

Abstract
The process of liver repair and regeneration following hepatic injury is complex and relies on a temporally coordinated integration of several key signaling pathways. Pathways activated by members of the CXC family of chemokines play important roles in the mechanisms of liver repair and regeneration through their effects on hepatocytes. However, little is known about the signaling pathways utilized by CXC chemokine receptors in hepatocytes. Here we review our current understanding of the pathways involved in both CXC chemokine receptor signaling in other cell types, most notably neutrophils, and similar pathways operant during hepatocyte proliferation/liver regeneration in order to formulate a basis for the function of CXC chemokine receptor signaling in hepatocytes.
Keywords: Liver injury, ischemia/reperfusion, hepatectomy, CXCR1, CXCR2
The phenomenon of liver regeneration has been promulgated for centuries. Greek mythology tells the stories of two different characters, Prometheus and Tityus, who were tortured by birds of prey, sent by the god Zeus to punish them, that ate from their livers every day, only to have them grow back to be eaten from the next day (1). Interestingly, the ancient Greeks held the liver in great regard, viewing it as the center of life, soul, and intelligence, therefore linking the regenerative capacity of the liver to the indestructibility of the soul (2). The wide breadth of liver pathology that effects populations today makes its regenerative capacity one of great interest. Despite the many advances in medicine over the years, and the ability of clinicians to provide support to failing organ systems such as the kidneys and lungs, liver failure is often lethal without transplantation. Similarly, traumatic injury to the liver as well as oncologic disease of the liver, both of which often necessitate resection, again bring the unique qualities of the liver into focus.
Liver regeneration involves numerous soluble mediators, growth factors, and metabolic factors that work to stimulate quiescent hepatocytes to enter into the cell cycle, replicate, and expand existing liver mass. A class of small proteins called chemokines play important roles in the processes of liver repair and regeneration. A better understanding of their various functions will have widespread implications, including the ability to minimize organ dysfunction and enhance graft survival rates after liver transplantation, allowance for utilization of livers from extended criteria donors, or performance of more aggressive surgical interventions for hepatic malignancies.
Basic Principles of Liver Regeneration
Hepatocytes possess the unique ability to proliferate upon appropriate stimulation, normally maintaining themselves in a stage of quiescence, known as the G0 phase. The molecular basis of liver regeneration is composed of three different phases, including a priming phase, a proliferative phase, and a termination phase (3). These phases have also been qualified as cytokine, growth factor and metabolic pathways, respectively, as it pertains to the factors predominantly mediating a particular phase (4). It is important to note that while it is conceptually easier to denote the sequence of events into “phases,” there is in fact a highly coordinated, synchronous schema of interactions between growth factors, cytokines and other mediators that allow the process of liver regeneration to occur (5). Cytokines are a key factor in stimulating quiescent hepatocytes from the G0 phase into the G1 phase. TNFα and IL-6, along with the transcription factors, STAT3 and NF-κB, required for the initiation of liver regeneration (4, 6). Through activation of STAT3 and NF-κB, target genes are transcribed which are important to hepatocyte proliferation. Growth factors, specifically hepatocyte growth factor (HGF) and epidermal growth factor (EGF), then drive the cell from G1 into the S phase of DNA replication (4). Arguably one of the most important mediators of liver regeneration is HGF, a 100kDa protein that was originally identified in 1984 (7, 8). A potent mitogen for hepatocyte growth, HGF is locally released and upregulated during the initiation of the regenerative process, cleaved from its inactive single-chain form into its active two-chain form by uPA (3).
Phospholipase Cγ1 (PLCγ1), phospholipase Cβ1 (PLCβ1), phospholipase D1 (PLD1), and phosphoinositide-3-kinase (PI3K) have been implicated in the mechanisms of hepatocyte proliferation immediately after HGF or EGF binding (9–12). PLCγ1 and PLCβ1 appear to play different roles in the regenerating liver, with PLCγ1 having more influence on the G2/M phase transition, and PLCβ1 seeming to trigger DNA replication (9). PLD1 may play a role in the activation of c-Jun/c-Fos transcription factors, further contributing to DNA synthesis (13). The HGF receptor, a c-met oncogene, has been shown to function through tyrosine kinase activity. However, Adachi, et al.(14), showed that pertussis-toxin sensitive G proteins were also involved in mitogen activated protein kinase (MAPK) activation and arachidonic acid release, specifically demonstrating that PLD activation was diminished to baseline levels in the presence of Gαi receptor complex inhibition. More recently, signaling through PI3K has been shown to be critical for the induction of cyclin D and DNA replication following HGF binding (12). Further downstream, MAPK-dependent production of arachidonic acid (AA) through PLA2 results in production of prostaglandins, further stimulating DNA synthesis (11). Prostaglandins, most significantly PGE2 and PGF2, are known to promote growth in hepatocytes (15). Conversely, during conditions in which hepatocytes may be stressed, activation of PLA2 and increased release of arachidonic acid may have a deleterious effect on hepatocytes (16). In the setting of hypoxic injury to hepatocytes, diminished ATP production leads to acidosis, therefore preventing activation of PLA2 until the return to physiologic pH during reperfusion, resulting in AA release and increased cell death (16, 17). In vivo studies have revealed that COX-2-dependent conversion of arachidonic acid to prostaglandins is crucial to the induction of protective mechanisms within the liver, and that COX-2 inhibition contributed to greater hepatotoxicity in the setting of carbon tetrachloride (CCl4) injury, perhaps indicating that the level of COX-2 following hepatic injury is important to recovery (18).
Experimental Models of Liver Injury, Repair, and Regeneration
Several different experimental models have been utilized to better understand the mechanisms by which liver regeneration occurs, with partial hepatectomy being the gold-standard. Although it is the most well studied model of liver regeneration to date, the hepatocytes which constitute the remnant liver after partial hepatectomy are not an accurate representation of the physiological scenario seen in many liver pathologies, in which hepatocytes are injured and/or stressed. Additional models of liver injury, such as acetaminophen toxicity, CCl4 injury, and ischemia/reperfusion injury represent a means to assess the reparative and regenerative mechanisms in stressed and injured hepatocytes. While differences exist in the timing of molecular and cellular events between the different models, hepatocytes remain the nidus of the regenerative process, stimulated by various soluble mediators released after injury (19).
Chemokines and their Receptors
The term chemokines describes a family of chemotactic cytokines originally described as mediators of immune cell trafficking and function (20, 21). Chemokines are a group of small (8–10 kD), basic, heparin-binding proteins that are secreted by leukocytes as well as various tissue cells (20, 22). While mainly involved in leukocyte chemoattraction, chemokines have also been implicated in other cellular activities, including regulation of angiogenesis, fibrosis, proliferation, cytotoxicity and apoptosis (23–26). The nomenclature for chemokines is based on the configuration of a conserved amino-proximal cysteine-containing motif (27). There are currently four branches of the chemokine family, CXC, CC, CX3C and C (where X is any amino acid). CC and CXC are the two major branches, whereas CX3C and C each have only one representative, consisting of fractalkine (CX3CL1) and lymphotactin (XCL1), respectively (28). The CC family is the largest, primarily involved in attracting mononuclear cells to sites of chronic inflammation, while members of the CXC family mediate the chemoattraction of neutrophils and monocytes to sites of acute inflammation (24). CXC chemokines can be further classified by the presence or absence of a Glu–Leu–Arg (ELR) amino acid motif in the amino terminus of the peptide. The ELR motif confers receptor-binding specificity (29, 30).
CXC chemokines exert their effects through the CXC chemokine receptors (CXCR) 1–6 (28). CXCR1 and CXCR2 bind specifically to CXC chemokines which contain the amino terminus sequence ELR (Glu-Leu-Arg) (Table 1) (23, 27). In addition to their leukocyte-chemoattractant properties, ELR+ CXC chemokines have been shown to have important roles in angiogenesis and cellular proliferation (25, 31, 32). CXCR1 and CXCR2 are expressed by neutrophils, monocytes, CD8+ T cells, epithelial cells and endothelial cells, as well as in hepatocytes (33–35). CXC chemokine receptors are heptahelical transmembrane G protein-coupled receptors (GPCR), with the individual components that make up each receptor contributing to their unique interactions. The external interface contributes to ligand specificity, and the other domains, including the transmembrane sequences, cytoplasmic loops and C-terminal domain, allow for receptor signaling and internalization (23).
Table 1.
CXC chemokines and their receptors.
| ELR+ CXC Chemokines Ligands for CXCR1 and CXCR2 |
ELR− CXC Chemokines Ligands for CXCR3-6 |
||||
|---|---|---|---|---|---|
| Chemokine | Old Nomenclature
|
Chemokine | Old Nomenclature
|
||
| Human | Mouse | Human | Mouse | ||
| CXCL1 | Gro-α | KC | CXCL4 | PF4 | PF4 |
| CXCL2 | Gro-β | MIP-2 | CXCL9 | MIG | MIG |
| CXCL3 | Gro-γ | DCIP-1 | CXCL10 | IP-10 | CRG-2 |
| CXCL5 | ENA-78 | LIX | CXCL11 | I-TAC | I-TAC |
| CXCL6 | GCP-2 | GCP-2 | CXCL12 | SDF-1 | SDF-1 |
| CXCL7 | NAP-2 | TCK-1 | CXCL13 | BCA-1 | BLC |
| CXCL8 | IL-8 | none | CXCL14 | BRAK | BRAK |
| CXCL15 | Lungkine | Lungkine | |||
| CXCL16 | none | none | |||
Contribution of CXC Chemokines to Liver Repair and Regeneration
CXC chemokines are known to be important mediators of the inflammatory cascade following hepatic injury, and also appear to have a dichotomous role in hepatocytes that may be related to their level of expression (36). For example, induction of CXC chemokines at relatively low levels is associated with liver repair and regeneration, whereas high expression levels have been associated with hepatotoxicity (35, 37–39). The impact of CXC chemokines on the regenerative capacity of the liver was first examined using an in vivo model of partial hepatectomy (37). Partial hepatectomy represents a clinically relevant model of hepatic resection, a procedure often performed due to trauma or malignancy. ELR+ CXC chemokines were found to be up-regulated after partial hepatectomy, and blockade of chemokines or CXCR2 resulted in diminished liver regeneration (37). Subsequent in vitro experiments demonstrated that hepatocytes treated with ELR+ CXC chemokines proliferated to a degree similar to that induced by HGF. These studies (35, 37–39) provided evidence that ELR+ CXC chemokines were important hepatocyte proliferative factors that functioned in vivo to promote liver regeneration after hepatectomy. However, as previously mentioned, the remnant liver after resection or hepatectomy, without Pringle maneuver, is comprised of unstressed hepatocytes. The role of CXC chemokines may be distinctly different in a setting in which hepatocytes are under significant stress.
This concept was first addressed in a murine model of hepatic ischemia/reperfusion (I/R) injury. Liver I/R represents a clinically relevant insult to the liver which produces significant inflammation and ultimately, parenchymal damage and organ dysfunction. There are two phases of liver injury following ischemia/reperfusion (39–41). The initial phase occurs during the first hours of reperfusion and is characterized by oxidant-induced hepatocellular injury and the induction of proinflammatory mediators (42–47). The later phase, which occurs many hours after reperfusion, is characterized by an intense inflammatory response culminating in the recruitment of activated neutrophils which directly injure hepatocytes via the release of oxidants and proteases (40, 48). The recruitment of neutrophils to the liver is CXC chemokine-dependent and thus these chemokines play a central role in the hepatic inflammatory response to I/R (49, 50).
Hepatocyte death following ischemia/reperfusion injury occurs through both apoptotic and necrotic mechanisms, with necrosis contributing most significantly after warm ischemia (51). Toxicity following ischemia/reperfusion injury is directly related to the generation and release of reactive oxygen species (ROS) by recruited neutrophils and resident Kupffer cells (52, 53). These ROS affect hepatocyte signal transduction and are directly cytotoxic. The c-Jun N terminal kinase (JNK) is a stress activated protein kinase member of the MAPK family which is activated by cytokines such as TNF-α and IL-1, as well as oxidants and other environmental stressors (54, 55). JNK has been shown to be activated following ischemia/reperfusion injury resulting in induction of both apoptosis and necrosis in hepatocytes (56, 57). Further study has specifically shown that with low levels of oxidative stress ERK1/2 and PKC/PKD based inhibition of JNK is protective in hepatocytes (58, 59). However, despite continued activation of ERK1/2 and PKC/PKD, high levels of oxidative stress overwhelm these protective mechanisms leading to sustained activation of JNK/AP-1 and resultant cell death (59, 60). Finally, Tsung, et al. (61) have demonstrated the importance of toll-like receptor 4 (TLR4) in recognizing endogenous damage associated molecular patterns (DAMPs) following ischemia/reperfusion injury, a process which appears to be of key importance in the overall pathogenesis of the inflammatory injury of warm hepatic I/R (61, 62). Interestingly, chimeric mice which lack non-parenchymal cell TLR4 were seen to be protected from I/R injury (61). These mice had diminished JNK and NF-κB activation, therefore implicating these intermediates in TLR4 signal transduction (61).
Liver recovery and repair after I/R injury in this model begins approximately 48 hours after reperfusion and is associated with increased expression of stathmin and marked hepatocyte proliferation (63). Liver repair and regeneration typically return the liver to its normal, homeostatic state within 5–7 days after reperfusion, depending on the severity of the injury. It is during this reparative/regenerative phase in which the function of CXC chemokines switches from a proinflammatory role to direct impingement on hepatocyte proliferation or death. Knockout of CXCR2, the primary receptor for ELR+ CXC chemokines in rodents, resulted in accelerated liver recovery associated with increased activation of NF-κB and STAT3 transcription factors resulting in increased hepatocyte proliferation (38). Antibody blockade of CXCR2 after induction of I/R injury had the same effect (38). These studies suggest that during the reparative/regenerative phase of I/R injury, ELR+ CXC chemokines have harmful effects which delay liver recovery.
These apparent harmful effects of ELR+ CXC chemokines is likely a result of specific signaling via CXCR2 in hepatocytes. While the presence and involvement of CXCR2 in murine models of hepatocyte injury and regeneration has been well characterized (37, 38), murine CXCR1 has only been recently identified (64, 65). Previous work has demonstrated that CXCR2 is constitutively expressed in hepatocytes (35), and may be up-regulated in the presence of certain cytokines (66). While CXCR2 and its ligands appear to play a key role in hepatocyte proliferation following partial hepatectomy, and hepatocyte toxicity following I/R injury or acetaminophen toxicity, the role of CXCR1 is less clear. CXCR1 is not constitutively expressed in the liver (64). This finding was recently confirmed, but CXCR1 was found to be induced in hepatocytes after I/R (34). Blockade and knockout of CXCR1 was found to result in delayed liver repair after I/R, although there were no observed changes in hepatocyte proliferation in vivo (34). While the effects of CXCR1 blockade or knockout on liver repair were not as striking as those observed with CXCR2, the findings suggest that CXCR1 has a divergent function in hepatocytes, compared to CXCR2.
While the stress level of the hepatocyte may alter its response to CXC chemokines, so may the concentration of available ligand. Following 70% partial hepatectomy, expression of ELR+ CXC chemokines increase approximately 5-fold (37), whereas after I/R they increase hundreds- to thousands-fold (38). In vitro, stimulation of primary hepatocytes with CXC chemokines has hepatoprotective effects at low concentrations and progressively cytotoxic effects at increasingly greater concentrations, effects which are specific to CXCR2 (34, 38). Adenoviral-mediated liver overexpression (>100-fold) of the CXC chemokine, keratinocyte-derived chemokine, has been shown to result in massive hepatocellular necrosis within 48 hours (39). Collectively, these studies suggest that moderate increases in CXCR2 ligands, as occurs after partial hepatectomy may promote liver regeneration, whereas much larger increases in expression of CXCR2 ligands, as occurs after I/R injury, may be hepatotoxic and/or oppose hepatocyte proliferation and regeneration.
CXCR1 and CXCR2 Signal Transduction Pathways in Neutrophils
CXCR1 and CXCR2 signaling pathways have been well characterized in neutrophils. While the expression of these receptors on hepatocytes has been demonstrated, the signaling pathways utilized to alter hepatocyte function are unclear. Understanding the signaling pathways utilized by CXCR1 and CXCR2 in neutrophils may offer valuable insights into how these receptors function in hepatocytes.
CXCR1 and CXCR2 receptors are G-protein coupled and involve the Gαi family, specifically Gαi2 and Gαi3, as well as Gα14 and Gα16 (67–69). Following G protein activation, release of the βγ subunit activates two key pathways involved in generating neutrophil end responses, namely PLCβ and PI3K. PLC-β2/β3 catalyzes the conversion of PIP2 into the second messengers DAG and IP3. DAG activates PKC and PLA2, and IP3 stimulates Ca2+ mobilization. Activation of PKC leads to translocation of p47phox to the plasma membrane, assembly of NADPH oxidase, and generation of the respiratory burst (70). PI3K activation catalyzes the production of PIP3. PI3K activity is central to several downstream pathways which influence neutrophil function, including Ras/Raf/MAPK and Akt, leading to neutrophil granule release, adhesion and chemotaxis, and respiratory burst (71, 72) (Figure 1). Additionally, expression and activation of the β2 integrin CD11b, important to neutrophil adhesion, is modulated by PKC alongside of MAPK. MAPK and PKC appear to function independently as discrete pathways, and both must be inhibited to completely abrogate integrin-dependent adhesion (73).
Figure 1.
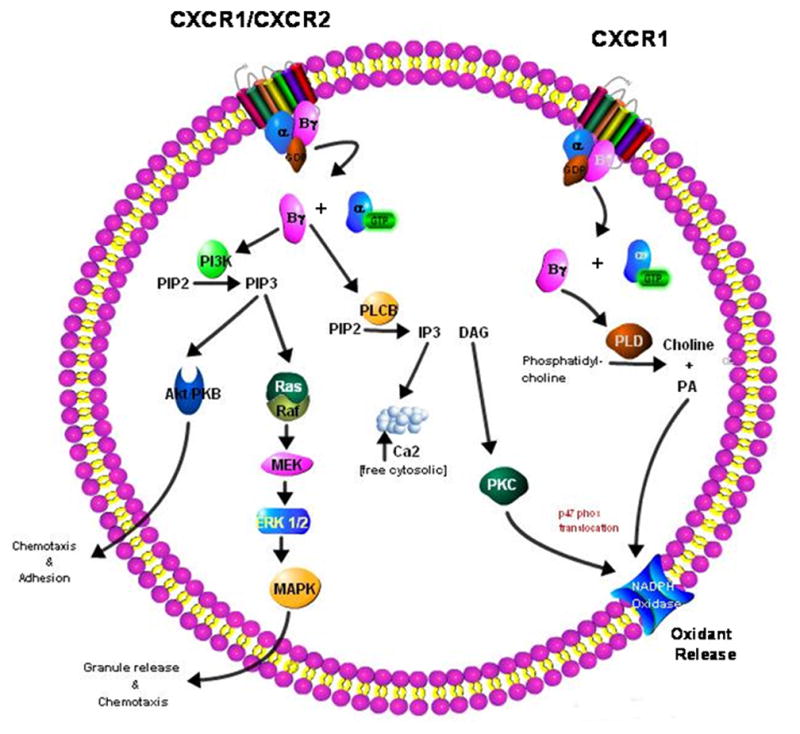
Signaling pathways of CXCR1 and CXCR2 in neutrophils.
CXCR1 and CXCR2 have intertwined yet diverse roles in neutrophil activation, and receptor-specific pathways have been identified for both receptors. Using receptor-specific antibodies, it was determined that CXCR1 was uniquely linked to activation of respiratory burst through the action of phospholipase D, known to catalyze the hydrolysis of phosphatidylcholine to phosphatidic acid and choline, thus allowing phosphatidic acid (along with DAG) to activate NADPH oxidase (74). Matrix metalloproteinase-9 (MMP-9) is present in the tertiary granules of neutrophils and can be released through ERK1/2 and PKC dependent pathways as a result of IL-8 stimulation. Blockade of CXCR2, but not CXCR1, diminished the release of MMP-9 in IL-8 treated neutrophils, suggesting a CXCR2 dependent system (75).
The differences between CXCR1 and CXCR2 may be further explored by examining the unique effect of ligand concentration, as well as their individual patterns of receptor internalization and desensitization. Previous work has demonstrated that IL-8 induces a time- and dose-dependent internalization of CXCR1 and CXCR2, with CXCR2 internalized more quickly than CXCR1, but CXCR1 recovering more quickly (76). Ligand concentration may play a role in receptor internalization, as it has recently been proposed that high ligand concentration may allow for receptor endocytosis via clarthrin-coated pits and subsequently act as a stop signal. This is in contrast to the generation of signaling for chemotaxis, which only requires low ligand concentrations, and does not result in receptor endocytosis (77). The concentration of chemoattractant needed to bring about chemotaxis versus granule enzyme release and respiratory burst also differ, with higher concentrations being necessary for the induction of granule release and respiratory burst (78). Neutrophils are known to exhibit a cross-desensitization effect in response to IL-8, and generation of these cross-desensitization signals may be related to the extent and length of activation of the receptor. Rapid phosphorylation of the carboxyl terminus of CXCR2 leads to receptor internalization and may prevent its ability to generate a cross-desensitization signal, indicating that CXCR1 may be responsible for this action (79). Collectively, this research indicates that the ability of CXCR1 and CXCR2 to generate certain signals is dependent upon ligand concentration and the length of receptor activation. While the CXCR1 and CXCR2 receptors in neutrophils certainly have commonalities, it is obvious that they have very distinct roles as well. Based on what is known about each receptor individually, it is possible that CXCR1 may have a more regulatory function, including a role in regulation of CXCR2 (33).
Proposed Signaling Pathways of CXCR1 and CXCR2 in Hepatocytes
Based on the current understanding of various signaling pathways operant in hepatocytes, and the known signal transduction pathways utilized by CXCR1 and CXCR2 in neutrophils, we propose a working hypothesis of signal transduction via CXCR1 and CXCR2 in hepatocytes. It is likely that hepatocyte CXCR1/2 are G-protein-coupled receptors (14, 67, 80). In addition, PI3K, PLC and PLD again all have significant roles in both neutrophil CXCR1/2 signaling as well as in hepatocyte proliferation, therefore it is probable that these elements are likewise a part of hepatocyte CXCR1/2 signaling (9–12, 72, 81). Downstream effectors in murine hepatocyte signal transduction including PKC, the MAPK ERK1/2, and PLA2 have been linked to both proliferative and cytotoxic effects, suggesting a potential role for these in hepatocyte CXC chemokine signaling. These mechanisms may involve significant cross-talk between receptor pathways, with CXCR2 signaling dominant early, and CXCR1 signaling playing a role later, after it the receptor is upregulated (33, 34).
We propose a system where signaling via CXCR2 induces a proliferative effect when low concentrations of ligand are present with little or no activity from CXCR1 signaling (Figure 2). As observed by Czaja, et al. (58–60), under conditions of a low stress environment, signaling through ERK1/2 and PKC is able to suppress JNK/AP-1 activation, therefore minimizing cytotoxic effects. Specifically, proliferative signals are likely mediated through MAPK/ERK1/2 activation, and PLC induced PLA2 stimulation of prostaglandin production, as has been seen during healthy hepatocyte proliferation following partial hepatectomy (11, 15). Conversely, when high ligand concentrations are present, we propose that CXCR2 predominantly activates toxicity pathways. In this scenario, it is probable that the high stress environment induced by an injury such as ischemia/reperfusion overwhelms the protective inhibition of ERK1/2 and PKC/PKD, leading to JNK/AP-1 activation and hepatocyte toxicity (58, 59). Additionally, in conditions of high chemokine concentrations and stressed/injured hepatocytes, PLA2 activation may lead to excessive production of arachidonic acid leading to further toxicity, and perhaps overwhelming any proliferative effect of prostaglandins (16). CXCR1 is also activated, providing some protective effects, and also exerting a regulatory effect on CXCR2 (34, 38).
Figure 2.

Proposed signaling pathways utilized by CXCR1 and CXCR2 in hepatocytes under conditions of low and high ligand availability.
Interestingly, some of the concentration-dependent effects seen in neutrophils parallel what has been observed in vivo in the liver (38, 50). The suggested cross-regulatory effect of CXCR1 on the expression of CXCR2 in neutrophils (33) may also be seen between CXCR1 and CXCR2 in hepatocytes as well, and further experimentation will be required in order to elucidate how these two receptors act in concert. Since we know that CXCR1 is inducible in the setting of ischemia/reperfusion, and that high concentrations of CXC chemokines are present in ischemia/reperfusion, it is conceivable that this receptor may have some kind of an impact on the induction of hepatocyte proliferation versus hepatocyte toxicity. Since the signaling mechanisms of hepatocyte CXCR1 and CXCR2 remain to be elucidated, there are likely other relevant pathways yet to be discovered that may prove to be significant in the regulation of liver pathology.
Acknowledgments
Financial Support: National Institutes of Health Grants DK56029 and AG025881 to ABL.
List of Abbreviations
- TNF-α
tumor necrosis factor α
- IL-6
interleukin-6
- STAT3
signal transducer and activator of transcription 3
- NF-κB
nuclear factor-κB
- HGF
hepatocyte growth factor
- EGF
epidermal growth factor
- PLCγ1
phospholipase Cγ1
- PLCβ1
phospholipase Cβ1
- PLD
phospholipase D
- PI3K
phosphoinositide-3-kinase
- MAPK
mitogen-activated protein kinase
- PLA2
phospholipase A2
- PGE2
prostaglandin E2
- PGF2
prostaglandin F2
- ATP
adenosine triphosphate
- AA
arachadonic acid
- COX-2
cyclo-oxygenase-2
- CCl4
carbon tetrachloride
- ELR
Glutamine-Leucine-Arginine
- GPCR
G protein coupled receptor
- I/R
ischemia/reperfusion
- ROS
reactive oxygen species
- JNK
c-Jun N-terminal kinase
- IL-1
interleukin-1
- ERK
extracellular signal related kinase
- PKC
protein kinase C
- PKD
protein kinase D
- AP-1
activator protein-1
- TLR4
toll-like receptor 4
- DAMP
damage associated molecular patterns
- PIP2
phosphatidylinositol diphosphate
- DAG
diacylglycerol
- IP3
inositol triphosphate
- NADPH
nicotinamide adenine dinucleotide phosphate
- PIP3
phosphatidylinositol triphosphate
- MMP-9
matrix metalloproteinase-9
- IL-8
interleukin-8
Contributor Information
Heather L. Van Sweringen, Email: lewishh@ucmail.uc.edu.
Nozomu Sakai, Email: sakainu@ucmail.uc.edu.
Amit D. Tevar, Email: tevara@ucmail.uc.edu.
Justin M. Burns, Email: burns2jn@ucmail.uc.edu.
Michael J. Edwards, Email: edwardm6@ucmail.uc.edu.
Alex B. Lentsch, Email: alex.lentsch@uc.edu.
References
- 1.Tiniakos DG, Kandilis A, Geller SA. Tityus: a forgotten myth of liver regeneration. J Hepatol. 53:357–361. doi: 10.1016/j.jhep.2010.02.032. [DOI] [PubMed] [Google Scholar]
- 2.Hesiod . Theogony-works and days. New York: Oxford University Press; 1988. [Google Scholar]
- 3.Zimmermann A. Regulation of liver regeneration. Nephrol Dial Transplant. 2004;19(Suppl 4):iv6–10. doi: 10.1093/ndt/gfh1034. [DOI] [PubMed] [Google Scholar]
- 4.Fausto N, Campbell JS, Riehle KJ. Liver regeneration. Hepatology. 2006;43:S45–53. doi: 10.1002/hep.20969. [DOI] [PubMed] [Google Scholar]
- 5.Michalopoulos GK. Liver regeneration. J Cell Physiol. 2007;213:286–300. doi: 10.1002/jcp.21172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Webber EM, Bruix J, Pierce RH, Fausto N. Tumor necrosis factor primes hepatocytes for DNA replication in the rat. Hepatology. 1998;28:1226–1234. doi: 10.1002/hep.510280509. [DOI] [PubMed] [Google Scholar]
- 7.Michalopoulos G, Houck KA, Dolan ML, Leutteke NC. Control of hepatocyte replication by two serum factors. Cancer Res. 1984;44:4414–4419. [PubMed] [Google Scholar]
- 8.Nakamura T, Nawa K, Ichihara A. Partial purification and characterization of hepatocyte growth factor from serum of hepatectomized rats. Biochem Biophys Res Commun. 1984;122:1450–1459. doi: 10.1016/0006-291x(84)91253-1. [DOI] [PubMed] [Google Scholar]
- 9.Albi E, Rossi G, Maraldi NM, Magni MV, Cataldi S, Solimando L, Zini N. Involvement of nuclear phosphatidylinositol-dependent phospholipases C in cell cycle progression during rat liver regeneration. J Cell Physiol. 2003;197:181–188. doi: 10.1002/jcp.10292. [DOI] [PubMed] [Google Scholar]
- 10.Nebigil CG. Suppression of phospholipase C beta, gamma, and delta families alters cell growth and phosphatidylinositol 4,5-bisphosphate levels. Biochemistry. 1997;36:15949–15958. doi: 10.1021/bi971721m. [DOI] [PubMed] [Google Scholar]
- 11.Adachi T, Nakashima S, Saji S, Nakamura T, Nozawa Y. Mitogen-activated protein kinase activation in hepatocyte growth factor-stimulated rat hepatocytes: involvement of protein tyrosine kinase and protein kinase C. Hepatology. 1996;23:1244–1253. doi: 10.1053/jhep.1996.v23.pm0008621160. [DOI] [PubMed] [Google Scholar]
- 12.Coutant A, Rescan C, Gilot D, Loyer P, Guguen-Guillouzo C, Baffet G. PI3K-FRAP/mTOR pathway is critical for hepatocyte proliferation whereas MEK/ERK supports both proliferation and survival. Hepatology. 2002;36:1079–1088. doi: 10.1053/jhep.2002.36160. [DOI] [PubMed] [Google Scholar]
- 13.Watanabe A, Nakashima S, Adachi T, Saji S, Nozawa Y. Changes in the expression of lipid-mediated signal-transducing enzymes in the rat liver after partial hepatectomy. Surg Today. 2000;30:622–630. doi: 10.1007/s005950070102. [DOI] [PubMed] [Google Scholar]
- 14.Adachi T, Nakashima S, Saji S, Nakamura T, Nozawa Y. Possible involvement of pertussis toxin-sensitive G protein in hepatocyte growth factor-induced signal transduction in cultured rat hepatocytes: pertussis toxin treatment inhibits activation of phospholipid signaling, calcium oscillation, and mitogen-activated protein kinase. Hepatology. 1997;26:295–300. doi: 10.1002/hep.510260207. [DOI] [PubMed] [Google Scholar]
- 15.Refsnes M, Dajani OF, Sandnes D, Thoresen GH, Rottingen JA, Iversen JG, Christoffersen T. On the mechanisms of the growth-promoting effect of prostaglandins in hepatocytes: the relationship between stimulation of DNA synthesis and signaling mediated by adenylyl cyclase and phosphoinositide-specific phospholipase C. J Cell Physiol. 1995;164:465–473. doi: 10.1002/jcp.1041640304. [DOI] [PubMed] [Google Scholar]
- 16.Harrison DC, Lemasters JJ, Herman B. A pH-dependent phospholipase A2 contributes to loss of plasma membrane integrity during chemical hypoxia in rat hepatocytes. Biochem Biophys Res Commun. 1991;174:654–659. doi: 10.1016/0006-291x(91)91467-q. [DOI] [PubMed] [Google Scholar]
- 17.Malhi H, Gores GJ, Lemasters JJ. Apoptosis and necrosis in the liver: a tale of two deaths? Hepatology. 2006;43:S31–44. doi: 10.1002/hep.21062. [DOI] [PubMed] [Google Scholar]
- 18.Reilly TP, Brady JN, Marchick MR, Bourdi M, George JW, Radonovich MF, Pise-Masison CA, et al. A protective role for cyclooxygenase-2 in drug-induced liver injury in mice. Chem Res Toxicol. 2001;14:1620–1628. doi: 10.1021/tx0155505. [DOI] [PubMed] [Google Scholar]
- 19.Fausto N, Riehle KJ. Mechanisms of liver regeneration and their clinical implications. J Hepatobiliary Pancreat Surg. 2005;12:181–189. doi: 10.1007/s00534-005-0979-y. [DOI] [PubMed] [Google Scholar]
- 20.Oppenheim JJ, Zachariae CO, Mukaida N, Matsushima K. Properties of the novel proinflammatory supergene “intercrine” cytokine family. Annu Rev Immunol. 1991;9:617–648. doi: 10.1146/annurev.iy.09.040191.003153. [DOI] [PubMed] [Google Scholar]
- 21.Miller MD, Krangel MS. Biology and biochemistry of the chemokines: a family of chemotactic and inflammatory cytokines. Crit Rev Immunol. 1992;12:17–46. [PubMed] [Google Scholar]
- 22.Baggiolini M. Chemokines in pathology and medicine. J Intern Med. 2001;250:91–104. doi: 10.1046/j.1365-2796.2001.00867.x. [DOI] [PubMed] [Google Scholar]
- 23.Mantovani A, Bonecchi R, Locati M. Tuning inflammation and immunity by chemokine sequestration: decoys and more. Nat Rev Immunol. 2006;6:907–918. doi: 10.1038/nri1964. [DOI] [PubMed] [Google Scholar]
- 24.Charo IF, Ransohoff RM. The many roles of chemokines and chemokine receptors in inflammation. N Engl J Med. 2006;354:610–621. doi: 10.1056/NEJMra052723. [DOI] [PubMed] [Google Scholar]
- 25.Strieter RM, Polverini PJ, Kunkel SL, Arenberg DA, Burdick MD, Kasper J, Dzuiba J, et al. The functional role of the ELR motif in CXC chemokine-mediated angiogenesis. J Biol Chem. 1995;270:27348–27357. doi: 10.1074/jbc.270.45.27348. [DOI] [PubMed] [Google Scholar]
- 26.Belperio JA, Keane MP, Arenberg DA, Addison CL, Ehlert JE, Burdick MD, Strieter RM. CXC chemokines in angiogenesis. J Leukoc Biol. 2000;68:1–8. [PubMed] [Google Scholar]
- 27.Luster AD. Chemokines--chemotactic cytokines that mediate inflammation. N Engl J Med. 1998;338:436–445. doi: 10.1056/NEJM199802123380706. [DOI] [PubMed] [Google Scholar]
- 28.Thorpe R, Bacon K, Baggiolini M, Broxmeyer H, Horuk R, Lindley I, Mantovani A, et al. Chemokine/chemokine receptor nomenclature. Cytokine. 2003;21:48–49. [Google Scholar]
- 29.Clark-Lewis I, Dewald B, Geiser T, Moser B, Baggiolini M. Platelet factor 4 binds to interleukin 8 receptors and activates neutrophils when its N terminus is modified with Glu-Leu-Arg. Proc Natl Acad Sci U S A. 1993;90:3574–3577. doi: 10.1073/pnas.90.8.3574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hebert CA, Vitangcol RV, Baker JB. Scanning mutagenesis of interleukin-8 identifies a cluster of residues required for receptor binding. J Biol Chem. 1991;266:18989–18994. [PubMed] [Google Scholar]
- 31.Yoshidome H, Lentsch AB, Cheadle WG, Miller FN, Edwards MJ. Enhanced pulmonary expression of CXC chemokines during hepatic ischemia/reperfusion-induced lung injury in mice. J Surg Res. 1999;81:33–37. doi: 10.1006/jsre.1998.5490. [DOI] [PubMed] [Google Scholar]
- 32.Colletti LM, Green ME, Burdick MD, Strieter RM. The ratio of ELR+ to ELR− CXC chemokines affects the lung and liver injury following hepatic ischemia/reperfusion in the rat. Hepatology. 2000;31:435–445. doi: 10.1002/hep.510310225. [DOI] [PubMed] [Google Scholar]
- 33.Stillie R, Farooq SM, Gordon JR, Stadnyk AW. The functional significance behind expressing two IL-8 receptor types on PMN. J Leukoc Biol. 2009;86:529–543. doi: 10.1189/jlb.0208125. [DOI] [PubMed] [Google Scholar]
- 34.Clarke C, Kuboki S, Sakai N, Kasten KR, Tevar AD, Schuster R, Blanchard J, et al. CXC chemokine receptor-1 is expressed by hepatocytes and regulates liver recovery after hepatic ischemia/reperfusion injury. Hepatology. 53:261–271. doi: 10.1002/hep.24028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hogaboam CM, Bone-Larson CL, Steinhauser ML, Lukacs NW, Colletti LM, Simpson KJ, Strieter RM, et al. Novel CXCR2-dependent liver regenerative qualities of ELR-containing CXC chemokines. FASEB J. 1999;13:1565–1574. doi: 10.1096/fasebj.13.12.1565. [DOI] [PubMed] [Google Scholar]
- 36.Clarke CN, Kuboki S, Tevar A, Lentsch AB, Edwards M. CXC chemokines play a critical role in liver injury, recovery, and regeneration. Am J Surg. 2009;198:415–419. doi: 10.1016/j.amjsurg.2009.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Colletti LM, Green M, Burdick MD, Kunkel SL, Strieter RM. Proliferative effects of CXC chemokines in rat hepatocytes in vitro and in vivo. Shock. 1998;10:248–257. doi: 10.1097/00024382-199810000-00004. [DOI] [PubMed] [Google Scholar]
- 38.Kuboki S, Shin T, Huber N, Eismann T, Galloway E, Schuster R, Blanchard J, et al. Hepatocyte signaling through CXC chemokine receptor-2 is detrimental to liver recovery after ischemia/reperfusion in mice. Hepatology. 2008;48:1213–1223. doi: 10.1002/hep.22471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stefanovic L, Stefanovic B. Mechanism of direct hepatotoxic effect of KC chemokine: sequential activation of gene expression and progression from inflammation to necrosis. J Interferon Cytokine Res. 2006;26:760–770. doi: 10.1089/jir.2006.26.760. [DOI] [PubMed] [Google Scholar]
- 40.Jaeschke H. Reactive oxygen and ischemia/reperfusion injury of the liver. Chem Biol Interact. 1991;79:115–136. doi: 10.1016/0009-2797(91)90077-k. [DOI] [PubMed] [Google Scholar]
- 41.Jaeschke H, Farhood A, Smith CW. Neutrophils contribute to ischemia/reperfusion injury in rat liver in vivo. FASEB J. 1990;4:3355–3359. [PubMed] [Google Scholar]
- 42.Wanner GA, Ertel W, Muller P, Hofer Y, Leiderer R, Menger MD, Messmer K. Liver ischemia and reperfusion induces a systemic inflammatory response through Kupffer cell activation. Shock. 1996;5:34–40. doi: 10.1097/00024382-199601000-00008. [DOI] [PubMed] [Google Scholar]
- 43.Colletti LM, Remick DG, Burtch GD, Kunkel SL, Strieter RM, Campbell DA., Jr Role of tumor necrosis factor-alpha in the pathophysiologic alterations after hepatic ischemia/reperfusion injury in the rat. J Clin Invest. 1990;85:1936–1943. doi: 10.1172/JCI114656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Husted TL, Blanchard J, Schuster R, Shen H, Lentsch AB. Potential role for IL-23 in hepatic ischemia/reperfusion injury. Inflamm Res. 2006;55:177–178. doi: 10.1007/s00011-006-0073-1. [DOI] [PubMed] [Google Scholar]
- 45.Kato A, Gabay C, Okaya T, Lentsch AB. Specific role of interleukin-1 in hepatic neutrophil recruitment after ischemia/reperfusion. Am J Pathol. 2002;161:1797–1803. doi: 10.1016/S0002-9440(10)64456-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lentsch AB, Kato A, Yoshidome H, McMasters KM, Edwards MJ. Inflammatory mechanisms and therapeutic strategies for warm hepatic ischemia/reperfusion injury. Hepatology. 2000;32:169–173. doi: 10.1053/jhep.2000.9323. [DOI] [PubMed] [Google Scholar]
- 47.Lentsch AB, Yoshidome H, Kato A, Warner RL, Cheadle WG, Ward PA, Edwards MJ. Requirement for interleukin-12 in the pathogenesis of warm hepatic ischemia/reperfusion injury in mice. Hepatology. 1999;30:1448–1453. doi: 10.1002/hep.510300615. [DOI] [PubMed] [Google Scholar]
- 48.Mavier P, Guigui B, Preaux AM, Rosenbaum J, Lescs MC, Zafrani ES, Dhumeaux D. In vitro toxicity of hydrogen peroxide against normal vs. tumor rat hepatocytes: role of catalase and of the glutathione redox cycle. Hepatology. 1988;8:1673–1678. doi: 10.1002/hep.1840080634. [DOI] [PubMed] [Google Scholar]
- 49.Colletti LM, Kunkel SL, Walz A, Burdick MD, Kunkel RG, Wilke CA, Strieter RM. Chemokine expression during hepatic ischemia/reperfusion-induced lung injury in the rat. The role of epithelial neutrophil activating protein. J Clin Invest. 1995;95:134–141. doi: 10.1172/JCI117630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lentsch AB, Yoshidome H, Cheadle WG, Miller FN, Edwards MJ. Chemokine involvement in hepatic ischemia/reperfusion injury in mice: roles for macrophage inflammatory protein-2 and KC. Hepatology. 1998;27:1172–1177. doi: 10.1002/hep.510270440. [DOI] [PubMed] [Google Scholar]
- 51.Gujral JS, Bucci TJ, Farhood A, Jaeschke H. Mechanism of cell death during warm hepatic ischemia-reperfusion in rats: apoptosis or necrosis? Hepatology. 2001;33:397–405. doi: 10.1053/jhep.2001.22002. [DOI] [PubMed] [Google Scholar]
- 52.Jaeschke H, Bautista AP, Spolarics Z, Spitzer JJ. Superoxide generation by Kupffer cells and priming of neutrophils during reperfusion after hepatic ischemia. Free Radic Res Commun. 1991;15:277–284. doi: 10.3109/10715769109105223. [DOI] [PubMed] [Google Scholar]
- 53.Jaeschke H, Bautista AP, Spolarics Z, Spitzer JJ. Superoxide generation by neutrophils and Kupffer cells during in vivo reperfusion after hepatic ischemia in rats. J Leukoc Biol. 1992;52:377–382. doi: 10.1002/jlb.52.4.377. [DOI] [PubMed] [Google Scholar]
- 54.Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239–252. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 55.Chan ED, Winston BW, Jarpe MB, Wynes MW, Riches DW. Preferential activation of the p46 isoform of JNK/SAPK in mouse macrophages by TNF alpha. Proc Natl Acad Sci U S A. 1997;94:13169–13174. doi: 10.1073/pnas.94.24.13169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Uehara T, Bennett B, Sakata ST, Satoh Y, Bilter GK, Westwick JK, Brenner DA. JNK mediates hepatic ischemia reperfusion injury. J Hepatol. 2005;42:850–859. doi: 10.1016/j.jhep.2005.01.030. [DOI] [PubMed] [Google Scholar]
- 57.Schwabe RF, Brenner DA. Mechanisms of Liver Injury. I. TNF-alpha-induced liver injury: role of IKK, JNK, and ROS pathways. Am J Physiol Gastrointest Liver Physiol. 2006;290:G583–589. doi: 10.1152/ajpgi.00422.2005. [DOI] [PubMed] [Google Scholar]
- 58.Czaja MJ, Liu H, Wang Y. Oxidant-induced hepatocyte injury from menadione is regulated by ERK and AP-1 signaling. Hepatology. 2003;37:1405–1413. doi: 10.1053/jhep.2003.50233. [DOI] [PubMed] [Google Scholar]
- 59.Czaja MJ. Cell signaling in oxidative stress-induced liver injury. Semin Liver Dis. 2007;27:378–389. doi: 10.1055/s-2007-991514. [DOI] [PubMed] [Google Scholar]
- 60.Singh R, Czaja MJ. Regulation of hepatocyte apoptosis by oxidative stress. J Gastroenterol Hepatol. 2007;22 (Suppl 1):S45–48. doi: 10.1111/j.1440-1746.2006.04646.x. [DOI] [PubMed] [Google Scholar]
- 61.Tsung A, Hoffman RA, Izuishi K, Critchlow ND, Nakao A, Chan MH, Lotze MT, et al. Hepatic ischemia/reperfusion injury involves functional TLR4 signaling in nonparenchymal cells. J Immunol. 2005;175:7661–7668. doi: 10.4049/jimmunol.175.11.7661. [DOI] [PubMed] [Google Scholar]
- 62.Zhai Y, Shen XD, O’Connell R, Gao F, Lassman C, Busuttil RW, Cheng G, et al. Cutting edge: TLR4 activation mediates liver ischemia/reperfusion inflammatory response via IFN regulatory factor 3-dependent MyD88-independent pathway. J Immunol. 2004;173:7115–7119. doi: 10.4049/jimmunol.173.12.7115. [DOI] [PubMed] [Google Scholar]
- 63.Barone S, Okaya T, Rudich S, Petrovic S, Tenrani K, Wang Z, Zahedi K, et al. Distinct and sequential upregulation of genes regulating cell growth and cell cycle progression during hepatic ischemia-reperfusion injury. Am J Physiol Cell Physiol. 2005;289:C826–835. doi: 10.1152/ajpcell.00629.2004. [DOI] [PubMed] [Google Scholar]
- 64.Fu W, Zhang Y, Zhang J, Chen WF. Cloning and characterization of mouse homolog of the CXC chemokine receptor CXCR1. Cytokine. 2005;31:9–17. doi: 10.1016/j.cyto.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 65.Fan X, Patera AC, Pong-Kennedy A, Deno G, Gonsiorek W, Manfra DJ, Vassileva G, et al. Murine CXCR1 is a functional receptor for GCP-2/CXCL6 and interleukin-8/CXCL8. J Biol Chem. 2007;282:11658–11666. doi: 10.1074/jbc.M607705200. [DOI] [PubMed] [Google Scholar]
- 66.Bone-Larson CL, Hogaboam CM, Evanhoff H, Strieter RM, Kunkel SL. IFN-gamma-inducible protein-10 (CXCL10) is hepatoprotective during acute liver injury through the induction of CXCR2 on hepatocytes. J Immunol. 2001;167:7077–7083. doi: 10.4049/jimmunol.167.12.7077. [DOI] [PubMed] [Google Scholar]
- 67.Wu D, LaRosa GJ, Simon MI. G protein-coupled signal transduction pathways for interleukin-8. Science. 1993;261:101–103. doi: 10.1126/science.8316840. [DOI] [PubMed] [Google Scholar]
- 68.Damaj BB, McColl SR, Mahana W, Crouch MF, Naccache PH. Physical association of Gi2alpha with interleukin-8 receptors. J Biol Chem. 1996;271:12783–12789. doi: 10.1074/jbc.271.22.12783. [DOI] [PubMed] [Google Scholar]
- 69.Schraufstatter IU, Chung J, Burger M. IL-8 activates endothelial cell CXCR1 and CXCR2 through Rho and Rac signaling pathways. Am J Physiol Lung Cell Mol Physiol. 2001;280:L1094–1103. doi: 10.1152/ajplung.2001.280.6.L1094. [DOI] [PubMed] [Google Scholar]
- 70.Nauseef WM, Volpp BD, McCormick S, Leidal KG, Clark RA. Assembly of the neutrophil respiratory burst oxidase. Protein kinase C promotes cytoskeletal and membrane association of cytosolic oxidase components. J Biol Chem. 1991;266:5911–5917. [PubMed] [Google Scholar]
- 71.Thelen M, Didichenko SA. G-protein coupled receptor-mediated activation of PI 3-kinase in neutrophils. Ann N Y Acad Sci. 1997;832:368–382. doi: 10.1111/j.1749-6632.1997.tb46265.x. [DOI] [PubMed] [Google Scholar]
- 72.Knall C, Young S, Nick JA, Buhl AM, Worthen GS, Johnson GL. Interleukin-8 regulation of the Ras/Raf/mitogen-activated protein kinase pathway in human neutrophils. J Biol Chem. 1996;271:2832–2838. doi: 10.1074/jbc.271.5.2832. [DOI] [PubMed] [Google Scholar]
- 73.Takami M, Terry V, Petruzzelli L. Signaling pathways involved in IL-8-dependent activation of adhesion through Mac-1. J Immunol. 2002;168:4559–4566. doi: 10.4049/jimmunol.168.9.4559. [DOI] [PubMed] [Google Scholar]
- 74.Jones SA, Wolf M, Qin S, Mackay CR, Baggiolini M. Different functions for the interleukin 8 receptors (IL-8R) of human neutrophil leukocytes: NADPH oxidase and phospholipase D are activated through IL-8R1 but not IL-8R2. Proc Natl Acad Sci U S A. 1996;93:6682–6686. doi: 10.1073/pnas.93.13.6682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chakrabarti S, Patel KD. Regulation of matrix metalloproteinase-9 release from IL-8-stimulated human neutrophils. J Leukoc Biol. 2005;78:279–288. doi: 10.1189/jlb.1004612. [DOI] [PubMed] [Google Scholar]
- 76.Chuntharapai A, Kim KJ. Regulation of the expression of IL-8 receptor A/B by IL-8: possible functions of each receptor. J Immunol. 1995;155:2587–2594. [PubMed] [Google Scholar]
- 77.Rose JJ, Foley JF, Murphy PM, Venkatesan S. On the mechanism and significance of ligand-induced internalization of human neutrophil chemokine receptors CXCR1 and CXCR2. J Biol Chem. 2004;279:24372–24386. doi: 10.1074/jbc.M401364200. [DOI] [PubMed] [Google Scholar]
- 78.Snyderman R, Pike MC. Chemoattractant receptors on phagocytic cells. Annu Rev Immunol. 1984;2:257–281. doi: 10.1146/annurev.iy.02.040184.001353. [DOI] [PubMed] [Google Scholar]
- 79.Richardson RM, Pridgen BC, Haribabu B, Ali H, Snyderman R. Differential cross-regulation of the human chemokine receptors CXCR1 and CXCR2. Evidence for time-dependent signal generation. J Biol Chem. 1998;273:23830–23836. doi: 10.1074/jbc.273.37.23830. [DOI] [PubMed] [Google Scholar]
- 80.Melien O, Christoffersen T, Sioud M. Evidence for the involvement of Gi2 in activation of extracellular signal-regulated kinases in hepatocytes. BMC Cell Biol. 2001;2:13. doi: 10.1186/1471-2121-2-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Knall C, Worthen GS, Johnson GL. Interleukin 8-stimulated phosphatidylinositol-3-kinase activity regulates the migration of human neutrophils independent of extracellular signal-regulated kinase and p38 mitogen-activated protein kinases. Proc Natl Acad Sci U S A. 1997;94:3052–3057. doi: 10.1073/pnas.94.7.3052. [DOI] [PMC free article] [PubMed] [Google Scholar]