

Abstract
Please cite this paper as: Easterbrook et al. (2011) Obese mice have increased morbidity and mortality compared to non‐obese mice during infection with the 2009 pandemic H1N1 influenza virus. Influenza and Other Respiratory Viruses 5(6), 418–425.
Background Obesity has been identified as an independent risk factor for severe or fatal infection with 2009 pandemic H1N1 influenza (2009 pH1N1), but was not previously recognized for previous pandemic or seasonal influenza infections.
Objectives Our aim was to evaluate the role of obesity as an independent risk factor for severity of infection with 2009 pH1N1, seasonal H1N1, or a pathogenic H1N1 influenza virus.
Methods Diet‐induced obese (DIO) and their non‐obese, age‐matched control counterparts were inoculated with a 2009 pH1N1, A/California/04/2009 (CA/09), current seasonal H1N1, A/NY/312/2001 (NY312), or highly pathogenic 1918‐like H1N1, A/Iowa/Swine/1931 (Sw31), virus.
Results Following inoculation with CA/09, DIO mice had higher mortality (80%) than control mice (0%) and lost more weight during infection. No effect of obesity on morbidity and mortality was observed during NY312 or Sw31 infection. Influenza antigen distribution in the alveolar regions of the lungs was more pronounced in DIO than control mice during CA/09 infection at 3 days post‐inoculation (dpi), despite similar virus titers. During CA/09 infection, localized interferon‐β and proinflammatory cytokine protein responses in the lungs were significantly lower in DIO than control mice. Conversely, serum cytokine concentrations were elevated in DIO, but not control mice following infection with CA/09. The effect of obesity on differential immune responses was abrogated during NY312 or Sw31 infection.
Conclusions Together, these data support epidemiologic reports that obesity may be a risk factor for severe 2009 pandemic H1N1 influenza infection, but the role of obesity in seasonal or highly virulent pandemic influenza infection remains unclear.
Keywords: IFN‐β, influenza, leptin, obesity, testosterone
Introduction
The prevalence of high calorie diets and increasingly sedentary lifestyles has given rise to increased rates of obesity and serious public health concerns worldwide. More than one‐third of the adult US population is obese, as defined by a body mass index (BMI) greater than 30. 1 Obese individuals are predisposed to developing serious medical conditions that increase their morbidity and mortality, including sleep apnea, cardiovascular disease, diabetes, and renal disease. 2 , 3 Many of these obesity‐associated co‐morbidities are known risk factors for severe and complicated influenza infections, and obesity was independently identified as a risk factor for severe or fatal 2009 pandemic H1N1 (2009 pH1N1) infection. 1 , 3 , 4 , 5 , 6 The minimal existing data are inconsistent, but from what does exist, a correlation between obesity and severity of disease during infection with seasonal influenza in humans has not been fully established. 7 , 8 , 10
Obesity has been linked to impaired immune function in both humans and rodents, whether during infection or in response to vaccination, but the relationship between the two still remains controversial. 9 , 10 , 11 , 12 , 13 In response to influenza infection, the expression of antiviral and proinflammatory cytokine mRNA is elevated in the lungs of both obese and control mice during infection with the mouse‐adapted virus strain, A/Puerto Rico/8/34/H1N1 (PR8), at 3–7 days post‐inoculation (dpi), but elevated immune responses are consistently more pronounced and developed earlier in control mice. 12 , 14 Furthermore, in obese mice, localized pulmonary macrophage, DC, and NK cell responses are reduced during infection with PR8, which may be an effect of the differential cytokine milieu. 9 , 10 , 12 The prevailing hypothesis is that prolonged overproduction of leptin by an excessive number of adipocytes in obese humans or animals causes leptin resistance. When leptin can no longer activate various immune pathways, the ability to respond to an infection may be compromised. 11 , 15
The diet‐induced obese (DIO) mouse is a more relevant model of typical human obesity than is genetically induced obesity. Male DIO C57BL/6 mice fed a high fat diet mimic polygenic human obesity, developing visceral adiposity, leptin resistance, and hyperleptinemia, as well as mild glucose intolerance and mild hyperglycemia. 12 , 16 , 17 , 18 , 19 The effects of obesity on influenza virus infection have only been evaluated in DIO mice in the past using the highly pathogenic, mouse‐adapted PR8 strain, which is the most common laboratory strain, but not the most biologically relevant strain for studying current human influenza infection. 9 , 10 , 12 , 14 DIO mice infected with a sub‐lethal dose of the mouse‐adapted PR8 have higher mortality than their non‐obese counterparts despite similar viral titers in the lungs. 12
Whether the effect of obesity on outcome of infection or immune response varies among infection with a high, moderate, or low pathogenicity influenza viruses has not been explored. Specifically, whether obesity has a differential effect on infection with current human pandemic, seasonal, or previous pandemic strains has been suggested in epidemiologic studies, but has yet to be evaluated in animal models. In this study, we compared the outcome of infection and the associated immune and hormone responses in DIO and non‐obese control mice infected with the 2009 pandemic H1N1, a recent seasonal H1N1, and a close relative to the 1918 pandemic virus. These data have important public health implications in improving our ability to identify susceptible populations during upcoming influenza seasons and future pandemics.
Materials and methods
Viruses
A/California/04/2009/H1N1 (CA/09) originates from an early patient sample during the 2009 novel H1N1 pandemic and was prepared using a standard reverse genetics system. 20 A/NY/312/2001/H1N1 (NY312) was obtained from the New York State Department of Health Wadsworth Center–Griffin Laboratory in Slingerlands, NY. A/Iowa/Swine/1931/H1N1 (Sw31) was obtained from Jack Bennick in the Laboratory of Viral Diseases, National Institute of Allergy and Infectious Diseases, National Institutes of Health. Sw31 originates from one of the first “classical” swine H1N1 virus isolates and can be regarded as a surrogate for 1918, as both are lethal at low doses in mice and cause similar pathology in mice and ferrets. 21 All viruses were passaged twice in MDCK cells in the presence of 1 μg/ml TPCK‐Trypsin (Sigma‐Aldrich, St Louis, MO, USA). Viral titers were determined by plaque assay in MDCK cells and expressed as plaque‐forming units per milliliter (pfu/ml) as previously described.
Mouse experiments
Groups of 20‐week‐old male C57/BL6 DIO or control mice (Jackson Labs, Bar Harbor, ME, USA) were housed 5/cage and acclimated for 2 weeks after arrival. Diet‐induced obese mice were fed a special diet with 60% kcal fat, and control mice were fed a diet with 10% kcal fat since the age of 6 weeks (Research Diets Inc, New Brunswick, NJ, USA). Mice were lightly anaesthetized with isofluorane supplemented with O2 (1·5 l/min) and inoculated intranasally with 2·5 × 105 pfu per mouse of CA/09, NY312, or Sw31 in 50 μl DMEM or vehicle alone. The dose was empirically determined based on previous studies, 20 , 21 and the experimental design was to model a range of pathogenicity in control mice. Survival and body weight were monitored for 14 days, and mice were humanely euthanized if more than 25% body weight was lost. Lungs were collected for viral titration and protein evaluation (n = 3 per group) at 1 and 3 dpi and pathologic examination (n = 2 per group) at 3 dpi. Blood was collected by cardiac puncture after euthanasia at 3 dpi, and serum was used for subsequent analyses. All experimental work was performed in an ABSL2 laboratory at the NIH, following approval of the animal safety protocol by the NIH Animal Care and Use Committee. This experiment was performed once.
Cytokine protein and influenza virus titers
Lung tissue was frozen at −80°C and was homogenized into suspension (10% w/v) in sterile L15 media with 1% anti‐microbial and anti‐fungal (Invitrogen, Carlsbad, CA, USA), and the supernatant was collected after centrifugation at 1500 g for 10 min for subsequent analysis. Cytokine protein was measured using the Bio‐Plex Pro mouse cytokine 8‐plex assay (Bio‐Rad, Hercules, CA, USA) and an ELISA kit for mouse IFN‐β (PBL Biomedical Laboratories, Piscataway, NJ, USA), according to the manufacturer’s protocol. Protein concentrations are expressed as fold change relative to protein concentrations in uninfected (mock‐infected) mice. Virus titers in the lungs were measured by plaque assay. 20
Hormone concentrations
Serum was diluted 1:10 in assay buffer to measure testosterone and leptin by enzyme immunoassay (EIA) according to the manufacturer’s protocol (Cayman Chemicals, Ann Arbor, MI, USA). Circulating concentrations of corticosterone typically are measured in serum, but are susceptible to immediate changes in response to stressors (e.g. animal handling), so fecal concentrations were used to more accurately compare corticosterone concentrations between DIO and control mice during influenza infection. 22 Fecal samples were homogenized in methanol at a ratio of 10 mg per 100 μl methanol to extract corticosterone. Samples were further diluted 1:100 in assay buffer, and corticosterone was measured by EIA (Cayman Chemicals).
Histopathological and immunohistochemical analyses
Following 24‐h fixation in 10% formaldehyde, inflated lung samples were embedded in paraffin, cut into 5‐μm sections, and mounted on positively charged slides (American HistoLabs, Gaithersburg, MD, USA). Influenza virus antigen distribution was evaluated by immunohistochemistry using goat polyclonal antibody to Influenza A HA protein (Abcam, Cambridge, MA, USA) and the manufacturer’s protocol, as described previously. 21 A single pathologist reviewed the histopathology using H&E‐stained slides and immunohistochemistry in a blinded fashion.
Statistical analyses
Differences in weight loss between DIO and control mice during infection were evaluated by manova. Specific comparisons of DIO and control mice for each virus were done using the student’s t‐test and within DIO or control mice by time point by anova (Graph Pad Prism, La Jolla, CA, USA). Mean differences were considered statistically significant if P < 0·05.
Results
Survival and weight loss
Diet‐induced obese mice that were infected with CA/09 lost more weight than the control mice and only 20% (1/5) survived infection, whereas 100% (5/5) of the control mice survived (Figure 1A,B). Both DIO and control mice did not lose any significant amount of weight during infection with NY312 and no deaths occurred. All mice that were infected with Sw31 lost weight at a similar rate and demonstrated 100% mortality (Figure 1A,B).
Figure 1.
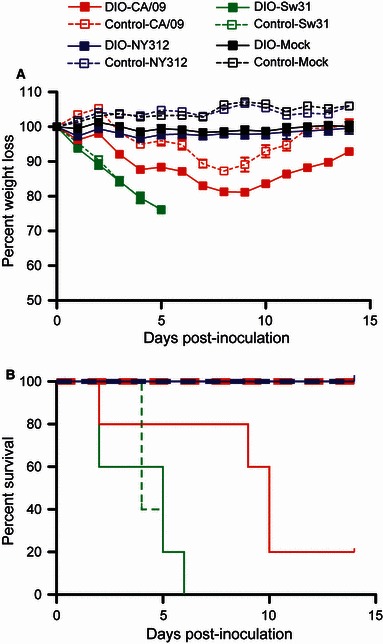
Diet‐induced obese mice had a higher mortality rate and lost more weight than non‐obese control mice during infection with 2009 pandemic H1N1 (CA/09). Mice were inoculated with 2·5 × 105 pfu per mouse CA/09, NY312, Sw31, or vehicle alone (mock) and were monitored daily for weight loss (A) and survival (B) for 14 days.
Lung pathology and virus antigen distribution
Both control (Figure 2A,B) and DIO mice (Figure 2C–F) infected with CA/09 at 3 dpi had multifocal bronchiolitis and mild alveolitis with viral antigen localized in the bronchiolar epithelium, alveolar epithelial cells, and alveolar macrophages. Increased foci of alveolitis and viral antigen were observed in the lungs of DIO compared to control mice (Figure 2C–F). Little pulmonary pathology was observed in mice infected with NY312, but antigen was found in bronchioles to a greater extent in the lungs of DIO than non‐obese control mice (Figure S1E–H). The lungs of both DIO and control mice displayed similar necrotizing bronchiolitis, moderate multifocal alveolitis, alveolar edema, hemorrhage, and extensive virus antigen distribution throughout the lungs 3 days after inoculation with Sw31 (Figure S1A–D).
Figure 2.
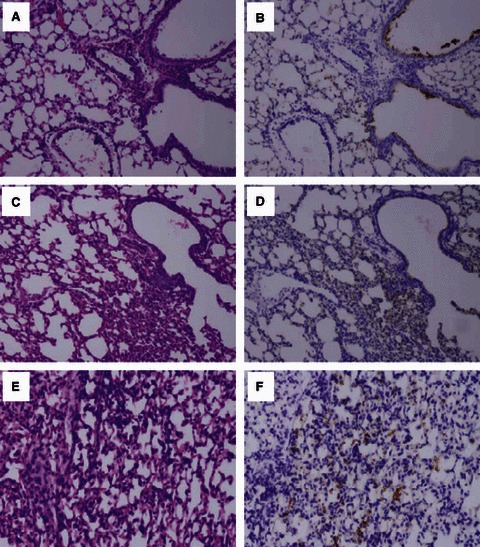
Pathology and virus antigen distribution was more pronounced in the alveolar regions of lungs in diet‐induced obese (DIO) than control mice infected with CA/09. Photomicrographs of hematoxylin and eosin–stained tissue sections and immunohistochemically stained sections to detect influenza viral antigen in the lungs of control (A,B) or DIO (C–F) mice infected with CA/09.
Lung virus titers
Virus titers in the lungs did not significantly differ between DIO and control mice for any of the viruses. The mean titers (±SEM) for CA/09 were below limits of detection at 1 dpi and 8·21 ± 3·05 × 103 pfu/g lung in DIO mice and 4·09 ± 1·14 × 103 pfu/g lung in control mice at 3 dpi. Virus titers during NY312 infection in the lungs of both DIO and control mice were below the limit of detection at 1 and 3 dpi. Virus titers during Sw31 infection were 7·80 ± 3·10 × 105 pfu/g lung in DIO mice and 4·20 ± 1·50 × 105 pfu/g lung in control mice at 1 dpi and 7·43 ± 0·72 × 104 pfu/g lung in DIO mice and 11·18 ± 1·03 × 104 pfu/g lung in control mice during Sw31 infection at 3 dpi.
Cytokine protein in the lungs
All cytokine and hormone data in influenza‐infected mice are presented as fold change from baseline concentrations in respective uninfected DIO or control mice. Control mice had elevated production of IFN‐β at 1 dpi and even more so at 3 dpi during CA/09 infection (P < 0·05), while DIO mice did not mount an appreciable IFN‐β response during CA/09 infection (Figure 3A). Diet‐induced obese mice that were inoculated with NY312 had elevated IFN‐β protein production at 1 dpi that was undetectable by 3 dpi, while control mice had only a minor elevation of IFN‐β protein during infection with NY312 (P < 0·05), but these differences were not significantly different between obese and control mice (Figure 3A). Both DIO and control mice infected with Sw31 mounted similar and strong IFN‐β responses 1 dpi that while still elevated were waning by 3 dpi (Figure 3A).
Figure 3.
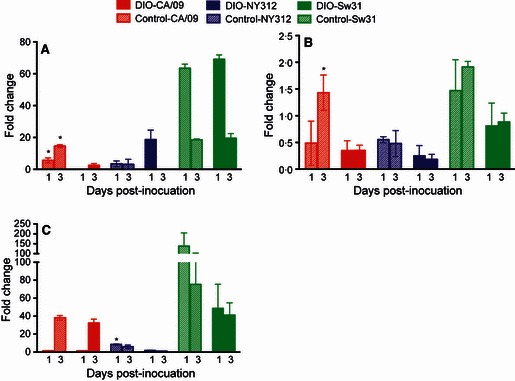
Diet‐induced obese (DIO) mice had impaired production of IFN‐β during infection with CA/09. At 1 and 3 dpi during infection with CA/09, NY312, Sw31, or vehicle alone, the amount of IFN‐β (A), TNF‐α (B), and IL‐5 (C) protein was measured in the lungs. The data are presented as fold change in DIO and control mice during infection from the concentrations in uninfected DIO or control mice, respectively. *Significantly different between DIO and control, P < 0·05.
Control mice had about a twofold higher production of TNF‐α protein (P < 0·05) in the lungs during CA/09 infection at 3 dpi than DIO mice (Figure 3B). TNF‐α protein tended to be reduced in the lungs of DIO mice during infection with CA/09 and NY312 (P = 0·07 for both; Figure 3B). The amount of IL‐5 protein in the lungs of DIO and control mice was similarly elevated 3 dpi during CA/09 and Sw31 infection, but was elevated only in control mice infected with NY312 (P < 0·05; Figure 3C). Diet‐induced obese and control mice had no detectable IFN‐γ in the lungs during infection or any difference between DIO and control mice in the production of IL‐2, IL‐10, or IL‐4 protein in the lungs during infection with CA/09, NY312, or Sw31.
Circulating cytokine concentrations
Serum concentrations of IL‐1β, TNF‐α, IFN‐γ, IL‐5, and IL‐10 were elevated from baseline concentrations in DIO, but not control mice during infection with CA/09, NY312, or Sw31 at 3 dpi (Figure 4A–C and data not shown). Circulating cytokine concentrations, however, were not significantly different between DIO and control mice infected with CA/09, NY312, or Sw31 at 3 dpi. Serum concentrations of IL‐2 and IL‐4 were below limits of detection.
Figure 4.
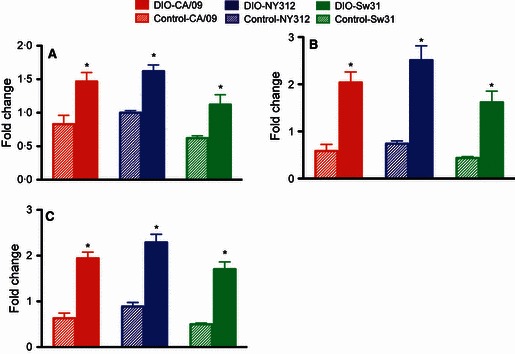
Circulating concentrations of proinflammatory cytokines were elevated in diet‐induced obese (DIO) mice during influenza infection. Three days following infection with CA/09, NY312, Sw31, or vehicle alone, the amount of TNF‐α (A), IL‐1β (B), and IFN‐γ (C) protein was measured in serum. The data are presented as fold change in influenza‐infected DIO and control mice from the concentrations in uninfected DIO or control mice, respectively. *Fold change is significantly different between DIO and control, P < 0·05.
Circulating hormone concentrations
Serum leptin was about five times higher at baseline in uninfected DIO mice as compared with uninfected control mice (1146 ± 360 versus 191 ± 31 pg/ml; P < 0·05). Leptin was elevated in control, but not DIO mice during infection with CA/09 at 3 dpi (P < 0·05; Figure 5A); concentrations in control and DIO mice, however, were similar (DIO: 457 ± 283 pg/ml and control: 508 ± 166 pg/ml). Leptin concentrations were not altered in control or DIO mice during infection with NY312 or Sw31 (Figure 5A).
Figure 5.
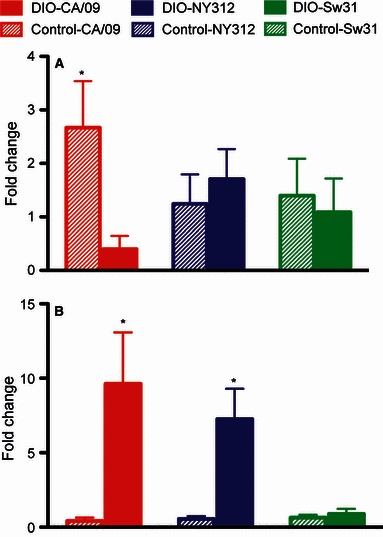
Leptin is elevated in non‐obese control mice and testosterone is elevated in diet‐induced obese (DIO) mice during infection with CA/09. Leptin (A) and testosterone (B) were measured by EIA in the serum of DIO and control mice infected with CA/09, NY312, Sw31, or vehicle alone at 3 dpi. Hormone concentrations are presented as fold change in DIO and control mice from the concentrations in their respective uninfected DIO or control mice. *Indicates that DIO and control differ significantly, P < 0·05.
Uninfected DIO and control mice had similar baseline serum concentrations of testosterone. Circulating testosterone was elevated at 3 dpi in DIO mice infected with CA/09 or NY312 (P < 0·05), but not Sw31 (Figure 5B). Testosterone concentrations in control mice were not altered by CA/09, NY312, or Sw31 infection (Figure 4B).
Baseline corticosterone concentrations were lower in uninfected DIO than control mice (DIO: 395 ± 77 pg/mg feces and control: 1269 ± 221 pg/mg feces; P < 0·05). Corticosterone concentrations were elevated to a greater extent in DIO mice 3 days after infection with CA/09 (threefold) and Sw31 (11‐fold) than control mice (no elevation and threefold during CA/09 and Sw31, respectively); concentrations of corticosterone, however, were similar between control and DIO mice during infection. There was no effect on corticosterone concentrations in DIO or control mice during NY312 infection.
Discussion
After infection with the 2009 pandemic H1N1 strain CA/09, obese mice had greater morbidity and much higher mortality than non‐obese mice. The significantly greater weight loss and more extensive pulmonary pathology observed in these mice suggest that obesity is playing a role in the pathogenesis of this H1N1 virus in these animals. This relationship between obesity and outcome of infection was much less pronounced during infection with a seasonal H1N1 (NY312) that has been described to cause minimal disease in mice, 20 as well as following a high‐dose infection with the highly virulent 1918‐like Sw31 21 virus.
Despite the difference in morbidity and mortality observed after infection with CA/09, DIO mice had similar titers of infectious virus isolated from the lungs. However, a difference in the distribution of virus antigen was noted, with more viral antigen in alveolar cells of the lungs during CA/09 infection in obese than non‐obese control mice. This suggests that increased virus spread to the alveolar epithelium in obese mice could be contributing to increased morbidity and mortality following infection with 2009 pandemic H1N1. Future studies of animals at later time points closer to time of death (i.e. 6–10 dpi) will be needed to further investigate the role of viral spread by evaluating clearance rates of virus in DIO and control mice.
A difference in the distribution of virus antigen was also observed in obese mice infected with a mild seasonal influenza (NY312), with more virus antigen observed in the tracheobronchial tree in the lungs of DIO mice. Obesity may play a role in enhanced spread of seasonal H1N1, but given the limited ability of this virus to replicate in the lungs of these animals, this may have little to no effect on outcome after infection. The effect of obesity on mortality and viral distribution was not observed in the animals after infection with the highly pathogenic Sw31, as all animals rapidly succumbed to infection and displayed severe pathology accompanied by a high virus load distributed throughout the lungs. These data suggest that when infected with such a high dose of a highly virulent virus, virulence rather than host factors, such as obesity, may play a more significant role in the outcome of infection.
The mechanisms mediating the differences in morbidity, mortality, and virus distribution in obese mice after infection with CA/09 are likely multifactorial, and it is likely that differences in localized and peripheral immune responses may play a role. DIO mice had reduced or abrogated IFN‐β, TNF‐α, and IL‐1β production in the lungs during CA/09 infection as compared to control mice, representing weakened localized innate antiviral and proinflammatory immune responses. Conversely, circulating concentrations of proinflammatory cytokines were elevated in DIO, but not control, mice during infection with CA/09 at 3 dpi. Thus, blunted localized responses may contribute to a reduced ability to limit viral replication, while elevated peripheral proinflammatory responses may contribute to enhanced symptoms of disease during CA/09 infection. These data are reasonably consistent with previous studies in which 2–8 times more IFN‐β, TNF‐α, IL‐1β, and IL‐6 mRNA were observed in the lungs of non‐obese control mice during PR8 influenza infection than DIO mice. 9 , 12 One major difference, however, is that significant type I IFN and proinflammatory mRNA expression responses are mounted in the lungs of DIO mice during infection with PR8 (approximately 40–300‐fold from baseline concentrations in uninfected DIO mice) 9 , 12 that were not observed here in the lungs of DIO mice infected with CA/09. Interestingly, DIO mice were able to mount a short‐lived type I IFN response in the lungs against the more mild seasonal H1N1 strain, NY312, and a comparable strong type I IFN and proinflammatory response in the lungs as compared to control mice during infection with the pathogenic H1N1 strain, Sw31. These data suggest that the localized but not peripheral immune responses during infection in DIO and control mice may differ between virus strains, virulence of the virus, or severity of infection. How CA/09 is signaling IFN‐β production differently than some other strains of influenza and the possible role of the different influenza NS‐1 type I IFN antagonists will require further investigation.
Another possible mediator of pathogenesis in obesity may be peripheral leptin, as elevated leptin concentrations observed in obese humans and rodents consistently lead to leptin resistance. 11 , 12 , 23 In addition to metabolic pathways, leptin also acts through the Jak/STAT signaling pathway, as do type I IFNs, to activate various immune responses. 15 The observed 2·5‐fold increase in leptin during CA/09 infection in non‐obese mice that can respond to leptin signaling may activate immune responses necessary to control infection. These data are consistent with previous studies that have demonstrated that baseline serum concentrations of leptin are higher in DIO than control mice, but after infection with PR8, DIO mice have reduced and control mice have about 2·5‐fold increased serum leptin. 12 The lack of a leptin response observed in DIO mice may have contributed to an insufficient immune response in the lungs after infection with CA/09, but because DIO mice are known to develop leptin resistance, it is not clear what role, if any, leptin is playing during influenza infection in DIO mice.
Obesity in humans has also been associated with reduced circulating testosterone, likely caused by the production of aromatase by adipocytes to convert testosterone to estrogens. 24 The significance of this during infection is unclear, but the administration of testosterone in vitro or in vivo to mice has been observed to cause an increase in influenza virus proliferation and reduced innate and proinflammatory responses during infection. 25 , 26 , 27 In this study, DIO and control mice did not have significantly different concentrations of circulating testosterone in the absence of infection, but testosterone was elevated in DIO as compared to non‐obese control mice during infection with CA/09 and NY312. How this may impact virus distribution in the lungs and overall outcome is unclear, but it may play a role in modulation of the immune response during infection.
Changes in circulating cortisol levels, although not different between obese and non‐obese people because of increased secretion offsetting elevated production, 28 may be another effect of obesity in mice that may play a role in mediating response to infection. 29 Lower baseline corticosterone levels were observed in uninfected DIO than uninfected control mice. During infection with CA/09, corticosterone was elevated in DIO mice, as well as in DIO and control mice infected with Sw31. These three groups of animals were the only groups to demonstrate mortality as an outcome of infection, suggesting that a corticosterone stress response may play a role during severe disease either as a contributing factor or consequence.
Obese mice suffered a more severe outcome after infection with the 2009 pandemic H1N1 than did non‐obese control mice. The differential effects of obesity observed after infection with a current pandemic H1N1, seasonal H1N1, and highly pathogenic H1N1 demonstrate that this effect may be highly variable and context dependent. Additionally, as pathogenicity varied among these three viruses that were inoculated at the same dose, further studies would be necessary to further tease apart the effects of obesity on the outcome of infection with these three viruses at doses that cause similar pathology in control mice (e.g. lower doses of the highly pathogenic Sw31 and higher doses of the seasonal H1N1, NY312), as the results may differ. Multiple mechanisms likely contribute to the effect of obesity on influenza pathogenesis, including several of the immune mechanisms evaluated here. Future studies should further evaluate the role of other immunological factors, hormonal immune regulation, and non‐immune, physiologic changes associated with hypoxia and stress, which may be affected by obesity and may limit the ability of an obese host to adequately respond to certain influenza infections.
These data correlate with the clinical observations made during the 2009 pandemic and continue to suggest that obesity can play a role in more severe or complicated influenza infections. Although it is unclear if the pandemic H1N1 virus will return or if seasonal influenza strains will dominate in future seasons, 30 public health initiatives should strongly consider that obese individuals are potentially at high risk for severe or complicated influenza infections. Future epidemiologic as well as translational studies should focus on teasing apart the effects of the comorbidities of obesity compared to obesity alone to better understand the role of obesity during influenza infection. Even if obesity itself is not an independent risk factor during most influenza infections, it nevertheless may predispose people to diseases that are known risk factors for influenza and measures should continue to be taken to reduce the prevalence of obesity internationally in an effort to mitigate the effects of influenza as well as many other diseases.
Supporting information
Figure S1. Pathology and virus antigen distribution did not differ during Sw31 infection in DIO and control mice, but NY312 antigen was more abundant in DIO than control mice.
Supporting info item
Acknowledgements
We thank the Comparative Medicine Branch (NIH/NIAID) for their assistance with animal studies. This work was supported by the Intramural Research Program of the NIH and the NIAID.
References
- 1. Flegal KM, Carroll MD, Ogden CL, Curtin LR. Prevalence and trends in obesity among US adults, 1999–2008. JAMA 2010; 303:235–241. [DOI] [PubMed] [Google Scholar]
- 2. Jain S, Kamimoto L, Bramley AM et al. Hospitalized patients with 2009 H1N1 influenza in the United States, April–June 2009. N Engl J Med 2009; 361:1935–1944. [DOI] [PubMed] [Google Scholar]
- 3. Louie JK, Acosta M, Winter K et al. Factors associated with death or hospitalization due to pandemic 2009 influenza A(H1N1) infection in California. JAMA 2009; 302:1896–1902. [DOI] [PubMed] [Google Scholar]
- 4. Gill JR, Sheng ZM, Ely SF, et al. Pulmonary pathologic findings of fatal 2009 pandemic influenza A/H1N1 viral infections. Arch Pathol Lab Med 2010; 134:235–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Morgan OW, Bramley A, Fowlkes A et al. Morbid obesity as a risk factor for hospitalization and death due to 2009 pandemic influenza A(H1N1) disease. PLoS ONE 2010; 5:e9694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vaillant L, La Ruche G, Tarantola A, Barboza P. Epidemiology of fatal cases associated with pandemic H1N1 influenza 2009. Euro Surveill 2009; 14 pii:19309. [DOI] [PubMed] [Google Scholar]
- 7. Fiore AE, Shay DK, Broder K et al. Prevention and control of seasonal influenza with vaccines: recommendations of the Advisory Committee on Immunization Practices (ACIP), 2009. MMWR Recomm Rep 2009; 58:1–52. [PubMed] [Google Scholar]
- 8. Kelly HA, Grant KA, Williams S, Fielding J, Smith D. Epidemiological characteristics of pandemic influenza H1N1 2009 and seasonal influenza infection. Med J Aust 2009; 191:146–149. [DOI] [PubMed] [Google Scholar]
- 9. Smith AG, Sheridan PA, Tseng RJ, Sheridan JF, Beck MA. Selective impairment in dendritic cell function and altered antigen‐specific CD8+ T‐cell responses in diet‐induced obese mice infected with influenza virus. Immunology 2009; 126:268–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Karlsson EA, Sheridan PA, Beck MA. Diet‐induced obesity impairs the T cell memory response to influenza virus infection. J Immunol 2010; 184:3127–3133. [DOI] [PubMed] [Google Scholar]
- 11. Marti A, Marcos A, Martinez JA. Obesity and immune function relationships. Obes Rev 2001; 2:131–140. [DOI] [PubMed] [Google Scholar]
- 12. Smith AG, Sheridan PA, Harp JB, Beck MA. Diet‐induced obese mice have increased mortality and altered immune responses when infected with influenza virus. J Nutr 2007; 137:1236–1243. [DOI] [PubMed] [Google Scholar]
- 13. Weber DJ, Rutala WA, Samsa GP, Bradshaw SE, Lemon SM. Impaired immunogenicity of hepatitis B vaccine in obese persons. N Engl J Med 1986; 314:1393. [DOI] [PubMed] [Google Scholar]
- 14. Buchweitz JP, Harkema JR, Kaminski NE. Time‐dependent airway epithelial and inflammatory cell responses induced by influenza virus A/PR/8/34 in C57BL/6 mice. Toxicol Pathol 2007; 35:424–435. [DOI] [PubMed] [Google Scholar]
- 15. Procaccini C, Lourenco EV, Matarese G, La Cava A. Leptin signaling: a key pathway in immune responses. Curr Signal Transduct Ther 2009; 4:22–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Collins S, Martin TL, Surwit RS, Robidoux J. Genetic vulnerability to diet‐induced obesity in the C57BL/6J mouse: physiological and molecular characteristics. Physiol Behav 2004; 81:243–248. [DOI] [PubMed] [Google Scholar]
- 17. Petro AE, Cotter J, Cooper DA, Peters JC, Surwit SJ, Surwit RS. Fat, carbohydrate, and calories in the development of diabetes and obesity in the C57BL/6J mouse. Metabolism 2004; 53:454–457. [DOI] [PubMed] [Google Scholar]
- 18. Rossmeisl M, Rim JS, Koza RA, Kozak LP. Variation in type 2 diabetes – related traits in mouse strains susceptible to diet‐induced obesity. Diabetes 2003; 52:1958–1966. [DOI] [PubMed] [Google Scholar]
- 19. Surwit RS, Kuhn CM, Cochrane C, McCubbin JA, Feinglos MN. Diet‐induced type II diabetes in C57BL/6J mice. Diabetes 1988; 37:1163–1167. [DOI] [PubMed] [Google Scholar]
- 20. Qi L, Kash JC, Dugan VG et al. Role of sialic acid binding specificity of the 1918 influenza virus hemagglutinin protein in virulence and pathogenesis for mice. J Virol 2009; 83:3754–3761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Memoli MJ, Tumpey TM, Jagger BW et al. An early ‘classical’ swine H1N1 influenza virus shows similar pathogenicity to the 1918 pandemic virus in ferrets and mice. Virology 2009; 393:338–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cavigelli SA, Monfort SL, Whitney TK, Mechref YS, Novotny M, McClintock MK. Frequent serial fecal corticoid measures from rats reflect circadian and ovarian corticosterone rhythms. J Endocrinol 2005; 184:153–163. [DOI] [PubMed] [Google Scholar]
- 23. Ueno N, Asakawa A, Inui A. Blunted metabolic response to fasting in obese mice. Endocrine 2007; 32:192–196. [DOI] [PubMed] [Google Scholar]
- 24. Saad F, Gooren LJ. The role of testosterone in the etiology and treatment of obesity, the metabolic syndrome, and diabetes mellitus type 2. J Obes 2011; pii:471584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kalter SS, Smolin HJ, McElhaney JM, Tepperman J. Endocrines and their relation to influenza virus infection. J Exp Med 1951; 93:529–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kalter SS, Stuart DC, Tepperman J. Alterations in rate of influenza virus proliferation produced by growth hormone and testosterone. Proc Soc Exp Biol Med 1950; 74:605–607. [DOI] [PubMed] [Google Scholar]
- 27. Klein SL, Passaretti C, Anker M, Olukoya P, Pekosz A. The impact of sex, gender and pregnancy on 2009 H1N1 disease. Biol Sex Differ 2010; 1:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Stewart PM, Boulton A, Kumar S, Clark PM, Shackleton CH. Cortisol metabolism in human obesity: impaired cortisone→cortisol conversion in subjects with central adiposity. J Clin Endocrinol Metab 1999; 84:1022–1027. [DOI] [PubMed] [Google Scholar]
- 29. Liu Y, Nakagawa Y, Wang Y et al. Reduction of hepatic glucocorticoid receptor and hexose‐6‐phosphate dehydrogenase expression ameliorates diet‐induced obesity and insulin resistance in mice. J Mol Endocrinol 2008; 41:53–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Morens DM, Taubenberger JK, Fauci AS. The 2009 H1N1 pandemic influenza virus: what next? MBio 2010; 1:e00211–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Pathology and virus antigen distribution did not differ during Sw31 infection in DIO and control mice, but NY312 antigen was more abundant in DIO than control mice.
Supporting info item