

Abstract
We describe extracellular interactions between fibronectin (Fn) and vascular endothelial growth factor (VEGF) that influence integrin-growth factor receptor crosstalk and cellular responses. In previous work, we found that VEGF bound specifically to fibronectin (Fn) but not vitronectin or collagens. Herein we report that VEGF binds to the heparin-II domain of Fn and that the cell-binding and VEGF-binding domains of Fn, when physically linked, are necessary and sufficient to promote VEGF-induced endothelial cell proliferation, migration, and Erk activation. Using recombinant Fn domains, the C-terminal heparin-II domain of Fn (type III repeats 13 to 14) was identified as a key VEGF-binding site. Mutation of the heparin-binding residues on FnIII13–14 abolished VEGF binding, and peptides corresponding to the heparin-binding sequences in FnIII13–14 inhibited VEGF binding to Fn. Fn fragments containing both the α5β1 integrin-binding domain (III 9 to 10) and the VEGF-binding domain (III 13 to 14) significantly enhanced VEGF-induced EC migration and proliferation and induced strong phosphorylation of the VEGF receptor and Erk. Neither the cell-binding or VEGF-binding fragment of Fn alone had comparable VEGF-promoting effects. These results suggest that the mechanism of VEGF/Fn synergism is mediated extracellularly by the formation of a novel VEGF/Fn complex requiring both the cell-binding and VEGF-binding domains linked in a single molecular unit. These data also highlight a new function for the Fn C-terminal heparin-binding domain that may have important implications for angiogenesis and tumor growth.
Keywords: endothelial cell differentiation, endothelial cell growth, endothelial cells, fibronectin, integrins, signal transduction, vascular endothelial growth factor, vascular endothelial growth factor receptors
Fibronectin (Fn) and vascular endothelial growth factor (VEGF or VEGF-A) are key regulators of blood vessel growth.1–3 Gene deletion studies have demonstrated that both Fn and VEGF, and their cognate receptors α5β1 and VEGF receptor-2 (VEGFR-2), are critical for vascular development.4–9 During vascular growth, endothelial cells (ECs) are recruited into a tightly controlled program of cell activation, gene expression, adhesion, motility, and proliferation.10,11 Crosstalk between integrins and growth factor receptors (receptor tyrosine kinases, RTKs) is a key part of this control process.12–16 EC responses to growth factors like VEGF are modulated by the outside-in signals conveyed from integrins, reflecting the conditions of the extracellular milieu. Hence, the local mixture of extracellular matrix (ECM) proteins as well as the type and density of integrins expressed by the cell may modulate its responses to growth factors. Integrin engagement by a specific matrix protein can transactivate VEGF receptors, as shown by Groopman and colleagues for fibronectin (Fn) and VEGF receptor-3, via α5β1,17 and by Moro et al for epidermal growth factor receptor.18 Others have shown that β1 activation by Fn can modulate VEGF responses,19 and that β3 activation may increase VEGF production in tumor cells.20 Conversely, occupancy of VEGFR-2 by VEGF can activate integrins. Byzova et al have shown that VEGF activates both αvβ3 and α5β1 on ECs.21 Finally, integrins and receptor tyrosine kinases (RTKs) share a number of important signaling molecules, including Src, FAK, and phosphatidylinositol 3-kinase (PI3K).13 Our previous work has focused on the specificity of interactions between Fn and VEGF, and their cognate receptors, α5β1 and VEGFR-2.22,23 We found VEGF bound to Fn and that platelets secreted VEGF/Fn complexes. In the presence of the ligands VEGF and Fn, their corresponding receptors coimmunoprecipitated. Fn, but not vitronectin, induced sustained mitogen-activated protein kinase (MAPK) activation in response to VEGF. Thus we predicted that the VEGF-enhancing activities of Fn were attributable, in part, to its binding to VEGF.
This present study was undertaken to more precisely map the binding site(s) for VEGF within the C-terminal regions of Fn and to determine the mechanisms of Fn-induced enhancement of VEGF activity. Using recombinant Fn fragments, and synthetic peptides, we have mapped the VEGF-binding site to the established heparin-II (Hep-II) site of Fn type III modules 13 to 14.24,25 This VEGF-binding domain of Fn was found to be necessary but, by itself, was not sufficient for the promotion of VEGF biological activity. Only bivalent Fn constructs encompassing both the α5β1 and VEGF-binding domains significantly promoted endothelial migration, proliferation, and sustained Erk phosphorylation. Purely costimulatory signaling (via occupancy of integrin and VEGFR-2 receptors) was a weaker influence on VEGF activity, because mixtures of VEGF with monovalent Fn fragments containing cell- and VEGF-binding domains showed less migration, proliferation, and no late phase of Erk phosphorylation.
Materials and Methods
Cell Culture
Human umbilical vein ECs (HUVECs) (Cascade Biologics) were maintained in MCDB-131 growth medium (MCDB-131 medium containing 5% FBS, 2 ng/mL basic fibroblast growth factor [bFGF], 10 ng/mL VEGF, and 10 μg/mL heparin). HUVECs between passage 3 to 8 were used for experiments.
cDNA Construction
Human Fn cDNA was obtained by RT-PCR using human liver mRNA and sequenced to verify no base changes during PCR amplification. The Fn type III modules were obtained by PCR amplification using Fn cDNA as template. We used the following nomenclature for describing the recombinant Fn constructs: rFnIII9–10 indicates recombinant Fn protein encompassing type III repeats 9 to 10; rFnIII9–10/12–14, a single protein encompassing type III repeats 9 to 14, omitting 11. A rFnIII12–14 with the heparin-binding sites on FnIII domains 13 and 14 mutated (Arg or Lys→Ser, termed “gag AC”) was obtained as previously described.26 For details on the construction of the rFn plasmids and their expression and purification, please see the online data supplement, available at http://circres.ahajournals.org.
EC Migration Assay
EC chemotactic migration to VEGF (10 ng/mL; upper chamber) was studied in the presence of different rFn fragments (0.2 μmol/L; bottom chamber). Ninety-six well ChemoTx microtiter plates (Neuro Probe Inc) were used, as previously described.22,27 Positive and negative controls included native Fn, vitronectin, or albumin all at 10 μg/mL, in the place of the tested rFn fragments.
Endothelial Proliferation Assay
Early-passage HUVECs (3000 cells/well) in 96-well plates were cultured in MCDB-131 medium containing fibronectin-depleted FBS (0.25%). Cells were incubated with albumin, VEGF alone (10 ng/mL), or VEGF with native Fn (10 μg/mL) or rFn fragments (0.2 μmol/L) for 72 hours. Heparin was used at a final concentration of 1 μg/mL. Cell growth was determined by using CyQuant reagent (Molecular Probes) according to the instructions of the manufacturer.
Western Blot Analyses
For analysis of Erk phosphorylation, 5×105 HUVECs were plated onto 60-cm2 culture dishes and incubated overnight in MCDB-131 growth medium. Cells were then washed twice and incubated for 4 hours in serum-free MCDB-131 medium. Cells were stimulated with VEGF (10 ng/mL) or VEGF with different Fn combinations. At different time points, cells were lysed and Erk phosphorylation detected as previously described.28 Signal densities were quantified by the NIH Image program, and the activation of phosphorylated protein was expressed as the fold increase over basal (unstimulated) levels, adjusted for total protein loading, measured from separate blots.
To measure VEGFR-2 phosphorylation, HUVECs (2×106 cells) were cultured for 48 hours in MCDB-131 growth medium. Before assay, cultures were washed and replaced with serum-free MCDB-131 containing ITS supplement (BD Bioscience) and incubated for a further 4 hours. Culture plates were then stimulated with a low dose of VEGF (1 ng/mL) in the absence or presence of rFNIII9–10/12–14 for 2 minutes at 37°C. Cell lysates were incubated with anti–VEGFR-2 rabbit monoclonal antibodies (Cell Signaling) for 16 hours at 4°C and immune complexes precipitated with protein G sepharose. Protein samples were reduced, separated by Bis-Tris PAGE gels, transferred to polyvinyl difluoride (PVDF) membranes, probed with anti-phospho VEGFR-2 antibodies and signals developed by chemiluminescence. Blots were stripped and reprobed with anti–VEGFR-2.
125I-VEGF Solid-Phase Binding Assays
The measurement of direct binding of 125I-VEGF to Fn fragments was performed as described previously.22 For peptide competition assays, the same binding assay methods were used, with recombinant C-terminal rFn III9–10/12–14 or rFnIII12–14 immobilized on the plates.
Surface Plasmon Resonance Analysis
Surface plasmon resonance analysis (SPR) binding experiments were conducted to measure the equilibrium binding parameters between VEGF165 and native fibronectin or rFnIII9–10/12–14. The detailed methods are described elsewhere.29 In brief, a 2-channel SPR670M apparatus (Moritex Corp) was used with a running buffer of PBS, 0.05% Tween-20 at a flow rate of 15 μL/min at room temperature. The matrix proteins were immobilized on the gold chip using N-hydroxysuccinimidyl coupling, sensograms recorded over a range of VEGF concentrations (25 to 800 nmol/L), and the kinetic binding parameters derived using the software of the manufacturer.
Statistical Analysis
Individual experiments were performed at least in triplicate, and the data presented are the means and SEM of at least 3 to 4 independent experiments, as noted. The Student’s t test was used to compare different treatments.
Results
Expression and Purification of Fibronectin Fragments
Figure 1 illustrates a schematic of the native fibronectin protein and the design of the recombinant fragments. Each protein was expressed and purified to homogeneity, as described in Materials and Methods. A Bis-Tris PAGE gel shows representative samples of the purified proteins.
Figure 1.

Schematic diagram of the domain structure of human Fn showing type I, type II, type III and their functions. Also diagramed are the recombinant Fn type III domains used in this study, and on the left is a representative SDS-PAGE gel of purified fragments.
VEGF Binding to Recombinant Fn Domains and Competition by Peptides
We first measured the concentration dependence of 125I-VEGF165 binding to different immobilized recombinant Fn fragments. These experiments were all conducted in the absence of exogenous heparin, indicating that heparin is not a required cofactor for VEGF/Fn binding. In preliminary experiments, the actual quantity of experimental proteins deposited on the plate was measured by protein assay, confirming that the final density of coating was comparable for all test proteins. As shown in Figure 2, VEGF bound rFnIII12–14 and rFnIII8–14 in a concentration-dependent manner. Binding to rFnIII8–11 was negligible. In subsequent binding assays, a coating concentration of 0.2 μmol/L was used for all proteins. The binding of VEGF to individual rFnIII domains was then evaluated. Figure 3A shows that VEGF does not bind significantly to any of the individual type III domains studied but did bind to all fragments containing FnIII13–14 in continuity (ie, native Fn, rFn8–14, rFn12–14). Figure 3A and 3B shows that the linkage of the α5β1 integrin-binding domain with rFnIII12–14 (as in the constructs rFnIII8–14 or III9–10/12–14) further enhanced VEGF binding when compared with rFnIII12–14 alone. The binding of VEGF to rFnIII8–14 and rFnIII9–10/12–14 when compared with native Fn was consistently higher, suggesting a higher affinity for these fragments. Surface plasmon resonance binding analysis was used to compare the VEGF affinities of rFnIII9 –10/12–14 to native Fn. The rFnIII9–10/12–14 fragment had a 43% lower dissociation constant (Kd, 73.5 versus 129.3 nmol/L), a faster on-rate and a smaller off-rate (supplemental Table I). Because the region of FnIII12–14 is known to be an important heparin-binding region,24,25,30 we wanted to determine whether the key heparin-binding residues of FnIII12–14 were required for binding VEGF. To that end, the construct rFnIII9–10/12–14gagAC was prepared, in which the lysines and arginines of repeats 13 and 14 were mutated to serine, as previously described.26 Figure 3B shows that the loss of these heparin-binding residues completely abolished VEGF binding.
Figure 2.
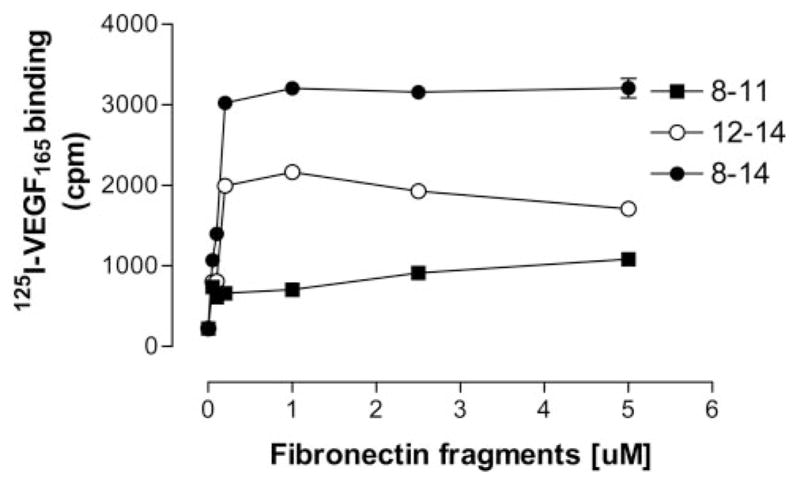
Concentration dependent 125I-VEGF binding to immobilized rFn fragments. 125I-VEGF was incubated for 2 hours on wells coated with increasing concentrations of rFn fragments. After washing, bound VEGF was eluted with 0.1 mol/L NaOH/1% SDS and radioactivity quantified in a gamma counter. Data are presented as the mean of triplicate determinations±SEM.
Figure 3.

Mapping the VEGF-binding site on C-terminal Fn. A and B, 125I-VEGF was incubated for 2 hours on wells coated with either individual or continuous rFn type III domains (0.2 μmol/L) (A) or wells coated with rFn molecules encompassing both the integrin (9 to 10) and heparin-binding domains (12 to 14) (B). GagAC denotes mutation of the heparin-binding domains on type III module 13 and 14. After washing, bound VEGF was eluted with 0.1 mol/L NaOH/1% SDS and radioactivity quantified in a gamma counter. Data are the means±SEM of 3 to 4 independent experiments performed in triplicate. Dotted lines are 95% confidence intervals for control binding to albumin.
Peptides (listed in the Table) encompassing the native heparin-binding regions in FnIII13–14 were also used as soluble competitors of 125I-VEGF binding to immobilized rFnIII12–14 or rFnIII9–10/12–14. Two other peptides were also studied: a truncated version containing the putative key heparin-binding site of FnIII13, as well as a well-characterized heparin-binding site from the A1 domain of von Willebrand factor (vWf)31 (Table). The results of peptide competition against VEGF binding to either rFnIII12–14, or rFnIII9–10/12–14 substrates were not significantly different, so the data were pooled. The Table shows their estimated IC50 (concentration that inhibits VEGF binding by 50%), and Figure 4 shows their inhibition curves. The most potent peptide was a heparin-binding region in FnIII14 (core motif is PRAR). Second and third in rank order were the heparin-binding peptides derived from FnIII13 (core motif PRRAR) and FnIII14 (IYVIALKNNQKSEPLIGRKKT), respectively. Truncating the heparin-binding domain of FnIII13 eliminated its ability to compete with the immobilized Fn fragment for binding VEGF. Figure 4B correlates the potency of each peptide competitor with its total number of basic residues. The peptide charge accounts for some, but not all, of the apparent affinity of the peptides for VEGF. The most potent Fn peptide bears the same net charge as the vWf heparin-binding peptide but has an IC50 that is 600 times lower. The other 2 Fn peptides that share the PRRAR or PRAR motif possess the same number of heparin-binding residues but differ in affinity by 3.5-fold.
Relative Inhibition of 125I-VEGF165 Binding by Heparin Binding Peptides
| Protein | Peptide Sequence | IC50 (μmol/L) |
|---|---|---|
| rFnIII13 | 1814 vspprrarvtdatettitiswrtktetitgfq | 43 |
| rFnIII13 (truncated) | 1814 pprrarv | >1000 |
| rFnIII14 | 1926 praritgyiikyekpgspprevvprprpgv | 0.4 |
| rFnIII14 | 1971 iyvialknnqksepligrkkt | 150 |
| von Willebrand factor | 568 kdrkrselrriasqvk | 250 |
Figure 4.

Peptide inhibition of VEGF binding to rFnIII9–10/12–14. 125I-VEGF/Fn binding assays were performed in the presence of the peptides listed in the Table. A, Results are expressed as the percentage of VEGF binding observed in the absence of competitor (100%) and are the means±SEM of 4 to 6 independent experiments performed in triplicate. B, IC50 (μmol/L) was interpolated for each peptide and plotted against the total number of basic residues/peptide.
Specificity of Fn Domains That Promote VEGF-Mediated EC Cell Migration and Proliferation
We hypothesized that the formation of a unique extracellular VEGF/Fn complex promotes VEGF biological activity, mediated in part by the bivalent capacity of Fn to simultaneously bind α5β1 and VEGF. Accordingly, we measured the migration and proliferation of HUVECs in response to VEGF, in the presence of different native and recombinant matrix proteins. Figure 5A shows that there was minimal cellular migration toward VEGF in the absence of a matrix component, or with the addition of rFnIII12–14 alone. Most of the proteins containing an integrin-binding domain (eg, vitronectin, rFn9–10, and rFn9–10/12–14gagAC) supported some migration to VEGF. In contrast, all bivalent matrices encompassing both the α5β1 and the VEGF-binding domains (FnIII8–14 and FnIII9–10/12–14) increased cell migration by 2- to 3-fold. To distinguish between simple costimulatory signaling through the independent ligands (ie, integrin and growth factor receptor), chemotactic migration to VEGF was also measured in the presence of equivalent concentrations of a mixture of the isolated domains of rFnIII9–10 and rFnIII12–14. This mixture did support a modicum of cell migration above baseline but did not increase migration to the extent induced by a bivalent Fn construct that linked both those domains.
Figure 5.

A, EC migration to VEGF in the presence of rFn fragments. HUVECs suspended in serum-free MCDB-131 medium containing 0.1% ovalbumin were seeded on to ChemoTx microtiter plates and migration assay performed as described in Materials and Methods. Experiments were performed in triplicate and results expressed as mean±SEM. The final bar (indicated by 9-10 + 12- 14) signifies treatment with a mixture of separate fragments of rFnIII9–10 and rFnIII12–14, each at 0.2 μmol/L. B, EC proliferation in response to VEGF and rFn fragments. HUVECs (3000 cells/well) in 96-well plates were incubated in MCDB131 medium containing Fn-depleted FBS (0.25%). Cells were incubated with VEGF alone (10 ng/mL), VEGF (V) with native Fn (5 ng/mL), or rFn fragments (0.2 μmol/L). In some experiments, heparin (hep) was added at a final concentration of 1 μg/mL. Cell growth was determined after 72 hours. Experiments were done in triplicate and results are expressed as mean±SEM.
We next examined whether rFn fragments promoted VEGF-induced HUVEC proliferation. As shown in Figure 5B, the extent of HUVEC growth in response to VEGF was enhanced in the presence of native Fn or rFnIII9–10/12–14, compared with the cell or VEGF-binding domains alone (rFnIII9–10 or rFnIII12–14). Addition of heparin (1 μg/mL) to the VEGF/Fn complex further augmented proliferation to the same extent heparin augmented proliferation of VEGF alone. A mixture of cell-binding and VEGF-binding fragments of Fn (at the same concentrations, 10 μg/mL) showed less increase in proliferation compared with the divalent fragment.
Modulation of VEGF-Mediated Signaling by Specific rFn Domains
Previously, our laboratory reported that VEGF/Fn complexes prolonged cell signaling induced by VEGF alone,22 suggesting that such extracellular complexes may influence the magnitude and duration of cell signaling. Therefore, we examined the time course and sensitivity of Erk signaling to VEGF and different Fn domains. As shown in Figure 6, VEGF combined with native Fn produced a higher early (5 minutes) peak in phosphorylation, as well as more sustained activation at 1.5 to 3 hours. In comparison with native Fn, the rFnIII9–10/12–14 induced an even higher early peak and a second peak of Erk activation at approximately 1.5 hour. A mixture of rFnIII9–10 and rFnIII12–14 (at the same individual concentrations as other fragments tested) did not induce any late Erk activation. When tested individually, the cell binding (rFnIII9–10) or VEGF-binding domains (rFnIII12–14) did not induce any late Erk activation. The divalent recombinant Fn fragment rFnIII9 –10/12–14, significantly enhanced VEGF-induced Erk phosphorylation even at very low VEGF concentrations, between 0.1 to 0.5 ng/mL (Figure 7A and 7B). We next examined the influence of rFnIII9–10/12–14 on the phosphorylation status of VEGFR-2 activated by a low dose of VEGF. Figure 7C shows that VEGF alone (1 ng/mL) did not significantly induce VEGFR-2 phosphorylation. However, in the presence of rFnIII9–10/12–14, significant VEGFR-2 phosphorylation was observed. Stimulation of cells with VEGF and either rFnIII9–10 or rFnIII12–14 did not show enhanced VEGFR-2 phosphorylation.
Figure 6.

HUVEC Erk phosphorylation in response to VEGF and Fn moieties. HUVECs were exposed to 10 ng/mL of VEGF165, combined with the indicated Fn proteins (0.2 μmol/L). At the time points, cells were harvested and processed to measure phospho-Erk and total Erk protein by Western blotting. Each point on the graph is the mean of data obtained from 4 to 5 separate experiments and quantified as described in Materials and Methods.
Figure 7.

rFnIII9–10/12–14 promotes Erk and VEGFR-2 phosphorylation at low VEGF concentration. A and B, HUVECs were exposed to varying concentrations of VEGF in the absence (A) or presence (B) of rFnIII9–10/12–14 (0.2 μmol/L). Erk phosphorylation was determined by Western blotting. A representative blot from 2 separate experiments is shown. C, HUVEC cultures were stimulated with a low dose of VEGF (1 ng/mL) in the absence or presence of recombinant Fn fragments (0.2 μmol/L). Cell lysates were immunoprecipitated with anti–VEGFR-2 rabbit monoclonal antibodies followed by Western blotting with anti-phosphoVEGFR-2 rabbit monoclonal antibodies. Blots were stripped and reprobed with anti–VEGFR-2 antibodies. Experiments were repeated twice with similar results.
Discussion
Based on our previous studies, we hypothesized that Fn may be a unique biological partner with VEGF: when Fn complexes with VEGF, their coordinated binding to their cognate receptors enhance the specific cellular responses to VEGF. These extracellular events might be an important step in modulating the complex signaling pathways that lead from receptor ligation to cellular response. In this report we have physically mapped a key Fn-binding site for VEGF to the fibronectin C-terminal domain within type III repeats 13 to 14 (the Hep-II domain). Previous studies have established this site as a heparin-binding domain.24,25,30 The Hep-II site is a major syndecan-binding site that plays a role in focal adhesion and stress fiber formation.32 This report reveals a novel function for the Hep-II region as a site for VEGF binding, as well as a modulator of VEGF activity. Narrowing the binding site further, we observed that a peptide obtained from the N-terminal part of FnIII14 was most effective at blocking VEGF binding, although other peptides representing heparin-binding sites were also inhibitory. Also, the heparin-binding properties of this Fn domain accounted for a significant portion of VEGF affinity. A heparin-mutant fragment did not bind VEGF, nor did it promote the biological activities of VEGF.
The second important finding from these experiments concerns the mechanism(s) of biological synergism between VEGF and Fn. We found that the VEGF-binding domain of Fn alone was not sufficient to enhance VEGF-mediated cell migration, proliferation, or growth factor signaling, even though it avidly bound VEGF. However, constructs of rFn in which the cell binding and VEGF-binding domains were physically linked dramatically enhanced these cellular responses, even more so than native Fn. rFn fragments containing only the cell-binding domain (FnIII9–10) did modestly enhance VEGF biological responses. These results may reflect the isolated contribution of the costimulatory signaling pathways that are known to exist between α5β1 and VEGFR-2 downstream to Erk. However, the current data suggest that these cooperative pathways are capable of a much more robust response when their receptors are occupied by a specialized VEGF/Fn complex. The rFnIII9–10/12–14 construct dramatically increased early Erk phosphorylation in response to VEGF, as well as inducing a major late peak in Erk activation at 1.5 hour. Thus, the coordinated extracellular modulation of these ligand/receptor interactions imposes more stringent directions on the intracellular signaling responses to VEGF.
Several lines of evidence suggest that the binding interaction between VEGF and the Hep-II domain are complex. VEGF did not bind to any single isolated immobilized type III repeat, yet VEGF bound to the Fn moieties containing the type III12–14 repeats in continuity. This suggests that a more complex tertiary structure within the Hep-II domain is necessary for VEGF binding. We observed that the constructs rFnIII8–14 and rFnIII9–10/12–14 bound more VEGF than rFnIII12–14 alone. If FnIII9–10 itself has no intrinsic VEGF-binding properties (as we observed), why should the larger construct that includes FIII9–10 bind VEGF more avidly? These larger constructs (FnIII8–14 and FnIII9–10/12–14) likely assume a conformation that more optimally displays the VEGF-binding sites. This is supported by the surface plasmon resonance studies, which showed that rFnIII9–10/12–14 has a higher affinity for VEGF than native Fn. The bivalent Fn constructs encompassing both the integrin and VEGF-binding sites were also more biologically effective than native Fn. This again may be attributable to conformational differences, or their lack of large portions of the N and C-terminal regions of native Fn, which may play a negative modulatory role on VEGF function.
The heparin-binding properties of the FnIII12–14 domain are clearly important, as illustrated by the failure of the heparin mutant fragment to bind or modulate VEGF activity. An alternative explanation for the behavior of the heparin mutant protein might be that the mutations altered the conformation of the Fn fragment. Weighing against that possibility are the studies of Mostafavi-Pour, which suggest that these mutations do not seriously alter the conformation of the protein.26 Also, conformational changes of the mutant (or wild-type recombinant) protein would not explain why peptides selected from these heparin-binding domains successfully competed with rFnIII9–10/12–14 for binding VEGF.
However, the peptide competition experiments also suggest that heparin-binding is not the entire VEGF/Fn binding story. The heparin-binding site in III13 is thought to be the dominant interactive site for heparins,24,25 yet its peptide domain was not the most potent at blocking VEGF binding, nor did its truncated form (the core heparin-binding residues) retain activity. Likewise, comparisons of peptide charge versus inhibitory potency suggest that nonelectrostatic, protein/protein interactions also account for VEGF/Fn binding. The vWf heparin-binding peptide, a highly charged, classical heparin-binding domain, had relatively low affinity for VEGF. This same vWf protein domain has been shown to interfere with cell adhesive strength and chemokinesis, presumably because of its interference with cell syndecan binding to the Hep-II domain of Fn.33 Thus, this vWf heparin-binding domain may mimic the Fn Hep-II region, from the viewpoint of cell surface syndecans, but not from the perspective of VEGF. Finally, exogenous heparin was not required for rFn-VEGF binding (Figures 2 and 3). Heparin had the same mild stimulatory effect on the biological activity of rFn/VEGF complexes as its effect on VEGF alone (which is well described).
A number of mechanisms have been suggested for integrin/growth factor synergism, many emphasizing pathways for intracellular signaling crosstalk. Signaling from α5β1 ligation (and syndecan engagement) can support or reinforce the downstream signaling from VEGFR-2 to Erk.22 Conversely, VEGFR-2 activation can lead to integrin activation.21 There exists some controversy as to whether the primary integrin synergizing with VEGF pathways is α5β1 or αvβ3.21,34 This may be attributable to the type of ECM protein the cells are exposed to when stimulated with VEGF. Our previous studies suggest that VEGF/Fn complexes primarily cause a physical association between VEGFR-2 and α5β1, but not αvβ3.22 Moreover, we have also shown that VEGF/Fn complexes, as well as hepatocyte growth factor/Fn complexes, exist and are released from thrombin stimulated degranulating platelets.22,35 These observations suggest that platelets may play a role in promoting angiogenesis. Recent studies by Kisucka et al have characterized the role platelets play in modulating angiogenesis in vivo.36 We also observed that blockade of α5β1 profoundly inhibits VEGF-mediated migration and differentiation of endothelial progenitor cells.22,23 These findings support the view espoused by Hynes and colleagues that α5β1 is a pivotal integrin for vascular development.6,37,38 The current experiments suggest that the mechanism of Fn-induced enhancement of VEGF activity arises from both the formation of an extracellular complex interacting with the cell surface receptors and the resulting promotion of costimulatory signaling from integrin and VEGF receptor. Simple engagement of integrin and growth factor receptors by free, independent ligands does not induce the same quality or quantity of signaling, especially the late sustained Erk phosphorylation. In support of our hypothesis, recent studies of a parallel system demonstrated that binding of basic fibroblast growth factor to fibrinogen is required for the enhancement of basic fibroblast growth factor induced angiogenesis.39
More work will be needed to identify the interactive sites within VEGF and to define the role of the CS-1 domain of Fn also remains to be elucidated in this context. Further understanding of the structure/function relationship of these synergisms should permit the development and optimization of Fn-derived constructs that specifically amplify (or inhibit) the biological actions of VEGF.
Supplementary Material
Acknowledgments
Sources of Funding
This research was supported by grants from the NIH (R01HL39903 and HL079182 to M.S.), the Department of Veterans Affairs Medical Research Service (to M.S.), the American Heart Association (to E.S.W.), and the Japan Science and Technology Agency (to Y.S.).
Footnotes
Disclosures
None.
References
- 1.Dvorak HF. Angiogenesis: update 2005. J Thromb Haemost. 2005;3:1835–1842. doi: 10.1111/j.1538-7836.2005.01361.x. [DOI] [PubMed] [Google Scholar]
- 2.Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669–676. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- 3.Hynes RO, Bader BL, Hodivala-Dilke K. Integrins in vascular development. Braz J Med Biol Res. 1999;32:501–510. doi: 10.1590/s0100-879x1999000500002. [DOI] [PubMed] [Google Scholar]
- 4.Carmeliet P, Ferreira V, Breier G, Pollefeyt S, Kieckens L, Gertsenstein M, Fahrig M, Vandenhoeck A, Harpal K, Eberhardt C, Declercq C, Pawling J, Moons L, Collen D, Risau W, Nagy A. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature. 1996;380:435–439. doi: 10.1038/380435a0. [DOI] [PubMed] [Google Scholar]
- 5.Ferrara N, Carver-Moore K, Chen H, Dowd M, Lu L, O’Shea KS, Powell-Braxton L, Hillan KJ, Moore MW. Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature. 1996;380:439–442. doi: 10.1038/380439a0. [DOI] [PubMed] [Google Scholar]
- 6.Francis SE, Goh KL, Hodivala-Dilke K, Bader BL, Stark M, Davidson D, Hynes RO. Central roles of alpha5beta1 integrin and fibronectin in vascular development in mouse embryos and embryoid bodies. Arterioscler Thromb Vasc Biol. 2002;22:927–933. doi: 10.1161/01.atv.0000016045.93313.f2. [DOI] [PubMed] [Google Scholar]
- 7.George EL, Baldwin HS, Hynes RO. Fibronectins are essential for heart and blood vessel morphogenesis but are dispensable for initial specification of precursor cells. Blood. 1997;90:3073–3081. [PubMed] [Google Scholar]
- 8.Shalaby F, Rossant J, Yamaguchi TP, Gertsenstein M, Wu XF, Breitman ML, Schuh AC. Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature. 1995;376:62–66. doi: 10.1038/376062a0. [DOI] [PubMed] [Google Scholar]
- 9.Yang JT, Rayburn H, Hynes RO. Embryonic mesodermal defects in alpha 5 integrin-deficient mice. Development. 1993;119:1093–1105. doi: 10.1242/dev.119.4.1093. [DOI] [PubMed] [Google Scholar]
- 10.Davis GE, Senger DR. Endothelial extracellular matrix: biosynthesis, remodeling, and functions during vascular morphogenesis and neovessel stabilization. Circ Res. 2005;97:1093–1107. doi: 10.1161/01.RES.0000191547.64391.e3. [DOI] [PubMed] [Google Scholar]
- 11.Folkman J, Shing Y. Angiogenesis. J Biol Chem. 1992;267:10931–10934. [PubMed] [Google Scholar]
- 12.Yamada KM, Even-Ram S. Integrin regulation of growth factor receptors. Nat Cell Biol. 2002;4:E75–E76. doi: 10.1038/ncb0402-e75. [DOI] [PubMed] [Google Scholar]
- 13.Eliceiri BP. Integrin and growth factor receptor crosstalk. Circ Res. 2001;89:1104–1110. doi: 10.1161/hh2401.101084. [DOI] [PubMed] [Google Scholar]
- 14.Miyamoto S, Teramoto H, Gutkind JS, Yamada KM. Integrins can collaborate with growth factors for phosphorylation of receptor tyrosine kinases and MAP kinase activation: roles of integrin aggregation and occupancy of receptors. J Cell Biol. 1996;135:1633–1642. doi: 10.1083/jcb.135.6.1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schwartz MA, Ginsberg MH. Networks and crosstalk: integrin signalling spreads. Nat Cell Biol. 2002;4:E65–E68. doi: 10.1038/ncb0402-e65. [DOI] [PubMed] [Google Scholar]
- 16.Sieg DJ, Hauck CR, Ilic D, Klingbeil CK, Schafer E, Damsky CH, Schlaepfer DD. Fak integrates growth-factor and integrin signals to promote cell migration. Nature Cell Biology. 2000;2:249–256. doi: 10.1038/35010517. [DOI] [PubMed] [Google Scholar]
- 17.Wang JF, Zhang XF, Groopman JE. Stimulation of beta 1 integrin induces tyrosine phosphorylation of vascular endothelial growth factor receptor-3 and modulates cell migration. J Biol Chem. 2001;276:41950–41957. doi: 10.1074/jbc.M101370200. [DOI] [PubMed] [Google Scholar]
- 18.Moro L, Venturino M, Bozzo C, Silengo L, Altruda F, Beguinot L, Tarone G, Defilippi P. Integrins induce activation of EGF receptor: role in MAP kinase induction and adhesion-dependent cell survival. EMBO J. 1998;17:6622–6632. doi: 10.1093/emboj/17.22.6622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Podar K, Tai Y, Lin BK, Narisimhan RP, Sattler M, Kijima T, Salgia R, Gupta D, Chauhan D, Anderson KC. Vascular endothelial growth factor-induced migration of multiple myeloma cells is associated with b1 integrin- and phosphatidylinositol 3-kinase-dependent PKCA activation. J Biol Chem. 2002;277:7875–7881. doi: 10.1074/jbc.M109068200. [DOI] [PubMed] [Google Scholar]
- 20.De S, Razorenova O, McCabe NP, O’Toole T, Qin J, Byzova TV. VEGF-integrin interplay controls tumor growth and vascularization. Proc Natl Acad Sci U S A. 2005;102:7589–7594. doi: 10.1073/pnas.0502935102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Byzova TV, Goldman CK, Pampori N, Thomas KA, Bett A, Shattil SJ, Plow EF. A mechanism for modulation of cellular responses to VEGF: activation of the integrins. Mol Cell. 2000;6:851–860. [PubMed] [Google Scholar]
- 22.Wijelath ES, Murray J, Rahman S, Patel Y, Ishida A, Strand K, Aziz S, Cardona C, Hammond WP, Savidge GF, Rafii S, Sobel M. Novel vascular endothelial growth factor binding domains of fibronectin enhance vascular endothelial growth factor biological activity. Circ Res. 2002;91:25–31. doi: 10.1161/01.res.0000026420.22406.79. [DOI] [PubMed] [Google Scholar]
- 23.Wijelath ES, Rahman S, Murray J, Patel Y, Savidge G, Sobel M. Fibronectin promotes VEGF-induced CD34 cell differentiation into endothelial cells. J Vasc Surg. 2004;39:655–660. doi: 10.1016/j.jvs.2003.10.042. [DOI] [PubMed] [Google Scholar]
- 24.Barkalow FJ, Schwarzbauer JE. Localization of the major heparin-binding site in fibronectin. J Biol Chem. 1991;266:7812–7818. [PubMed] [Google Scholar]
- 25.Kimizuka F, Taguchi Y, Ohdate Y, Kawase Y, Shimojo T, Hashino K, Kato I, Sekiguchi K, Titani K. Production and characterization of functional domains of human fibronectin expressed in Escherichia coli. J Biochem (Tokyo) 1991;110:284–291. doi: 10.1093/oxfordjournals.jbchem.a123572. [DOI] [PubMed] [Google Scholar]
- 26.Mostafavi-Pour Z, Askari JA, Whittard JD, Humphries MJ. Identification of a novel heparin-binding site in the alternatively spliced IIICS region of fibronectin: roles of integrins and proteoglycans in cell adhesion to fibronectin splice variants. Matrix Biol. 2001;20:63–73. doi: 10.1016/s0945-053x(00)00131-1. [DOI] [PubMed] [Google Scholar]
- 27.Ishida A, Murray J, Saito Y, Kanthou C, Benzakour O, Shibuya M, Wijelath ES. Expression of vascular endothelial growth factor receptors in smooth muscle cells. J Cell Physiol. 2001;188:359–368. doi: 10.1002/jcp.1121. [DOI] [PubMed] [Google Scholar]
- 28.Nishibe T, Parry G, Ishida A, Aziz S, Murray J, Patel Y, Rahman S, Strand K, Saito K, Saito Y, Hammond WP, Savidge GF, Mackman N, Wijelath ES. Oncostatin M promotes biphasic tissue factor expression in smooth muscle cells: evidence for Erk-1/2 activation. Blood. 2001;97:692–699. doi: 10.1182/blood.v97.3.692. [DOI] [PubMed] [Google Scholar]
- 29.Suda Y, Arano A, Fukui Y, Koshida S, Wakao M, Nishimura T, Kusumoto S, Sobel M. Immobilization and clustering of structurally defined oligosaccharides for sugar chips: an improved method for surface plasmon resonance analysis of protein-carbohydrate interactions. Bioconjugate Chem. 2006 doi: 10.1021/bc0600620. In press. [DOI] [PubMed] [Google Scholar]
- 30.Lyon M, Rushton G, Askari J, Humphries M, Gallagher J. Elucidation of the structural features of heparan sulfate important for interaction with the hep-2 domain of fibronectin. J Biol Chem. 2000;275:4599–4606. doi: 10.1074/jbc.275.7.4599. [DOI] [PubMed] [Google Scholar]
- 31.Sobel M, Soler DF, Kermode JC, Harris RB. Localization and characterization of a heparin binding domain peptide of human von Willebrand factor. J Biol Chem. 1992;267:8857–8862. [PubMed] [Google Scholar]
- 32.Woods A, McCarthy JB, Furcht LT, Couchman JR. A synthetic peptide from the COOH-terminal heparin-binding domain of fibronectin promotes focal adhesion formation. Mol Biol Cell. 1993;4:605–613. doi: 10.1091/mbc.4.6.605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chon JH, Chaikof EL. A von Willebrand factor-derived heparin-binding peptide regulates cell-substrate adhesive strength and chemokinesis behavior. Biochim Biophys Acta. 2002;1542:195–208. doi: 10.1016/s0167-4889(01)00181-1. [DOI] [PubMed] [Google Scholar]
- 34.Soldi R, Mitola S, Strasly M, Defilippi P, Tarone G, Bussolino F. Role of alphavbeta3 integrin in the activation of vascular endothelial growth factor receptor-2. EMBO J. 1999;18:882–892. doi: 10.1093/emboj/18.4.882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rahman S, Patel Y, Murray J, Patel KV, Sumathipala R, Sobel M, Wijelath ES. Novel hepatocyte growth factor (HGF) binding domains on fibronectin and vitronectin coordinate a distinct and amplified Metintegrin induced signalling pathway in endothelial cells. BMC Cell Biol. 2005;6:8. doi: 10.1186/1471-2121-6-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kisucka J, Butterfield CE, Duda DG, Eichenberger SC, Saffaripour S, Ware J, Ruggeri ZM, Jain RK, Folkman J, Wagner DD. Platelets and platelet adhesion support angiogenesis while preventing excessive hemorrhage. Proc Natl Acad Sci U S A. 2006;103:855–860. doi: 10.1073/pnas.0510412103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hynes RO. A reevaluation of integrins as regulators of angiogenesis. Nat Med. 2002;8:918–921. doi: 10.1038/nm0902-918. [DOI] [PubMed] [Google Scholar]
- 38.Taverna D, Hynes RO. Reduced blood vessel formation and tumor growth in alpha5-integrin-negative teratocarcinomas and embryoid bodies. Cancer Res. 2001;61:5255–5261. [PubMed] [Google Scholar]
- 39.Sahni A, Khorana AA, Baggs RB, Peng H, Francis CW. FGF-2 binding to fibrin(ogen) is required for augmented angiogenesis. Blood. 2006;107:126–131. doi: 10.1182/blood-2005-06-2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.