

Abstract
Controlling the equilibria between backbone cis- and trans-amides in peptoids, or N-substituted glycine oligomers, constitutes a significant challenge in the construction of discretely folded peptoid structures. Through the analysis of a set of monomeric peptoid model systems, we have developed new and general strategies for controlling peptoid conformation that utilize local noncovalent interactions to regulate backbone amide rotameric equilibria, including n→π*, steric, and hydrogen bonding interactions. The chemical functionalities required to implement these strategies are typically confined to the peptoid side chains, preserve chirality at the side chain N-α-carbon known to engender peptoid structure, and are fully compatible with standard peptoid synthesis techniques. Our examinations of peptoid model systems have also elucidated how solvents affect various side chain-backbone interactions, revealing fundamental aspects of these noncovalent interactions in peptoids that were largely uncharacterized previously. As validation of our monomeric model systems, we extended the scope of this study to include peptoid oligomers and have now demonstrated the importance of local steric and n→π* interactions in dictating the structures of larger, folded peptoids. This new, modular design strategy has guided the construction of peptoids containing 1-naphthylethyl side chains, which we show can be utilized to effectively eliminate trans-amide rotamers from the peptoid backbone, yielding the most conformationally homogeneous class of peptoid structures yet reported in terms of amide rotamerism. Overall, this research has afforded a valuable and expansive set of design tools for the construction of both discretely folded peptoids and structurally-biased peptoid libraries, and should shape our understanding of peptoid folding.
Introduction
The family of oligomers comprising poly-N-substituted glycines, or “peptoids”, was originally conceived to address the early demands of combinatorial chemistry for efficient library synthesis.1, 2 This legacy is manifest in the peptoid submonomer synthesis method, which is capable of delivering sizable and structurally diverse peptoid libraries derived from a continually expanding pool of commercially available primary amines.3 As such, thousands of peptoid monomers are now readily accessible via this protocol, rivaling if not exceeding the number of amino acids available for the construction of analogous peptide libraries. Resistance to proteolysis4 and rapid cellular uptake5 have further rendered peptoids attractive candidates for biological applications. Screening of peptoid libraries, both random and focused, has yielded numerous bioactive compounds, including antimicrobial agents,6 neutralizers of bacterial endotoxins,7 lung surfactants,8 drug delivery vehicles,9 and ligands for pharmaceutically relevant targets such as the HDM2 protein,10 G-protein-coupled receptors,11 and the SH3 domains of signaling proteins.12 Apart from biological applications, screening of peptoids has also produced new DNA drag tags,13 chemical sensors,14 and organocatalysts.15, 16
The decoration of discretely folded oligomeric scaffolds with chemical functionality in a combinatorial fashion has proven an attractive strategy for library focusing.17–19 The design of such libraries requires a thorough understanding of the folding propensities of the chosen oligomeric scaffolds. This view, in part, has motivated enormous efforts to decipher the relationships between sequence and secondary structure in biopolymers such as peptides and proteins. These relationships have already been used to efficiently bias peptide libraries20 and have also greatly benefited the understanding of synthetic foldamers, but compared to peptides and proteins, the folding of peptoids has not been as extensively studied. Indeed, considering the tremendous potential demonstrated by peptoids in the short time since their inception, the intramolecular interactions that direct their folding remain largely unclear, even in comparison to other foldamer systems such as β-peptides.21 A deeper understanding of the peptoid folding process would certainly contribute to the rational design of peptoid structures competent for a wide range of applications, and likely increase the utility of future peptoid libraries.
Early research on peptoid secondary structure revealed the propensity of peptoids to adopt helical structures analogous to the cis-amide polyproline I (PPI) helix.22–24 N-α-chiral side chains were shown to promote the folding of these structures in both solution25 and the solid state,26 despite the lack of main chain chirality and secondary amide hydrogen bond donors crucial to the formation of many α-peptide secondary structures (Figure 1). Although computational studies initially suggested that steric interactions between N-α-chiral aromatic side chains and the peptoid backbone primarily dictated helix formation,27 both intra- and intermolecular aromatic stacking interactions have also been proposed to participate in stabilizing such helices.23, 25, 27 Other classes of peptoid side chains have been designed to introduce dipole-dipole, hydrogen bonding, and electrostatic interactions that stabilize the peptoid helix, although their development typically has not encompassed scrutinizing the individual noncovalent interactions involved.24, 28–30 The majority of these strategies rely on incorporation of N-α-chiral centers in order to impart helix handedness. However, a facile equilibration between the cis- and trans-conformations of the tertiary backbone amides often counters these organizing influences by giving rise to significant populations of both rotamers, leading to disruption and fraying of peptoid helices, especially at the termini.25 The inherent flexibility of peptoids in general can lead to coexisting and potentially inter-converting conformers in solution, making analysis of peptoid structure less than straightforward.25 Moreover, this conformational heterogeneity complicates the de novo design of new peptoid secondary structures.
Figure 1.

Comparison of the peptoid and α-peptide primary structures.
A recent report describing a distinctive threaded loop secondary structure adopted by several peptoid homononamers composed of N-α-chiral amide side chains has provided new insights into how peptoid amide isomerization might be controlled.31 Notably, this loop structure contains both cis- and trans-amides, and is stabilized by hydrogen bonds between the peptoid backbone and the N-terminal ammonium group. These hydrogen bonds are apparently of sufficient strength as to overwhelm the helical propensities resulting from local interactions between the side chains and the backbone. This finding — that a relatively small number of noncovalent interactions can stabilize both cis- and trans-amides within the backbones of large peptoid structures — suggests that strategically positioned noncovalent interactions could be harnessed to rationally control amide isomerism and generate new peptoid structural motifs. In this context, peptoids stand apart from typical peptides and other secondary amide-based foldamers in that their tertiary amide backbones are devoid of hydrogen bond donors, creating a unique opportunity to rationally control the distribution of these donors by selection of appropriate hydrogen bonding side chains. Such engineered hydrogen bonding networks could conceivably act as templates for both inter- and intramolecular structural assembly. Alternatively, the elimination of hydrogen bonding motifs in peptoids would greatly facilitate investigations of other more subtle noncovalent interactions that, while relatively weak individually, might collectively exert significant influences on peptoid structure.
We have undertaken a research program focused on characterizing the roles of hydrogen bonding and other noncovalent interactions in directing peptoid amide isomerism, and thereby, peptoid folding in general. The recent focus of these studies has been on small, peptoid model systems. The principal objective of these studies was to isolate and characterize only the local interactions between peptoid side chains and the backbone, in the hope that this restriction might yield highly modular and easily applicable design criteria for controlling the structures of larger peptoid oligomers. We recently reported the initial findings of these studies, which showed that n→π* interactions can regulate peptoid amide isomerism in monomeric peptoid model systems.32, 33 However, the roles that these interactions might play in determining the folded structures of actual oligomeric peptoids remained unexplored. Here, we present a comprehensive overview of our investigations in this area to date that encompass a much wider range of side chain structures, noncovalent interaction classes, and solvents. We have developed several new and general strategies for controlling peptoid conformation that utilize local noncovalent interactions to regulate backbone amide isomerism. The chemical functionalities required to implement these strategies typically are confined to the peptoid side chains, preserve the chirality of the side chain at the N-α-carbon, and are fully compatible with standard peptoid synthesis techniques. Our examinations of peptoid model systems have also elucidated how various side chain-backbone interactions are affected by solvent, revealing fundamental aspects of these noncovalent interactions in peptoids that were largely uncharacterized previously in the literature. As validation of these monomeric model systems, we extended the scope of the study to include longer peptoid oligomers that demonstrate the importance of such noncovalent interactions within the context of larger, structured peptoids. This modular approach to peptoid design culminated in the installation of 1-napthylethyl side chains into a series of peptoid oligomers, which were found to virtually suppress trans-amide rotamers in the backbone and consequently should prove to be valuable structure-promoting elements in peptoids. Indeed, our preliminary structural characterization suggests that these oligomers represent the most conformationally defined, acyclic cis-peptoids yet reported. Collectively, this work has afforded a valuable new set of approaches for the rational design of both folded peptoids and structurally biased peptoid libraries, and should contribute to the growing understanding of peptoid folding.
Experimental Section
General
All reagents were purchased from commercial sources (Alfa-Aesar, Aldrich, Advanced ChemTech, and Acros) and used without further purification. Solvents were purchased from commercial sources (Aldrich and J. T. Baker) and used as is, with the exception of dichloromethane (CH2Cl2) and n-hexane, which were distilled immediately prior to use. Thin layer chromatography (TLC) was performed on silica gel 60 F254 plates (E-5715-7, EMD). SiliaFlash® P60 silica gel (40–63 μm, Silicycle) was used for manual flash column chromatography.34 High-pressure flash chromatography (HPFC) was performed using prepacked silica gel columns (KP-SIL, Biotage) on a Biotage SP system.
1H NMR (500 MHz), 13C NMR (125 MHz), and 19F NMR (470 MHz) spectra were recorded on either a Varian Unity or an Inova 500 MHz spectrometer in deuterated solvents. 1H NMR (300 MHz), 13C NMR (75 MHz), and 19F NMR (282 MHz) spectra were recorded on either a Bruker AC-300 or a Varian MercuryPlus 300 spectrometer in deuterated solvents. Chemical shifts are reported in parts per million (ppm, δ) using tetramethyl silane (TMS) as a reference (0.0 ppm), unless otherwise noted. Couplings are reported in hertz. Nuclear Overhauser effect spectroscopy (NOESY), gradient double quantum filtered correlation spectroscopy (gDQCOSY), and heteronuclear single quantum coherence adiabatic (HSQCAD) NMR experiments were performed on a Varian Inova 500 or 600 MHz spectrometer, and were referenced to solvent. The data were processed using the Varian VNMR software package (v. 6.1C) and visualized using SPARKY software.35
Electrospray ionization (ESI) mass spectra were obtained on a Waters (Micromass) LCT™ spectrometer equipped with a time-of-flight analyzer. Samples were dissolved in methanol and sprayed with a sample cone voltage of 20. Electron impact (EI) mass spectra were obtained on a Waters (Micromass) AutoSpec™ spectrometer equipped with a magnetic sector. The samples were placed in a glass capillary and inserted directly into the source, and the resulting vapor was bombarded with 70 eV electrons. Perfluorokerosene (PFK) was used for calibration. Matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectra were obtained on a Bruker REFLEX II spectrometer equipped with a 337 nm laser and a reflectron. In positive ion mode, the acceleration voltage was 25 kV.
Reversed-phase high performance liquid chromatography (RP-HPLC) was performed using a Shimadzu system equipped with an SCL-10Avp controller, a LC-10AT pump, an FCV-10ALvp solvent mixer, and an SPD-10MAvp UV/vis diode array detector. A Restek Premier C18 column (5 μm, 4.6 mm × 250 mm) was used for all analytical RP-HPLC work. A Vydac protein & peptide C18 column (10 μm, 22 mm × 250 mm) was used for all preparative RP-HPLC work. Standard RP-HPLC conditions were as follows: flow rates were 1 mL/min for analytical separations and 9 mL/min for preparative separations; mobile phase A = 0.1% trifluoroacetic acid (TFA) in water; mobile phase B = 0.1% TFA in acetonitrile. Purities were determined by integration of peaks with UV detection at 220 nm.
LC-MS data were obtained using a Shimadzu LCMS-2010 equipped with two LC-10ADvp pumps, an SCL-10Avp controller, an SIL-10ADvp autoinjector, an SPD-M10Avp UV/vis diode array detector, and a single quadrupole analyzer (for ESI). A Supelco 15 cm × 2.1 mm C18 wide-pore column was used for all LC-MS work. Standard RP-HPLC conditions for LC-MS were as follows: flow rate = 200 μL/min; mobile phase A = 0.4% formic acid in water; mobile phase B = 0.2% formic acid in acetonitrile.
Attenuated total reflectance (ATR)-IR spectra were recorded with either a Bruker Tensor 27 spectrometer outfitted with a single reflection MIRacle Horizontal ATR by Pike Technologies and a ZnSe crystal with a spectral range of 20,000 to 650 cm−1, or a Bruker Equinox 55 spectrometer outfitted with a single reflection MIRacle Horizontal ATR and a Ge crystal with a spectral range of 5500 to 600 cm−1. Circular dichroism (CD) spectra were obtained on an Aviv 62A DS spectropolarimeter with Aviv CDS software.
Synthesis and characterization of peptoid model systems 1–32
All peptoid model systems were synthesized in solution according to previously reported procedures.32 Peptoid model systems 1–15 and 30 have been characterized previously.32 Full characterization data for peptoid model systems 16–29, 31, and 32 are provided in the Supporting Information.
Synthesis and characterization of peptoid oligomers 33–38
Peptoids 33–38 were synthesized using microwave-assisted solid-phase methods on Rink-amide-derivatized polystyrene resin as previously reported.36, 37 The crude peptoids were purified by preparative RP-HPLC and characterized by MS to confirm their identity (see Table 1). See Supporting Information for HPLC data for peptoids 33–38.
Table 1.
Structures, purities, and calculated and observed masses for peptoid pentamers 33–38.
| peptoid pentamer | monomer sequence (amino to carboxy terminus)a | % purityb | calculated mass | observed massc |
|---|---|---|---|---|
| 33 | Ac-(spe)2-snp-(spe)2-CONH2 | 95 | 909.4 | 932.1 [M+Na+]+ |
| 34 | Ac-(spe)5-CONH2 | 99 | 864.5 | 887.1 [M+Na+]+ |
| 35 | Ac-(sfe-spe)2-spe-CONH2 | 99 | 1044.4 | 1067.1 [M+Na+]+ |
| 36 | Ac-(sfe)5-CONH2 | 99 | 1314.2 | 1337.1 [M+Na+]+ |
| 37 | Ac-(s1npe)2-CONH2 | 99 | 481.2 | 504.1[M+Na+]+ |
| 38 | Ac-(s1npe)3-CONH2 | 98 | 692.3 | 715.4 [M+Na+]+ |
See text for definitions of peptoid monomer abbreviations.
Determined by integration of the HPLC trace with UV detection at 220 nm.
Mass spectrometry data were acquired using either ESI or MALDI-TOF techniques.
Determination of cis/trans rotamer ratios (Kcis/trans) for the peptoid model systems
Each model peptoid was dissolved in the desired solvent to give a 15 mM solution. 1H NMR spectra were acquired using 16 scans and a relaxation delay of 25 s. 1D-NOESY spectra were acquired to verify previously reported guidelines for distinguishing cis- and trans-amide rotamers.31, 33 In cases where these guidelines appeared to be inapplicable, additional 1D-NOESY spectra were acquired to distinguish cis- and trans-amide rotamers. Cis/trans ratios were calculated by averaging the integration ratios for at least two sets of rotamer-related peaks. The cis/trans ratios of the hydrophobic model peptoid 4 were statistically identical at 1, 5, 15, 60, and 180 mM concentrations in the polar solvent CD3CN, and the cis/trans ratios of the more hydrophilic model peptoid 27 at 1, 5, 10, 15, 25, 50, and 75 mM were statistically identical in the nonpolar solvent CDCl3, suggesting that solvophobic aggregation is insignificant under the conditions examined in this study.
NMR analyses for peptoid model systems 27 and 30
Peptoids 27 and 30 were dissolved in CD3CN to give 15 mM solutions and analyzed by NOESY as detailed below. The experiments were performed on a Varian Inova 600 MHz spectrometer using a 5 mm hcn probe.
NOESY experiments on 27 in CD3CN were performed at 24 °C using the following parameter values: mix time = 700 ms; spectral width = 8000 Hz; number of transients (nt) = 16; number of increments (ni) = 64. The number of points (np) was 2048, and 192 points were obtained by linear prediction in the f1 dimension. Square cosine window functions were applied in both dimensions. The spectra were zero-filled to generate f1 × f2 matrices of 4096 × 4096 points. Parameters for NOESY experiments on 30 in CD3CN were reported previously. 32
NMR analyses for peptoid oligomers 33–38
Peptoids 33–38 were dissolved in CD3CN to give 15 mM solutions and analyzed by gDQCOSY and HSQCAD NMR experiments as detailed below. The experiments were performed on a Varian Inova 600 MHz spectrometer using a 5 mm hcn probe. The gDQCOSY spectra were used for assignment purposes, and the HSQCAD NMR spectra were subsequently integrated to give the Kcis/trans values in Table 6. Assignments of methyne cis and trans peaks were based on previously observed trends in chemical shift,37 and verified by NOESY NMR, unless otherwise noted.
Table 6.
Polypeptoid structures 33–38 and Kcis/trans values for each residue type, as determined by 1H-COSY and HSQCAD NMR in CD3CN at 24°C.
| spe | snp | sfe | s1npe | overallb | ||
|---|---|---|---|---|---|---|
| entry | structure | Kcis/trans | Kcis/trans | Kcis/trans | Kcis/trans | Kcis/trans |
| 33 | Ac-(spe)2-snp-(spe)2-CONH2 | -- a | -- | 2.1 | ||
| 34 | Ac-(spe)5-CONH2 | 1.5 | 1.5 | |||
| 35 | Ac-(sfe-spe)2-spe-CONH2 | 0.9 | 5.6 | 1.7 | ||
| 36 | Ac-(sfe)5-CONH2 | 2.8 | 2.8 | |||
| 37 | Ac-(s1npe)2-CONH2 | 7.0 | 7.0 | |||
| 38 | Ac-(s1npe)3-CONH2 | 9.7 | 9.7 |
Data not available.
Weighted average of Kcis/trans for all residues.
gDQCOSY NMR experiments were performed for peptoids 33–36 at 24 °C using the following parameter values: Spectral widths were 5323, 5287, 4727, and 6611 Hz, respectively. The numbers of transients (nt) were 8, 16, 16, and 16, respectively. The relaxation delays (d1) were 1.20, 1.12, 1.68, and 2.30, respectively. The numbers of increments (ni) were 300, 300, 300, and 256, respectively. The number of points (np) was 4096, with 1200 points obtained by linear prediction in the f1 dimension. Square cosine window functions were applied in both dimensions. The spectra were zero-filled to generate f1 × f2 matrices of 8192 × 8192 points.
HSQCAD NMR experiments38 were performed for peptoids 33–38 at 24 °C using the following parameter values: Spectral widths were 5323, 5212, 4727, 6611, 5914, and 5914 Hz, respectively, in the 1H dimension. Spectral widths were 28787, 28684, 30234, 36199, 30948, and 25983 Hz, respectively, in the 13C dimension. The relaxation delays (d1) were 1.50, 0.84, 1.68, 2.30, 3.36, and 2.94, respectively. The number of transients (nt) were 8, 8, 8, 8, 2, and 2, respectively. The number of increments (ni) was 256, with 1200 points obtained by linear prediction in the f1 dimension. The number of points was 2048. Square cosine window functions were applied in both dimensions. The spectra were zero-filled to generate f1 × f2 matrices of 8192 × 2048 points.
CD analyses for peptoids 26, 37, and 38
Peptoid stock solutions were prepared by dissolving at least 2 mg of each peptoid in spectroscopic grade acetonitrile. The stock solutions then were diluted with spectroscopic grade acetonitrile to the desired concentration (~60 μM) by mass using a high-precision balance (Mettler-Toledo XS105). CD spectra were obtained in a square quartz cell (path length 0.1 cm) at 25 °C using a scan rate of 100 nm/min, with five averaged scans per spectrum. The spectrum of an acetonitrile blank was subtracted from the raw CD data, and the resulting data were plotted using Kaleidagraph software (v. 4.03).
Results and Discussion
Design and synthesis of peptoid model systems
We designed monomeric peptoid model systems that mimicked the local environments of isolated amide side chains within a peptoid oligomer in order to dissect short-range (i· · ·i or i· · ·i±1) interactions. Since we were primarily interested in elucidating the impact of side chain structure on backbone amide isomerism, our initial monomeric models consisted of two tertiary amides positioned opposite the side chain, as in a typical peptoid backbone. We developed two straightforward syntheses of these diamide models (Scheme 1) that were easily adapted for the installation of other C-terminal and N-terminal capping groups besides amides, such as esters.32 This flexibility of design permitted us to probe the role of either terminus in any side chain-backbone interaction of interest in our model system.
Scheme 1.

The generic synthetic routes (eqs 1 and 2) used in the construction of the peptoid model systems. Reagents and conditions: a. 0.9 equiv. R1NH2, 1 equiv. triethylamine, CH2Cl2, 0 °C, 60 min. b. 0.85 equiv. R2NH2, DMF, 0–24 °C, 12 h. c. 4 equiv. (CH3CO)2O, 2.1 equiv. (i-Pr)2EtN, CH2Cl2, 25 °C, 30 min. d. 2.1 equiv. (CH3CO)2O, CH2Cl2, 25 °C, 30 min. e. 2 equiv. NaH, 1.2 equiv. CH3I, DMF, 0 °C, 5 min. Details of representative syntheses of these compound classes have been reported previously.32
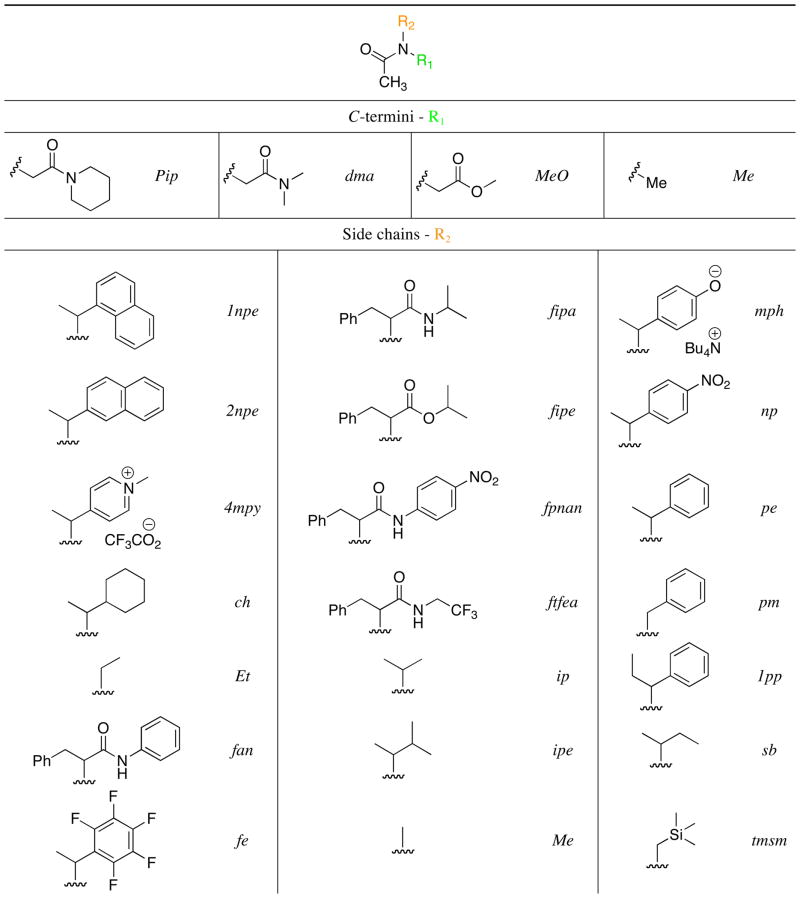
The peptoid model system side chains and terminal capping groups examined in this study are shown in Table 2. N-α-chiral amide side chains are heavily represented, as these have been extensively investigated previously and found to promote peptoid helix and loop folding (see above). In addition to this consideration, side chain functionalities were selected to explore the effects of four key types of noncovalent interactions on peptoid amide cis/trans equilibria: (1) n→π* interactions between an amide and an aromatic ring (n→π*Ar), (2) n→π* interactions between two carbonyls (n→π*C=O), (3) side chain-backbone steric interactions, and (4) side chain-backbone hydrogen bonding interactions. Our analyses of each type of interaction in a series of model systems are described sequentially below. The cis/trans ratio of the N-terminal amide in each model system was determined by 1H NMR and NOESY and used to assess the influence of the noncovalent interactions on peptoid amide isomerism.
Table 2.
Structure of the generic peptoid model system (top), and the structures and abbreviated names of the C-termini (R1) and side chains (R2) examined in this study.a
|
Single stereocenters do not influence Kcis/trans in these systems; enantiomeric excesses were therefore not determined. The cis-rotamer was defined as that which orients the side chain and the oxygen atom on the same side of the amide bond.
Investigations of the impact of n→π*Ar interactions on peptoid amide rotamer equilibria
In earlier work, we observed that the incorporation of electron-deficient aromatic rings into peptoids could have a profound impact on their structure.37,39 Analysis of related, small model systems suggested that these structural perturbations arise in part from an interaction between a lone pair of the proximal amide oxygen and a π* orbital of the electron-deficient aromatic ring, that is, an n→π*Ar interaction (also referred to as an lp-π interaction).40, 41 This type of interaction has been loosely defined by a carbonyl oxygen-to-ring centroid distance between 2.8 and 3.8 Å, and a dihedral angle between the aryl plane and the carbonyl plane (X2C=O) of 90° or less.42 We reasoned that an interaction between an N-α-chiral aromatic peptoid side chain and the nearest backbone carbonyl would easily meet these geometrical constraints and, if operative, would exclusively stabilize the cis-rotamer of the participating amide, increasing Kcis/trans (Figure 2). We tested this possibility by constructing peptoid model systems 1–3 and examining them using 1H NMR, initially focusing on the acetamide cis/trans ratios obtained in acetonitrile (CD3CNKcis/trans), since this solvent has been commonly used in structural studies of peptoids (Figure 3, Table 3).22, 23, 25, 37 Furthermore, this solvent minimizes inter-amide interactions that can also affect Kcis/trans; these interactions and their solvent-dependence are discussed in the next section.
Figure 2.

Left: The n→π*Ar interaction (indicated by the red arrow) proposed to increase Kcis/trans for peptoid backbone amides. Right: Newman projection depicting the n→π*Ar interaction.
Figure 3.

Structures of the peptoid model systems (1–15) designed to probe n→π* interactions.
Table 3.
Peptoid model system structures 1–15, N-terminal acetamide cis/trans ratios in various solvents (Kcis/trans),a and corresponding free energy differences (ΔGcis/trans).b Standard deviations (SD) for Kcis/trans were typically <10%.
| entry | R1 | R2 | CD3OD | CD3CN | CDCl3 | C6D6 | AVGKcis/trans | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Kcis/trans | ΔG (kcal/mol) | Kcis/trans | ΔG (kcal/mol) | Kcis/trans | ΔG (kcal/mol) | Kcis/trans | ΔG (kcal/mol) | ||||
| 1 | Pip | fe | 2.46 | −0.53 | 3.84 | −0.79 | 1.60 | −0.28 | 2.07 | −0.43 | 2.49 |
| 2 | Pip | np | 1.84 | −0.36 | 3.43 | −0.73 | 1.53 | −0.25 | 1.59 | −0.28 | 2.10 |
| 3 | Pip | pe | 1.26 | −0.14 | 2.04 | −0.42 | 0.70 | 0.21 | 1.03 | −0.02 | 1.26 |
| 4 | Pip | ch | 0.72 | 0.20 | 1.22 | −0.12 | 0.31 | 0.69 | 0.37 | 0.59 | 0.66 |
| 5 | Me | np | 2.83 | −0.61 | 3.27 | −0.70 | 4.20 | −0.85 | 4.31 | −0.86 | 3.65 |
| 6 | Me | pe | 1.95 | −0.40 | 2.12 | −0.44 | 1.78 | −0.34 | 2.46 | −0.53 | 2.08 |
| 7 | Me | ch | 1.23 | −0.12 | 1.30 | −0.15 | 1.00 | 0.00 | 1.34 | −0.18 | 1.22 |
| 8 | Me | 4mpy | 6.79 | −1.31 | 7.82 | −1.22 | >10 | <−1.36 | -- c | -- | >8.20 |
| 9 | Me | mph | 1.82 | −0.35 | 1.25 | −0.13 | 1.69 | −0.31 | -- | -- | 1.59 |
| 10 | MeO | np | 1.07 | −0.04 | 1.05 | −0.03 | 1.34 | −0.17 | 1.20 | −0.11 | 1.17 |
| 11 | MeO | pe | 0.75 | 0.17 | 0.67 | 0.24 | 0.66 | 0.25 | 0.68 | 0.22 | 0.69 |
| 12 | MeO | ch | 0.38 | 0.58 | 0.29 | 0.73 | 0.31 | 0.70 | 0.32 | 0.67 | 0.33 |
| 13 | dma | np | 1.54 | −0.25 | 3.06 | −0.66 | 1.12 | −0.07 | 1.20 | −0.11 | 1.73 |
| 14 | dma | pe | 1.13 | −0.07 | 1.69 | −0.31 | 0.54 | 0.36 | 0.70 | 0.21 | 1.02 |
| 15 | dma | ch | 0.71 | 0.20 | 0.58 | 0.33 | 0.25 | 0.82 | 0.42 | 0.52 | 0.49 |
Determined by integrating 1H NMR spectra of 15 mM solutions at 24 °C.
ΔG=-RTln(Kcis/trans).
Data not available.
We hypothesized that model compounds 1 and 2 would exhibit higher cis/trans ratios than 3, since electron-withdrawing groups on aromatic rings are known to increase the strength of n→π*Ar interactions.41 In accord with our hypothesis, we observed a nearly two-fold increase in CD3CNKcis/trans for the compounds containing the electron-deficient benzyl side chains fe and np (1 and 2) compared to the control compound containing the unsubstituted benzyl side chain pe (3) (Table 3). Replacement of the aromatic ring with a cyclohexyl ring (cy, to yield 4) lowered CD3CN Kcis/trans, as expected. Although these model systems validated the possibility of ni→πi+1*Ar interactions in a typical peptoid, an ni→πi*Ar interaction involving the C-terminal amide could not be excluded. We thus constructed control compounds 5–7, in which the C-terminal amide was replaced with a methyl group (Figure 3). This series of compounds exhibited nearly identical CD3CN Kcis/trans values to the piperidinyl series (2–4), suggesting that ni→πi*Ar interactions do not significantly regulate peptoid amide isomerism, and establishing that the C-terminal amide has little impact on CD3CN Kcis/trans in these systems (Table 3). We also synthesized formally charged model systems (pyridinium 8 (4mpy) and phenolate 9 (mph), Figure 3) to further explore the possibility of controlling Kcis/trans by tuning the electronics of N-α-chiral aromatic side chains. As expected, the presence of mph in 9 substantially demoted the cis-amide rotamer relative to pe in 6, while the 4mpy side chain in 8 strongly promoted the cis-amide. Comparison of NOESY NMR data for model compounds 6 and 8 indicated that the conformation in which the aromatic ring and the amide carbonyl are eclipsed is significantly more populated by 8 than by 6 (Figure 4), implying that carbonyl-aromatic interactions dictate Kcis/trans in these systems.
Figure 4.

Left: Structures of model compounds 6 and 8. Right: Newman projections depicting the most populated conformations for cis-6 and cis-8, as indicated by the NOEs shown: red=stronger for 8 v. 6, black=identical for 6 and 8.
Our previously reported calculations of Mulliken atomic charges suggested that electrostatic interactions between the atoms directly participating in the n→π*Ar interaction do not contribute considerably to Kcis/trans in this class of peptoid model systems (5, 6, 8, and 9).32, 43 On the contrary, the calculations indicated that Kcis/trans correlates closely with the LUMO energy, and even more strikingly with the LUMO density at the carbon in the aromatic ring connected to the side chain. We sought to decouple, as much as possible, the contributions of electrostatic and stereoelectronic influences on n→π*Ar interactions to complement these computational data. Solvent modulation is a useful approach for probing noncovalent interactions,44–46 even though its impact on a particular interaction of interest can be somewhat obscured by solvent-charge screening and/or by perturbations elsewhere in the molecule. We thus proceeded to determine Kcis/trans values for the structurally simplest series — the methyl-capped compounds 5–7 — in a range of solvents (Table 3).
Kcis/trans for compound 7 (ch), which served as a baseline indicator of solvent effects on Kcis/trans in the absence of n→π* interactions, generally tracked with solvent polarity but displayed only slight sensitivity to the choice of solvent (Table 3). The aromatic side chain of 6 (pe) did not significantly sensitize Kcis/trans to solvent polarity compared to 7, as indicated by the similar (differing by less than 0.1 kcal) differences in ΔGcis/trans observed between solvents for 6 and 7. However, 5 (np) diverged significantly (ΔGcis/trans differing by more than 0.2 kcal) from this trend, exhibiting a larger Kcis/trans in CDCl3. Chloroform would not be expected to enhance withdrawal of electron density from the aromatic ring by the nitro group (thereby lowering the aryl LUMO energy and increasing Kcis/trans), as the resulting polarization would be less stabilized by such a nonpolar solvent. Therefore, this divergence may derive from electrostatic/dipole-dipole interactions between the nitro group and the acetamide that reinforce the n→π*Ar interaction in CDCl3. These electrostatic interactions would likely be more effectively screened in more polar solvents such as methanol; however, CD3ODΔ Gcis/trans for 5 remained significantly lower than for 6, and ΔGcis/trans for 5 and 6 were similarly affected by switching the solvent from acetonitrile to methanol, suggesting that electrostatic interactions are less important than the n→π*Ar orbital interactions in this case. Indeed, even in aqueous solution, we observed that the cis-amide rotamer of 8 (4mpy) is dominant (D2OKcis/trans = 3.92), demonstrating that the side chain-backbone interactions in these highly polarized systems are robust, even in strongly polar solvents that would be expected to significantly screen electrostatic interactions. This finding suggests that the inclusion of these charged aromatic monomers could produce water-soluble peptoid helices with enhanced conformational stability relative to pe homooligomers; such scaffolds could be valuable for biological applications. The impact of n→π*Ar interactions in general on the structures of peptoid oligomers is examined further in the last section of this paper.
Investigations of the impact of n→π*C=O interactions on peptoid amide rotamer equilibria
Having scrutinized n→π*Ar interactions in model systems containing a single amide, we turned our attention to diamide peptoid model systems that might more accurately reproduce the local environment experienced by a peptoid monomer unit within a larger peptoid oligomer. As discussed above, we observed that the presence of a piperidinyl amide at the C-terminus did not significantly perturb CD3CN Kcis/trans for the N-terminal acetamide relative to the single amide models (e.g., 2–4 vs. 5–7). Nonetheless, we were intrigued by recent literature reports by Raines, Wennemers, and co-workers of n→π*C=O interactions between tertiary prolyl amides and their considerable importance in stabilizing the trans-amides of polyproline II (PPII) helices.47–50 Similar to the n→π*Ar interactions discussed above, these interactions are characterized by donation of electron density from an amide carbonyl lone pair into the π* orbital of an adjacent carbonyl. Due to geometric and stereoelectronic differences between the π* orbitals of aromatic rings and amides, a somewhat more restricted set of geometric constraints has been demarcated for n→π*C=O interactions (Oi−1…Ci=Oi distance ≤ 3.2 Å, angle = 109° ± 10°; Figure 5)47 relative to n→π*Ar interactions. These constraints preclude cis-amides from acting as donors in i· · ·i+1 n→π*C=O interactions along the backbone, and n→π*C=O interactions are thus observed to exclusively stabilize trans-amide donors in polyprolines.
Figure 5.

Left: The n→π*C=O interaction (indicated by the red arrow) proposed to reduce Kcis/trans for the donating amide in peptoids. Right: Newman projection depicting the n→π*C=O interaction
The strong similarity between the tertiary amide backbones of peptoids and polyprolines caused us to speculate that these n→π*C=O interactions could also be exploited to stabilize trans-amides in peptoids, providing a design strategy complementary to the cis-amide-stabilizing n→π*Ar interactions described above. Previous studies have shown that esters are excellent acceptors for these n→π*C=O interactions due to significantly reduced π-electron delocalization into the carbonyl π* orbital.33, 51, 52 This reduction of electron donation into the carbonyl lowers the energy of the π* orbital, enhancing n→π*C=O interactions in esters relative to amides, just as removing substituents that donate electrons into aromatic rings (e.g., 9 vs. 6 above) enhances n→π*Ar interactions. We hypothesized that amides disfavoring π-electron delocalization might also enhance n→π*C=O interactions, albeit to a lesser extent than esters. To test this hypothesis and probe for the presence of n→π*C=O interactions within peptoids in general, we constructed model systems possessing methyl esters and dimethylamides at the C-terminus (10–15, Figure 3). As noted in the previous section, piperidinyl amide-capped compounds 2–4 did not exhibit any evidence of these interactions in acetonitrile, giving nearly identical CD3CN Kcis/trans values to those of the methyl-capped compounds 5–7. However, we expected that the dimethylamide would be somewhat less capable of stabilizing positive charge at the nitrogen compared to the piperidinyl amide, and thus might participate in an n→π*C=O interaction with the N-terminal acetamide. Analogous to the studies above, we also examined these systems in a range of solvents (Table 3).
We were pleased to observe that the nature of the C-terminal cap dramatically modulated the acetamide cis-trans equilibrium, with the ester group (in 10–12) producing the largest reductions in Kcis/trans relative to the methyl- and piperidinyl-amide-capped systems (2–4), as predicted. Although stabilization of the trans-amide in the dimethylamide systems (13–15) relative to compounds 2–4 was modest, the former demonstrated the feasibility of inter-amide n→π*C=O interactions within larger peptoid oligomers. We were especially intrigued by the large increases in ΔGcis/trans observed upon decreasing the solvent polarity for the piperidinyl and dimethyl amide series, relative to the ester series. Since previous studies have shown that n→π*C=O interactions are primarily hyperconjugative rather than electrostatic in nature,53, 54 we hypothesized that the greater solvent sensitivity of ΔGcis/trans for the amides (2–4, 13–15) versus the esters (10–12) may be due to reduced charge separation within the more polarizable piperidinyl and dimethylamides in nonpolar solvents. Such suppression of amide resonance would be expected to increase the strength of n→π*C=O interactions by lowering the energy of the π* orbital.33 Although some debate over the significance of amide resonance has emerged,55, 56 the greater polarizability of amides, defined here as their capacity to accommodate internal charge separation via resonance, is commonly cited to explain their significantly higher rotational barriers (~18 kcal/mol) compared to esters (~10 kcal/mol) and carbamates (~15 kcal/mol),57–60 Indeed, the greater sensitivity of amide rotational barriers to solvent polarity, compared to that of carbamates, has been attributed to the less polarizable “ester-type resonance” in the latter.59 In the context of this argument, the absence of n→π*C=O interactions for compounds 2–4 in acetonitrile can be ascribed to the high degree of amide polarization facilitated by the both the piperidinyl substituent and the polar solvent.
Interestingly, the amide-capped systems (2–4, 13–15) showed a marked increase in ΔGcis/trans in methanol as compared to acetonitrile, deviating from the solvent polarity trend above, while ΔGcis/trans for the methyl- and ester-capped systems (5–7, 10–12) remained relatively constant in both solvents. This implies that amide-amide n→π*C=O interactions, but not amide-ester interactions or isolated amide rotameric equilibria, are strongly affected by solute-solvent hydrogen bonding in these systems. Since esters are known to be poorer hydrogen bond acceptors than amides,61 these data suggest that hydrogen bonds to the amide acceptors of n→π*C=O interactions may enhance these interactions. Further corroboration of these hypotheses in the context of peptoids containing amide and ester side chains is described later in this paper.
Investigations of the impact of steric interactions on peptoid amide rotamer equilibria
Steric interactions play an extremely important role in peptoid folding by constraining the rotations of bonds within the peptoid backbone. For example, it is these interactions that likely transmit chirality from the N-α-chiral peptoid side chains to the backbone, imparting “handedness” to the peptoid helix.27 Steric interactions in peptoids are expected to be highly robust in that they are relatively unaffected by solvation.24 This property also greatly simplifies their computational simulation and facilitates their utilization for rational design applications, as aptly illustrated by the successful computational prediction of the peptoid helical structure.27 Although computations predict that steric interactions restrict all three peptoid backbone torsions (φ, ψ, and ω),27 the present study focuses on the roles of these interactions in regulating peptoid amide isomerism (ω) by systematically probing the steric profiles of peptoid side chains in a series of model systems typically containing a C-terminal piperidinyl amide (16–26; Figure 6, Table 4).
Figure 6.

Structures of the peptoid model systems (16–26) designed to probe steric interactions.
Table 4.
Peptoid model system structures 16–26, N-terminal acetamide cis/trans ratios in various solvents (Kcis/trans),a and corresponding free energy differences (ΔGcis/trans).b Solvents are listed in order of decreasing polarity. Standard deviations (SD) for Kcis/trans did not exceed 10%.
| CD3OD | CD3CN | CDCl3 | C6D6 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| entry | R1 | R2 | Kcis/trans | ΔG (kcal/mol) | Kcis/trans | ΔG (kcal/mol) | Kcis/trans | ΔG (kcal/mol) | Kcis/trans | ΔG (kcal/mol) | AVGKcis/trans |
| 16 | Pip | Me | 0.42 | 0.52 | 0.60 | 0.30 | 0.15 | 1.10 | 0.17 | 1.04 | 0.34 |
| 17 | Pip | Et | 0.47 | 0.45 | 0.66 | 0.25 | 0.17 | 1.05 | 0.20 | 0.95 | 0.38 |
| 18 | Pip | ip | 0.64 | 0.26 | 1.09 | −0.05 | 0.40 | 0.54 | 0.47 | 0.44 | 0.65 |
| 19 | Pip | sb | 0.64 | 0.26 | 1.05 | −0.03 | 0.33 | 0.66 | 0.48 | 0.43 | 0.63 |
| 20 | Pip | ipe | 0.67 | 0.23 | 1.18 | −0.10 | 0.33 | 0.65 | 0.48 | 0.43 | 0.67 |
| 21 | Pip | tmsm | 1.61 | −0.28 | 1.91 | −0.38 | 0.48 | 0.44 | 0.78 | 0.14 | 1.20 |
| 22 | Pip | 1pp | 1.67 | −0.30 | 2.20 | −0.47 | 0.90 | 0.06 | 1.23 | −0.12 | 1.50 |
| 23 | Me | pm | 1.49 | −0.24 | 1.68 | −0.31 | 1.30 | −0.15 | -- c | -- c | -- c |
| 24 | Pip | pm | 0.62 | 0.28 | 1.15 | −0.09 | 0.24 | 0.84 | 0.25 | 0.82 | 0.57 |
| 25 | Pip | 2npe | 1.55 | −0.26 | 2.21 | −0.47 | 0.86 | 0.09 | 1.20 | −0.11 | 1.46 |
| 26 | Pip | 1npe | 4.63 | −0.72 | 6.27 | −1.08 | 2.60 | −0.56 | 4.07 | −0.83 | 4.39 |
Determined by integrating 1H NMR spectra of 15 mM solutions at 24 °C.
ΔG=-RTln(Kcis/trans).
Data not available.
In general, increasing the steric bulk of the peptoid side chain in the model systems should increase the population of the cis-amide isomer, since the relatively small carbonyl oxygen, as compared to the acetamide methyl group, can more easily accommodate steric crowding.27 Comparison of the ΔGcis/trans values within each solvent demonstrates that these systems largely conform to this paradigm. The solvent-dependent trends observed largely mirror those of piperidinyl amides 1–4 (Table 3) and are consistent with the action of n→π* interactions, as discussed in the previous sections. The sole exception is compound 23, which lacks a C-terminal carbonyl and exhibits little solvent-dependent change in Kcis/trans, as expected. We observed that complete removal of all N-α-substituents (i.e., Me, 16) produced a very low average Kcis/trans value for the solvents examined (AVGKcis/trans), in keeping with this steric argument (Table 4) and in agreement with previous studies of similar N,N-dialkyl amides.62 Although a single appended methyl group (Et, 17) gave a similarly low value for AVGKcis/trans, further substitution at the N-α-carbon (ip, 18) increased AVGKcis/trans by two-fold, again commensurate with side chain steric bulk. In contrast, increasing the steric bulk at the N-β-carbon, as with sb (19) and ipe (20), did not have any significant effect on AVGKcis/trans, giving values similar to those observed for ip (18) and the cyclohexyl side chain (ch, 4; Table 3). We also examined a (trimethylsilyl)methyl side chain (tmsm, 21) as part of an effort to determine whether a lack of steric bulk at the N-α-position might be compensated for by an even larger increase in bulk at the N-β-position relative to the ipe side chain. Although the Kcis/trans values for 21 were indeed higher than those of the aliphatic side chain model systems, analysis of this system is complicated by the possibility of dative oxygen-silicon bonding between the side chain silyl group and the acetamide oxygen. We suggest that this class of interactions could also be useful for controlling amide isomerism in peptoids, and is worthy of study in the future.
Turning our attention to the sterics of aromatic side chains, we appended a methyl group to the well-characterized pe side chain to determine if such modifications could be used to increase Kcis/trans (1pp, 22; Table 4). As expected, we observed a modest (20%) increase in AVGKcis/trans relative to pe (3); this increase was nearly identical in each solvent tested, strongly suggesting a steric origin for the increase. Similar reasoning explains the lower Kcis/trans values determined for the N-benzyl compounds pm-Me (23) and pm-Pip (24), as compared to the pe-Me (6) and pe-Pip (3) systems containing an N-α-methyl group (Table 2). Intriguingly, the absence of an N-α-methyl group in the side chain of pm-Pip (24), in conjunction with the presence of a piperidinyl amide at the C-terminus, especially promotes the trans-acetamide, since Kcis/trans for pm-Pip (24) is dramatically lower (by 50%, on average) than that for pm-Me (23) in all solvents examined, while pe-Me (6) and pe-Pip (3) exhibit more similar Kcis/trans values.
In an effort to more dramatically regulate amide isomerism using steric interactions, we constructed model systems containing naphthylethyl side chains (2npe-Pip, 25 and 1npe-Pip, 26; Table 4). We conjectured that the expanded size of these side chains could multiply steric interactions with nearby atoms, thereby restricting bond rotations. Furthermore, the electronic perturbation resulting from expansion of the aromatic ring system might simultaneously engender strong n→π*Ar interactions. Although the model system containing the 2-substituted naphthyl group (25) displayed only a slight increase in Kcis/trans compared to the related aromatic side chain pe (in 3), the 1-substituted naphthyl group (in 26) largely suppressed the trans-acetamide rotamer. Preliminary molecular mechanics calculations suggest that steric interactions between the naphthyl side chain and the backbone of 26 restrict rotation of the naphthyl group such that n→π*Ar interactions stabilizing the cis-rotamer are favored; these steric interactions are calculated to be significantly less severe in 25 (see Supporting Information.) The strong cis-rotameric preference elicited by the 1-naphthylethyl side chain, if representative of that within a polypeptoid, suggests that this side chain could prove exceptionally useful for the construction of peptoid helices. We return to this possibility below.
Investigations of the impact of hydrogen bonding interactions on peptoid amide rotamer equilibria
Hydrogen bonds are ubiquitous in peptide and protein structures, and are also essential to the folding of many non-natural foldamer classes.21 Peptoids offer the distinctive opportunity to engineer the locations of all hydrogen bond donors via side chain selection, while conserving structural elements that promote folding. The strength of typical hydrogen bonds in organic solvents (up to 3 kcal/mol) relative to other noncovalent interactions makes them attractive architectural elements, and they have been exploited previously to stabilize peptoid structures (e.g., the threaded loop, vide supra).24, 28, 31 We therefore undertook a study of a set of peptoid model systems containing side chains capable of hydrogen bonding to the peptoid backbone (27–32; Figure 7).
Figure 7.

Structures of the peptoid model systems (27–32) designed to probe hydrogen bonding interactions.
We commenced our investigations of hydrogen bonding by examining the primary amide side chain fipa (27), derived from phenylalanine (Figure 7). We initially considered the possibility that a hydrogen bond could form between this side chain amide and the N-terminal acetamide, stabilizing the cis-acetamide isomer (Figure 8A), even though such γ-turn motifs are rare in peptides.63 However, Kcis/trans for 27 (Table 5) increased in the hydrogen bonding solvent methanol and decreased in the nonpolar solvent chloroform, implying that a side chain-acetamide hydrogen bond does not significantly affect the rotameric ratio of 27. Furthermore, the downfield chemical shift of the amide side chain proton in the trans-acetamide relative to the cis-acetamide in the 1H NMR spectrum of 27 (in acetonitrile and chloroform) suggests that any hydrogen bonds formed are stronger in the trans-acetamide than in the cis-acetamide (see Supporting Information).64
Figure 8.

Possible noncovalent interactions affecting Kcis/trans for 27. A) Direct hydrogen bond (red dashes) stabilizing the cis-acetamide. B) Backbone-side chain n→π*C=O interaction (green arrow) stabilizing the cis-acetamide. C) Backbone-backbone n→π*C=O interaction (blue arrow) stabilizing the trans-acetamide.
Table 5.
Peptoid model system structures 27–32, N-terminal acetamide cis/trans ratios in various solvents (Kcis/trans),a and corresponding free energy differences (ΔGcis/trans).b Solvents are listed in order of decreasing polarity. Standard deviations (SD) for Kcis/trans did not exceed 10%, except for 30 in CD3CN (SD = 20%).
| CD3OD | CD3CN | CDCl3 | ||||||
|---|---|---|---|---|---|---|---|---|
| entry | R1 | R2 | Kcis/trans | ΔG (kcal/mol) | Kcis/trans | ΔG (kcal/mol) | Kcis/trans | ΔG (kcal/mol) |
| 27 | Pip | fipa | 1.80 | −0.35 | 1.56 | −0.26 | 0.51 | 0.40 |
| 28 | Pip | ftfea | 2.77 | −0.60 | 3.26 | −0.70 | 0.86 | 0.09 |
| 29 | Pip | fan | 3.33 | −0.71 | 4.57 | −0.9 | 0.58 | 0.32 |
| 30 | Pip | fpnan | -- c | -- | ≥10 | ≤ −1.36 | -- | -- |
| 31 | Pip | fipe | 1.39 | −0.19 | 2.73 | −0.59 | 0.82 | 0.12 |
| 32 | Me | fipe | 2.44 | −0.53 | 2.58 | −0.56 | 2.57 | −0.56 |
Determined by integrating 1H NMR spectra of 15 mM solutions at 24 °C.
ΔG=-RTln(Kcis/trans).
Data not available.
We reasoned that modulating the strength of the putative hydrogen bonds in the peptoid model systems might inform our understanding of how they affect Kcis/trans in these systems. To this end, we examined additional model systems in which the secondary amide substituent on the side chain was systematically altered (28–30; Figure 7, Table 5). (We note that NMR spectroscopy did not detect any trans-rotamers (as defined in Table 2) of the side chain secondary amides of this series.) The CD3CN Kcis/trans values obtained increased concomitantly with the expected acidity of the secondary amide proton; however, a direct hydrogen bond to the cis-acetamide seemed unlikely (Figure 8A; see discussion of 27 above). Based on these observations and NOESY data for 27 and 30,32 we hypothesized that a hydrogen bond preferentially forms between the side chain and the piperidinyl amide instead, leaving Kcis/trans to be determined by the relative energies of competing backbone-side chain (Figure 8B; favoring cis) and backbone-backbone (Figure 8C; favoring trans) n→π*C=O interactions. Convincing solid state evidence for this hypothesis was provided by our previously reported X-ray crystal structure of the fpnan system (30), which exhibits both the hydrogen bond and the n→π*C=O interaction shown in Figure 8B.32 The strong (≥10:1) preference for the cis-acetamide in acetonitrile, coupled with the NOESY data, also corroborates this model for stabilization of the cis-acetamide. As with the previously examined systems containing the C-terminal piperidinyl amide (vide supra), the CD3OD Kcis/trans values for 27–29 and 30 were typically lower than the CD3CNKcis/trans values, while CDCl3 Kcis/trans values were uniformly low as compared to the other solvents examined, consistent with stronger backbone-backbone n→π*C=O interactions (Figure 8C) in this solvent. This solvent trend suggested that the tertiary piperidinyl amide, being more polarizable than the side chain secondary amide, might be more sensitive to solvent polarity and could out-compete the side chain amide as the acceptor of the n →π *C=O interaction with the acetamide in chloroform; this solvent-dependent, divergent behavior of secondary and tertiary amides has been reported previously.65
In order to further evaluate this theory, we replaced the amide hydrogen bond donor in the side chain with an ester (31, Table 5), an excellent acceptor of n→π*C=O interactions (see above). As expected, CD3CN ΔGcis/trans significantly decreased (by 0.33 kcal/mol) versus amide 27, confirming that any hydrogen bonding that occurs is not required for stabilizing the cis-acetamide, and further supporting our hypothesis that backbone-to-side chain n→π *C=O interactions (Figure 8B) stabilize the cis-acetamide rotamer in this series. Moreover, ΔGcis/trans for 31 in chloroform also decreased by ~0.3 kcal/mol compared to 27, signifying that this backbone-side chain n→π*C=O interaction is mostly unaffected by solvent polarity per se, again in line with expectations for the relatively non-polarizable ester (vide supra). However, ΔGcis/trans values for amides 29 and 30 decreased by varying amounts in the hydrogen bonding solvent methanol relative to acetonitrile, and were generally lower than for ester 31, even though esters are typically better acceptors of n→π*C=O interactions. These findings, in conjunction with the X-ray crystal structure of 30, suggest that a hydrogen bond can stabilize a conformation (observed in the solid state) that predisposes the backbone and side chain amides to participate in an n→π*C=O interaction (Figure 8B). In this respect, the hydrogen bond appears to perform a similar function to that of the steric interactions in 1-naphthylethyl system 26, in that it serves to enforce a conformational bias that engenders an n→π interaction. The significance of this hydrogen bond is underscored by the fact that a significant decrease of ΔGcis/trans in methanol vs. acetonitrile is observed only for the compounds with amide side chains, which would be susceptible to hydrogen bond disruption by the solvent. (Note, solvent-mediated enhancement of backbone-backbone n→π*C=O interactions in methanol, such as that observed for 2–4 and 13–15 above, could also play a role in lowering ΔGcis/trans.) On the other hand, strengthening of the backbone-backbone n→π*C=O interactions (Figure 8C) due to reduced polarization of the piperidinyl amide in chloroform again appears to be the major factor contributing to the reduction of Kcis/trans in chloroform versus acetonitrile for 28–30 (vide supra). Further confirmation of this finding was obtained by examining 32, which lacked the piperidinyl amide cap and therefore exhibited uniformly higher (as compared to 31) and nearly identical Kcis/trans values for 32 in the three solvents examined, including chloroform. Indeed, the nearly identical CD3CNΔGcis/trans values for 31 and 32 indicate that backbone-backbone n→π *C=O interactions (Figure 8C) are virtually nonexistent in acetonitrile. These data also eliminate the possibility of significant n→π *C=O interactions between the backbone piperidinyl amide and the side chain ester.
Overall, the data acquired for this series of model systems (27–32) demonstrate that this class of side chain amide hydrogen bond donors does not directly interact with the N-terminal amide to dictate Kcis/trans (Figure 8A). Rather, our results suggest that solvent-dependent hydrogen bonding and amide polarization affect the energies of the competing backbone-side chain (Figure 8B, favoring cis) and backbone-backbone (Figure 8C, favoring trans) n→π*C=O interactions that determine Kcis/trans. In light of the data above, incorporation of amide side chains into peptoids could constitute a highly effective strategy for promoting cis-amides in the peptoid backbone in polar solvents, as evidenced by the large cis/trans ratios exhibited by acetanilide 29 (~5:1), and especially 30 (≥10:1), in acetonitrile. Intriguingly, this same side chain class might also be used to construct new and optically active, trans-amide peptoid secondary structures in nonpolar solvents, since its capacity to stabilize trans-amides in chloroform rivals the other side chains in this report.30 In general, the wide range of Kcis/trans values attainable using side chain amides, coupled with their synthetic accessibility via standard coupling techniques, suggests that this side chain class could be extremely useful for rationally tuning amide isomerism in the peptoid backbone.
Studies of Peptoid Oligomers Derived from the Model Systems
The peptoid model systems discussed above were designed to investigate interactions between peptoid side chains and the backbone, and thus typically consisted of a single side chain flanked by two backbone carbonyls. In a peptoid oligomer, however, each backbone carbonyl is in turn typically surrounded by two peptoid side chains, which may act cooperatively or competitively to influence amide rotameric equilibria. Therefore, peptoids containing at least two side chains could validate a role for the noncovalent interactions discussed above in directing amide isomerism within larger peptoid oligomers. To this end, we synthesized several polypeptoids (33–38, Figure 9) that were designed to probe these noncovalent interactions within peptoid oligomers, and analyzed their 1H-COSY and HSQCAD NMR spectra in CD3CN to determine backbone amide cis/trans ratios (Table 6).
Figure 9.

Structures of the polypeptoids (33–37) designed to examine noncovalent interactions in peptoid oligomers.
At the outset, we were most interested in examining n→π*Ar interactions in polypeptoids, since these were expected to promote peptoid helix formation by stabilizing backbone cis-amides. We therefore incorporated a single snp residue (the S-enantiomer of the np side chain) within an spe pentamer at the central position away from the termini, in an attempt to minimize interference with its cis/trans ratio by hydrogen bonding to the C-terminus and to more accurately simulate the structural environment within longer peptoids. We were gratified to observe that this snp side chain, which exhibited a Kcis/trans of ~3.3 in the methyl (5) and piperidinyl (2) model systems, significantly increased the population of cis-amides in 33 relative to the spe homopentamer 34 (Table 6). We subsequently examined two pentamers containing the sfe residue (35 and 36), which exhibited excellent resolution of signals by residue type in their HSQCAD spectra.37 Once again, we found that our model systems were qualitatively predictive of Kcis/trans in peptoid 35, with the sfe residues exhibiting an even higher (46%) CD3CN Kcis/trans value than the corresponding model system (1), and a six-fold higher CD3CNKcis/trans than the spe residues. Interestingly, the overall cis/trans ratios for the homopentamers 34 and 36 were found to be somewhat lower than those of the corresponding monomeric model systems. Since previous studies have demonstrated that hydrogen bonding at peptoid termini in related systems can profoundly affect conformation, especially by locally promoting trans-amides,23, 25, 31, 37 we suggest that hydrogen bonding at the termini of these pentamers may have a similar impact.66 This assertion is supported by the low Kcis/trans values obtained for the spe residues of the mixed oligomer 35, two of which constitute the terminal residues. Notwithstanding such hydrogen bonding-derived effects, these findings demonstrate that n→π*Ar interactions can indeed regulate amide isomerism at internal residues of N-α-chiral aromatic polypeptoids, and can be used to rationally control peptoid structure.
We next constructed homooligomers composed of (S)-1-naphthylethyl (s1npe) side chains (37 and 38, Table 6), since the consistently high Kcis/trans values obtained using this side chain suggested that it could prove particularly useful for constructing robust peptoid helices. We were pleased to observe that the very high Kcis/trans observed for the monomeric system (26; 6.27) in acetonitrile was amplified in the s1npe homooligomers in a length-dependent manner, delivering a CD3?CN Kcis/trans of 7.0 and 9.7 in the dimer (37) and trimer (38), respectively – to our knowledge, the highest overall amide cis/trans ratios observed for any acyclic peptoid oligomers to date. This length-dependent amplification of Kcis/trans is indicative of cooperatively stabilized structure — a hallmark of well-folded peptoids — although the nature of such cooperative effects has not been examined extensively so far.23, 67
To gain additional insight into the structures of 37 and 38, we obtained their circular dichroism (CD) spectra in acetonitrile. The CD spectra of 37, and of 38 in particular, contained very intense extrema (Figure 10), especially near 220 nm, which has been associated with the presence of cis-amides23, 31 and helical peptoids in general.25, 31 Comparison of these spectra with those of the corresponding optically active model system (S-26) reveals a striking length-dependent increase in ellipticity per amide, again suggestive of a cooperatively folded secondary structure.29 In conjunction with the NMR data, these findings establish that 1npe is highly effective for inducing local cis-amide conformations in polypeptoids. Moreover, a reduction in the number of correlations in the HSQCAD NMR (see Supporting Information) and concomitant increase in CD signal strength data with increasing oligomer length, as observed in these systems, have been cited as evidence for increasing conformational homogeneity as a function of oligomer length.26 Given that spe pentamer 34 has been shown to be more structured than the analogous spe trimer,23 the significantly higher (more than 6-fold) overall Kcis/trans measured for 1npe trimer 38 as compared to spe pentamer 34 (Table 6) suggests that incorporation of 1npe may permit the construction of extremely robust peptoid helices. Indeed, the intensity of the CD signal at 220 nm for trimer 38 is more than twice that reported for pentamer 3437 and its enantiomer.23 In light of these data, a much higher degree of side chain diversity than was previously possible in designed peptoid helices may be achievable by using the 1npe side chain to reduce the number of residues dedicated to maintaining structural integrity.22
Figure 10.

CD spectra of Ac-s1npe-Pip (S-26), Ac-(s1npe)2-CONH2 (37), and Ac-(s1npe)3-CONH2 (38) at 60 μM concentration in acetonitrile. Spectra were collected at 25 °C.
Summary
This comprehensive study has revealed several new strategies for controlling peptoid amide isomerism using noncovalent interactions, including n→π*, steric, and hydrogen bonding interactions. These strategies were developed through the design, synthesis, and characterization of small, model peptoid systems, which were shown to be largely predictive of the rotameric equilibria of the backbone amides in N-α-chiral aromatic peptoid oligomers, an important and often utilized class of peptoids. One of the most significant findings of this study is that cis-amides in the peptoid backbone can be exclusively promoted by select N-α-chiral acetanilide and N-1-naphthylethyl side chains, putatively by n →π *C=O/hydrogen bonding interactions and n→π *Ar/steric interactions, respectively. In the former case, the relative polarizabilities of the side chain- versus backbone-amides appear to give rise to the dramatic solvent effects observed, with nonpolar solvents reversing the cis-amide preference in the peptoid backbone. This phenomenon evokes an intriguing new possibility for peptoid structural design entailing the rational tuning of backbone- and side chain-amide electronics to stabilize either cis- or trans-amides, potentially generating novel peptoid structures that can also fold in a solvent-dependent manner.28 The subtle and solvent-dependent interplay of n→π*, steric, and hydrogen bonding interactions in peptoids has not been previously investigated; these findings could thus provide a framework for further development of new peptoid design strategies.
The application of one of the new design tactics reported herein – the incorporation of N-1-naphthylethyl side chains into peptoids – yielded peptoid oligomers that exhibited unprecedented conformational homogeneity. In this context, the structure-promoting elements presented here should prove especially useful for constructing well-folded peptoid scaffolds with an expanded number of side chain sites available for rational or combinatorial diversification. In addition to elucidating aspects of the regulation of peptoid amide isomerism via noncovalent interactions, these findings may also be useful in other foldamer regimes employing tertiary amides. As a whole, this work significantly expands the available repertoire of peptoid design strategies, and should enable the construction of both familiar and as of yet uncharacterized peptoid architectures.
Supplementary Material
Acknowledgments
We thank the NSF (CHE-0449959), ONR (N000140710255), Burroughs Wellcome Fund, Research Corporation, Dreyfus Foundation, Johnson & Johnson, and 3M for financial support of this work. H.E.B is an Alfred P. Sloan Foundation fellow. S.A.F. acknowledges the ACS Division of Medicinal Chemistry for a pre-doctoral fellowship (sponsored by Sanofi-Aventis) and Pfizer Global R&D for a Diversity in Organic Chemistry Fellowship. Support for the NMR facilities at UW-Madison by the NIH (1 S10 RR13866-01 and 1 S10 RR08389-01) and the NSF (CHE-0342998 and CHE-9629688) is gratefully acknowledged. We thank Professors Ronald Raines and Samuel Gellman for many contributive discussions, and Dr. Grant Geske for computational experiments.
Footnotes
Supporting Information Available: Full characterization data for model peptoids, HPLC data for peptoid pentamers, NMR spectra for model systems and pentamers, computational data, and a complete author list for the reference in footnote 1. This material is available free of charge via the Internet at http://pubs.acs.org.
References and Notes
- 1.Simon RJ, et al. Proc Natl Acad Sci U S A. 1992;89:9367–9371. doi: 10.1073/pnas.89.20.9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Figliozzi GM, Goldsmith R, Ng SC, Banville SC, Zuckermann RN. Methods Enzymol. 1996;267:437–447. doi: 10.1016/s0076-6879(96)67027-x. [DOI] [PubMed] [Google Scholar]
- 3.Zuckermann RN, Kerr JM, Kent SBH, Moos WH. J Am Chem Soc. 1992;114:10646–10647. [Google Scholar]
- 4.Miller SM, Simon RJ, Ng S, Zuckermann RN, Kerr JM, Moos WH. Bioorg Med Chem Lett. 1994;4:2657–2662. [Google Scholar]
- 5.Kwon YU, Kodadek T. J Am Chem Soc. 2007;129:1508–1509. doi: 10.1021/ja0668623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Patch JA, Barron AE. J Am Chem Soc. 2003;125:12092–12093. doi: 10.1021/ja037320d. [DOI] [PubMed] [Google Scholar]
- 7.Mora P, Masip I, Cortes N, Marquina R, Merino R, Merino J, Carbonell T, Mingarro I, Messeguer A, Perez-Paya E. J Med Chem. 2005;48:1265–1268. doi: 10.1021/jm040834i. [DOI] [PubMed] [Google Scholar]
- 8.Wu CW, Seurynck SL, Lee KYC, Barron AE. Chem Biol. 2003;10:1057–1063. doi: 10.1016/j.chembiol.2003.10.008. [DOI] [PubMed] [Google Scholar]
- 9.Wender PA, Mitchell DJ, Pattabiraman K, Pelkey ET, Steinman L, Rothbard JB. Proc Natl Acad Sci U S A. 2000;97:13003–13008. doi: 10.1073/pnas.97.24.13003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hara T, Durell SR, Myers MC, Appella DH. J Am Chem Soc. 2006;128:1995–2004. doi: 10.1021/ja056344c. [DOI] [PubMed] [Google Scholar]
- 11.Zuckermann RN, Martin EJ, Spellmeyer DC, Stauber GB, Shoemaker KR, Kerr JM, Figliozzi GM, Goff DA, Siani MA, Simon RJ. J Med Chem. 1994;37:2678–2685. doi: 10.1021/jm00043a007. [DOI] [PubMed] [Google Scholar]
- 12.Nguyen JT, Porter M, Amoui M, Miller WT, Zuckermann RN, Lim WA. Chem Biol. 2000;7:463–473. doi: 10.1016/s1074-5521(00)00130-7. [DOI] [PubMed] [Google Scholar]
- 13.Haynes RD, Meagher RJ, Won JI, Bogdan FM, Barron AE. Bioconjugate Chem. 2005;16:929–938. doi: 10.1021/bc0496915. [DOI] [PubMed] [Google Scholar]
- 14.Reddy MM, Kodadek T. Proc Natl Acad Sci U S A. 2005;102:12672–12677. doi: 10.1073/pnas.0501208102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pirrung MC, Park K, Tumey LN. J Comb Chem. 2002;4:329–344. doi: 10.1021/cc010083v. [DOI] [PubMed] [Google Scholar]
- 16.Maayan G, Ward MD, Kirshenbaum K. Proc Natl Acad Sci U S A. 2009;106:13679–13684. doi: 10.1073/pnas.0903187106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Houghten RA, Pinilla C, Giulianotti MA, Appel JR, Dooley CT, Nefzi A, Ostresh JM, Yu Y, Maggiora GM, Medina-Franco JL, Brunner D, Schneider J. J Comb Chem. 2008;10:3–19. doi: 10.1021/cc7001205. [DOI] [PubMed] [Google Scholar]
- 18.Le GT, Abbenante G, Becker B, Grathwohl M, Halliday J, Tometzki G, Zuegg J, Meutermans W. Drug Discov Today. 2003;8:701–709. doi: 10.1016/s1359-6446(03)02751-x. [DOI] [PubMed] [Google Scholar]
- 19.Trabocchi A, Menchi G, Guarna F, Machetti F, Scarpi D, Guarna A. Synlett. 2006:331–353. [Google Scholar]
- 20.Herman RE, Badders D, Fuller M, Makienko EG, Houston ME, Jr, Quay SC, Johnson PH. J Biol Chem. 2007;282:9813–9824. doi: 10.1074/jbc.M610722200. [DOI] [PubMed] [Google Scholar]
- 21.Gellman SH. Acc Chem Res. 1998;31:173–180. [Google Scholar]
- 22.Wu CW, Sanborn TJ, Huang K, Zuckermann RN, Barron AE. J Am Chem Soc. 2001;123:6778–6784. doi: 10.1021/ja003154n. [DOI] [PubMed] [Google Scholar]
- 23.Wu CW, Sanborn TJ, Zuckermann RN, Barron AE. J Am Chem Soc. 2001;123:2958–2963. doi: 10.1021/ja003153v. [DOI] [PubMed] [Google Scholar]
- 24.Sanborn TJ, Wu CW, Zuckermann RN, Barron AE. Biopolymers. 2002;63:12–20. doi: 10.1002/bip.1058. [DOI] [PubMed] [Google Scholar]
- 25.Armand P, Kirshenbaum K, Goldsmith RA, Farr-Jones S, Barron AE, Truong KT, Dill KA, Mierke DF, Cohen FE, Zuckermann RN, Bradley EK. Proc Natl Acad Sci U S A. 1998;95:4309–4314. doi: 10.1073/pnas.95.8.4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu CW, Kirshenbaum K, Sanborn TJ, Patch JA, Huang K, Dill KA, Zuckermann RN, Barron AE. J Am Chem Soc. 2003;125:13525–13530. doi: 10.1021/ja037540r. [DOI] [PubMed] [Google Scholar]
- 27.Armand P, Kirshenbaum K, Falicov A, Dunbrack RL, Jr, Dill KA, Zuckermann RN, Cohen FE. Fold Des. 1997;2:369–375. doi: 10.1016/S1359-0278(97)00051-5. [DOI] [PubMed] [Google Scholar]
- 28.Shin SBY, Kirshenbaum K. Org Lett. 2007;9:5003–5006. doi: 10.1021/ol702207n. [DOI] [PubMed] [Google Scholar]
- 29.Kirshenbaum K, Barron AE, Goldsmith RA, Armand P, Bradley EK, Truong KT, Dill KA, Cohen FE, Zuckermann RN. Proc Natl Acad Sci U S A. 1998;95:4303–4308. doi: 10.1073/pnas.95.8.4303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shah NH, Butterfoss GL, Nguyen K, Yoo B, Bonneau R, Rabenstein DL, Kirshenbaum K. J Am Chem Soc. 2008;130:16622–16632. doi: 10.1021/ja804580n. [DOI] [PubMed] [Google Scholar]
- 31.Huang K, Wu CW, Sanborn TJ, Patch JA, Kirshenbaum K, Zuckermann RN, Barron AE, Radhakrishnan I. J Am Chem Soc. 2006;128:1733–1738. doi: 10.1021/ja0574318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gorske BC, Bastian BL, Geske GD, Blackwell HE. J Am Chem Soc. 2007;129:8928–8929. doi: 10.1021/ja071310l. [DOI] [PubMed] [Google Scholar]
- 33.Hodges JA, Raines RT. Org Lett. 2006;8:4695–4697. doi: 10.1021/ol061569t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Still WC, Kahn M, Mitra A. J Org Chem. 1978;43:2923–2925. [Google Scholar]
- 35.Goddard TD, Kneller DG. SPARKY, v. 3.110. University of California; San Francisco: [Google Scholar]
- 36.Gorske BC, Jewell SA, Guerard EJ, Blackwell HE. Org Lett. 2005;7:1521–1524. doi: 10.1021/ol0502984. [DOI] [PubMed] [Google Scholar]
- 37.Gorske BC, Blackwell HE. J Am Chem Soc. 2006;128:14378–14387. doi: 10.1021/ja065248o. [DOI] [PubMed] [Google Scholar]
- 38.Boyer RD, Johnson R, Krishnamurthy K. J Magn Reson. 2003;165:253–259. doi: 10.1016/j.jmr.2003.08.009. [DOI] [PubMed] [Google Scholar]
- 39.Fowler SA, Luechapanichkul R, Blackwell HE. J Org Chem. 2009;74:1440–1449. doi: 10.1021/jo8023363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Toth G, Koever KE, Murphy RF, Lovas S. J Phys Chem B. 2004;108:9287–9296. [Google Scholar]
- 41.Gamez P, Mooibroek TJ, Teat SJ, Reedijk J. Acc Chem Res. 2007;40:435–444. doi: 10.1021/ar7000099. [DOI] [PubMed] [Google Scholar]
- 42.Egli M, Sarkhel S. Acc Chem Res. 2007;40:197–205. doi: 10.1021/ar068174u. [DOI] [PubMed] [Google Scholar]
- 43.Qian X, Xu X, Li Z, Frontera A. Chem Phys Lett. 2003;372:489–496. [Google Scholar]
- 44.Madison V, Kopple KD. J Am Chem Soc. 1980;102:4855–4863. [Google Scholar]
- 45.Cubberley MS, Iverson BL. J Am Chem Soc. 2001;123:7560–7563. doi: 10.1021/ja015817m. [DOI] [PubMed] [Google Scholar]
- 46.Hill DJ, Moore JS. Proc Natl Acad Sci U S A. 2002;99:5053–5057. doi: 10.1073/pnas.072642799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hinderaker MP, Raines RT. Protein Sci. 2003;12:1188–1194. doi: 10.1110/ps.0241903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bretscher LE, Jenkins CL, Taylor KM, DeRider ML, Raines RT. J Am Chem Soc. 2001;123:777–778. doi: 10.1021/ja005542v. [DOI] [PubMed] [Google Scholar]
- 49.Hodges JA, Raines RT. J Am Chem Soc. 2003;125:9262–9263. doi: 10.1021/ja035881z. [DOI] [PubMed] [Google Scholar]
- 50.Kumin M, Sonntag LS, Wennemers H. J Am Chem Soc. 2007;129:466–467. doi: 10.1021/ja067148o. [DOI] [PubMed] [Google Scholar]
- 51.Neuvonen H, Neuvonen K, Koch A, Kleinpeter E, Pasanen P. J Org Chem. 2002;67:6995–7003. doi: 10.1021/jo020121c. [DOI] [PubMed] [Google Scholar]
- 52.Bromilow J, Brownlee RTC, Craik DJ, Fiske PR, Rowe JE, Sadek M. J Chem Soc, Perkin Trans. 1981;2:753–759. [Google Scholar]
- 53.DeRider ML, Wilkens SJ, Waddell MJ, Bretscher LE, Weinhold F, Raines RT, Markley JL. J Am Chem Soc. 2002;124:2497–2505. doi: 10.1021/ja0166904. [DOI] [PubMed] [Google Scholar]
- 54.Choudhary A, Gandla D, Krow GR, Raines RT. J Am Chem Soc. 2009;131:7244–7246. doi: 10.1021/ja901188y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wiberg KB, Laidig KE. J Am Chem Soc. 2002;109:5935–5943. [Google Scholar]
- 56.Wiberg KB. Origin of the Amide Rotational Barrier. In: Greenberg A, Breneman CM, Liebman JF, editors. The Amide Linkage: Structural Significance in Chemistry, Biochemistry, and Materials Science. Wiley-IEEE; New York: 2002. pp. 33–46. [Google Scholar]
- 57.Eberhardt ES, Loh SN, Hinck AP, Raines RT. J Am Chem Soc. 1992;114:5437–5439. doi: 10.1021/ja00039a072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nakanishi H, Fujita H, Yamamoto O. Bull Chem Soc Jpn. 1978;51:214–218. [Google Scholar]
- 59.Cox C, Lectka T. J Org Chem. 1998;63:2426–2427. doi: 10.1021/jo9800863. [DOI] [PubMed] [Google Scholar]
- 60.Kemnitz CR, Loewen MJ. J Am Chem Soc. 2007;129:2521–2528. doi: 10.1021/ja0663024. [DOI] [PubMed] [Google Scholar]
- 61.Gordy W, Stanford SC. J Chem Phys. 1941;9:204–214. [Google Scholar]
- 62.Stewart WE, Siddall TH. Chem Rev. 1970;70:517–551. [Google Scholar]
- 63.Yang D, Li W, Qu J, Luo SW, Wu YD. J Am Chem Soc. 2003;125:13018–13019. doi: 10.1021/ja036136p.and references therein
- 64.Wagner G, Pardi A, Wuethrich K. J Am Chem Soc. 1983;105:5948–5949. [Google Scholar]
- 65.Jordan T, Mukerji I, Wang Y, Spiro TG. J Mol Struct. 1996;379:51–64. [Google Scholar]
- 66.We note that the ammonium groups at the N-termini of the analogous, unacylated peptoids (see reference 37) would also be expected to form strong hydrogen bonds that could account for the similar overall Kcis/trans values observed for these systems.
- 67.Lee BC, Zuckermann RN, Dill KA. J Am Chem Soc. 2005;127:10999–11009. doi: 10.1021/ja0514904. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.