

Abstract
The melanocortin MC4 receptor is a potential target for the development of drugs for both obesity and cachexia. Melanocortin MC4 receptor ligands known thus far are orthosteric agonists or antagonists, however the agonists, in particular, have generally exhibited unwanted side effects. For some receptors, allosteric modulators are expected to reduce side-effect profiles. To identify allosteric modulators of the melanocortin MC4 receptor, we created HEK293 cell lines coexpressing the human melanocortin MC4 receptor and a modified luciferase-based cAMP sensor. Monitoring luminescence as a readout of real-time intracellular cAMP concentration, we demonstrate this cell line is able to report melanocortin agonist responses, as well as inverse agonist response to the physiological AgRP peptide. Based on the MC4R-GLO cell line, we developed an assay that was shown to meet HTS standards (Z’=0.50). A pilot screen run on the Microsource Spectrum compound library (n= 2,000) successfully identified 62 positive modulators. This screen identified predicted families of compounds: β2AR agonists –the β2AR being endogenously expressed in HEK293 cells-, an adenylyl cyclase activator and finally a distribution of phosphodiesterase (PDE) inhibitors well characterized or recently identified. In this last category, we identified a structural family of coumarin-derived compounds (imperatorin, osthol and prenyletin), along with deracoxib, a drug in veterinary use for its COX2 inhibitory properties. This latter finding unveiled a new off-target mechanism of action for deracoxib as a PDE inhibitor. Overall, these data are the first report of an HTS for allosteric modulators for a Gs protein coupled receptor.
1. Introduction
The melanocortin circuitry of the CNS is a critical component of the adipostat (Cone, 2005). Activation of these circuits inhibits food intake and stimulates energy expenditure and thus the melanocortin MC4 receptor has been a target of the major pharmaceutical companies for the development of drugs for the treatment of common obesity (Wikberg and Mutulis, 2008). However, the first clinical trials of potent melanocortin MC4 receptor agonists failed due to pressor activity (Greenfield et al., 2009). Severe early onset obesity due to defective melanocortin signaling is linked, in up to 5% of cases, with non-synonymous coding mutations causing haploinsufficiency of the melanocortin MC4 receptor (Farooqi and O'Rahilly, 2006). It would not be unusual to expect that 10–30% of early onset childhood obesity may thus result from defective melanocortin signaling, assuming melanocortin MC4 receptor promoter mutations and mutations in additional genes in the pathway may ultimately be discovered. In the general population, these mutations are present at a frequency of around 0.6 % (Calton et al., 2009; Hinney et al., 2006). The majority of these mutations disrupt trafficking of receptors to the cell surface, rather than affinity for ligand (Govaerts et al., 2005). In contrast to common obesity, treatment of severe obesity due to melanocortin receptor haploinsufficiency may involve returning melanocortin MC4 receptor signaling levels to normal, without causing unwanted pressor activity, suggesting a possible application for allosteric modulators of the melanocortin MC4 receptor.
Alternative approaches in other receptor systems based on development of allosteric ligands provided promising results relative to orthosteric agonist agents (Conn et al., 2009; Kenakin; May et al., 2007). Due to their mechanism of action, allosteric molecules should display agonism in a more physiological temporo-spatial pattern and may have an increased selectivity amongst melanocortin receptor subtypes. Given the rather unique pharmacological profile of melanocortin MC4 receptor involving the physiological expression of both agonists (proopiomelanocortin products) and inverse agonists (agouti-related protein, AgRP) (Cone, 2005), one might speculate that a variety of compounds targeting allosteric(s) site(s) on melanocortin MC4 receptor might be identified.
So far, most cAMP assays in use are static, and based on the accumulation of cAMP in the presence of a PDE blocker to enhance sensitivity. These static assays preclude the study of any complex time-dependent pattern of response. Live cell real-time cAMP imaging techniques based on downstream cAMP targets such as PKA (Zhang et al., 2001), EPAC (DiPilato et al., 2004) or cyclic nucleotide-gated channels (Rich et al., 2001) are just emerging (Willoughby and Cooper, 2008). Based on these indirect cAMP readouts, to our knowledge, only a single high-throughput screen was documented using a PDE blocker (Titus et al., 2008). So far, no highly sensitive real-time high-throughput screening was documented, therefore precluding the identification of allosteric modulators by HTS. We therefore developed an assay of the human melanocortin MC4 receptor function based on real time cAMP detection, and validated this assay for high-throughput screening using a pilot screen designed to detect allosteric modulators.
2. Material and methods
2.1 Creation of the Human MC4R-GLO Cell Line
Human HEK293 cells were cotransfected with a plasmid encoding the human melanocortin MC4 receptor cDNA (pCDNA3.1 vector) and with a plasmid encoding an engineered cAMP sensitive luciferase (pGLO sensor™ - 20FcAMP plasmid, Promega) by the Lipofectamin method (Invitrogen). These cells were grown in 90% minimum essential medium (MEM), 10% fetal bovine serum (FBS), geneticin (700 µg/ml) and hygromycin B (200 µg/ml). Resistant clones were isolated, expanded and selected for their ability to respond to α-MSH. Briefly, the day before the experiment stable clones seeded in a 384 well plate in 10 µL of culture medium without antibiotics were incubated by adding 10 µL of the substrate containing media (GloSensor™ cAMP assay, Promega) diluted at 4% in CO2-independent medium (Gibco). The luminescence was recorded before and after injection of the drugs (α-MSH, forskolin or vehicle) for 15 min to obtain the maximal luminescent responses on a Spectramax M5 (Molecular Devices) plate reader (100 msec integration). The selected positive clones responding to both α-MSH and forskolin were further compared together to select a representative clone that was further characterized. Similar methods were used to develop a clone of cells expressing the human β2 adrenergic receptor (β2AR-GLO line), used to counterscreen compounds for receptor specificity
2.2 High-Throughput Screening on the hMC4R-GLO Line
HTS assay design- All assays were performed in the Vanderbilt Institute of Chemical Biology High-Throughput Screening Facility. 10,000 MC4R-GLO cells suspended in 10 µL of medium (MEM medium with 2% FBS without antibiotics) were plated in 384 well, black wall, poly-D- lysine coated optical bottom plates (BD Biosciences) by means of a liquid handler (Multidrop Combi, Thermo Fisher) and incubated overnight in the cell incubator at 37°C, 5% CO2. Exactly two h prior to the drug activation experiment, each individual cell plate received 10 µL of the GLO reagent at 4% in CO2-independent medium by means of the Bravo liquid dispenser (Agilent) and returned to the cell incubator. A few minutes prior to the end of the incubation period, the plate is transferred at 37°C in the Functional Drug Screening System 6000 (FDSS, Hamamatsu, Bridgewater, NJ) plate reader for a simultaneous luminescence recording of all 384 individual wells for 15 min (3 s integration time) and drug injections. Test compounds were transferred to daughter plates using an Echo acoustic plate reformatter (Labcyte, Sunnyvale, CA) and then diluted into CO2 independent medium using a Combi to generate a 50 µM stock solution. α-MSH, the orthosteric endogenous melanocortin MC4 receptor ligand, was diluted into CO2 independent medium to generate a 0.75 nM stock solution and loaded in a daughter plate. The drug injection protocol selected on the FDSS6000 for this allosteric screen protocol is as follows: 1) 5 µL injection of compound at 10 µM final concentration at time 5 min and 2) 5 µL injection of α-MSH at EC20 (0.15 nM final) at time 10 min. The overall assay protocol was automated using the instruments noted above integrated with a Thermo Fisher F3 robotic arm (Thermo Fisher Scientific) under the control of a Polara scheduler (Thermo Fisher Scientific). FDSS data were analyzed by means of a custom analysis application and were associated with unique compound identifiers based on liquid handler transfer logs and plate barcode readings captured by the Echo and by Polara.
2.3 Spectrum collection HTS screening
To validate our assay, a library of 2,000 compounds (Spectrum Collection, Microsource) composed of active drugs (50%), natural products (30%) and other bioactive components (20%) was run on our HTS melanocortin MC4 receptor assay. This library was assayed using a potentiator screen (EC20 injection protocol). Potentiator “hits” were selected by comparing the amplitude of the responses at the time of EC20 addition plus and minus test compounds. To confirm “hits”, initial concentration-response curve experiments were performed on serial dilutions of the compounds (1:3) transferred in daughter plates using the Echo instrument. These dilutions were again applied in the two injection protocol of the FDSS followed by α-MSH injection at EC20. Subsequent confirmation of concentration-response parameters on selected compounds were performed using independent serial dilutions from dry compounds, and data from multiple independent experiments were integrated and fit using a four-point logistical equation in Prism (GraphPad Software, Inc., San Diego, CA).
2.4. MAPK Phosphorylation Assay
MC4R-GLO cells were plated in 12 well culture dishes in DMEM/F12 supplemented with 5% fetal bovine serum. Cells were incubated in serum-free medium over night before the experiment. Cells were then treated with the indicated concentration of α-MSH or vehicle for 5 min at 37°C in the presence or absence of 10 µM deracoxib or imperatorin. Dishes were then placed on ice and cells were scraped in 1x LDS-PAGE sample buffer (invitrogen) with 100mMDTT. Proteins were resolved by SDS-PAGE, and phospho-ERK1/2 and total ERK1/2 were identified by immunoblotting with antibodies from Cell Signaling Technology (Beverly, MA) at 1:2500 dilution (mouse anti-total-ERK1/2 and rabbit anti-phospho-ERK1/2). Membranes were imaged using the Odyssey scanner (Li-Cor) to detect the fluorescently labeled secondary antibodies (anti-mouse 700 and anti-rabbit 800 from Li-Cor).
2.5. PDE Activity Assay
PDE activity assays were performed by addition of human recombinant PDE4D3, kindly provided by Dr. Marco Conti, to a reaction mixture (100 µl) containing 40 mM MOPS, pH 7, 0.8 mM EGTA, 15 mM magnesium acetate, 2 mg/ml bovine serum albumin (Sigma-Aldrich), and [3H]cAMP (Amersham) (0.075µM/assay tube) as substrate, as described in detail elsewhere (Blount et al., 2006). To determine IC50 values for various inhibitors, increasing concentrations of the respective inhibitors were diluted in the PDE assay mixture and then added to the assay; n=4 experiments for deracoxib and imperatorin; n=2 for rolipram.
3. Results
3.1. A Cell Line Expressing a cAMP-dependent Luminescent Reporter Monitors Human Melanocortin MC4 Receptor Signaling in Real Time
To monitor the cAMP response of the human melanocortin MC4 receptor in living cells, we generated clones of HEK293 cells stably coexpressing the human melanocortin MC4 receptor and a modified luciferase that carries the cAMP binding domain from the RIIβB subunit of cAMP dependent protein kinase (pGloSensor™, Promega). cAMP binding is required for the activity of this modified luciferase enzyme. Double transfectants were selected for further experiments based on their response to forskolin, as a measure of expression of the reporter, and to the melanocortin MC4 receptor agonist α-MSH, demonstrating the expression of a functional human melanocortin MC4 receptor at cell surface. All experiments were done using the hMC4R-GLO clone 4-10-02. As expected, this cell line displayed a strong luminescence response to α-MSH (Fig.1A). The response peaked after 5 minutes, and could be further augmented by treatment with the adenylyl cyclase activator forskolin, or inhibited by addition of the melanocortin MC4 receptor antagonist, SHU9119 (Hruby et al., 1995).
Fig. 1. Real-time monitoring of human melanocortin MC4 receptor signaling.
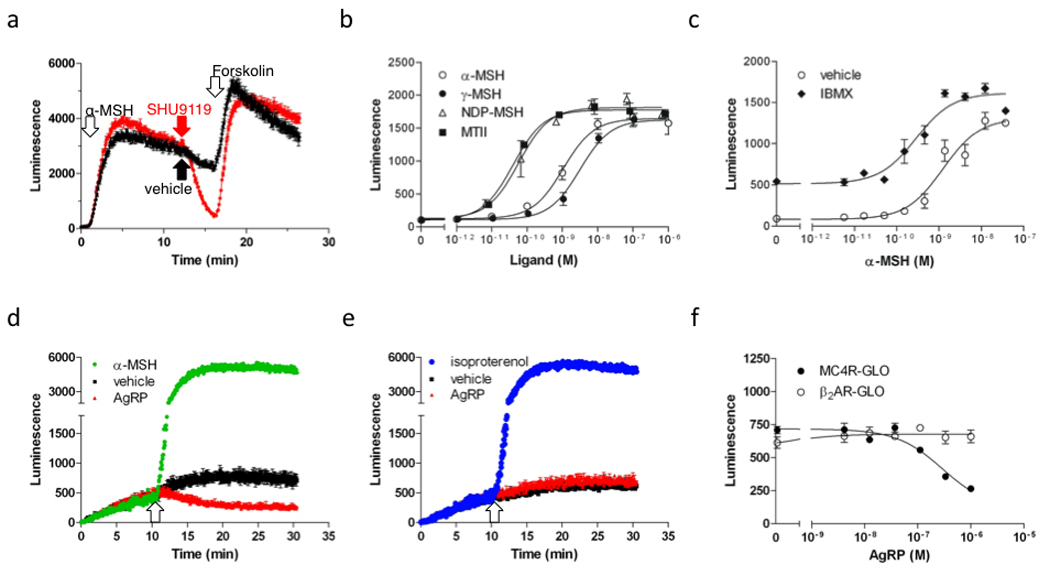
A, Real-time measurement of cAMP production in the MC4R-GLO cell line, as reported by luciferase luminescence, following successive treatments with the melanocortin agonist α-MSH (10 nM), the antagonist SHU9119 (1 µM) (in red trace) or vehicle (black trace) and the adenylyl cyclase activator forskolin (1 µM), at the times indicated by arrows. Luminescence recording was performed in 384 well plates using a FDSS6000 plate reader at 37°C for 26 min. Luminescence is reported as the average of quadruplicates ±SEM for a representative experiment. B, Measurement of real-time cAMP production in response to increasing concentrations of the melanocortin agonists α-MSH, γ-MSH, NDP-α-MSH and MT-II in the MC4R-GLO cell line. EC50 of α-MSH, γ-MSH, NDP- α MSH and MT-II were 2.0 ±0.8, 3.6 ±0.3, 0.063 ±0.003 and 0.035 ±0.005 nM, respectively. C, Effect of IBMX on the α-MSH-stimulated cAMP response in the MC4R-GLO cell line. cAMP response of MC4R-GLO cells incubated with increasing concentration of α-MSH after pretreatment with vehicle or IBMX (100 µM) for 5 min time. The EC50 of α-MSH in cells treated with vehicle is 0.84 ±0.30 nM whereas it is 0.19 ±0.06 nM after IBMX treatment. As expected, IBMX causes an increase in both the basal concentration of cAMP and the maximal response to α-MSH. D–E, Real-time recording of cAMP production in MC4R-GLO cells (D) or β2AR-GLO cells treated with the indicated ligands: AgRP (300 nM), α-MSH (30 nM), isoproterenol (300 nM). Ligands were added at the time indicated by the arrow. The assay was performed at 28°C to limit the activity of phosphodiesterases. F, Measurement of AgRP inverse agonism (as shown in panel d and e). MC4R-GLO and β2AR-GLO cells were incubated with increasing concentration of AgRP in the absence of melanocortin agonist. Basal cAMP concentration, as reported by the luminescence, was measured. The IC50 of AgRP on melanocortin MC4 receptor constitutive activity is 189 ±55 nM. As expected, no effect of AgRP was measured on the cAMP concentration in the β2AR-GLO cell line. Results shown are representative of at least three independent experiments performed in triplicates or more. Results are presented as average ±SEM.
α-MSH, γ-MSH, NDP-MSH and MT-II, a selection of natural and synthetic melanocortin agonists, recapitulated their order of potency from previously reported EC50 values (2.0 ± 0.8 nM, 3.6 ± 0.3 nM, 0.06 ± 0.003 nM, 0.04 ± 0.01 nM, respectively) documented in cAMP accumulation assays (Fig 1b) (Hruby et al., 1995; Mountjoy et al., 1994). Because the cAMP production in response to these peptides was previously measured in the presence of isobutylmethylxanthine (IBMX), a non-selective PDE inhibitor, the effect of IBMX on the concentration response curve of α-MSH on our cell line was assessed using a two injection protocol separated by a 5 minute lapse (Fig. 1C). Interestingly, IBMX (first injection) induced a potent and sustained increase in luminescence from 82 to 513 RLU (relative light units) during the lapse before the injection of α-MSH (second injection) consistent with a PDE-inhibition-mediated cAMP accumulation in the MC4R-GLO cell line (Fig 1c). As expected, both the potency and the efficacy of α-MSH in the presence of IBMX (EC50= 0.19 ± 0.06 nM, Emax= 1615 RLU) were significantly greater than with vehicle (EC50= 0.84 ± 0.30 nM, Emax= 1310 RLU).
Characterization of mutations in the human melanocortin MC4 receptor associated with morbid obesity that reduce constitutive activity of the receptor but not agonist binding or activation argue that human melanocortin MC4 receptor constitutive activity is physiologically important (Srinivasan et al., 2004). However, thus far no real time assay was used to characterize constitutive activity of the receptor, or the pharmacological properties of the endogenous melanocortin inverse agonist AgRP. The sensitivity of our assay was therefore optimized to accurately record constitutive activity, even at the low basal concentration of cAMP, by incubating the MC4R-GLO cell line at 28°C instead of 37°C, thus decreasing the activity of the endogenous PDEs. In these experimental conditions, performed in the absence of melanocortin agonist, the basal cAMP concentration as reported by the luminescent signal was decreased significantly after addition of AgRP (Fig. 1D). No decrease in signal was detected after addition of AgRP using a cell line expressing both pGLO and the β2-adrenergic receptor (Fig. 1E), supporting the specificity of AgRP as an inverse agonist of the melanocortin MC4 receptor. Finally, our concentration-response curves for AgRP attest a potency of approximately 200 nM (IC50) for this inverse agonist (Fig. 1F).
3.2. The hMC4R-GLO assay is compatible with high throughput screening
To test whether the hMC4R-GLO cell line is compatible with HTS standards on 384 well plates, a process of assay validation was undertaken on the FDSS6000 plate reader (Hamamatsu). As a result of this process, a statistical analysis of a checkerboard distribution of α-MSH in the wells was performed to determine the maximal resolution of the assay (Fig. 2A). When repeated three times, an average Z’ factor of 0.50 was obtained, meeting the recommended criteria for HTS compatiblity (Zhang et al., 1999).
Fig. 2. The hMC4R-GLO assay is compatible with high-throughput drug screening.
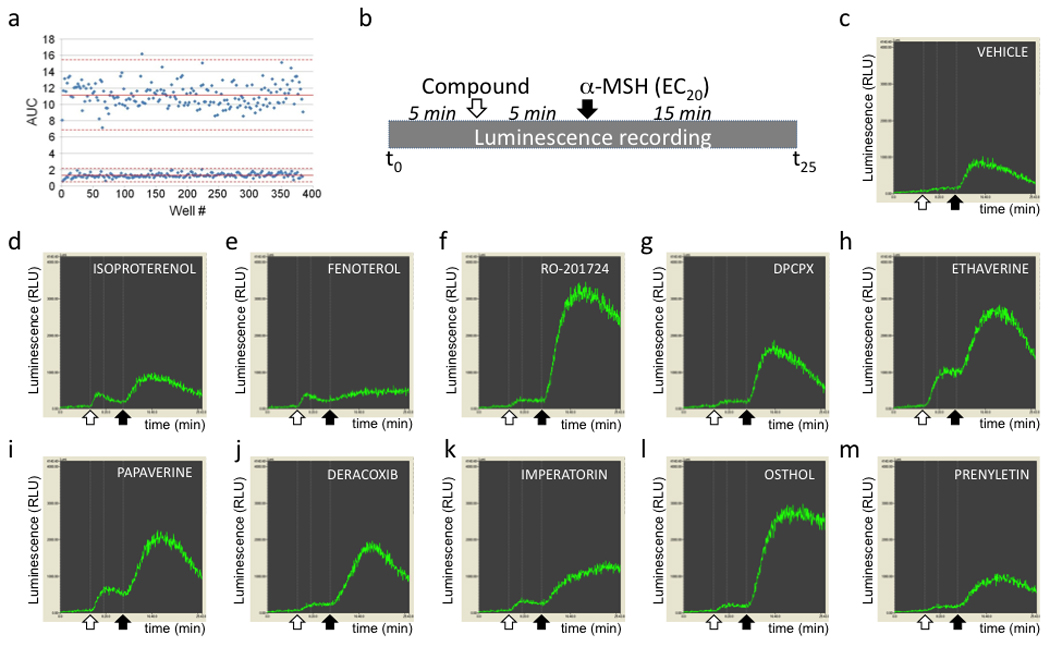
A, Calculation of the Z’ value for the pGLO cAMP assay. MC4R-GLO cells were plated in a 384 well plate. Vehicle or α-MSH (300 nM) were added in the wells in an alternating fashion, so that no two adjacent cells-containing wells were treated with agonist. cAMP-induced luminescence was measured and the Z’ value, representing the separation between the signal from vehicle treated cells and from agonist treated cells, was calculated based on the mean (solid lines) and mean ± 3 SD (broken lines) for each control population. Shown is a representative experiment. Z’ = 0.50 ±0.025. B, Schematic representing the protocol used for the pilot screen study performed on the spectrum library. The time of addition of compound (10 µM) or α-MSH (EC20) are indicated by arrows. C–M, Examples of cAMP response patterns in response to the indicated compounds (D–M). The white arrows show the time of addition of the compound and the black arrow the time of α-MSH addition. The responses illustrated in those graphs are different from the profile obtained with vehicle (C), qualifying the listed compounds as “hits”.
3.3. An HTS Assay for Human Melanocortin MC4 Receptor Allosteric Modulators: Pilot Screen Results
In order to identify positive allosteric modulators of the human melanocortin MC4 receptor by HTS, we used the MC4R-GLO cell line seeded on 384 well plates and a two injection protocol with a luminescence reading every 3 s for 25 minutes (Fig. 2B). This protocol includes baseline reading (for 5 min), either vehicle or test compound injection followed by a 5 min reading (10 µM final nominal concentration) and finally a submaximal dose (EC20) of the melanocortin agonist α-MSH followed by a 15 min reading.
To test the ability of our HTS assay to pick positive modulators from a library of compounds, we ran a pilot screen on the 2,000 compounds of the Spectrum collection composed of drugs on the market (50%), natural products (30%), and other bioactive compounds (20%). The recorded luminescent trace resulting from each compound was compared to the signal from cell-containing wells injected with vehicle. Compounds associated with a different pattern of response or differing in their amplitude of response, compared to the nearest surrounding wells, were considered as primary hits; representative examples of hits, along with a typical vehicle response, are shown in Fig. 2C to 2M. A broad hit list was made including compounds with moderate effects. In our simplified classification (Table 1), any pattern of response elevated over vehicle controls, at the time of injection of the compound (agonist effect) or at the time of injection of α-MSH (modulators) were considered as positive hits (n=62); traces lower than vehicle controls were considered as inhibitors (n=60).
Table 1.
Results of the MC4R-GLO screen run on the 2,000 compounds of Spectrum collection.
| Luminescent response | n | % |
|---|---|---|
| Positive modulators | 62 | 3.1% of library |
| PDE inhibitor | 7 | 11% of positive hits |
| Adrenergic agonist | 10 | 16% of positive hits |
| Other documented functiona | 26 | 42% of positive hits |
| No named functiona | 19 | 31% of positive hits |
| Inhibitors | 60 | 3.0% of library |
| Miscellaneous functiona | 40 | 67% of inhibitors |
| No named functiona | 20 | 33% of inhibitors |
according to the Microsource Spectrum compound library
As predicted, two pharmacological families are overrepresented: β2 adrenergic receptor agonists: n=10 (16%), and PDE inhibitors: n=7 (11%); β2 adrenergic receptor agonists were anticipated since HEK293 cells endogenously express the β2 adrenergic receptor. The cAMP response to β2 adrenergic receptor agonists is characterized in all cases by a moderate “agonist like” response shortly after addition of the compound (Fig. 2D and 2E), therefore fully consistent with the activation of endogenous β2 adrenergic receptor. Some of the compounds known to have PDE inhibitory activity like Ro20–1724 and DPCPX mainly elicited a strong potentiation of the α-MSH signaling response (Fig. 2F and 2G) whereas others like ethaverine hydrochloride, papaverine hydrochloride, vardenafil hydrochloride, dipyridamole and IBMX were able, in addition to the potentiation on MC4R signaling, to raise cAMP concentration by themselves. This “agonist” type of profile is therefore consistent with our previous observation obtained with IBMX (Fig. 1C), and is likely due to the accumulation of the basal level of cAMP produced in the resting cells. Also, as expected, the only adenylyl cyclase activator of the library –colforsin– generated the strongest signal (not shown). Unexpectedly, the potent nonsteroidal anti-inflammatory cyclooxygenase-2 (COX2) inhibitor deracoxib strongly potentiated α-MSH-mediated cAMP production (Fig. 2J) whereas the other three COX2 inhibitors of this compound library (celecoxib, rofecoxib or valdecoxib) did not. Among the miscellaneous positive hits without a specified function (n=19), according to the Spectrum datasheet (http://www.msdiscovery.com/spectrum.html), different families of compounds from plants were overrepresented, in particular flavonoids and coumarins. Remarkably, in the latter category, the two structurally homologous compounds imperatorin and osthol strongly potentiated melanocortin MC4 receptor signaling (Fig. 2). This molecular homology, also shared with the less potent compound prenyletin, define a clear structural family based on a coumarin backbone with a prenyl group (Fig. 3A–C). Finally, the present screen focusing on potentiators also identified 60 possible inhibitors whose analysis is beyond the scope of this study.
Fig. 3. Structures of a subset of positive modulators identified with the pilot screen.
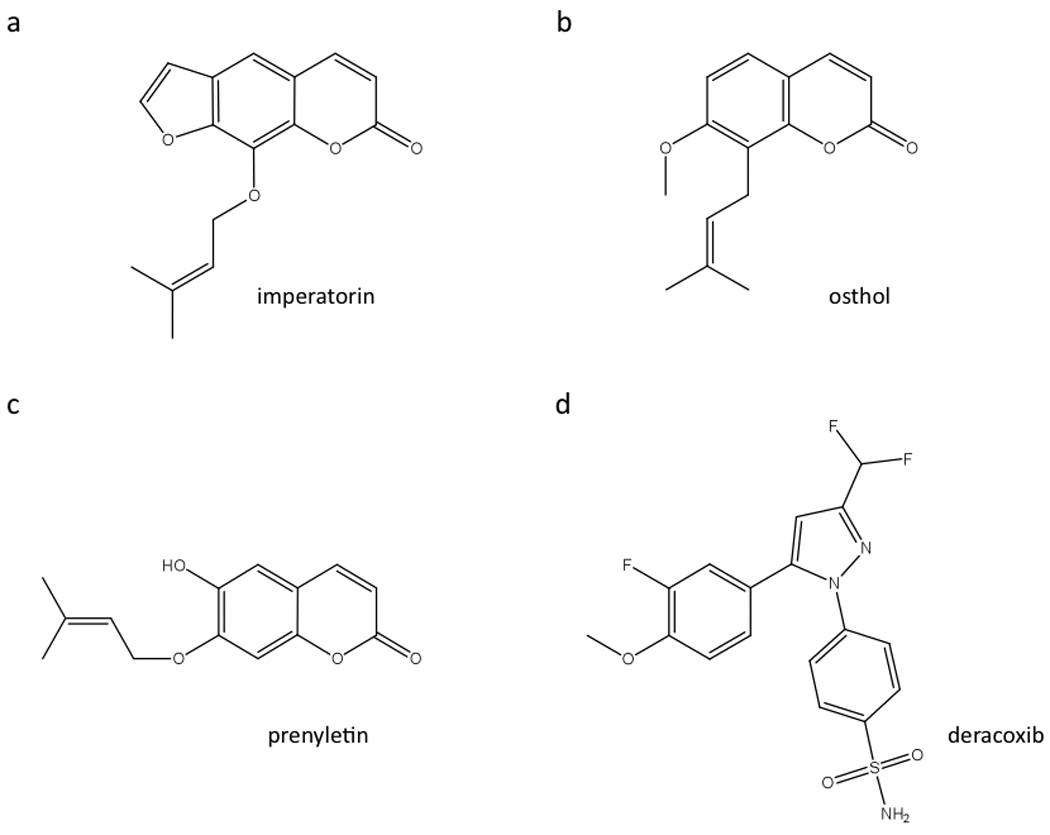
A–C, Imperatorin, osthol and prenyletin share a coumarin backbone and an identical prenyl group clearly defining a structural family. D, Deracoxib, a known COX2 inhibitor, is the only compound of its class to be identified as a “hit” in the screen using the Spectrum Collection.
3.4. Structure-function Relationships of the Hits Selected from the Spectrum Collection in the MC4R-GLO Line
To confirm the hits picked during the screen and to evaluate the potency of a selection of positive hits, concentration-response curves of the most promising compounds were performed in the presence of α-MSH at EC20 (Fig. 4A). Among these hits, osthol is clearly the most potent compound (EC50= 59 nM). The potencies of the other compounds tested are closer to the micromolar range). To test if COX2 inhibitors, other than deracoxib, would potentiate α-MSH signaling, we ran the same assay with celecoxib and rofecoxib from a new batch. As shown in Fig. 4B, only deracoxib showed this property, therefore fully recapitulating the results of the pilot screen. In order to further characterize the mechanism of action of both deracoxib and a representative compound with the coumarin-derived structure, we tested the effect of deracoxib and imperatorin, obtained from new batches from a commercial source, on α-MSH-mediated cAMP production. A commercial source for osthol was not available. In the MC4R-GLO line, the α-MSH induced cAMP response was significantly potentiated by the presence of 10 µM imperatorin or deracoxib (EC50= 0.12 nM and 0.21 nM, respectively) relative to vehicle (EC50= 0.97 nM), thus corresponding to an 8 fold increase in melanocortin MC4 receptor signaling with imperatorin and a 5 fold increase with deracoxib (Fig. 4C).
Fig. 4. Pharmacological properties of a selected subset of “hits”.
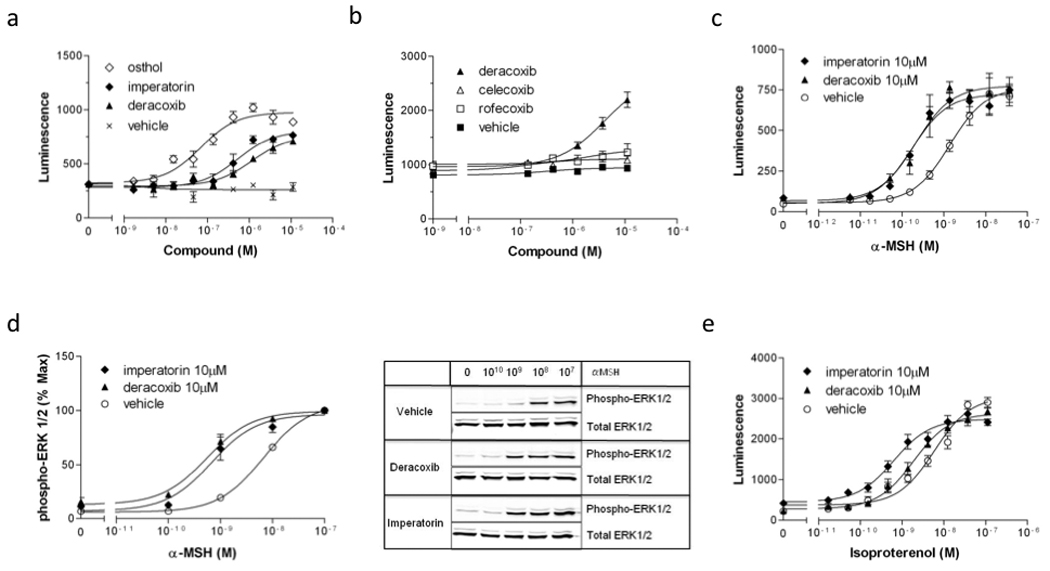
A–B, cAMP production in MC4R-GLO cells incubated with increasing concentrations of osthol, imperatorin, deracoxib or other COX2 inhibitors like celecoxib and rofecoxib, in the presence of α-MSH (EC20). Osthol, imperatorin and deracoxib increased the α-MSH-stimulated cAMP response with an EC50 of 59 ±21, 344 ±98 and 835 ±309 nM, respectively (A). Celecoxib and rofecoxib had no potentiating effect on the α-MSH-stimulated cAMP response (B), supporting the idea that the mechanism of potentiation by deracoxib is independent from its COX2 inhibitory properties. C, Concentration-response curves of α-MSH-stimulated cAMP production in the presence of vehicle, imperatorin (10 µM) or deracoxib (10 µM) in MC4R-GLO cells. The EC50 of α-MSH alone was 0.97 ±0.76 nM. In the presence of imperatorin or deracoxib the EC50 were 0.12 ±0.05 and 0.21 ±0.13 nM, respectively. D, ERK1/2 phosphorylation in MC4R-GLO cells in response to increasing concentrations of α-MSH in the presence or absence of imperatorin (10 µM) or deracoxib (10 µM). EC50 were 6.7 ±0.04 nM for α-MSH alone, 0.45 ±0.07 nM in the presence of deracoxib (10 µM) and 0.55 ±0.09 nM with imperatorin. E, Specificity of imperatorin and deracoxib. Isoproterenol-stimulated cAMP production in the β2AR-GLO cell line in the presence or absence of imperatorin or deracoxib. The EC50 of isoproterenol alone was 6.7 ±2.8 nM whereas it was 0.9 ±0.4 in the presence of imperatorin and 2.0 ±0.7 nM, in the presence of deracoxib. Results shown are representative of three independent experiments performed in triplicates.
To determine if imperatorin and deracoxib can potentiate signaling pathways downstream of melanocortin MC4 receptor other than the cAMP pathway, we measured the potency of α-MSH to activate the extracellular signal-regulated kinases 1 and 2 (ERK1/2) in the presence or absence of those compounds in the same cell line (Fig. 4D). We show that both imperatorin and deracoxib significantly increase the potency of α-MSH to induce ERK1/2 phosphorylation. The EC50 of α-MSH-mediated ERK1/2 activation was 0.55 nM in the presence of imperatorin and 0.45 nM in the presence of deracoxib compared to 6.7 nM with vehicle. This result provides evidence that even though the screen is designed to look exclusively at cAMP signaling, the hits identified can modulate multiple signaling networks downstream of the melanocortin MC4 receptor.
3.5. The Effect of Imperatorin and Deracoxib is Not Specific to Human Melanocortin MC4 Receptor
In order to test if imperatorin or deracoxib specifically modulate the human melanocortin MC4 receptor, we recorded cAMP production in HEK293 cells stably expressing both the pGLO reporter and the human β2 adrenergic receptor (β2AR-GLO line). In this assay, we looked at the ability of compounds to increase the dose response to isoproterenol, a β2 adrenergic agonist. Similar to their effect on α-MSH-mediated cAMP production through the melanocortin MC4 receptor, imperatorin and deracoxib increased isoproterenol potency to promote cAMP signaling through the β2 adrenergic receptor (Fig. 4E). Overall, these results are consistent with a shared mechanism of action of both compounds on the two Gs coupled receptors, inhibition of PDEs. This hypothesis is supported by the fact that similar experiments done in the presence of the well characterized PDE4 inhibitor rolipram (not shown) yields very comparable results to those obtained with imperatorin and deracoxib. None of the 62 positive hits showed specificity for the human melanocortin MC4 receptor, which was not surprising given that most of the compounds in the Spectrum collection were specifically developed against other targets (not shown).
3.6. PDE Inhibition: a Possible Mechanism of Action for Imperatorin and Deracoxib
Just recently, a yeast-based screening assay focusing on murine PDE inhibitors identified imperatorin and osthol as PDE inhibitors (Ivey et al., 2008); on the other hand, deracoxib was not documented as a PDE inhibitor. Since the subtypes of PDEs endogenously expressed in HEK293 cells are members mainly of the PDE3 and PDE4 family (Lynch et al., 2005) and most of the PDE inhibitor compounds from the spectrum library identified as hits during the screen were PDE4 inhibitors, we tested the potency of deracoxib and imperatorin to inhibit PDEs. To this end, a cell-free assay using a human recombinant PDE4D3, a member of the most highly expressed PDE family in HEK293 cells, PDE4D. Using this assay measuring the inhibition of PDE4D3 activity by rolipram, as a positive control, we show that both rolipram and deracoxib are strong PDE4 inhibitors with a Ki of 0.4 µM and 3.6 µM respectively, and that imperatorin displays no or weak inhibition at the same enzyme with a Ki of 100 µM (Table 2).
Table 2.
PDE assay based on PDE4D3 and [3H]cAMP. Used as a control, rolipram is a potent selective PDE4 inhibitor.
| Compound | IC50 (µM) |
|---|---|
| deracoxib | 3.6 ± 0.5 |
| imperatorin | 100 ± 20 |
| rolipram | 0.4 ± 0.15 |
In this study we show that IBMX potently increased the basal cAMP concentration in the MC4R-GLO cell line (Fig. 1C), and that different PDE inhibitors tested in the pilot screen can induce different patterns of response in the absence of agonist (Fig. 2). Those results suggest that the studied cell lines (MC4R-GLO and β2AR-GLO) or other pGLO reporter expressing cell lines could be efficiently used to sensitively monitor real time activity of endogenous PDEs. To verify this hypothesis, cAMP-dependent luminescence in the MC4R-GLO cell line was monitored before and after addition of imperatorin, deracoxib, or other known PDE inhibitors (Fig. 5). As expected, the non-selective PDE inhibitor IBMX caused a larger rise in intracellular cAMP than the more specific PDE4 inhibitor rolipram. Interestingly, imperatorin and deracoxib both increased cAMP levels.
Fig. 5. Real time monitoring of PDE activity using the MC4R-GLO cell line.
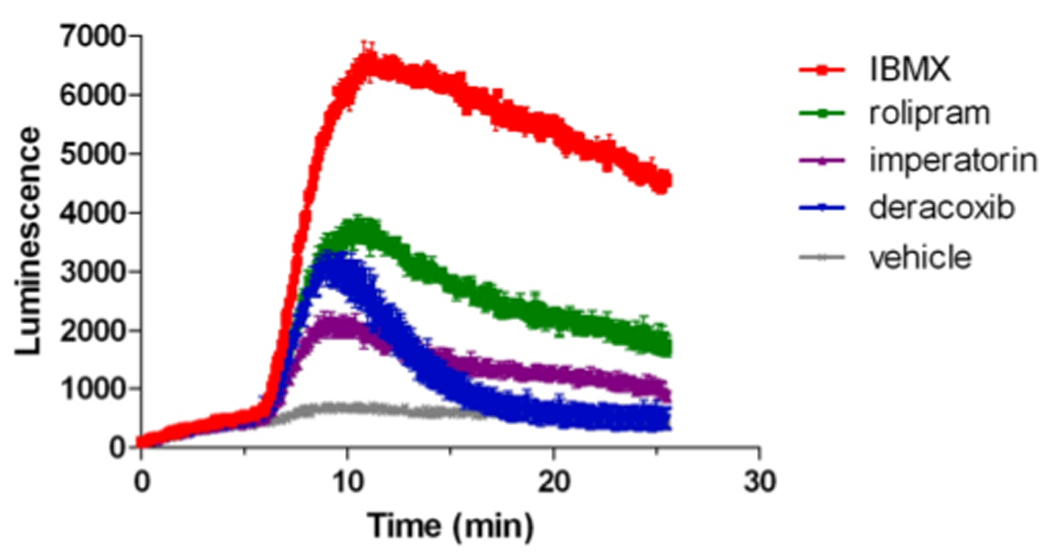
After recording of the baseline cAMP concentration in the MC4R-GLO cell line, as reported by luciferase activity, cells were treated with vehicle or with the indicated PDE inhibitors (100 µM). Changes in intracellular cAMP concentration over time following treatment were monitored for 20 minutes.
4. Discussion
We document here a cell line expressing a modified cAMP-sensitive luciferase allowing real time detection of cAMP concentrations resulting from human melanocortin-4 receptor signaling. In this cell line, the known melanocortin agonists and the physiological inverse agonist (AgRP) exert their expected activities. Furthermore, the sensitivity of this technique further allows one to monitor real time activity of endogenous PDEs. The sensitivity of this assay also allowed us to examine the inverse agonist activity of AgRP in real time in the absence of PDE inhibitors, and thus to calculate the IC50, ~200nM, for this activity. The divergence in IC50 values for the inverse agonist activity of AgRP versus the inhibition of α-MSH binding, reported as 1.9nM (McNulty et al., 2005), argues for two binding sites for AgRP on the melanocortin MC4 receptor.
A pilot drug screen run in a high throughput format on the Spectrum library of compounds (n= 2,000) successfully identified 62 positive modulators, including some very potent compounds that included expected hits. Most importantly, we identified a clear structural family sharing a similar coumarin-derived structure (imperatorin, osthol and prenyletin) and unveiled a new secondary site of action for deracoxib, a drug previously known as a COX2 inhibitor.
Several criteria suggest that the assay reported here is suitable for the identification of allosteric modulators of the human melanocortin MC4 receptor by HTS. First, this cAMP sensitive human melanocortin MC4 receptor-expressing cell line responds in real time to known endogenous agonists (α-MSH, γ-MSH, NDP-MSH, MT-II) and the endogenous inverse agonist AgRP. Second, the statistical Z’ analysis of the resolution of this assay is compatible with a HTS format (Zhang et al., 1999). Third, within our pilot screen, we not only identified some predicted hits (known PDE inhibitors, β2-adrenoceptor agonists and adenylyl cyclase activator), but most importantly, identified original active compounds more potent than well characterized PDE inhibitors, such as osthol. Our pilot allosteric screen, the first report based on a real-time, live-cell cAMP luminescence imaging technique, discloses original data and demonstrated its ability to point out individual agonist and/or modulator effects based on the pattern of response of each compound of the Spectrum library. Such data were not possible in previously used cAMP assays, essentially end point measurements performed in the presence of a PDE inhibitor.
One technical complexity of the assay involves the responsiveness of the background cell line to ligands targeting endogenously expressed receptors such as β2 adrenergic agonists. Most β2 adrenergic agonists present in the library showed an agonist response after compound addition (n=10). However, the signal intensity of β2 adrenergic agonists was low, therefore consistent with the lower endogenous expression level compared to the ectopically expressed human melanocortin MC4 receptor. Use of a counterscreen with the β2AR-GLO cell line described here can be used to quickly remove β2 adrenergic agonists or other non-melanocortin ligands from a candidate hit list.
Compounds that are PDE4 specific gave the highest signal in our assay (Ro-201724 or DPCPX), in agreement with the known pattern of PDE expression in the HEK293 cells (Lynch et al., 2005). However compounds with another documented cAMP specificity, PDE10A (papaverine, ethaverine), or even cGMP specificity PDE5 (vardenafil) showed up as hits, suggesting some level of cross specificity with endogenously expressed PDEs. Finally, a PDE3 inhibitor (cilostazol) did not give any increase in luminescence, therefore supporting that PDE4 is the major contributor of cAMP turnover in HEK293 cells, in agreement with a recent observation using a PKA assay (Stefan et al., 2007).
In addition to picking well characterized PDE inhibitors, we identified natural compounds derived from different classes of flavonoids (nobiletin, genistein, chalcones) and strikingly, a structurally homogenous coumarin-derived family of three members (osthol, imperatorin and prenyletin). Although flavonoids are known to be PDE inhibitors in different systems (Peluso, 2006), a yeast-based HTS screen focusing on murine PDE2A, 4A and 4B also identified a collection of coumarins, furanocoumarins and furanochromone, including both imperatorin and osthol, as PDE inhibitors (Ivey et al., 2008). In their study, the authors documented a significant PDE inhibitory activity for osthol (PDE2A and 4A) and for imperatorin (PDE4A and 4B). These observations fit with our data obtained based on our cAMP sensitive HEK293 cell line (Lynch et al., 2005). In addition, our data demonstrated no significant activity of imperatorin against a PDE4D isoform, the highly expressed PDE4 subtype in HEK293 (Lynch et al., 2005), therefore suggesting a relative selectivity of imperatorin within the PDE4 family. More generally, our results demonstrate that PDE inhibitors might be picked in our HTS assay. Interestingly, osthol and imperatorin are extracted from many medicinal plants, such as Cnidium monnieri and Angelica archangelica, respectively, which are widely used in traditional Chinese medicine. In addition, these molecules are depicted with miscellaneous biological functions including vasorelaxation of corpus cavernosum (Chen et al., 2000), increased cell differentiation in osteoblasts (Kuo et al., 2005), anticonvulsant (Luszczki et al., 2009), antidiabetic (Liang et al., 2009) vascular vasodilation (He et al., 2007), as well as an activity that might be associated with increased melanocortin signaling, reduction in liver steatosis (Ogawa et al., 2007). Together with the data of Ivey et al (Ivey et al., 2008), our observations strongly support a mechanism based on PDE inhibition that could account at least for some of the previously depicted properties of these compounds.
PDEs, a complex family of proteins (Conti and Beavo, 2007), play a key role in the temporo-spatial pattern of response of cAMP dependent events in the cell due to their unique role in cAMP degradation (Baillie, 2009). PDE4 isoforms, encoded by four genes (PDE4A, B, C and D) are expressed in many tissues, including the brain, in a wide but differential expression pattern (Cherry and Davis, 1999; Perez-Torres et al., 2000). These genes have non-redundant roles in endocrine cells, inflammation and the CNS (Ariga et al., 2004; Hansen et al., 2000; Jin et al., 1999; Zhang et al., 2002; Zhang et al., 2008; Zhang et al., 2009). Based on these properties, PDE4 blockers are considered as promising drug targets. Non-selective PDE4 inhibitors have demonstrated their efficacy in the treatment of chronic obstructive pulmonary disease in humans (Spina, 2008) and more selective compounds are being looked at for a wide range of CNS diseases; side effects are documented however (Menniti et al., 2006). Based on our pilot screen data, we anticipate that a significant number of the non-melanocortin MC4 receptor specific hits that will be picked in our ongoing large HTS could identify novel leads as PDE4 inhibitors. Interestingly, the prospect to discover subtype-selective inhibitors amongst PDE4 with improved tolerance was just illustrated (Burgin et al.), therefore providing new avenues for modulating cAMP-based signaling in CNS diseases. The data shown raise the possibility that obesity resulting from deficient melanocortin MC4 receptor signaling might be treated with a well-tolerated subtype-selective PDE4 inhibitor.
COX2 inhibitors are a major drug class for inflammatory disease states that also show promising anticancer properties. The balance between efficacy and safety is a sensitive issue in this therapeutic class that led to abundant research on secondary targets (Marnett, 2009). The identification of a documented COX2 inhibitor drug (deracoxib) in our cAMP based screen, was quite unexpected. Our concentration-response curve experiments confirmed an effect of deracoxib in two different pathways (PKA and MAPK) and further demonstrated that this effect was not melanocortin MC4 receptor specific. Direct activity of deracoxib in a PDE4D subtype enzyme assay was also demonstrated. This new pharmacological property for deracoxib raises questions regarding its specificity in vivo as a drug. None of the other COX2 inhibitors we have tested so far showed this PDE4 antagonist activity. Among these, celecoxib and deracoxib are structurally the closest: both are sulfonamide COX2 inhibitors that also share an identical pyrazole heterocycle. They differ at three positions, one on the pyrazole heterocycle and two on the aromatic group: 3-(difluoromethyl)-5-(3-fluoro-4-methoxyphenyl) for deracoxib and 3-(trifluoromethyl) −5-(4-methylphenyl) for celecoxib, therefore supporting a rather narrow functional specificity that will need further characterization. Second, deracoxib is a COX2 selective inhibitor drug approved for veterinary use in post-operative pain relief or for the long-term treatment of arthritis. So far, despite the fact that the drug was documented as rather well tolerated at normal doses, one cannot exclude that this newly identified property participates in some of the side effects of the drug (Case et al.; Roberts et al., 2009). Most importantly, this drug should therefore be used with care when a second medication needs to be coadministrated due to very likely drug-drug interactions resulting from PDE inhibition. Overall, the discovery of this new mechanism of action sheds light on the structure-function relationships within this important family of drugs, and may provide a basis for avoiding second site activity.
In summary, we describe a cell line that can be used for real-time monitoring of human melanocortin MC4 receptor signaling, based on a very sensitive cAMP detection system that exhibits a proper pharmacological response to known melanocortin ligands. The feasibility of running a high-throughput drug screen on this cell line was demonstrated by the success of a pilot screen in identifying relevant hits and in particular unveiling a new mechanism of action for a drug previously known as a COX2 inhibitor. While no melanocortin MC4 receptor specific allosteric modulators were identified in the screen of the small Spectrum collection of existing drugs, the data shown here argue that this screen should be effective in identifying such compounds from larger collections of drug-like molecules.
ACKNOWLEDGMENTS
We thank Dr. Larry Marnett (Vanderbilt) for providing celecoxib and rofecoxib, and Dr. Marco Conti (UCSF) for recombinant human PDE4D3. This study was supported by National Institutes of Health [Grant DK70332 to RDC and JP], and the Bristol-Myers Squibb Foundation (Freedom to Discover Unrestricted Metabolic Diseases research Grant. JP is a recipient of the Sabbatical Leave Programme from the European Society for Paediatric Endocrinology, supported by E. Lilly Co.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors declare no conflict of interest.
Contributor Information
Jacques Pantel, Department of Molecular Physiology and Biophysics, Vanderbilt University School of Medicine, Nashville, TN, USA 37232.
Savannah Y. Williams, Department of Molecular Physiology and Biophysics, Vanderbilt University School of Medicine, Nashville, TN, USA 37232
Dehui Mi, Department of Pharmacology, Vanderbilt University School of Medicine, Nashville, TN, USA 37232; Department of Vanderbilt Institute of Chemical Biology, Vanderbilt University School of Medicine, Nashville, TN, USA 37232.
Julien Sebag, Departments of Molecular Physiology and Biophysics, Vanderbilt University School of Medicine, Nashville, TN, USA 37232.
Jackie D. Corbin, Department of Molecular Physiology and Biophysics, Vanderbilt University School of Medicine, Nashville, TN, USA 37232
C. David Weaver, Department of Pharmacology, Vanderbilt University School of Medicine, Nashville, TN, USA 37232; Department of Vanderbilt Institute of Chemical Biology, Vanderbilt University School of Medicine, Nashville, TN, USA 37232.
Roger D. Cone, Departments of Molecular Physiology and Biophysics, Vanderbilt University School of Medicine, Nashville, TN, USA 37232
References
- Ariga M, Neitzert B, Nakae S, Mottin G, Bertrand C, Pruniaux MP, Jin SL, Conti M. Nonredundant function of phosphodiesterases 4D and 4B in neutrophil recruitment to the site of inflammation. J Immunol. 2004;173:7531–7538. doi: 10.4049/jimmunol.173.12.7531. [DOI] [PubMed] [Google Scholar]
- Baillie GS. Compartmentalized signalling: spatial regulation of cAMP by the action of compartmentalized phosphodiesterases. FEBS J. 2009;276:1790–1799. doi: 10.1111/j.1742-4658.2009.06926.x. [DOI] [PubMed] [Google Scholar]
- Blount MA, Zoraghi R, Ke H, Bessay EP, Corbin JD, Francis SH. A 46-amino acid segment in phosphodiesterase-5 GAF-B domain provides for high vardenafil potency over sildenafil and tadalafil and is involved in phosphodiesterase-5 dimerization. Mol. Pharmacol. 2006;70:1822–1831. doi: 10.1124/mol.106.028688. [DOI] [PubMed] [Google Scholar]
- Burgin AB, Magnusson OT, Singh J, Witte P, Staker BL, Bjornsson JM, Thorsteinsdottir M, Hrafnsdottir S, Hagen T, Kiselyov AS, Stewart LJ, Gurney ME. Design of phosphodiesterase 4D (PDE4D) allosteric modulators for enhancing cognition with improved safety. Nat Biotechnol. 28:63–70. doi: 10.1038/nbt.1598. [DOI] [PubMed] [Google Scholar]
- Calton MA, Ersoy BA, Zhang S, Kane JP, Malloy MJ, Pullinger CR, Bromberg Y, Pennacchio LA, Dent R, McPherson R, Ahituv N, Vaisse C. Association of functionally significant Melanocortin-4 but not Melanocortin-3 receptor mutations with severe adult obesity in a large North American case-control study. Hum Mol Genet. 2009;18:1140–1147. doi: 10.1093/hmg/ddn431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case JB, Fick JL, Rooney MB. Proximal duodenal perforation in three dogs following deracoxib administration. J Am Anim Hosp Assoc. 46:255–258. doi: 10.5326/0460255. [DOI] [PubMed] [Google Scholar]
- Chen J, Chiou WF, Chen CC, Chen CF. Effect of the plant-extract osthole on the relaxation of rabbit corpus cavernosum tissue in vitro. J Urol. 2000;163:1975–1980. [PubMed] [Google Scholar]
- Cherry JA, Davis RL. Cyclic AMP phosphodiesterases are localized in regions of the mouse brain associated with reinforcement, movement, and affect. J Comp Neurol. 1999;407:287–301. [PubMed] [Google Scholar]
- Cone RD. Anatomy and regulation of the central melanocortin system. Nat Neurosci. 2005;8:571–578. doi: 10.1038/nn1455. [DOI] [PubMed] [Google Scholar]
- Conn PJ, Christopoulos A, Lindsley CW. Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nat Rev Drug Discov. 2009;8:41–54. doi: 10.1038/nrd2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti M, Beavo J. Biochemistry and physiology of cyclic nucleotide phosphodiesterases: essential components in cyclic nucleotide signaling. Annu Rev Biochem. 2007;76:481–511. doi: 10.1146/annurev.biochem.76.060305.150444. [DOI] [PubMed] [Google Scholar]
- DiPilato LM, Cheng X, Zhang J. Fluorescent indicators of cAMP and Epac activation reveal differential dynamics of cAMP signaling within discrete subcellular compartments. Proc Natl Acad Sci U S A. 2004;101:16513–16518. doi: 10.1073/pnas.0405973101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farooqi S, O'Rahilly S. Genetics of obesity in humans. Endocr Rev. 2006;27:710–718. doi: 10.1210/er.2006-0040. [DOI] [PubMed] [Google Scholar]
- Govaerts C, Srinivasan S, Shapiro A, Zhang S, Picard F, Clement K, Lubrano-Berthelier C, Vaisse C. Obesity-associated mutations in the melanocortin 4 receptor provide novel insights into its function. Peptides. 2005;26:1909–1919. doi: 10.1016/j.peptides.2004.11.042. [DOI] [PubMed] [Google Scholar]
- Greenfield JR, Miller JW, Keogh JM, Henning E, Satterwhite JH, Cameron GS, Astruc B, Mayer JP, Brage S, See TC, Lomas DJ, O'Rahilly S, Farooqi IS. Modulation of blood pressure by central melanocortinergic pathways. N Engl J Med. 2009;360:44–52. doi: 10.1056/NEJMoa0803085. [DOI] [PubMed] [Google Scholar]
- Hansen G, Jin S, Umetsu DT, Conti M. Absence of muscarinic cholinergic airway responses in mice deficient in the cyclic nucleotide phosphodiesterase PDE4D. Proc Natl Acad Sci U S A. 2000;97:6751–6756. doi: 10.1073/pnas.97.12.6751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He JY, Zhang W, He LC, Cao YX. Imperatorin induces vasodilatation possibly via inhibiting voltage dependent calcium channel and receptor-mediated Ca2+ influx and release. Eur J Pharmacol. 2007;573:170–175. doi: 10.1016/j.ejphar.2007.06.043. [DOI] [PubMed] [Google Scholar]
- Hinney A, Bettecken T, Tarnow P, Brumm H, Reichwald K, Lichtner P, Scherag A, Nguyen TT, Schlumberger P, Rief W, Vollmert C, Illig T, Wichmann HE, Schafer H, Platzer M, Biebermann H, Meitinger T, Hebebrand J. Prevalence, spectrum, and functional characterization of melanocortin-4 receptor gene mutations in a representative population-based sample and obese adults from Germany. Journal of Clinical Endocrinology and Metabolism. 2006;91:1761–1769. doi: 10.1210/jc.2005-2056. [DOI] [PubMed] [Google Scholar]
- Hruby VJ, Lu D, Sharma SD, Castrucci AL, Kesterson RA, Al-Obeidi FA, Hadley ME, Cone RD. Cyclic lactam α-melanotropin analogues of Ac-Nle4-c[Asp4,D-Phe7, Lys10]α-MSH(4–10)-NH2 with bulky aromatic amino acids at position 7 show high antagonist potency and selectivity at specific melanocortin receptors. J. Med. Chem. 1995;38:3454–3461. doi: 10.1021/jm00018a005. [DOI] [PubMed] [Google Scholar]
- Ivey FD, Wang L, Demirbas D, Allain C, Hoffman CS. Development of a fission yeast-based high-throughput screen to identify chemical regulators of cAMP phosphodiesterases. J Biomol Screen. 2008;13:62–71. doi: 10.1177/1087057107312127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin SL, Richard FJ, Kuo WP, D'Ercole AJ, Conti M. Impaired growth and fertility of cAMP- specific phosphodiesterase PDE4D-deficient mice. Proc Natl Acad Sci U S A. 1999;96:11998–12003. doi: 10.1073/pnas.96.21.11998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin T. Collateral efficacy in drug discovery: taking advantage of the good (allosteric) nature of 7TM receptors. Trends Pharmacol Sci. 2007;28:407–415. doi: 10.1016/j.tips.2007.06.009. [DOI] [PubMed] [Google Scholar]
- Kuo PL, Hsu YL, Chang CH, Chang JK. Osthole-mediated cell differentiation through bone morphogenetic protein-2/p38 and extracellular signal-regulated kinase 1/2 pathway in human osteoblast cells. J Pharmacol Exp Ther. 2005;314:1290–1299. doi: 10.1124/jpet.105.085092. [DOI] [PubMed] [Google Scholar]
- Liang HJ, Suk FM, Wang CK, Hung LF, Liu DZ, Chen NQ, Chen YC, Chang CC, Liang YC. Osthole, a potential antidiabetic agent, alleviates hyperglycemia in db/db mice. Chem Biol Interact. 2009;181:309–315. doi: 10.1016/j.cbi.2009.08.003. [DOI] [PubMed] [Google Scholar]
- Luszczki JJ, Wojda E, Andres-Mach M, Cisowski W, Glensk M, Glowniak K, Czuczwar SJ. Anticonvulsant and acute neurotoxic effects of imperatorin, osthole and valproate in the maximal electroshock seizure and chimney tests in mice: a comparative study. Epilepsy Res. 2009;85:293–299. doi: 10.1016/j.eplepsyres.2009.03.027. [DOI] [PubMed] [Google Scholar]
- Lynch MJ, Baillie GS, Mohamed A, Li X, Maisonneuve C, Klussmann E, van Heeke G, Houslay MD. RNA silencing identifies PDE4D5 as the functionally relevant cAMP phosphodiesterase interacting with beta arrestin to control the protein kinase A/AKAP79- mediated switching of the beta2-adrenergic receptor to activation of ERK in HEK293B2 cells. J Biol Chem. 2005;280:33178–33189. doi: 10.1074/jbc.M414316200. [DOI] [PubMed] [Google Scholar]
- Marnett LJ. The COXIB experience: a look in the rearview mirror. Annu Rev Pharmacol Toxicol. 2009;49:265–290. doi: 10.1146/annurev.pharmtox.011008.145638. [DOI] [PubMed] [Google Scholar]
- May LT, Leach K, Sexton PM, Christopoulos A. Allosteric modulation of G protein-coupled receptors. Annu Rev Pharmacol Toxicol. 2007;47:1–51. doi: 10.1146/annurev.pharmtox.47.120505.105159. [DOI] [PubMed] [Google Scholar]
- McNulty JC, Jackson PJ, Thompson DA, Chai B, Gantz I, Barsh GS, Dawson PE, Millhauser GL. Structures of the agouti signaling protein. J Mol Biol. 2005;346:1059–1070. doi: 10.1016/j.jmb.2004.12.030. [DOI] [PubMed] [Google Scholar]
- Menniti FS, Faraci WS, Schmidt CJ. Phosphodiesterases in the CNS: targets for drug development. Nat Rev Drug Discov. 2006;5:660–670. doi: 10.1038/nrd2058. [DOI] [PubMed] [Google Scholar]
- Mountjoy KG, Mortrud MT, Low MJ, Simerly RB, Cone RD. Localization of the melanocortin-4 receptor (MC4-R) in neuroendocrine and autonomic control circuits in the brain. Mol. Endo. 1994;8:1298–1308. doi: 10.1210/mend.8.10.7854347. [DOI] [PubMed] [Google Scholar]
- Ogawa H, Sasai N, Kamisako T, Baba K. Effects of osthol on blood pressure and lipid metabolism in stroke-prone spontaneously hypertensive rats. J Ethnopharmacol. 2007;112:26–31. doi: 10.1016/j.jep.2007.01.028. [DOI] [PubMed] [Google Scholar]
- Peluso MR. Flavonoids attenuate cardiovascular disease, inhibit phosphodiesterase, and modulate lipid homeostasis in adipose tissue and liver. Exp Biol Med (Maywood) 2006;231:1287–1299. doi: 10.1177/153537020623100802. [DOI] [PubMed] [Google Scholar]
- Perez-Torres S, Miro X, Palacios JM, Cortes R, Puigdomenech P, Mengod G. Phosphodiesterase type 4 isozymes expression in human brain examined by in situ hybridization histochemistry and[3H]rolipram binding autoradiography. Comparison with monkey and rat brain. J Chem Neuroanat. 2000;20:349–374. doi: 10.1016/s0891-0618(00)00097-1. [DOI] [PubMed] [Google Scholar]
- Rich TC, Tse TE, Rohan JG, Schaack J, Karpen JW. In vivo assessment of local phosphodiesterase activity using tailored cyclic nucleotide-gated channels as cAMP sensors. J Gen Physiol. 2001;118:63–78. doi: 10.1085/jgp.118.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts ES, Van Lare KA, Marable BR, Salminen WF. Safety and tolerability of 3-week and 6-month dosing of Deramaxx (deracoxib) chewable tablets in dogs. J Vet Pharmacol Ther. 2009;32:329–337. doi: 10.1111/j.1365-2885.2008.01043.x. [DOI] [PubMed] [Google Scholar]
- Spina D. PDE4 inhibitors: current status. Br J Pharmacol. 2008;155:308–315. doi: 10.1038/bjp.2008.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan S, Lubrano-Berthelier C, Govaerts C, Picard F, Santiago P, Conklin BR, Vaisse C. Constitutive activity of the melanocortin-4 receptor is maintained by its N-terminal domain and plays a role in energy homeostasis in humans. J Clin Invest. 2004;114:1158–1164. doi: 10.1172/JCI21927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefan E, Aquin S, Berger N, Landry CR, Nyfeler B, Bouvier M, Michnick SW. Quantification of dynamic protein complexes using Renilla luciferase fragment complementation applied to protein kinase A activities in vivo. Proc Natl Acad Sci U S A. 2007;104:16916–16921. doi: 10.1073/pnas.0704257104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titus S, Neumann S, Zheng W, Southall N, Michael S, Klumpp C, Yasgar A, Shinn P, Thomas CJ, Inglese J, Gershengorn MC, Austin CP. Quantitative high-throughput screening using a live-cell cAMP assay identifies small-molecule agonists of the TSH receptor. J Biomol Screen. 2008;13:120–127. doi: 10.1177/1087057107313786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wikberg JE, Mutulis F. Targeting melanocortin receptors: an approach to treat weight disorders and sexual dysfunction. Nat Rev Drug Discov. 2008;7:307–323. doi: 10.1038/nrd2331. [DOI] [PubMed] [Google Scholar]
- Willoughby D, Cooper DM. Live-cell imaging of cAMP dynamics. Nat Methods. 2008;5:29–36. doi: 10.1038/nmeth1135. [DOI] [PubMed] [Google Scholar]
- Zhang HT, Huang Y, Jin SL, Frith SA, Suvarna N, Conti M, O'Donnell JM. Antidepressant-like profile and reduced sensitivity to rolipram in mice deficient in the PDE4D phosphodiesterase enzyme. Neuropsychopharmacology. 2002;27:587–595. doi: 10.1016/S0893-133X(02)00344-5. [DOI] [PubMed] [Google Scholar]
- Zhang HT, Huang Y, Masood A, Stolinski LR, Li Y, Zhang L, Dlaboga D, Jin SL, Conti M, O'Donnell JM. Anxiogenic-like behavioral phenotype of mice deficient in phosphodiesterase 4B (PDE4B) Neuropsychopharmacology. 2008;33:1611–1623. doi: 10.1038/sj.npp.1301537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Ma Y, Taylor SS, Tsien RY. Genetically encoded reporters of protein kinase A activity reveal impact of substrate tethering. Proc Natl Acad Sci U S A. 2001;98:14997–15002. doi: 10.1073/pnas.211566798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JH, Chung TD, Oldenburg KR. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- Zhang R, Maratos-Flier E, Flier JS. Reduced adiposity and high-fat diet-induced adipose inflammation in mice deficient for phosphodiesterase 4B. Endocrinology. 2009;150:3076–3082. doi: 10.1210/en.2009-0108. [DOI] [PMC free article] [PubMed] [Google Scholar]