4049/jimmunol.174.4.2258. [DOI] [PubMed] [Google Scholar]
- 40.Burgess JK, Ceresa C, Johnson SR, et al. Tissue and matrix influences on airway smooth muscle function. Pulm Pharmacol Ther. 2009;22:379–87. doi: 10.1016/j.pupt.2008.12.007. [DOI] [PubMed] [Google Scholar]
- 41.Hirst SJ, Twort CH, Lee TH. Differential effects of extracellular matrix proteins on human airway smooth muscle cell proliferation and phenotype. Am J Respir Cell Mol Biol. 2000;23:335–44. doi: 10.1165/ajrcmb.23.3.3990. [DOI] [PubMed] [Google Scholar]
- 42.Lu B, Pang PT, Woo NH. The yin and yang of neurotrophin action. Nat Rev Neurosci. 2005;6:603–14. doi: 10.1038/nrn1726. [DOI] [PubMed] [Google Scholar]
- 43.Damera G, Tliba O, Panettieri RA., Jr Airway smooth muscle as an immunomodulatory cell. Pulm Pharmacol Ther. 2009;22:353–9. doi: 10.1016/j.pupt.2008.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hotta K, Emala CW, Hirshman CA. TNF-alpha upregulates Gialpha and Gqalpha protein expression and function in human airway smooth muscle cells. Am J Physiol. 1999;276:L405–11. doi: 10.1152/ajplung.1999.276.3.L405. [DOI] [PubMed] [Google Scholar]
- 45.Gosens R, Meurs H, Bromhaar MM, et al. Functional characterization of serum- and growth factor-induced phenotypic changes in intact bovine tracheal smooth muscle. Br J Pharmacol. 2002;137:459–66. doi: 10.1038/sj.bjp.0704889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pelaia G, Renda T, Gallelli L, et al. Molecular mechanisms underlying airway smooth muscle contraction and proliferation: implications for asthma. Respir Med. 2008;102:1173–81. doi: 10.1016/j.rmed.2008.02.020. [DOI] [PubMed] [Google Scholar]
- 47.Sweeney M, McDaniel SS, Platoshyn O, et al. Role of capacitative Ca2+ entry in bronchial contraction and remodeling. J Appl Physiol. 2002;92:1594–602. doi: 10.1152/japplphysiol.00722.2001. [DOI] [PubMed] [Google Scholar]
- 48.Trian T, Benard G, Begueret H, et al. Bronchial smooth muscle remodeling involves calcium-dependent enhanced mitochondrial biogenesis in asthma. J Exp Med. 2007;204:3173–81. doi: 10.1084/jem.20070956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pani B, Ong HL, Brazer SC, et al. Activation of TRPC1 by STIM1 in ER-PM microdomains involves release of the channel from its scaffold caveolin-1. Proc Natl Acad Sci U S A. 2009;106:20087–92. doi: 10.1073/pnas.0905002106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Clarke DL, Clifford RL, Jindarat S, et al. TNFalpha and IFNgamma synergistically enhance transcriptional activation of CXCL10 in human airway smooth muscle cells via STAT-1, NF-kappaB, and the transcriptional coactivator CREB-binding protein. J Biol Chem. 285:29101–10. doi: 10.1074/jbc.M109.0999952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lahiri T, Moore PE, Baraldo S, et al. Effect of IL-1beta on CRE-dependent gene expression in human airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2002;283:L1239–46. doi: 10.1152/ajplung.00231.2001. [DOI] [PubMed] [Google Scholar]
- 52.Finkbeiner S. CREB couples neurotrophin signals to survival messages. Neuron. 2000;25:11–4. doi: 10.1016/s0896-6273(00)80866-1. [DOI] [PubMed] [Google Scholar]
- 53.White TA, Xue A, Chini EN, et al. Role of TRPC3 in tumour necrosis factor-alpha enhanced calcium influx in human airway myocytes. Am J Respir Cell Mol Biol. 2006;35:243–51. doi: 10.1165/rcmb.2006-0003OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen H, Tliba O, Van BesienCR, et al. TNF-alpha modulates murine tracheal rings responsiveness to G-protein-coupled receptor agonists and KCl. J Appl Physiol. 2003;95:864–72. doi: 10.1152/japplphysiol.00140.2003. [DOI] [PubMed] [Google Scholar]
- 55.Amrani Y, Chen H, Panettieri RA., Jr Activation of tumour necrosis factor receptor 1 in airway smooth muscle: a potential pathway that modulates bronchial hyper-responsiveness in asthma. Respir Res. 2000;1:49–53. doi: 10.1186/rr12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dhawan P, Richmond A. A novel NF-kappa B-inducing kinase-MAPK signaling pathway up-regulates NF-kappa B activity in melanoma cells. J Biol Chem. 2002;277:7920–8. doi: 10.1074/jbc.M112210200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kim JM, Oh YK, Lee JH, et al. Induction of proinflammatory mediators requires activation of the TRAF, NIK, IKK and NF-kappaB signal transduction pathway in astrocytes infected with Escherichia coli. Clin Exp Immunol. 2005;140:450–60. doi: 10.1111/j.1365-2249.2005.02804.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Malinin NL, Boldin MP, Kovalenko AV, Wallach D. MAP3K-related kinase involved in NF-kappaB induction by TNF, CD95 and IL-1. Nature. 1997;385:540–4. doi: 10.1038/385540a0. [DOI] [PubMed] [Google Scholar]
- 59.Clarke DL, Clifford RL, Jindarat S, et al. TNFalpha and IFNgamma synergistically enhance transcriptional activation of CXCL10 in human airway smooth muscle cells via STAT-1, NF-kappaB, and the transcriptional coactivator CREB-binding protein. J Biol Chem. 2010;285:29101–10. doi: 10.1074/jbc.M109.0999952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.McKay S, Hirst SJ, Haas MB, et al. Tumour necrosis factor-alpha enhances mRNA expression and secretion of interleukin-6 in cultured human airway smooth muscle cells. Am J Respir Cell Mol Biol. 2000;23:103–11. doi: 10.1165/ajrcmb.23.1.3765. [DOI] [PubMed] [Google Scholar]
- 61.Shore SA. Direct effects of Th2 cytokines on airway smooth muscle. Curr Opin Pharmacol. 2004;4:235–40. doi: 10.1016/j.coph.2004.01.008. [DOI] [PubMed] [Google Scholar]